

Abstract
As more genomes are sequenced, there is an increasing need for automated first-pass annotation which allows timely access to important genomic information. The Ensembl gene-building system enables fast automated annotation of eukaryotic genomes. It annotates genes based on evidence derived from known protein, cDNA, and EST sequences. The gene-building system rests on top of the core Ensembl (MySQL) database schema and Perl Application Programming Interface (API), and the data generated are accessible through the Ensembl genome browser (http://www.ensembl.org). To date, the Ensembl predicted gene sets are available for the A. gambiae, C. briggsae, zebrafish, mouse, rat, and human genomes and have been heavily relied upon in the publication of the human, mouse, rat, and A. gambiae genome sequence analysis. Here we describe in detail the gene-building system and the algorithms involved. All code and data are freely available from http://www.ensembl.org.
Recent years have seen the release of huge amounts of sequence data from genome sequencing centers. However, these raw sequence data are most valuable to the laboratory biologist when provided along with quality annotation of the genomic sequence. This information can be the starting point for planning experiments, interpreting SNPs, inferring the function of gene products, predicting regulatory sites for gene expression, and so on. The currently agreed `gold standard' for the annotation of eukaryotic genomes is that made by a human being. Manual annotation is based on information derived from sequence homology searches and the results of various ab initio gene prediction methods. `Gold standard' annotation of large genomes such as mouse and human is slow and labor-intensive, taking large teams of annotators years to complete. As a result, the annotation can almost never be entirely up-to-date and free of inconsistencies (as the annotation process usually begins before the sequencing process is complete). Hence, an automated annotation system is desirable, because it is a relatively rapid process that allows frequent updates to accommodate new data. To meet this need, we produced the Ensembl annotation system by observing how annotators build gene structures, and by condensing this process into a set of rules.
Ensembl was conceived in three parts: as a scalable way of storing and retrieving genome-scale data, as a Web site for genome display, and as an automatic annotation method based on a set of heuristics. It was initially written for the draft human genome (International Human Genome Sequencing Consortium 2001) which was sequenced clone by clone, but has also been successfully used for whole-genome shotgun assemblies such as mouse (Waterston et al. 2002), rat (Rat Genome Sequencing Project Consortium 2004), and Anopheles gambiae (Holt et al. 2002). Although the storage and display parts of Ensembl have been used for many genomes, the automatic annotation has been used for human, mouse, rat, mosquito, Fugu rubripes, zebrafish, and C. briggsae. All these annotations can be found at http://www.ensembl.org.
RESULTS AND DISCUSSION
In this section we first describe the procedure developed in Ensembl for predicting gene structures, and then present details of specific gene builds.
Gene Prediction Procedure
Automated genome annotation commonly commences with running various stand-alone analyses. In the case of Ensembl, this initial stage of computation is known as the `raw compute' (Potter et al. 2004). The analyses conducted include RepeatMasker (A. Smit and P. Green, unpubl.), Genscan (Burge and Karlin 1997), tRNAscan (Lowe and Eddy 1997), eponine (Down and Hubbard 2002), and, importantly, homology searches using BLAST (Altschul et al. 1997). The results of these analyses are stored in the Ensembl database and are displayed in the Ensembl Web site. A similar approach is used in other genome browsers; see, for example, Karolchik et al. (2003). However, Ensembl takes these types of analyses one step further and provides a set of gene annotations based on them. Our aim is to produce a set of predicted gene structures to which we can link extra biological information such as gene family information, expression data, and gene ontologies.
As with the Ensembl analysis pipeline (Potter et al. 2004), the Ensembl gene build software is comprised of a set of Runnables and RunnableDBs. In addition to these, we have a set of classes that provide utility methods for some of the complex manipulations and checks that are necessary during the gene-build process. We have designed these classes to be as modular as possible so that the analyses and algorithms are readily reusable. This makes the code very powerful, as the tools can be quickly incorporated into new solutions for genomic analysis.
The efficient analysis of large genomes (over 3 Gb of DNA for human) presents a challenge when we consider that the average memory capacity of a reasonably priced compute farm machine is currently around 1 gigabyte. Many of the programs we use run most efficiently with about 200 kb of sequence. For clone-based genome assemblies (e.g., human), this is conveniently the size of a full-length clone, and in these cases the raw compute pipeline runs the analyses on individual clones (or the contigs that make up a clone for unfinished sequence).
We employ two approaches to cope with the large scale of the annotation process. One approach is to reduce the genomic search space by starting our analysis with fast approximate scans of the entire genome. This allows us to apply accurate and computationally expensive analyses on relatively short sequences. The second approach is to run some of the steps in the gene build on pieces of chromosome sequence, typically 1–5 Mb, which we call slices. The Ensembl API allows manipulation of the resulting features at either the clone, contig, or assembly (i.e., chromosome) level, thus seamlessly joining the clones and their features back together (Stabenau et al. 2004). The API can retrieve features from the database (e.g., exons, genes) that span the junction of two slices. Similarly, the API can correctly store genes that fall off the end of a slice and exons that map to the junction of two assembly contigs. In order to avoid feature duplication at slice boundaries, we use the convention of dropping those that fall off the lower coordinate end of the slice.
Whole-genome shotgun assemblies (WGS) are more challenging. The first assembly of a new genome, or one that has low coverage, can contain many small unassembled contigs. In this case a dummy assembly is created so that we can again run the gene build on regular-sized slices.
A variety of ab initio gene predictors have been developed that use purely genomic sequence for their gene structure prediction. Manual annotators can use predictions from these programs to confirm gene structures, using BLAST evidence to support the exon predictions. Ab initio prediction does have its place in the Ensembl annotation system, as described below. However, the tendency of even the best methods, such as Genscan (Burge and Karlin 1997), to overpredict genes, and to miss small exons (Burset and Guigo 1996) compels us to temper its use with other approaches. BLAST (Altschul et al. 1990, 1997) is a powerful tool for locating protein and cDNA sequences in the genome, but it is not suitable for predicting gene structures. BLAST simply detects homology and has no model for splice sites and hence exon boundaries. For these reasons, BLAST alone cannot be used to annotate genes. A more complex gene-building strategy is needed, where homology results are extended and augmented such that even an incomplete prediction can yield an accurate gene structure.
We combine evidence from various sources into our predictions. For instance, information derived from protein homologies is generally combined with information from other data sources to derive a full transcript structure. This need to meaningfully combine data from independent analyses adds a level of complexity to the automatic annotation process. Our gene-build procedure is currently biased towards protein-based prediction, as we concentrate on predicting genes that have valid translations.
