

Abstract
Prognosis for patients suffering from T-ALL is still very poor and new strategies for T-ALL treatment are urgently needed. Our study shows potent anti-leukemic effects of the myxobacterial V-ATPase inhibitor Archazolid A. Archazolid A reduced growth and potently induced death of leukemic cell lines and human leukemic samples. By inhibiting lysosomal acidification, Archazolid A blocked activation of the Notch pathway, however, this was not the mechanism of V-ATPase inhibition relevant for cell death induction. In fact, V-ATPase inhibition by Archazolid A decreased the anti-apoptotic protein survivin. As underlying mode of action, this work is in line with recent studies from our group demonstrating that Archazolid A induced S-phase cell cycle arrest by interfering with the iron metabolism in leukemic cells. Our study provides evidence for V-ATPase inhibition as a potential new therapeutic option for T-ALL.
Keywords: Archazolid, leukemia, natural compounds
INTRODUCTION
T-cell acute lymphoblastic leukemia (T-ALL) is an aggressive hematopoietic malignancy. Despite advances in understanding of the molecular basis of T-ALL and intensified treatment regimens that have improved outcome of therapy, some patients fail conventional chemotherapy and T-ALL remains fatal in 20% of children and more than 50% of adults [1]. Along this line, new therapeutic approaches are urgently needed to improve patient prognosis.
Activating mutations in Notch1 occur in more than 50% of T-ALLs, highlighting Notch1 as key player in T-ALL [2, 3]. In fact, constitutive activation of Notch1 signaling represents the most prominent oncogenic pathway in T-cell transformation [1]. Therefore, targeted therapies to inhibit the Notch1 pathway have been developed. γ-secretase inhibitors (GSIs) that prevent Notch1 activating cleavage have been widely tested in preclinical models and clinical trials. However, the role of Notch in leukemia is still not entirely clear: on the one hand, it was shown that Notch signaling promotes cell survival and apoptosis resistance in leukemia [4, 5] and that Notch blockade by γ-secretase inhibition exerts pro-apoptotic effects in leukemia [6]. On the other hand, activation of Notch or of the Notch downstream target Hes1 inhibits leukemia development, growth and survival [7-9]. Furthermore, GSIs have shown only modest and temporary responses in leukemia therapy. These controversial results about Notch in leukemia demand for better understanding of the mechanisms that contribute to Notch signaling in leukemia in order to develop novel strategies to inhibit Notch1 signaling with alternative mechanism different from GSIs as potential promising approaches for T-ALL therapy.
Besides Notch activation at the plasma membrane where ligand binding initiates y-secretase dependent cleavage of the Notch receptor, and subsequently NICD generation and translocation to the nucleus, Notch signaling in part depends on endocytosis. Recent reports showed that Notch can be activated in endosomes: in drosophila, Notch signaling is abolished when endosomal entry is blocked but is enhanced when endosomal retention is increased [10]. Moreover, in drosophila, acidification of endosomes by the V-ATPase is required for Notch activation [11].
Along this line, we hypothesized that inhibition of V-ATPase might be a promising strategy for T-ALL treatment. V-ATPase is an ATP-dependent proton pump that regulates pH homeostasis by translocating protons across membranes. The main function of V-ATPase is to regulate the acidification of intracellular compartments like lysosomes [12, 13]. V-ATPases are multisubunit heteromeric protein complexes with two functional domains: the V1 domain that is composed of eight subunits (A-H) mediates ATP hydrolysis and the V0 domain that is composed of five subunits (a, c, c’’, d, e) regulates proton translocation [14]. The V-ATPase is localized at endosomes and lysosomes and is essential for endocytotic processes, receptor internalization and recycling, and finally lysosomal degradation [13]. Therefore, V-ATPase is implicated in fundamental physiological processes, like neurotransmitter uptake, renal acidification, bone resorption or sperm maturation and is associated with pathologic conditions including osteopetrosis, renal tubular acidosis and disease-related processes such as entry of toxins and viruses [13].
