

Abstract
The Ensembl pipeline is an extension to the Ensembl system which allows automated annotation of genomic sequence. The software comprises two parts. First, there is a set of Perl modules (“Runnables” and “RunnableDBs”) which are `wrappers' for a variety of commonly used analysis tools. These retrieve sequence data from a relational database, run the analysis, and write the results back to the database. They inherit from a common interface, which simplifies the writing of new wrapper modules. On top of this sits a job submission system (the “RuleManager”) which allows efficient and reliable submission of large numbers of jobs to a compute farm. Here we describe the fundamental software components of the pipeline, and we also highlight some features of the Sanger installation which were necessary to enable the pipeline to scale to whole-genome analysis.
The Ensembl pipeline was first devised, three years ago, out of a need to be able to perform large-scale automated annotation of genomic sequence: in particular, that of the mouse and human genomes. To this end an analysis system was developed that would allow many different algorithms—such as RepeatMasker, BLAST, and Genscan—to be run quickly and efficiently across entire genomes. Additionally, the genomes of other organisms— such as the rat and zebrafish—were in the first stages of sequencing and were soon to require similar analysis.
Annotation is the process by which the information, `hidden' in the genomic sequence, is extracted; this is necessary for the key features of the genome, the genes, to be identified. It can take place in one of two ways, either by a fully automated method (such as used by Ensembl) or by manual annotation; each has its own relative merits and drawbacks. Clearly, human interpretation of the raw analysis by manual annotators gives the highest-quality data and most accurate gene structures. However, the process is by its nature slow, and annotators may produce conflicting interpretations of the analysis. On the other hand, fully automated prediction of gene structures has the advantage of being fast, does not require a team of trained annotators, and will process the raw analysis consistently; although it can under-predict both the number of genes and the number of alternative transcripts. However, automatic annotation is particularly attractive for newly sequenced genomes where biologists need to know gene locations but may not have the resources to manually an-notate the genome.
Prior to the development of the current analysis pipeline, the Sanger Institute ran a sequence analysis system based on AceDB (Durbin and Mieg 1991; http://www.acedb.org) and flat files. Although this sufficed as the human genome was being sequenced relatively slowly and annotated on a clone-by-clone basis, it was clear from the work on chromosome 22 that a system based on a large number of flat files would not scale to the analysis of whole genomes. A relational database, however, should be capable of delivering the performance required.
The system described herein evolved from necessity and was not designed from the outset as a generic solution for genome annotation. It has been extremely successful and is used in-house to produce automated gene predictions for human, mouse, rat, zebrafish, fly, worm, and fugu (Curwen et al. 2004). The Vertebrate Genome Analysis group (vega.sanger.ac.uk) uses a modified Ensembl analysis pipeline to produce high-quality annotation of human chromosomes 6, 9, 10, and 13.
At the time the Ensembl pipeline was developed there was nothing publicly available for large-scale genome analysis. Since that time, a number of other systems have been made available which use a pipeline framework in order to annotate sequence data. FlyBase (Berkeley) uses BOP (Mungall et al. 2002) to provide baseline annotations for their manual curation efforts. The National Center for Biotechnology Information (NCBI) has its own pipeline for full annotation of whole genomes (http://www.ncbi.nlm.nih.gov/genome/guide/build.html#annot), as does the UCSC group (Kent et al. 2002). Biopipe (Hoon et al. 2003), a system influenced by the Ensembl pipeline, is a generic system for large-scale bioinformatics analysis. Unlike the Ensembl pipeline, this has the advantage of not being restricted to a single storage and retrieval system for the input and output data. Smaller pipeline systems also exist for annotation of ESTs or individual clones. These systems include Genescript (Hudek et al. 2003) and ASAP (Glasner et al. 2003).
RESULTS AND DISCUSSION
Software Decisions
Much of bioinformatics centers on analysis of sequences and thus involves a lot of manipulation of text strings. This is partly the reason that the Perl programming language has been popular among the bioinformatics community. Although it is not a perfect solution, we chose to use Perl as our development language. The pros and cons of this decision are detailed elsewhere (Stabenau et al. 2004).