A very important decision in the annotation process is the choice of data sources for homology searches. We generally use species-specific protein and cDNA data. However, in addition, we want to take advantage of data from other species, but this should not take priority over the species-specific data in the annotation. This methodology is reflected in the logic of the automatic annotation process as described below, and requires that extra care be taken in managing data sources.
The Ensembl gene-build process can be briefly described as follows. Species-specific proteins and cDNAs are first placed in the genome to create transcript models. Proteins from other species are then used to locate transcripts which have not been found previously. Protein- and cDNA-based transcripts are combined to obtain transcripts with untranslated region (UTR) information. Redundant transcript structures are eliminated and genes are created using the protein- and cDNA-based transcripts. We now give a more thorough explanation of the different steps. An overview of the current Ensembl gene build is given in Figure 1.
Figure 1.
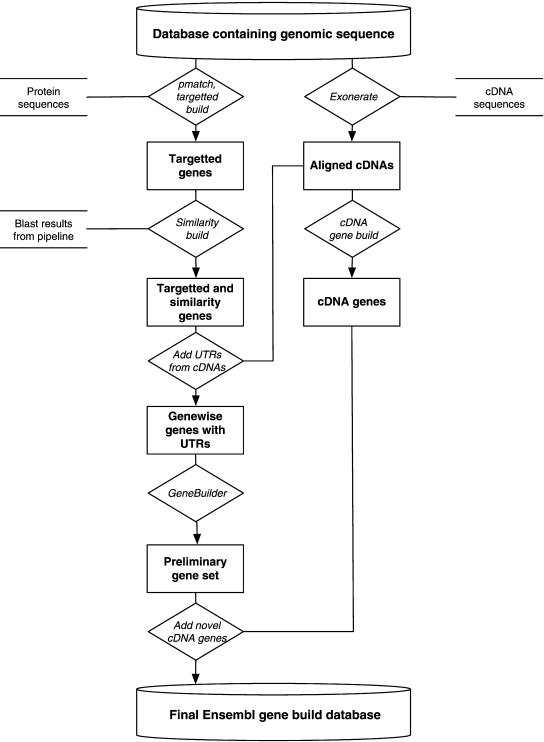
Overview of Ensembl Gene Build. Most genes are predicted using the sequences of known proteins aligned to the genome using genewise (Targetted and Similarity builds). UTR sequences for these genes are derived from the alignment of cDNAs to the genomic sequence (Exonerate, cDNA Gene Build). Transcripts created in this manner are then clustered to form genes (GeneBuilder). Finally, novel genes supported solely by cDNA evidence are added to the gene set, which is written to the database.
Targetted Protein Alignments
Following the initial raw computes, the first stage of the genebuild process proper is to place known proteins and full-length cDNAs derived for the genome of interest to their most likely position on the genome. This involves aligning these sequences such that they have correct splice sites and coherent translations. Here we describe the protein alignment process, known as the Targetted stage. The alignment of cDNAs is described in the section below entitled “cDNA Alignments.”
We place known proteins onto the genome using pmatch (R. Durbin, unpubl.), and construct transcript structures for them using genewise (Birney et al. 2004). Our protein sources are the genome-specific proteome sets from SWISS-PROT/TrEMBL (Boeckmann et al. 2003) and RefSeq (Pruitt et al. 2000). For the human build on assembly NCBI33, this combined source comprised 48,176 protein sequences (a redundant set) and for mouse assembly NCBI30 we had 33,605 protein sequences.
Pmatch is a fast, exact matching program for aligning protein sequences with either protein or DNA sequence; it looks for identical hits of at least 20 residues and then extends these until there is no longer an exact match. It has no splice-site model. We use it to find the rough genomic extent of the matches of the set of known proteins and the genome. In practice, pmatch gives us one or more hits for each protein-coding exon. These hits are pieced together to determine the rough genomic extent of the matches, while making sure that the exons remain on the same strand and are colinear with respect to the protein sequence.
We run pmatch first at the chromosomal level, rejecting any processed match that covers less than 25% of the parent protein. Then we take the best-in-genome match (based on protein coverage) plus any matches that fall within 2% of this. For human NCBI33, 87% of the proteins had a single best-in-genome match.
Pmatch is only the initial step in aligning proteins, but it does get us over the first hurdle, which is to reduce our alignment space for each protein from 3 Gb down to about 1 Mb.
We use genewise (Birney et al. 2004) to produce the final protein alignments. This program aligns at the protein level, allowing for splice sites and frameshifts, and in some respects is ideal for genome annotation—alignment at the protein level guarantees that our predicted genes will code for protein, a splice-site model allows the alignment to jump over introns, and tolerance of frameshifts allows for sequencing or assembly errors, so that we can annotate draft or low-coverage assemblies. Unfortunately the major drawback with genewise is speed; aligning a single 400-residue protein to a 100-kb DNA sequence takes on average 300 CPU sec. Even narrowing down the location of every protein aligned to 100 kb (currently around 40,000 for the human genome), genewise would take at least 6 CPU months. Thus we further reduce the search space by positioning individual exons using BLAST. We run BLAST between the piece of genomic sequence found by pmatch and the protein sequence. Each BLAST hit is padded with 200 bp to provide sequence around the splice sites. The genomic sequences of the padded BLAST hits are joined together to form a miniature genomic sequence containing only exon sequence and a small amount of intronic sequence, which we call a miniseq (Fig. 2). This procedure can reduce a 50-kb gene to under 2 kb, and the alignment of 40,000 proteins to a 3-Gb genome using genewise becomes a tractable problem. Using this three-step process (rough gene positioning, rough exon positioning, final alignment), all human proteins can be aligned to the genome in a few hours on 400 CPUs. There is, however, still the possibility of missing very small features or predictions, because we are not using all of the genomic sequence.
Figure 2.