Recent reports indicate important functions of V-ATPase in tumors. Augmented expression of V-ATPase in cancer cells was demonstrated to contribute to metastasis, survival and growth of cancer cells [15-18]. Localization of V-ATPase at the plasma membrane was associated with invasiveness of various types of cancer including breast, pancreatic, prostate, and melanoma cancer cells [14, 19-21]. V-ATPase was shown to localize to the plasma membrane in sarcoma cells as well and was elucidated as a promising target for Ewing sarcoma, osteosarcoma, chondrosarcoma or rhabdomyosarcoma [22, 23] as well as glioblastoma [24, 25]. Along this line, V-ATPase inhibition could represent a promising approach for tumor therapy. Although only few V-ATPase inhibitors have been described so far, their V-ATPase binding properties and mode of action are well-investigated. Amongst others, a class of natural compounds - the Archazolids - inhibits V-ATPase activity by binding to the V-ATPase V0 subunit [26-28] that is responsible for proton transport across the membrane [29]. Archazolids are macrolides that have originally been isolated from the myxobacterium Archangium gephyra [30], and are available also by chemical synthesis [31, 32]. Archazolids have attracted attention as highly potent V-ATPase inhibitors that exert promising anti-tumor effects [15-18, 33-36].
Because Notch signaling activation in part depends on endocytosis [10, 11, 37] and V-ATPase has therefore been linked with the Notch pathway [35, 38], we hypothesized that V-ATPase inhibition might represent an alternative option to target leukemic cells. Therefore, we had a closer look on the functional effects and the mechanism of action, including the Notch pathway and cellular stress response, of the V-ATPase inhibitor Archazolid A in leukemic cells.
RESULTS
V-ATPase in leukemic cells
First, we analyzed the expression of the V-ATPase subunits in different leukemic cell lines including the T-ALL cell lines Jurkat and CEM, the AML cell line HL60, and the CML cell line K562 in comparison to non-tumor primary human PBMCs. Most of the V-ATPase subunits were comparably expressed in non-tumor PBMCs, Jurkat, CEM, and HL60 cells and some subunits were increased in K562 cells (Table 1). Immunostainings show that subunit c ATP6V0C which is targeted by Archazolid A, is localized to the lysosomal system and to the plasma membrane of leukemic cells (Figure 1A). As V-ATPase is essential for endo-lysosomal function, we analyzed the size of the endo-lysosomal compartment. In fact, the size of the endosomal compartment was increased in leukemic cell lines compared to non-tumor primary human peripheral blood mononuclear cells (PBMCs) (Figure 1B). This set of data suggests a potential function of V-ATPase in leukemia.
Table 1. mRNA expression of V-ATPase subunits of the V1 domain (A-H) and the V0 domain (a, c, c’’, d, e) is shown in human leukemic cell lines related to non-tumor primary human PBMCs.
| V-ATPase subunit | S-Jurkat | CEM | K562 | HL60 |
|---|---|---|---|---|
| ATP6V1A | 0.70±0.07 | 1.41±0.11 | 2.83±0.47 | 0.70±0.05 |
| ATP6V1B1 | 0.43±0.16 | 1.16±0.60 | 1.24±1.13 | 0.36±0.19 |
| ATP6V1B2 | 0.52±0.07 | 0.63±0.10 | 0.88±0.10 | 0.68±0.12 |
| ATP6V1C1 | 0.90±0.06 | 0.93±0.09 | 2.59±0.22 | 0.83±0.09 |
| ATP6V1C2 | 0.79±0.21 | 0.48±0.11 | 0.75±0.15 | 34.28±15.71 |
| ATP6V1D | 0.96±0.01 | 1.19±0.14 | 1.87±0.05 | 0.24±0.01 |
| ATP6V1E1 | 1.00±0.05 | 0.97±0.01 | 3.43±0.44 | 0.89±0.06 |
| ATP6V1E2 | 0.51±0.05 | 0.37±0.03 | 0.65±0.06 | 0.17±0.01 |
| ATP6V1F | 1.12±0.27 | 1.64±0.34 | 5.27±0.39 | 1.00±0.06 |
| ATP6V1G1 | 1.00±0.17 | 1.06±0.06 | 1.61±0.13 | 0.60±0.04 |
| ATP6V1G2 | 0.12±0.03 | 1.00±0.14 | 0.11±0.03 | 0.06±0.01 |
| ATP6V1G3 | 0.18±0.07 | 0.69±0.25 | 0.10±0.00 | 0.11±0.04 |
| ATP6V1H | 0.85±0.04 | 1.16±0.06 | 2.32±0.08 | 0.66±0.04 |
| ATP6V0A1 | 0.48±0.01 | 1.07±0.06 | 10.67±0.87 | 0.60±0.05 |
| ATP6V0A2 | 0.78±0.03 | 0.66±0.21 | 1.55±0.10 | 0.30±0.04 |
| ATP6V0A3 | 0.01±0.00 | 0.11±0.01 | 0.37±0.24 | 0.68±0.07 |
| ATP6V0A4 | 0.82±0.31 | 1.46±0.23 | 1.26±0.71 | 0.83±0.28 |
| ATP6V0B | 1.53±0.12 | 1.47±0.14 | 4.78±0.53 | 1.46±0.49 |
| ATP6V0C | 0.67±0.06 | 0.81±0.07 | 2.39±0.36 | 0.90±0.10 |
| ATP6V0D1 | 0.38±0.03 | 0.44±0.04 | 1.26±0.12 | 0.86±0.09 |
| ATP6V0D2 | 0.10±0.04 | 0.19±0.17 | 9.09±4.84 | 18.38±5.61 |
| ATP6V0E1 | 0.34±0.08 | 0.72±0.16 | 0.84±0.03 | 0.62±0.04 |
| ATP6V0E2 | 1.97±0.08 | 2.62±0.15 | 0.54±0.05 | 0.07±0.01 |
Figure 1. V-ATPase in leukemic cell lines.