Based on our previous experience with storing sequences and output data in flat files, it was imperative that we moved to a relational database system. We ultimately chose MySQL to store our sequence and analysis data, as it provided the speed we needed for Web site access to whole-genome data, and also as it was freely available under the GNU Public Licence (GPL).
In order to make the best use of our computing resources we required a robust batch queuing system, capable of handling large numbers of jobs. At Sanger, we chose LSF (Load Sharing Facility, http://www.platform.com/products/LSF) from Platform Computing. The reasons for this choice are outlined elsewhere (Cuff et al. 2004). The code is structured to allow a different scheduler to be substituted with minimal effort.
Although much raw analysis can be automated, there are many situations when a one-off analysis is needed without the need for batch processing. It therefore made sense to write modules that could be used in a stand-alone way without the restriction that they had to be used within a pipeline framework. This requirement led to the analysis software being split into two parts. The first (described below in the section entitled “Runnables and RunnableDBs”) deals solely with the running of the individual analyses (RepeatMasker, BLAST, Genscan, etc.) and parsing the output. The second part (described in the section “The RuleManager”) deals with the automated running, in the correct order, of the many analyses that constitute the pipeline, keeping track of those that have run successfully, while also coping with problems such as job failures. Figure 1 shows how these pieces fit together. Before discussing the Perl modules, we describe the way in which input and output data are stored and handled.
Figure 1.
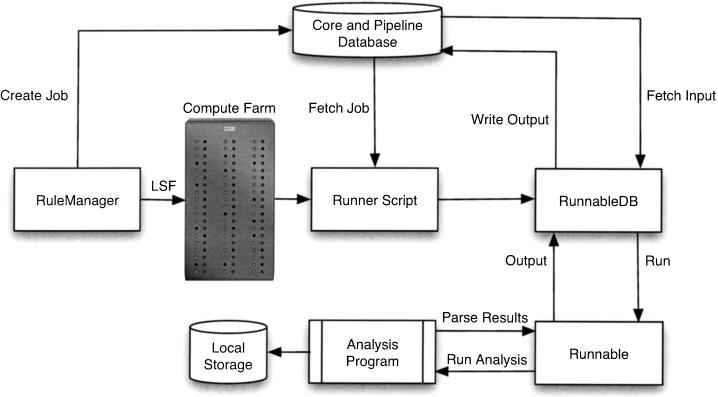
Ensembl pipeline system overview: The RuleManager uses LSF to submit analysis jobs to the compute farm. When an individual job starts executing on a remote node, the Runner script fetches the job information from the database and recreates the Job object. This in turn creates a RunnableDB and calls the appropriate methods (fetch_input, run, write_output, etc.) to run the analysis.
Data Access Architecture
Most current bioinformatics tools are based on flat files for input and output. As we do not have access to the source code for all of these tools, we have to base our system on these files. This is another reason the Perl language was chosen: The regular expression functions are particularly appropriate for parsing the output of external programs. We would like to conceal as much of the interaction with the file system as possible. To achieve this the pipeline only uses flat files locally on the execution node: Input data are retrieved directly from a database, and the output data are written back the same way.
Another issue that needed to be addressed was the source of input data and the final destination of output data. Analysis on a genome scale is often done on large farms of computers with independent jobs running on separate compute nodes. Any particular analysis typically relies on input data (DNA sequence) and library data (such as BLAST databases) and writes output, all of which are flat files. As the library data can often be sequence databases of several hundred megabytes, it was decided that all files associated with each job should be local to the machine that is running the process. The data architecture is described below in more detail.
Library Data
Library data includes all of the BLAST database files and other run-time files such as RepeatMasker libraries and Genscan matrices. These are distributed over the compute farm so that they are local to each node. This avoids the problems of simultaneous accesses to the same file and of sending large files multiple times over the network.