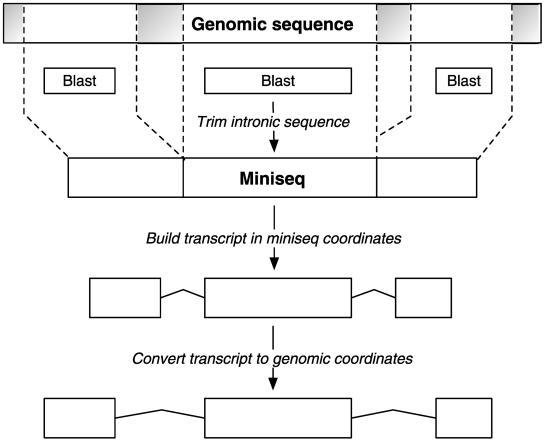
The Miniseq: We use a miniseq representation of genomic sequence in various stages of the gene build in order to reduce search space and increase processing speed. We BLAST a sequence of interest against a genomic region and pad the resulting hits with 200 bp. We then join the padded hits together to form a “mini genomic” sequence containing only exon sequence plus a small amount of intron sequence.
This miniseq approach forms a core part of the gene build and is not genewise-specific—it has been used within Ensembl to speed up the performance of other programs that would otherwise be too slow for whole-genome use, including est_genome (Mott 1997) and genomewise (Birney et al. 2004).
We compare the predicted translation with the parent protein, and reject any single-exon predictions matching fewer than 80% of the parent's residues, whereas multi-exon predictions must match at least 25% of the residues. We also check the lengths of introns and split predicted transcripts at introns exceeding 200 kb if the coverage of the parent protein is less than 95%. We reject any resultant single-exon transcripts, as we have observed that long introns are often either the first or last introns in a genewise prediction. All of these parameters are configurable.
Similarity Alignments
We next turn to proteins from other organisms for the stage known as the Similarity gene build. This is analogous to the Targetted stage (and indeed, many of the same software objects are reused) except that in this case the initial placement of proteins comes from the raw compute pipeline BLAST analysis. The pipeline uses BLAST to match ab initio predicted peptides against protein databases. We screen the raw BLAST hits for those which do not overlap a previously constructed Targetted transcript, and then re-BLAST promising proteins against the appropriate genomic region. We use this second set of BLAST hits to construct the miniseq, and run genewise as before.
We use different thresholds for the Similarity genes than for the Targetted genes. We reject any prediction whose translation matches less than 70% of the parent protein. Transcripts are split at introns longer than 10 kb unless the parent protein coverage exceeds 90%. We also reject any prediction with more than 60% of low-complexity sequence in its translation, assessed using Seg (Wootton and Federhen 1996).
DNA Alignments
In parallel with the protein alignments, we align all full-length cDNAs from an organism to its genomic sequence. Our sequence sources vary; for the human genome we use cDNA sequences from EMBL (Stoesser et al. 1997) and RefSeq (Pruitt et al. 2000), whereas for the mouse genome we additionally use the FANTOM2 data set (Okazaki et al. 2002). For the human build on NCBI33, we aligned 86,918 cDNAs.
We use Exonerate (G. Slater, unpubl.), which rapidly aligns cDNAs (and ESTs) to the genome in one step. Exonerate efficiently handles a large set of sequences and includes various models for aligning splice sites, combining speed and accuracy.
The standard thresholds used for the alignments are 90% coverage and 97% identity. For each cDNA match that exceeds these thresholds, we take the alignment with the best coverage and any other match within 2% of this one. These alignments are sorted by the number of exons they contain and by their coverage. If the best alignment is spliced, any subsequent unspliced alignment is rejected as being a potential processed pseudogene. From the human cDNA data set, 842 cDNAs which exceeded the threshold values were rejected as potential processed pseudogenes. The 86,918 cDNAs that were aligned to the NCBI33 human genome assembly were distributed in 97,166 distinct alignments. For 19,457 human cDNAs, Exonerate found an alignment but the coverage was below the chosen thresholds.
UTR Attachment
Now we use the information within cDNA-based transcripts to add UTRs to the protein-based genewise predictions obtained from the Targetted and Similarity stages. These are used to generate a consensus transcript structure that consists of a 5′ UTR, a coding region, and a 3′ UTR. The protein data sets contain both full-length sequences and some fragmented sequences that overlap with them. In order to avoid adding UTRs to a fragment instead of a full-length sequence (which could lead to an incorrect prediction having a short ORF with an extremely long UTR), we first sort the genewise transcript predictions by length, considering both genomic extent and total exon length. We then pair each of the genewise predictions with a cDNA prediction, allowing each cDNA to be matched with a single, long genewise prediction. An individual genewise prediction may at this stage be paired with more than one cDNA.
We compare the 5′ genewise exon to each of the exons in a cDNA transcript and call a match if: (1) The end of the 5′ genewise exon exactly coincides with the end of one of the cDNA exons, and either (2) the cDNA exon starts upstream of the genewise exon, or (3) the cDNA exon starts downstream of the genewise exon, and the matching cDNA exon is not the first in the prediction—that is, there are potential spliced UTR exons.
A similar procedure compares the 3′ terminal genewise exon with the cDNA, considering exon start coordinates. We do not require a cDNA prediction to extend both 5′ and 3′ UTRs. Single-exon genewise predictions match any cDNA which entirely encloses them within one of its exons. Various examples of matching cDNA and genewise structures are shown in Figure 3.
Figure 3.
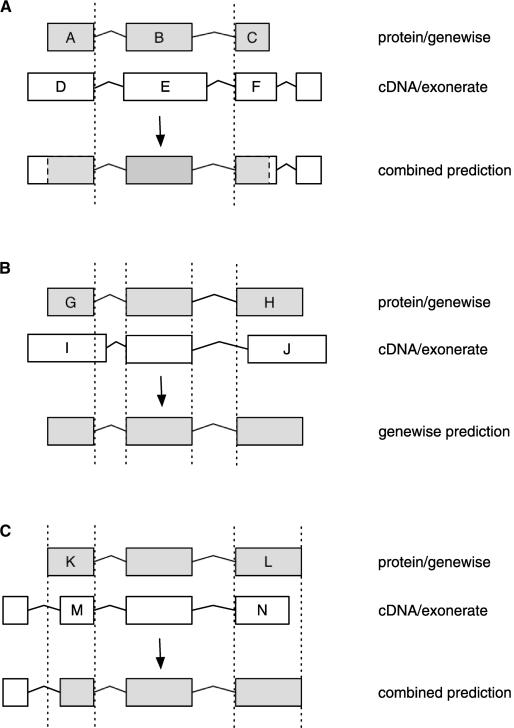
Rules for adding UTRs to genewise predictions: (A) Simplest case: Ends of exons A and D coincide, thus exon A is extended to include the UTR and the translation start is maintained. Starts of exons C and F coincide, thus UTR exons are added and the translation stop is maintained. The coordinates of genewise-derived exon B are used in preference over exon F. (B) cDNA prediction rejected: Neither the ends of exons G and I nor the starts of exons H and J coincide, so the genewise-predicted structure is unmodified. (C) cDNA prediction with short exons: The ends of the exons K and M and the starts of exons L and N coincide. Even though K is shorter than M, it is not the first exon of the cDNA prediction and is thus retained. However, N is shorter than L and there are no additional exons, so it is rejected.