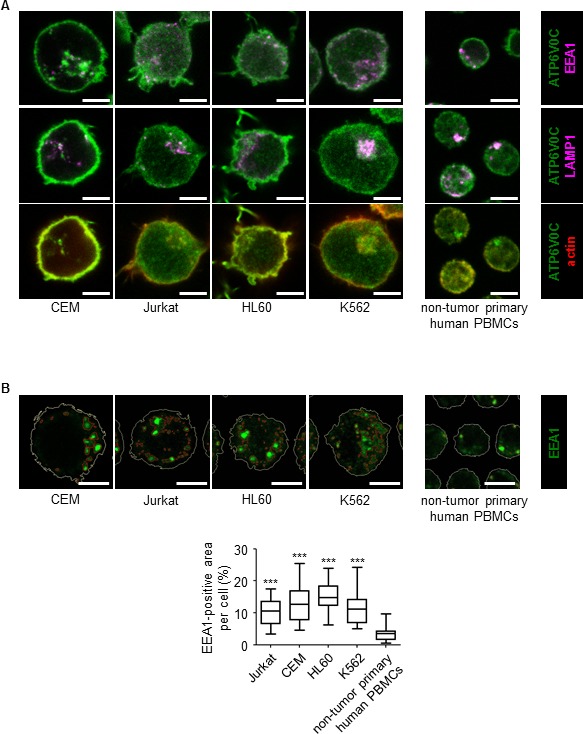
A. V-ATPase localization in leukemic cell lines is shown. Immunostaining of non-tumor primary human peripheral blood mononuclear cells (PBMCs) and leukemic cell lines (Jurkat, CEM, HL60, K562) for V-ATPase c-subunit (ATP6V0C, green) together with EEA1 (magenta), LAMP1 (magenta) and actin (red) is shown. Scale bar 5 μm. B. The size of the endo-lysosomal compartment in leukemic cell lines is shown. Staining of non-tumor primary human PBMCs and leukemic cell lines (Jurkat, CEM, HL60, K562) for EEA1 (green) is shown. Scale bar 5 μm. The white line labels the cell border. The red lines label the endosome area (EEA1-positive area). Endosome area per cell was calculated and is shown in the graph. One-Way ANOVA, Tukey's post test, ***p ≤ 0.001 (compared to non-tumor primary PBMCs).
V-ATPase inhibition by Archazolid A impairs growth and induces death of leukemic cell lines
Archazolid A inhibited V-ATPase activity in leukemic cells as shown by staining of lysosomes with a pH-sensitive fluorescence dye (LysoTracker) (Figure 2A). Archazolid A impaired proliferation of leukemic cell lines Jurkat (EC50 0.56 nM) and CEM (EC50 0.51 nM) (Figure 2B, 2C). In line, clonogenic growth of Jurkat and CEM cells was reduced by V-ATPase inhibition (Figure 2D, 2E).
Figure 2. Archazolid A inhibits growth of leukemic cell lines.