Input Data
Input data (which are generally DNA sequence) are stored in a relational database. The piece of sequence needed for each analysis is given by a unique identifier, which could be a sequence accession or a chromosomal region. The sequence is retrieved from the database using this identifier by the remote execution node, which then writes any necessary input files to local storage. After the job has completed, these temporary input files are deleted. This process eliminates any NFS mounting of disks.
Output Data
Output is written locally on each execution node into temporary files. It is then parsed into Perl objects and written back to the database. The output files are deleted from the host machine once the job is finished. We have designed our databases to store data as compactly as possible. Sequence alignments are stored using a shorthand notation called a “cigar string” (Stabenau et al. 2004) to denote where the insertions and deletions occur within it. This can reduce a 5-Mb BLAST output file down to a few hundred kilobytes with no loss of sensitivity.
Runnables and RunnableDBs
Each analysis has its own Perl module which takes as input several Perl objects containing the data to be analyzed along with any parameters needed by the program. Output is also in the form of Perl objects which can be of several types, for example, simple features, homology features, or more complicated objects such as genes. The Ensembl Perl feature types are discussed else-where (Stabenau et al. 2004). These modules are called Runnables and inherit from a simple interface:
- query
input sequence object
- run
run the program
- output
return the output objects
It is relatively straightforward to implement new Runnables, with a minimal amount of new code required to parse the program output and create the appropriate Perl objects. This simplicity is essential if the pipeline is to be easily extended to incorporate new analyses and algorithms.
As the Runnables use Ensembl objects for input and output, they can be combined to form more elaborate analyses. The example below shows how to create and execute BLAST and Genscan Runnables to compare human DNA sequence to vertebrate RNA and to check whether the resulting hits are coding or noncoding. As the Runnables are based on Bioperl, data input is simplified by the use of the Bio::SeqIO modules for reading sequence data from flat files.
use Bio::SeqIO; use strict; use Bio::EnsEMBL::Pipeline::Runnable::Blast; use Bio::EnsEMBL::Pipeline::Runnable::Genscan; my $query_seq = Bio::SeqIO→new( -file ⇒ `human_seq.fa', -format ⇒ `fasta' )→next_seq; my $blast = Bio::EnsEMBL::Pipeline::Runnable::Blast→new( `-query' ⇒ $query_seq, `–program' ⇒ `wublastn', `–database' ⇒ `embl_vertRNA', `–threshold_type' ⇒ `SCORE', `–threshold' ⇒200 ); $blast→run; # The Blast Runnable outputs objects called align features # which contain the coordinates and the sequence of each hit. for each my $af ($blast→output) { $af→attach_seq($query_seq); my $prediction = $af→feature1→seq; my $genscan = Bio::EnsEMBL::Pipeline::Runnable::Genscan→new( -query ⇒ $prediction, -matrix ⇒ `HumanIso.smat', ); $genscan→run; # The existence of a genscan result indicates that the BLAST # hit has coding regions. Don't need to do anything with # the genscan result—just print out details of the hit if ($genscan→output) { print join(“ “, $af→start, $af→hseqname, “coding”), “\n”; } }
The previous example reads its data from a file and outputs features to the screen. In the pipeline we typically want to read data from a database and write the results back to a database, but without sacrificing the usability of the Runnables as stand-alone modules. This led to a second set of modules, called RunnableDBs, that have extra methods to read from, and write to, a database:
- input_id
unique string identifying input DNA sequence (such as a clone accession)
- analysis
holds information about the analysis
- fetch_input
read input from an Ensembl database into Perl objects
- run
the appropriate runnable
- write_output
write output [Perl objects] to an Ensembl database
Each Runnable has an associated RunnableDB to read and write data, although one RunnableDB could use more than one Runnable.