The best matching cDNA is chosen for each genewise result based on exon overlap and, if necessary, the extent of shared genomic overlap between the two. The cDNA and genewise transcripts are now combined, giving preference to the genewise predicted ORF/translation coordinates—internal exons are taken from the genewise prediction. The exceptions to this rule are cases where the cDNA exon coordinates did not precisely match the 5′/3′ terminal genewise exons. This can occur as genewise sometimes fails to align a very short terminal coding region to the right exon. The translation must be then recalculated to take into account the corrected splice sites, and this is achieved using genomewise (Birney et al. 2004).
The combined predictions are stored in a clean database. Additionally, if no matching cDNA is found for a genewise prediction, we re-store the unmodified genewise prediction. This is important in preventing loss of supporting evidence, as at this stage we store only one transcript per predicted gene. The final GeneBuilder combines all transcripts belonging to the same gene and will transfer supporting evidence from any partial transcripts that may have been subsumed by full-length transcripts.
Final GeneBuilder
Finally, we need to cluster all the predicted transcripts and use them to create Ensembl genes. For many organisms we use only the genewise/cDNA combined predictions, though we can bring in transcripts built from ab initio methods if required (see below). The clustering process consists of a number of stages:
All transcripts are clustered by genomic overlap. Within each cluster, we prune redundant transcripts. First, we sort by both total exonic length and translation length so that the ordering is as follows: long translation + UTR > long translation > short translation + UTR > short translation. Then, shorter transcripts with exon structure redundant with a longer transcript are subsumed: if all the pairs of adjacent exons within a transcript overlap, the supporting evidence is transferred from the shorter to the longer transcript, and the shorter one is discarded. We use pairs of exons rather than individual exons for this comparison to make sure that we do not reject alternative variants. If a cluster contains only single-exon predictions, we retain the longest one and subsume the others into it.
We then cluster transcripts into genes considering exon overlap. Transcripts are placed into clusters such that each of the transcripts has one exon overlapping with an exon from at least one of the other transcripts in the cluster. Transcripts which lie entirely within the introns of other transcripts are thus clustered separately.
Occasionally, clusters contain very large numbers of transcripts, so we select the best transcripts for each cluster—defaulting to 10 transcripts per cluster, and giving priority totranscripts with long translations and UTRs over transcripts with just a short translation. Very few clusters are affected by this; in the most recent human build (NCBI33) only 44 clusters were involved.
The final transcript set is reclustered into genes, in case there were gene splits after the previous step. We remove shared duplicate exons from each transcript cluster, after first transferring the supporting evidence. For each gene we store a unique set of exons, and for each exon we store all the supporting evidence from each of the transcripts where the exon appears.
In recent builds we have used less evidence from ab initio methods, because the number of genes derived from this evidence in previous builds was small. As described in the accompanying Ensembl pipeline paper (Potter et al. 2004), we use BLAST to search ab initio predicted peptides against various DNA and protein sequence databases. This approach both improves search speed (by drastically reducing the genomic search space), and increases sensitivity (through the increased statistical power afforded by searching with BLAST amino acid sequences). The disadvantage is that significantly homologous sequences may be missed if they are not predicted by ab initio methods.
Using these pre-computes, we generate putative transcripts in the following way: We construct exon pairs from predicted adjacent exons if they are supported by BLAST evidence that spans the intron and has consistent coordinates (i.e., the pieces of evidence neither overlap by a large amount, nor have an excessive gap between them which might indicate a missing intermediate exon). Exon pairs are then recursively linked into transcripts which can be clustered together with genewise/cDNA-based transcripts as described above. Commonly, we use Genscan for ab initio prediction in human, mouse, and rat, but the system is equally applicable to other methods such as FgenesH (Solovyev et al. 1995) and genefinder (P. Green, unpubl.).
In order to exploit any cDNA information which has not yet been used, we extend the gene build using a module known as the GeneCombiner. Aligned cDNAs are merged into a minimal set of nonredundant spliced variants using the ClusterMerge algorithm (Eyras et al. 2004) to generate a set of genes based solely on cDNAs. These are merged with the genes from the GeneBuilder, giving priority to the protein-based set: all of the protein-based genes are kept, and cDNA-based transcripts are added if they represent a new gene locus. In order to increase alternative splicing annotation, we can optionally add cDNA-based transcripts if they represent a new variant in a protein-based gene locus: we test for an instance of exon-skipping and/or alternative 5′/3′ exons with respect to the protein-based transcripts.
Assigning a Gene Identifier
We use pmatch to compare our predicted peptide set with the set of SWISS-PROT and RefSeq proteins used for the gene build. We take the best match for each peptide as long as it is above 50% identity, and use it to assign an identifier to each predicted transcript. We are improving the process to take into account possible sequence ambiguities, as in the case of two transcripts which have different exon structure but very similar translations.
Correctly Predicting Tandemly Repeated Genes
Tandemly repeated genes can perturb the gene build, primarily because homology-searching programs often distinguish poorly between multiple high-scoring matches. As a result, miniseqs used in gene building can sometimes contain all or parts of more than one gene. If these genes are tandemly repeated or otherwise highly similar, genewise can mix their exons, as it tries to predict the gene that best matches the input protein sequence.
In order to rectify this, before a miniseq is passed to genewise we use a simple algorithm to decide whether it may contain multiple homologous genes. Essentially, the algorithm uses BLAST match information of the input protein with the miniseq to look for multiple copies of the input protein exons. If a sufficient proportion of duplicate exons are detected, the algorithm attempts to split the miniseq into separate fragments in order to isolate each gene or repeated segment. If the repeated units within the miniseq can be resolved into multiple complete genes, separate genewise runs are performed on each fragment of the starting miniseq. If the fragments do not form sets of whole genes, they are considered to be derived from a single gene with repetitive exons. Using this strategy we have built correct genes in regions containing high numbers of tandemly repeated genes, such as the HLA region of human chromosome 6. This method has also been successfully applied to the zebrafish genome in regions where genes from the same family are close together (e.g., the TCR-α locus and the MHC-2 region).