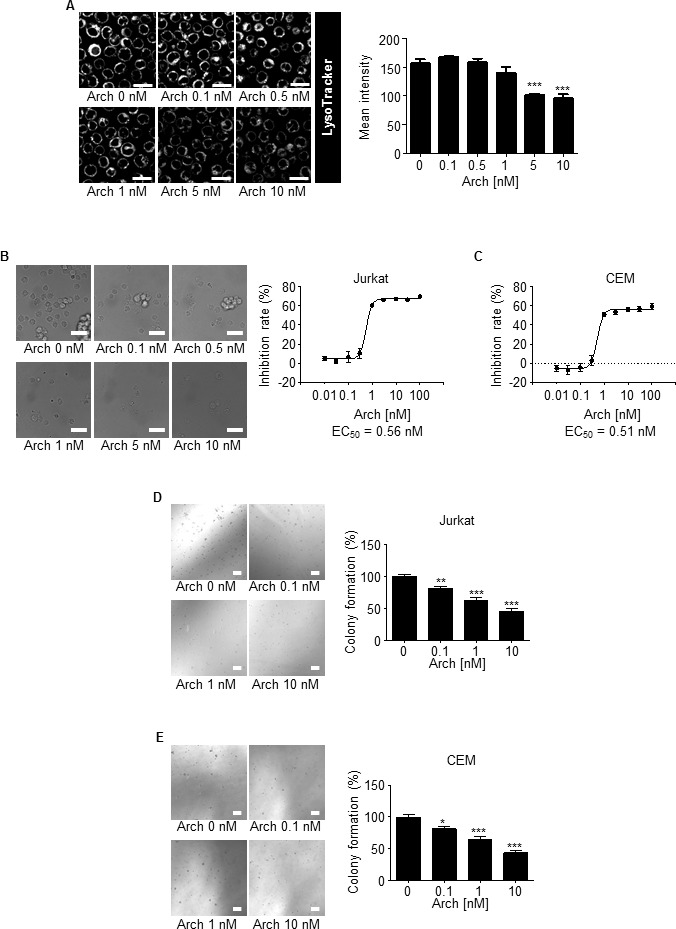
A. Archazolid A inhibits lysosome acidification. Stainings of Jurkat cells treated with Archazolid A (Arch, 0, 0.1, 0.5, 1, 5, 10 nM, 24h) with the pH-sensitive LysoTracker are shown. n = 3. Scale bar 20 μm. Quantification of LysoTracker staining is displayed (***p ≤ 0.001, One-Way ANOVA, Tukey, n = 3). B., C. Archazolid A inhibits the proliferation of leukemic cells. Inhibition rates of proliferation of Jurkat B. and CEM cells C. after treatments with Archazolid A (Arch) at indicated concentrations for 72h are shown. EC50 is indicated. n = 3. Scale bar 50 μm. D., E. Archazolid A inhibits clonogenic growth. Colony formation of Jurkat D. and CEM cells E. after treatments with Archazolid A (Arch) at indicated concentrations is shown. Scale bar 100 μm. One-Way ANOVA, Tukey's post test, *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, n = 3.
Moreover, as shown by Nicoletti assay (Figure 3A, 3B) and Annexin V staining (Figure 3C), Archazolid A potently induced death of leukemic cell lines. In line with a previous report from our group [15], Archazolid A induced cleavage of procaspase-3, procaspase-9, and PARP, increased the pro-apoptotic protein BNIP3, and decreased the anti-apoptotic protein Bcl-XL in leukemic cells (Figure 3D). Moreover, the pan-caspase inhibitor QVC-OPh partially rescued Archazolid A induced apoptosis (Figure 3E). This suggests that apoptosis by Archazolid A is at least partially mediated via the mitochondrial pathway.
Figure 3. Archazolid A induces death of leukemic cell lines.