In the Ensembl pipeline, these methods read and write to an Ensembl database but could easily work with another system or use flat files for small analyses. The input id method contains a unique string identifying the piece of data to run the analysis on, such as a clone accession. As the RunnableDBs do not know anything about the job submission system that created them, they can be used for stand-alone analysis. An example using the RunnableDB modules (based on the first example) is shown below.
use strict; use Bio::EnsEMBL::Pipeline::DBSQL::DBAdaptor; use Bio::EnsEMBL::Pipeline::RunnableDB::Blast; # the DBAdaptor encapsulates the connection to an Ensembl # database my $dbh = Bio::EnsEMBL::Pipeline::DBSQL::DBAdaptor→new( # DB connection parameters ); # An object representing a BLAST against a protein database. # Swall is a local equivalent of the SPTR nonredundant protein # database. my $analysis = $dbh→get_AnalysisAdaptor→fetch_by_logic_ name(“Swall”); # A BLAST run: # – fetch data from the input Ensembl database, # – data identified by input_id (a clone accession) # – extra parameters specified in the analysis retrieved above my $blast = Bio::EnsEMBL::Pipeline::RunnableDB::Blast→new( –db ⇒ $dbh, –input_id ⇒ `AZ123456.1.1.200000', –analysis ⇒ $analysis ); $blast→fetch_input; $blast→run; $blast→write_output;
There is no flat file access in this code at all. The input data are fetched from a database and written back to a database. The only flat file generation is done inside the BLAST Runnable, and these files are discarded after the job is complete.
The RuleManager
RuleManager is the script which is responsible for automatically running all analyses in the pipeline. It loops over all available input sequences and submits those analyses for which the dependencies are satisfied to the farm, or runs them locally if this is required. A flow diagram for this code is shown in Figure 2.
Figure 2.
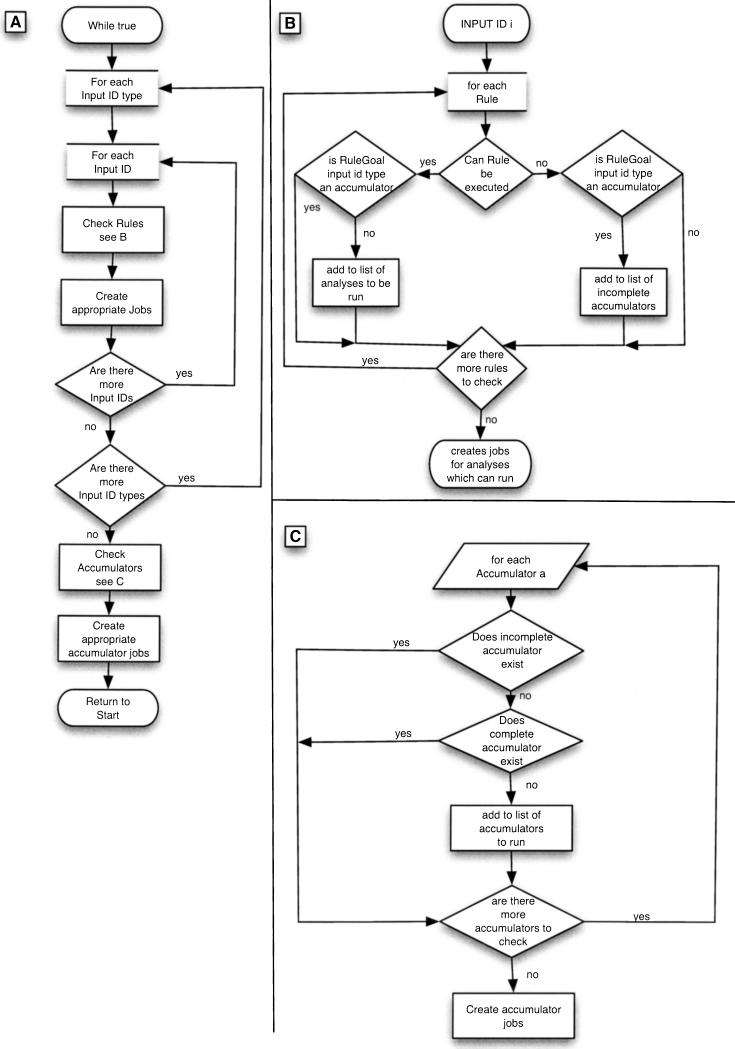
The central loop of the RuleManager. The procedure the code goes though while submitting jobs to the compute farm is shown. (A) The main loop during which the RuleManager processes all of the input IDs, submits jobs which can run, processes the accumulators, and marks analyses which have completely finished. (B) The process an individual input ID undergoes; checking the input ID against each of the rules and submitting jobs for those rules which can be executed. Accumulators are a specific type of analysis which the RuleManager script recognizes and is able to deal with appropriately. (C) Accumulator analyses mark other analyses which need to be all complete on every possible input id of their type before any dependent analyses can be executed. This panel shows how the RuleManager processes the accumulators at the end of each loop. Those accumulators which both haven't been marked as incomplete when the input IDs were being processed and aren't already complete are submitted to the system.