EST Gene Build
Expressed sequence tags (ESTs) are notorious for their variable quality. They are single-read sequences and thus prone to sequencing error. Additionally, the libraries from which they are derived can often be contaminated with genomic sequence which cannot be detected by an automatic annotation system. Finally, they are generally around 400 bp long, and thus a single EST is unlikely to cover an entire gene. For these reasons we have less confidence in genes built from ESTs, so we build them separately from the main gene build.
The Ensembl EST gene build process involves two steps. First, ESTs for the genome of interest are aligned to the genomic sequence using either Exonerate (G. Slater, unpubl.) or BLAT (Kent 2002). These programs are both capable of searching whole chromosomes or even genomes in a modest amount of CPU time. However, their memory usage is directly related to the number of hits they find, so we process the EST sequences in chunks of 300–500 sequences. With the total human division of dbEST (Boguski et al. 1993) being presently around 5.5 million ESTs, roughly 15,000 separate jobs must be computed. Fortunately, both Exonerate and BLAT are extremely fast. With 400 CPUs, this computation can be performed in less than three days (Exonerate) or around 36 h (BLAT). The variable quality of EST sequence leads us to employ conservative match criteria. We screen for the best-in-genome match for each EST, which must exhibit not worse than 97% identity with the genomic sequence over not less than 90% of its length. In general, only about half of the starting EST data set will meet these criteria, although this is highly dependent on EST quality.
The second stage is to use the aligned ESTs to build gene structures using the ClusterMerge algorithm (Eyras et al. 2004). Translations are then predicted using genomewise (Birney et al. 2004) with parameters set such that the exon boundaries are not modified. The ESTs are useful in determining possible alternative splicing of the predicted genes (Eyras et al. 2004).
The methods used in predicting the Ensembl EST gene set are fully described elsewhere (Eyras et al. 2004).
Processed Pseudogene Tagging
Processed pseudogenes result from reverse transcription of a mature mRNA and reinsertion into the genomic sequence. We have developed a system to detect potential processed pseudogenes among the Ensembl transcript predictions. We test the Ensembl predicted transcripts for:
lack of introns, that is, single-exon transcripts
presence of a poly A tail downstream of the disrupted open reading frame
absence of methionine at the start of the predicted translation
frameshifts—genewise can align a protein to a potential processed pseudogene by introducing frameshifts to avoid inframe stops, though these can also be due to sequencing errors and/or errors in the protein sequence used to predict the structure.
whether the supporting evidence is found spliced elsewhere in the genome—processed pseudogenes represent an unspliced copy of a functional transcript
whether there is sequence similarity in homologous regions in other species—according to Hillier et al. (2003), most of the detectable processed pseudogenes have appeared after speciation, hence they are independently integrated in the genome and therefore unlikely to have any sequence similarity in homologous regions. We use homology at the genomic level rather than gene orthology for this assessment.
We found that the strongest signals for processed pseudogenes are for single-exon predictions with in-frame stop codons, which are based on protein evidence that is spliced elsewhere in the genome and that have no sequence similarity in the homologous region in mouse and rat. Human gene predictions are therefore tagged as processed pseudogenes if they fulfill these three properties.
We tagged 962 pseudogenes out of 24,261 gene predictions in the NCBI33-based human gene build. We only tag genes with a single transcript, because a candidate that overlaps with a functional transcript is unlikely to have arisen from retrotransposition. As genes in Ensembl are classified as a set of transcripts having exon overlap, our method will detect pseudogenes even if they fall in the intron of a functional gene. The detected pseudogene set is underrepresented, as very stringent filters are applied in the process of gene prediction, including the rejection of transcripts with in-frame stops. In the future we plan to turn this process around and actively look for pseudogenes in various genomes.
At present we do not attempt to classify nonprocessed pseudogenes, but this is an active area of research in the group.
Annotation of Expression Data
For the human genome we link the Ensembl predictions to the eVoc expression vocabulary (Kelso et al. 2003). We add this information in the following way: eVoc links an EST identifier to the leaves of the vocabulary trees via its library name. We use aligned ESTs to link the Ensembl predictions to the vocabularies. We carry out a coordinate-based comparison between the ESTs and the Ensembl transcripts and link each Ensembl transcript to every EST which has a splicing structure compatible with it, following the same compatibility rules and comparison methods of Eyras et al. (2004). We usually allow a 6-bp mismatch at exon boundaries, whereas for terminal exons we allow any mismatch at the external boundaries. The results are stored as a link between Ensembl transcripts and EST identifiers, which is then used to link to the eVoc database (Kelso et al. 2003). The results of this analysis can be retrieved from EnsMart (Kasprzyk et al. 2004) and will be soon available through the Ensembl Web browser, http://www.ensembl.org.
Gene Build Results
Ensembl has a large and powerful compute farm, described by Cuff et al. (2004), and as a result the various stages of the build are now completed in days rather than weeks. For example, the Targetted and Similarity stages of the NCBI33 human build were each mostly complete within 48–72 h, and subsequent stages took only a few hours each. However, there are always a few jobs that take longer to run—for example, the many transcript variants of the 700-kD human protein titin always cause problems for genewise in the Targetted build, and some regions of the genome have thousands of BLAST hits to be processed during the Similarity build. Additionally, the gene-build code is under continuous development, inevitably in parallel with preparing data for release; these timings do not reflect the need to adjust parameters and rerun stages. Other time-consuming steps include visual inspection of the predictions at each stage of the build, extensive quality control of the final gene set, and setting up and checking multiple build databases (typically one per interim stage plus the final release database). These stages are essential if we are to produce a final database which is in a fit state to be handed over to the Web and EnsMart (Kasprzyk et al. 2004) teams for release. Also, with such a large farm and so much data there are always inevitably hardware issues, power cuts, and so on to contend with. Currently we expect to complete the gene build on a large genome such as human or mouse in 3–4 weeks.
Human Build on NCBI33 Assembly
From a starting point of 48,176 human proteins, 42,589 proteins were placed at one location in the genome, 3173 at two locations, and 781 at three locations. In addition, 492 proteins (1%) could not be located at all due to missing genomic sequence or insufficient coverage of the placed protein. The final gene build step resulted in 23,299 genes containing 32,035 transcripts. Of these, 270 (0.84%) were built solely from cDNAs, 6219 (19%) were built from human proteins with no UTR attachment, and 2983 (9%) were built from nonhuman proteins with no UTR attachment. Of the combined transcripts with UTRs, there were 21,889 (68%) built from human protein and cDNA, and 674 (2%) built from nonhuman protein and cDNA. We estimate that around 70% of our predictions are full-length (start with ATG, end with TAA/TAG/TGA). Of the transcripts built, 962 were tagged as pseudogenes.