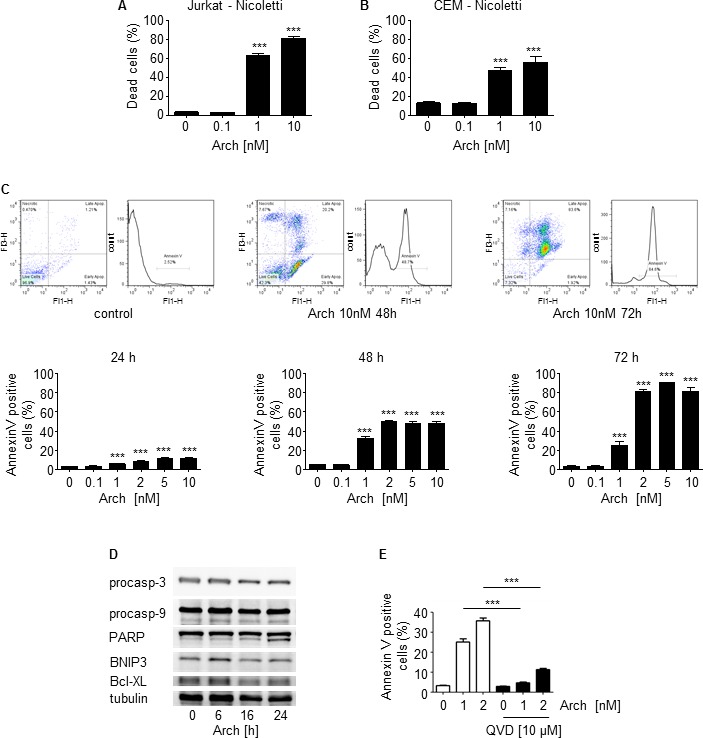
A., B. Apoptosis rate determined by Nicoletti assay of Jurkat A. and CEM cells B. after treatments with Archazolid A (Arch) at indicated concentrations for 72h is shown. One-Way ANOVA, Tukey's post test, ***p ≤ 0.001, n = 3. C. Pictures display Annexin V staining of Jurkat cells after treatments with Archazolid A (Arch). Bar graphs show the apoptosis rate determined by Annexin V staining of Jurkat cells after treatments with Archazolid A (Arch) at indicated concentrations for 24h, 48h, and 72h. One-Way ANOVA, Tukey's post test, ***p ≤ 0.001, n = 3. D. Immunoblots of Jurkat cells treated with Archazolid A (10 nM) for the indicated times for procaspase-3 (procasp-3), procaspase-9 (procasp-9), PARP, BNIP3, and Bcl-XL are shown. The immunoblot for tubulin indicates equal loading. n = 3. E. Annexin V/PI staining of cells treated with Archazolid A at indicated concentrations for 48h and with/without the pan-caspase inhibitor QVD-OPh (QVD) at 10 μM for 48h is shown. One-Way ANOVA, Tukey's post test, ***p ≤ 0.001, n = 3.
V-ATPase inhibitor Archazolid A induces death of primary human leukemic cells
To analyze the potential therapeutic relevance of V-ATPase inhibition by Archazolid A, patient derived xenograft (PDX) leukemic cells were studied. Clinical characteristics are listed in Table 2. PDX cells enable repetitive in vitro testing on viable patient-derived cells, by passaging primary tumor cells in severely immuno-compromised mice. These cells resemble the primary patient cells to a very high extent [39, 40]. In accordance with cell culture experiments, Archazolid A reduced viability (Figure 4A) and induced death of PDX human leukemic cell samples from different patients which was again shown by PI exclusion assays (Figure 4B) and Annexin V staining (Figure 4C). Moreover, Archazolid A induced cleavage of procaspase-3 in PDX samples (Figure 4D). This set of data demonstrates that V-ATPase inhibition by Archazolid A exerts anti-leukemic properties. Because Archazolid A did not induce cell death of non-tumor primary human PBMCs (Figure 4E) it might represent an option for anti-leukemic treatment.
Table 2. Clinical characteristics of patients from which the PDX cells have been generated are shown.
| number | Type of Leukemia | Disease stage | sex | age | cytogenetics |
|---|---|---|---|---|---|
| ALL-169 | cALL | diagnosis | m | pediatric | unknown |
| ALL-233 | pre B ALL | diagnosis | m | pediatric | t(2;15)(p13;q15) |
| ALL-256 | cALL | unknown | pediatric | +8, t(9;22)(q34;q11) | |
| ALL-363 | pre B ALL | diagnosis | m | adult | BCR/ABL translocation |
| AML-372 | AML | relapse | m | adult | Complex, including −17 |
| AML-412 | AML | diagnosis | f | adult | CN, FLT3-ITD, NPM1+ |
| ALL-435 | pre B ALL | unknown | pediatric | t(11;19) |
Figure 4. Archazolid A induces cell death in human patient derived xenograft (PDX) samples.