In creating and submitting analyses, the RuleManager creates Perl objects called Jobs. Each Job comprises an analysis which specifies what is being run, an input ID which identifies the input sequence for the analysis, paths to files which contain the job output from the batch scheduler, the farm submission identifier, and a status history of the job.
Dependencies between analyses, of which there are three types, are defined by objects called Rules. The first type is used for those analyses which do not depend on any previous analysis having been completed. For example, a piece of sequence can be repeat-masked independently of any other analysis. The second type is for those analyses which require prior completion of one or more other analyses on the same input sequence. For example, in order to run BLAST on a piece of sequence, it would normally have to be repeat-masked first. The last type of dependency is where the analysis must wait until all analyses it depends on are complete on all possible input sequences before it can start. For example, the second stage of the gene build—“similarity genewise”—can only be started once all of the protein BLASTs are complete for every contig in the database (see Fig. 3).
Figure 3.
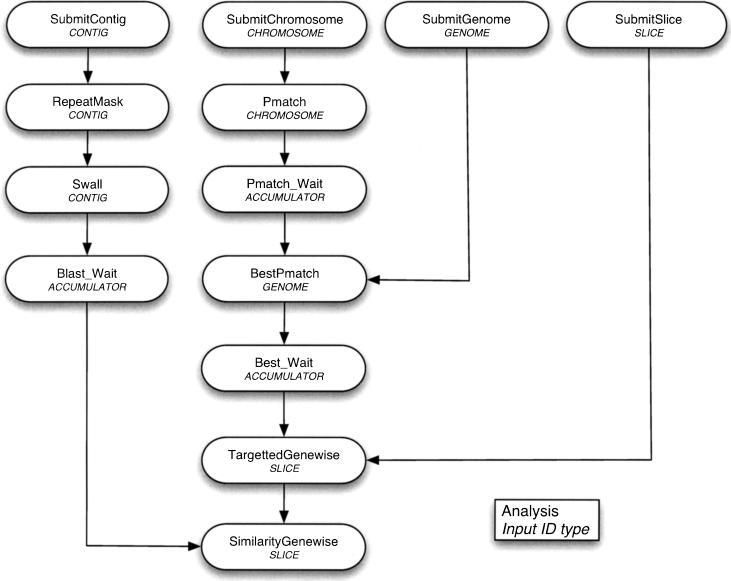
Pipeline control flow. This figure provides an example of the dependencies which can exist within the system. Analyses which have one dependency must maintain input id type, but analyses which have multiple dependencies can alter their input id type if an `accumulator' analysis is used. (See the move from Swall to Similarity_genewise.)
Each analysis has an `input ID type' which specifies the type of input sequence used in that analysis. These can include types such as `contig' or `slice' (a chromosomal region). There is one reserved type called `accumulator.' If an analysis has this type, the RuleManager will recognize that it must wait for all of the analyses it depends on to be complete on each of their potential input IDs before it can be started. This is used between stages of the gene build where one analysis needs to have completed over the whole genome before the next can start.