Mouse Build on NCBI30 Assembly
The last gene build on the mouse genome resulted in 24,948 genes containing 32,911 transcripts. No genes were built solely from cDNAs, as this process was not introduced at the time. Similarly to the human build, the majority of the transcripts (14,912 or 45%) were built from mouse proteins combined with UTRs from cDNAs; 1387 (4%) were built from non-mouse proteins with UTRs, 4589 (14%) were built from mouse protein only, and 9385 (29%) were built from non-mouse protein. The number that were built from similarity confirmation of Genscan ab initio predictions was 2368 (7%). As this build was completed before the human NCBI33 build, no pseudogenes were tagged.
Comparison to Manual Annotation
We compared the predicted Ensembl genes for the human NCBI33 assembly with the set of manual annotations for chromosomes 6 (Mungall et al. 2003) and 13 (A. Dunham, in prep.) from the HAVANA group at the Sanger Institute, and chromosome 14 (Heilig et al. 2003) from Genoscope, all available at http://vega.sanger.ac.uk. Chromosomes 6 and 13 were chosen for being the most recent available annotations, whereas 14 was chosen for serving as a reference point as it was annotated using less similar methods. We did not compare to chromosome 20 (Deloukas et al. 2001), as the annotations were made less recently and thus from older data sources. We only considered genes of type Known and Novel-CDS for the comparison (Known genes are supported by a cDNA or a protein and have a LocusLink or GDB entry; Novel-CDS genes are supported by spliced ESTs or by similarity to another gene and have an unambiguous ORF), as the evidence they are based upon is the closest to the evidence used for the Ensembl predictions. Genes predicted as pseudogenes by Ensembl were not considered in the comparisons. A difficulty in this comparison is that the annotation of chromosome 6 is not based on assembly NCBI33, and 113 Known and 34 Novel-CDS genes could not be transferred. Thus we compared Ensembl genes with annotated genes that could be transferred to NCBI33, namely, a total of 1497 Known and 473 Novel-CDS genes.
We carried out comparisons at the gene, transcript, exon, and base-pair level. At the gene level (Table 1), a gene was considered as found if the genomic extent of the gene had some overlap with at least one Ensembl gene. This represents a rough estimate of correctly predicted gene loci.
Table 1.
Gene-Level Comparison to Manual Annotations
| Sn | Sp | |
|---|---|---|
| chr13 | 0.90 | 0.74 |
| chr14 | 0.92 | 0.77 |
| chr6 | 0.94 | 0.72 |
Genes are compared according to the genomic extent, from where we draw gene-pairs. This represents a rough estimate of found gene loci.
We use methods described by Eyras et al. (2004) for taking into account the high degree of alternative splicing, especially at the exon level, and for calculating the possible transcript pairs for every gene pair formed by genomic overlap. The annotations contain an average of slightly over three transcripts per gene. In contrast, Ensembl genes have about 1.3 transcripts per gene. Thus sensitivity at the transcript level is bound to be low, though the specificity is on average 0.73. We therefore consider the fraction of those transcript pairs which had all exons paired up, disregarding exon boundaries (Table 2).
Table 2.
Transcript-Level Comparison to Manual Annotations
| No exons unpaired | 1 exon unpaired | 2 exons unpaired | No exons unpaired (coding) | 1 exon unpaired (coding) | 2 exons unpaired (coding) | |
|---|---|---|---|---|---|---|
| chr13 | 55.5% | 18.09% | 11% | 65% | 15% | 6% |
| chr14 | 52.41% | 22.08% | 8.12% | 62% | 18% | 6% |
| chr6 | 60.88% | 18.49% | 8.40% | 70% | 16% | 5% |
Isoforms from either gene in the gene-pairs are paired up according to the best transcript alignment. A transcript is considered found if there is an alignment with a predicted transcript which has no better alignment with the other annotated transcripts. Also given is the percentage of the transcript pairs that have 0, 1, and 2 exons unpaired.
At the exon level, we restrict the exon comparisons to the transcript pairs: we calculate the percentage of exact exon matches from the exon pairs within transcript pairs, and the sensitivity and specificity of exact exon matches within the transcript pairs. In Table 3 we present these results for all exons and for coding exons only. Finally, for the comparison at the basepair level (Table 4), we compared the genomic extent of both predicted and annotated exons. We give values for all exons, and also for coding exons only.
Table 3.
Exon Level Per Transcript-Pair Comparison to Manual Annotations
| Exact pairs | Sn | Sp | Exact pairs (coding) | Sn (coding) | Sp (coding) | |
|---|---|---|---|---|---|---|
| chr13 | 82% | 0.73 | 0.78 | 93% | 0.83 | 0.90 |
| chr14 | 80% | 0.69 | 0.77 | 90% | 0.78 | 0.88 |
| chr6 | 80% | 0.73 | 0.76 | 92% | 0.85 | 0.89 |
Only exons that are part of a transcript-pair are compared with each other. An annotated exon is considered found if there is at least one predicted exon with exact matching boundaries in a given transcript-pair. The second and fourth columns give the percentage of exact exon matches from the total number of exon-pairs formed within the transcript-pairs for all-exons and for coding exons, respectively. The sensitivity and specificity values are also given. The values are higher for coding exons, which indicates that UTR annotation remains a difficult problem.
Table 4.
Base-Pair Level Comparison to Manual Annotations
| Sn | Sp | Sn (coding) | Sp (coding) | |
|---|---|---|---|---|
| chr13 | 0.64 | 0.87 | 0.90 | 0.85 |
| chr14 | 0.70 | 0.86 | 0.89 | 0.84 |
| chr6 | 0.72 | 0.73 | 0.94 | 0.72 |
We took the projections of the exon predictions and annotations over the genomic sequence and compare the overlap of both. The first two columns give the results for the sensitivity and specificity when all the exons are considered; the other two columns give these values for coding-exons only.