All information about running jobs, rules, and completed analyses is stored in the database. This means that at any point we can see what status all existing jobs have, as well as which jobs have completed. The pipeline uses six tables in addition to those of a standard Ensembl database. Three are filled out before the pipeline is started: rule_goal and rule_conditions, to define dependencies, and input_id_type_analysis, to link analyses with input ID types. The others are filled as the pipeline is running: job and job_status track the status of all current jobs; input_id_analysis records each successful analysis.
The job submission system also has the facility to group a number of jobs together and send them to the batch queue as a single entity. These are distinct jobs as far as the pipeline is concerned, but execute sequentially within the same batch job. This is used for very short jobs which do not use the queuing system efficiently when submitted individually.
As well as submitting jobs, the RuleManager monitors both the number of pending jobs in the batch queues and the length of time each job has been running. If there are too many pending jobs, the RuleManager will sleep for a preset period of time: A large number of pending jobs puts undue strain on the batch scheduling daemon and doesn't increase throughput. If a job has been running for too long, normally 24 h, it will be killed. Finally, the script checks for jobs which have failed. All sections of code within the pipeline which could conceivably fail (such as calls to the “run” method of a RunnableDB) are enclosed within a Perl “eval” construct which allows jobs which have terminated prematurely to be identified. All jobs which have failed fewer than a set number of times are automatically resubmitted. The RuleManager can also be run in different modes to allow testing and small runs to be carried out independent of any job scheduling system.
Configuration
There is no hard coding of variables within the pipeline code. Though some configuration for the Runnables is still present in the analysis table in the database (such as parameters for BLAST), most run-time parameters are now placed in a set of configuration files. These are written as Perl modules which export variables into the calling package (e.g., a Runnable). There are three modules which provide basic configuration required by all pipeline installations:
General
This contains a few `system-wide' options, such as the default location of binaries, temporary directories, and the location of job output files.
BatchQueue
This holds parameters relevant to submitting jobs to a queuing system, such as the name of the scheduler, default batch queue, and the maximum number of pending jobs allowed. It also holds per-analysis parameters, such as resource requirements which can be used to restrict particular analyses to run only on certain farm nodes.
BLAST
The main purpose of this configuration file is to provide a Perl regular expression so that the Runnable can correctly retrieve the hit identifier from the fasta header reported in the BLAST output.
Some analyses, such as those used in the gene build, have additional configuration files. Example files for all configuration modules are given in the distribution (with the suffix “.example”).
Installation and Availability
The Ensembl pipeline software and schema are available for download via anonymous checkout through CVS from http://cvsweb.sanger.ac.uk. Detailed information on download and installation is available as cvs module ensembl-doc.
Application to Large Genomes
The analysis pipeline is used within Ensembl for annotation of a number of species including human, mouse, rat, zebrafish, and Fugu. Currently, it is only used for the first three stages of data preparation: raw compute, gene build, and protein annotation. The sections below describe the analyses carried out for the raw compute and the protein annotation. The Ensembl gene build is described elsewhere (Curwen et al. 2004).
Raw Compute
The raw computes fall into two categories. The first is the analyses which provide annotation needed by the gene-build stages: RepeatMasker, Genscan, dust (low-complexity sequence), and BLAST. Secondly there are other prediction programs that provide genomic features of interest: CpG islands, transcription start sites (Down and Hubbard 2002), TRF (tandem repeats; Benson 1999), e-PCR for STS markers (Schuler 1997), and tRNAscan (Lowe and Eddy 1997).
A large part of the analysis is run on small units of sequence called contigs. In the case of finished sequence, these would normally be whole clones (except where a finished clone has been fragmented in the assembly due to the presence of contaminants). For unfinished sequence, these will be pieces of contiguous sequence. The reason for running on such small pieces of sequence is because these analyses are generally fairly slow. Ideally, batch jobs should take between 10 min and 1 h to complete in order to make the most efficient use of the compute farm.