Some of the overpredicted genes overlap with pseudogene annotations: our current pseudogene tagging method did not detect these cases. These predictions are usually based on fragmented proteins. Others overlap with one of the other annotated gene types which we did not consider in the comparison: Novel-transcript, and Putative (for a detailed description of these gene types, see Deloukas et al. 2001). These annotations are based on ESTs, and the ORF is ambiguous or absent; we do not include nontranslating genes in the core gene build, and the discrepancy is probably due to the difference in the data sources. For example, the annotators use nonhuman cDNA data to annotate human sequence, and they combine human and nonhuman ESTs with protein and cDNA data to produce some gene structures, whereas the automatic Ensembl gene build uses only human cDNAs for predicting cDNA-based gene structures, and does not include EST data in the core gene set.
Almost all of the 143 missed genes and the missed transcript variants are based on EST evidence, though some are cDNA-based. We do not combine the EST gene build with the main Ensembl gene build and thus do not include these cases in our gene set. On the other hand, 46% of the missed genes are covered by the ESTGene set. Note that we use a best-in-genome approach in the EST mapping, which the annotators do not use. We also miss some pseudogenes. These are usually based on a protein that we used to annotate a coding gene elsewhere in the genome but we did not align it in the location of the pseudogene, presumably because it fell below our matching thresholds.
Building Genes on Virgin Genomes
The gene build was designed and tested with human data. It must be tuned for any new organisms that are analyzed, and as such has a number of configuration files controlling the various stages. For the mouse and rat genomes, few modifications were necessary, but less closely related organisms require more intervention.
Caenorhabditis briggsae
For the most recent C. briggsae build (March 03), we used the WormBase (Harris et al. 2003) protein set for the Targetted build, BLASTs against SwissProt and TrEMBL (Boeckmann et al. 2003) for the Similarity build, and cDNAs and ESTs from C. briggsae and C. elegans. Genefinder (P. Green, unpubl.) was the source of ab initio predictions.
The nature of genes in C. briggsae required us to alter the genewise parameters. The median intron length for human is 1502 bp (estimated from the Ensembl genes for the human assembly NCBI33) but for C. briggsae it is only 54 bp. Therefore we increased the genewise gap extension penalty from the standard two to 10 to prevent prediction of long exons. We also reduced the maximum allowed intron size from 20,000 to 5000 for the Targetted build and from 10,000 to 2500 in the Similarity build. Because we were using cDNAs and ESTs from C. elegans, we reduced the percentage identity threshold from 97% to 80%, while leaving the coverage filter at 90%.
This gene build produced 11,884 genes and 14,713 transcripts, just over half the expected gene number of 19,507. We believe this is because most of the genes were based on similarity to C. elegans proteins, and the evolutionary distance between the two species is too far (110 mya) for a full gene set to be predicted.
Anopheles gambiae
We also had to modify the gene build when analyzing Anopheles gambiae (Holt et al. 2002; Mongin et al. 2004); only a few gene families have been described, particularly those involved in odor reception and Plasmodium falciparum: Anopheles gambiae interactions, and as such there are few Anopheles protein, EST, or cDNA sequences available. Moreover, the closest dipteran is Drosophila melanogaster, which is 250 mya (Gaunt and Miles 2002).
Because our first Anopheles gene build produced a limited set of genes, we adjusted build parameters, concentrating mainly on the Similarity build. We used lower-scoring BLAST hits to select proteins to be used with genewise (cut-off score 125 instead of 200) and required that the predicted genewise translation cover at least 40% of the parent protein (instead of 70%). As a result, we produced an extremely large number of hits which were then sorted and filtered based on genome coverage, reducing the hits ∼10-fold. We used 40,000 Anopheles gambiae EST sequences in the EST gene build (Eyras et al. 2004), and combined genes from the Similarity and EST gene builds to produce the final set of 14,653 (Anopheles release 2).
Some issues are still to be resolved:
False positives. Anopheles repeats are not yet well described, leading us to include a few transposons and low-complexity genes in our gene set.
False negatives. Because the closest organism is 250 mya, we estimate that the Similarity build currently misses 20% of the genes.
Incomplete gene structure. As 5′ and 3′ exons are less conserved, they may be missed.
To reduce the number of false positives, a new set of Anopheles repeats has been created, and the ab initio gene predictor SNAP (I. Korf, unpubl.), has been trained using selected Anopheles ESTGenes.
Future Directions
Future developments will improve the annotations in two directions. As new genomes are sequenced, methods of gene prediction based on comparative analysis will be incorporated, including the use of gene orthology to improve our predictions. This will help us to find both genes in virgin genomes and new genes in well studied genomes. We also plan to extend our annotation to incorporate other biological features of clear interest for the research community, including noncoding mRNAs, nonprocessed pseudogenes, regulatory elements and transcription start sites, and antisense transcripts, and we plan to make these results available through the Web site.
Acknowledgments
We thank the users of our Web site and data sets and the developers on our mailing lists for much useful feedback and discussion. We also thank the zebrafish informatics group at the Sanger Institute, and in particular Kerstin Jekosch, for their feedback on the gene build system. We particularly acknowledge the Singapore members of the Fugu annotation project, the finished analysis group, and the annotation team at the Wellcome Trust Sanger Institute. Ensembl is funded principally by the Wellcome Trust with additional funding from EMBL and NIH-NIAID.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.1858004.