Some analyses can be run more efficiently on larger regions (from 1 Mb up to a whole chromosome). For these cases, a new RunnableDB is written (usually with `Slice' prepended to the name), with modifications to the fetch_input method (to retrieve a Slice object, rather than a Contig object) and the write_output method (to map the results back to contig coordinates). The latter is straightforward to implement, as the Ensembl API (Stabenau et al. 2004) has methods for transforming between coordinate systems. Table 1 summarizes the analyses along with the size of input sequence on which they are run.
Table 1.
Principal Analyses of the'Raw Compute' for the Human Genome With the Size of Input Sequence Used
| Analysis | Input sequence size |
|---|---|
| CpG island prediction | chromosome |
| RepeatMasker | contig |
| Dust (low-complexity repeats) | chromosome |
| TRF (tandem repeats) | contig |
| Eponine (transcription start site prediction) | 1-Mb slice |
| Genscan | contig |
| e-PCR (STS markers) | 1-Mb slice |
| tRNAscan | contig |
| BLAST vs. Swall* | contig |
| BLAST vs. Unigene* | contig |
| BLAST vs. EMBL Vertebrate RNA* | contig |
BLAST analyses are only run on the peptides predicted by Genscan and not on the full genomic sequence. This is done to speed up the analysis
Note that to save time, the BLAST analyses (marked * in the table) are only run on the predicted peptide sequences produced by gene predictors (Genscan in this case). This is a limitation, but without this short-cut these analyses would take far too long to complete.
The Ensembl compute farm currently comprises over 1000 nodes, though the maximum number of jobs allowed by each user is restricted to 400 to limit database contention problems. Nevertheless, the above pipeline can be run over 2.85 Gb of sequence in 7–10 d.
Protein Annotation
Following the gene build, the predicted proteins are assigned Interpro domains (Mulder et al. 2003) using the protein annotation pipeline. This process is almost identical to running the raw computes. The analyses undertaken can be divided into two categories: the Interpro components (Pfam, Prints, Prosite, and Profile) and other common protein annotations (Tmhmm [Krogh et al. 2001], ncoil, Seg [Wan and Wootton 2000], and Signal peptide [Nielsen et al. 1997]).
Again, different analyses have different run times and, as before, different sizes of input data need to be used. Table 2 summarizes the analyses run for the protein annotation and gives a description and the size of the protein input used.
Table 2.
Principal Analyses of the Human Genome Protein Annotation
| Analysis | Description | Input chunk size |
|---|---|---|
| Pfam | Interpro component | 100 sequences |
| Prints | Interpro component | 100 sequences |
| ScanProsite | Interpro component | whole protein data set |
| ProfileScan | Interpro component | 100 sequences |
| Tmhmm | Transmembranes | 100 sequences |
| ncoil | Coiled coils | 100 sequences |
| sigp | Signal peptide | 100 sequences |
| Seg | Low-complexity | whole protein data set |
Final Remarks
The pipeline works very well in its current role of high-throughput analysis of large amounts of sequence data. Recent modifications to the pipeline have included the introduction of `accumulator' analyses which allow all the dependencies for a full gene build to be specified at once. A gene build can now be performed from scratch with little manual intervention.
Designing a pipeline system which simultaneously manages to be highly flexible, efficient, and easy to use is not straightforward. However, we believe that the simplicity of the system, which shows good performance and scalability, far outweighs any of its shortcomings. There are certainly stages in the data preparation for an Ensembl release which could be automated but have not, in the past, been suitable applications for the pipeline. However, changes introduced recently should improve this and allow much more of the Ensembl release process to be automated.
As with any piece of software, there is inevitably a learning curve associated with using it. We provide documentation which gives detailed instructions for installation and running the pipe-line. These are maintained in CVS module ensembl-doc so that they can be kept up to date.
Acknowledgments
We thank the users of our Web site and the developers on our mailing lists for much useful feedback and discussion. We particularly acknowledge the Singapore members of the Fugu annotation project and the annotation team at the Wellcome Trust Sanger Institute. The Ensembl project is principally funded by the Wellcome Trust with additional funding from EMBL.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.1859804.
References
- Benson, G. 1999. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 27: 573-580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuff, J.A., Coates, G.M.P., Cutts, T.J.R., and Rae, M. 2004. The Ensembl computing architecture. Genome Res. (this issue). [DOI] [PMC free article] [PubMed]
- Curwen, V., Eyras, E., Andrews, D.T., Clarke, L., Mongin, E., Searle, S., and Clamp, M. 2004. The Ensembl automatic gene annotation system. Genome Res. (this issue). [DOI] [PMC free article] [PubMed]
- Down, T.A. and Hubbard, T.J.P. 2002. Computational detection and location of transcription start sites in mammalian genomic DNA. Genome Res. 12: 458-461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbin, R. and Mieg, T. 1991. A C. elegans Database. Documentation, code and data available from anonymous FTP servers at http://lirmm.lirmm.fr, cele.mrc-lmb.cam.ac.uk and ncbi.nlm.nih.gov.
- Glasner, J.D., Liss, P., Plunkett III, G., Darling, A., Prasad, T., Rusch, M., Byrnes, A., Gilson, M., Biehl, B., Blattner, F.R., et al. 2003. ASAP, a systematic annotation package for community analysis of genomes. Nucleic Acids Res. 31: 147-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoon, S., Ratnapu, K.K., Chia, J.-M., Kumarasamy, B., Xiao, J., Clamp, M., Stabenau, A., Potter, S., Clarke, L., and Stupka, E. 2003. Biopipe: A flexible framework for protocol-based bioinformatics analysis. Genome Res. 13: 1904-1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudek, A.K., Cheung, J., Boright, A.P., and Scherer, S.W. 2003. Gene-script: DNA sequence annotation pipeline. Bioinformatics 19: 1177-1178. [DOI] [PubMed] [Google Scholar]
- Kent, W.J., Sugnet, C.W., Furey, T.S., Roskin, K., Pringle, T.H., Zahler, A.M., and Haussler, D. 2002. The human genome browser at UCSC. Genome Res. 12: 996-1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh, A., Larsson, B., von Heijne, G., and Sonnhammer, E.L. 2001. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 305: 567-580. [DOI] [PubMed] [Google Scholar]
- Lowe, T.M. and Eddy, S.R. 1997. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25: 955-964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulder, N.J., Apweiler, R., Attwood, T.K., Bairoch, A., Barrell, D., Bateman, A., Binns, D., Biswas, M., Bradley, P., Bork, P., et al. 2003. The InterPro Database, 2003 brings increased coverage and new features. Nucleic Acids Res. 31: 315-318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mungall, C.J., Misra, S., Berman, B.P., Carlson, J., Frise, E., Harris, N., Marshall, B., Shu, S., Kaminker, J.S., Procknik, S.E., et al. 2002. An integrated computational pipeline and database to support whole genome sequence annotation. Genome Biol. 3: 1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen, H., Engelbrecht, J., Brunak, S., and von Heijne, G. 1997. A neural network method for identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Int. J. Neural Syst. 8: 581-599. [DOI] [PubMed] [Google Scholar]
- Schuler, G.D. 1997. Sequence mapping by electronic PCR. Genome Res. 7: 541-550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stabenau, A., McVicker, G., Melsopp, C., Proctor, G., Clamp, M., and Birney, E. 2004. The Ensembl core software libraries. Genome Res. (this issue). [DOI] [PMC free article] [PubMed]
- Wan, H. and Wootton, J.C. 2000. A global compositional complexity measure for biological sequences: AT-rich and GC-rich genomes encode less complex proteins. Comput. Chem. 24: 71-94. [DOI] [PubMed] [Google Scholar]
WEB SITE REFERENCES
- http://www.acedb.org; AceDB.
- http://www.platform.com/products/LSF; Load Sharing Facility.
- http://www.ncbi.nlm.nih.gov/genome/guide/build.html#annot; NCBI's annotation pipeline.
- http://vega.sanger.ac.uk; the Vertebrate Genome Annotation database.
- http://cvsweb.sanger.ac.uk; Public CVS repository for the Ensembl software.