References
- Altschul, S.F., Gish, W., Miller, W., Myers, E.W., and Lipman, D.J. 1990. Basic local alignment search tool. J. Mol. Biol. 215: 403-410. [DOI] [PubMed] [Google Scholar]
- Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D.J. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25: 3389-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birney, E., Clamp, M., and Durbin, R. 2004. Genewise and genomewise. Genome Res. (this issue). [DOI] [PMC free article] [PubMed]
- Boeckmann, B., Bairoch, A., Apweiler, R., Blatter, M.C., Estreicher, A., Gasteiger, E., Martin, M.J., Michoud, K., O'Donovan, C., Phan, I., et al. 2003. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 31: 365-370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boguski, M.S., Lowe, T.M., and Tolstoshev, C.M. 1993. dbEST–Database for expressed sequence tags. Nat. Genet. 4: 332-333. [DOI] [PubMed] [Google Scholar]
- Burge, C. and Karlin, S. 1997. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 268: 78-94. [DOI] [PubMed] [Google Scholar]
- Burset, M. and Guigo, R. 1996. Evaluation of gene structure prediction programs. Genomics 34: 353-367. [DOI] [PubMed] [Google Scholar]
- Cuff, J.A., Coates, G.M.P., Cutts, T.J.R., and Rae, M. 2004. The Ensembl computing architecture. Genome Res. (this issue). [DOI] [PMC free article] [PubMed]
- Deloukas, P., Matthews, L.H., Ashurst, J., Burton, J., Gilbert, J.G., Jones, M., Stavrides, G., Almeida, J.P., Babbage, A.K., Bagguley, C.L., et al. 2001. The DNA sequence and comparative analysis of human chromosome 20. Nature 414: 865-871. [DOI] [PubMed] [Google Scholar]
- Down, T.A. and Hubbard, T.J.P. 2002. Computational detection and location of transcription start sites in mammalian genomic DNA. Genome Res. 12: 458-461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyras, E., Caccamo, M., Curwen, V., and Clamp, M. 2004. ESTGenes: Alternative splicing from ESTs in Ensembl. Genome Res. (this issue). [DOI] [PMC free article] [PubMed]
- Gaunt, M.W. and Miles, M.A. 2002. An insect molecular clock dates the origin of the insects and accords with paleontological and biogeographic landmarks. Mol. Biol. Evol. 19: 748-761. [DOI] [PubMed] [Google Scholar]
- Harris, T.W., Lee, R., Schwarz, E., Bradnam, K., Lawson, D., Chen, W., Blasier, D., Kenny, E., Cunningham, F., Kishore, R., et al. 2003. WormBase: A cross-species database for comparative genomics. Nucleic Acids Res. 31: 133-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilig, R., Eckenberg, R., Petit, J.-L., Fonknechten, N., Da Silva., C., Cattolico, L., Levy, M., Barbe, V., de Berardinis., V., Ureta-Vidal, A., et al. 2003. The DNA sequence and analysis of human chromosome 14. Nature 421: 601-607. [DOI] [PubMed] [Google Scholar]
- Hillier, L.W., Fulton, R.S., Fulton, L.A., Graves, T.A., Pepin, K.H., Wagner-McPherson, C., Layman, D., Maas, J., Jaeger, S., Walker, R., et al. 2003. The DNA sequence of human chromosome 7. Nature 424: 157-164. [DOI] [PubMed] [Google Scholar]
- Holt, R.A., Subramanian, G.M., Halpern, A., Sutton, G.G., Charlab, R., Nusskern, D.R., Wincker, P., Clark, A.G., Ribeiro, J.M.C., Wides, R., et al. 2002. The genome sequence of the malaria mosquito Anopheles gambiae. Science 298: 129-149. [DOI] [PubMed] [Google Scholar]
- International Human Genome Sequencing Consortium. 2001. Initial sequencing and analysis of the human genome. Nature 409: 860-921. [DOI] [PubMed] [Google Scholar]
- Karolchik, D., Baertsch, R., Diekhans, M., Furey, T.S., Hinrichs, A., Lu, Y.T., Roskin, K.M., Schwartz, M., Sugnet, C.W., Thomas, D.J., et al. 2003. The UCSC Genome Browser Database. Nucleic Acids Res. 31: 51-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasprzyk, A., Keefe, D., Smedley, D., London, D., Spooner, W., Melsopp, C., Hammond, M., Rocca-Serra, P., Cox, T., and Birney, E. 2004. EnsMart: A generic system for fast and flexible access to biological data. Genome Res. 14: 160-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelso, J., Visagie, J., Theiler, G., Christoffels, A., Bardien, S., Smedley, D., Otgaar, D., Greyling, G., Jongeneel, C., McCarthy, M., et al. 2003. evoc: A controlled vocabulary for unifying gene expression data. Genome Res. 13: 1222-1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent, W.J. 2002. BLAT–The BLAST-like alignment tool. Genome Res. 12: 656-664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe, T.M. and Eddy, S.R. 1997. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25: 955-964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mongin, E., Louis, C., Holt, R.A., Birney, E., and Collins, F.H. 2004. The Anopheles gambiae genome: An update. Trends Parasitol. 20: 49-52. [DOI] [PubMed] [Google Scholar]
- Mott, R. 1997. Est_genome: A program to align spliced DNA sequences to unspliced genomic DNA. Comput. Appli. Biosci. 13: 477-478. [DOI] [PubMed] [Google Scholar]
- Mungall, A.J., Palmer, S.A., Sims, S.K., Edwards, C.A., Ashurst, J.L., Wilming, L., Jones, M.C., Horton, R., Hunt, S.E., Scott, C.E., et al. 2003. The DNA sequence and analysis of human chromosome 6. Nature 425: 775-776. [DOI] [PubMed] [Google Scholar]
- Okazaki, Y., Furuno, M., Kasukawa, T., Adachi, J., Bono, H., Kondo, S., Nikaido, I., Osato, N., Saito, R., Suzuki, H., et al. 2002. Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNAs. Nature 420: 563-573. [DOI] [PubMed] [Google Scholar]
- Potter, S.C., Clarke, L., Curwen, V., Keenan, S., Mongin, E., Searle, S.M.J., Stabenau, A., Storey, R., and Clamp, M. 2004. The Ensembl analysis pipeline. Genome Res. (this issue). [DOI] [PMC free article] [PubMed]
- Pruitt, K.D., Katz, K.S., Sicotte, H., and Maglott, D.R. 2000. Introducing RefSeq and LocusLink: Curated human genome resources at the NCBI. Trends Genet. 16: 44-47. [DOI] [PubMed] [Google Scholar]
- Solovyev, V.V., Salamov, A.A., and Lawrence, C.B. 1995. Identification of human gene structure using linear discriminate functions and dynamic programming. In Proceedings of the Third International Conference on Intelligent Systems for Molecular Biology, 3: 367-375. [PubMed] [Google Scholar]
- Stabenau, A., McVicker, G., Melsopp, C., Proctor, G., Clamp, M., and Birney, E. 2004. The Ensembl core software libraries. Genome Res. (this issue). [DOI] [PMC free article] [PubMed]
- Stoesser, G., Sterk, P., Tuli, M.A., Stoehr, P.J., and Cameron, G.N. 1997. EMBL nucleotide sequence database. Nucleic Acids Res. 25: 7-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterston, R.H., Lindblad-Toh, K., Birney, E., Rogers, J., Abril, J.F., Agarwal, P., Agarwala, R., Ainscough, R., Alexandersson, M., An, P., et al. 2002. Initial sequencing and comparative analysis of the mouse genome. Nature 420: 520-562. [DOI] [PubMed] [Google Scholar]
- Wootton, J.C. and Federhen, S. 1996. Analysis of compositionally biased regions in sequence databases. Methods Enzymol. 266: 554-571. [DOI] [PubMed] [Google Scholar]