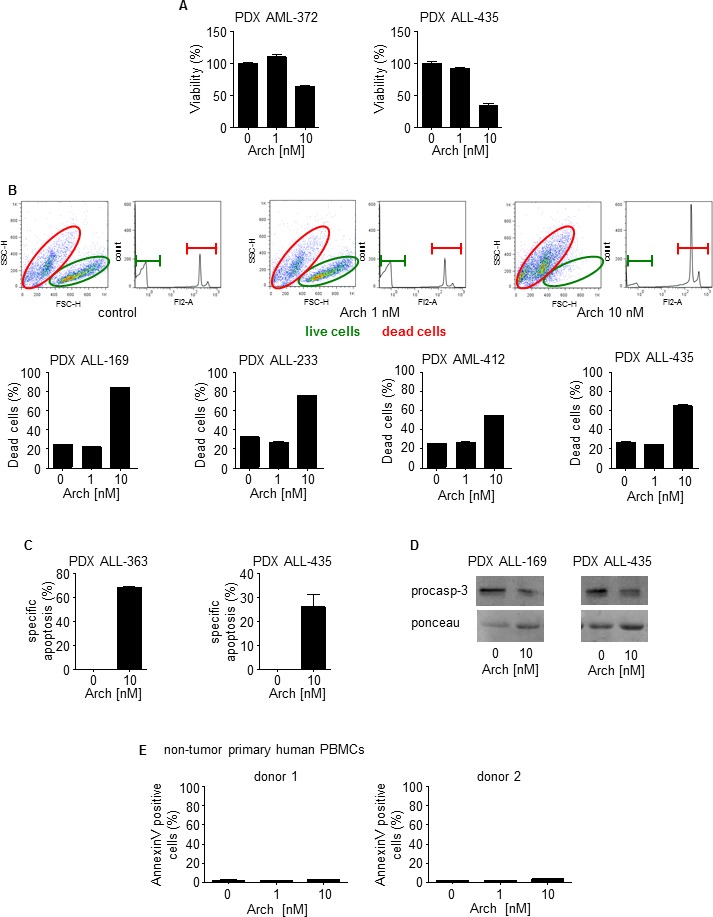
A. Viability of leukemic PDX samples with/without treatment with Archazolid A (Arch) for 72h at indicated concentrations is shown. B. PI exclusion staining of leukemic PDX samples with/without treatment with Archazolid A (Arch) for 48h at indicated concentrations is shown. Upper panels display dot plots and histograms of PDX leukemic cells from one respective patient (PDX ALL-169). Dead cells are stained by PI and are marked in red. Live cells without PI staining are displayed in green. Lower panels show apoptosis rate of PDX leukemic cells treated with Archazolid A (Arch) at indicated concentrations. C. The specific apoptosis rate determined by Annexin V/PI staining of PDX cells after treatments with Archazolid A (Arch) at indicated concentrations for 48h is shown. D. Immunoblots of PDX samples treated with Archazolid A (10 nM, 48h) for procaspase-3 (procasp-3) are shown. Ponceau staining indicates equal loading. E. Archazolid A does not induce cell death in non-tumor primary human PBMCs. Apoptosis rate determined by Annexin V/PI staining and of non-tumor primary human PBMCs (FACS analysis with gating for lymphocytes) of two different donors treated with Archazolid A (Arch) at indicated concentrations for 48h is shown.
V-ATPase inhibition by Archazolid A addresses Notch1 signaling in leukemic cells, but the Notch pathway is not responsible for Archazolid A induced cell death
In order to evaluate the mechanism of Archazolid A to induce leukemic cell death, the Notch pathway gained our attention. Expression of the Notch1 downstream target Hes1 was reduced by Archazolid A as well as by the y-secretase inhibitor (GSI) Dibenzazepine (DBZ) that served as positive control for Notch signaling inhibition (Figure 5A). Archazolid A reduced levels of Notch1 intracellular domain (NICD) and the Notch1 downstream target c-myc (Figure 5B). As expected, DBZ treatment decreased NICD and c-myc as well. NICD expression revealed that the Notch1 pathway was active in PDX samples (Figure 5C). Archazolid A-mediated decrease of NICD (Figure 5C) proved that Archazolid A addressed the Notch1 pathway in PDX cells. Archazolid A induced cell death in these PDX samples as well (Figure 4C and 10A), suggesting sensitivity of leukemic cells with Notch1 pathway activation towards Archazolid A. Moreover, Archazolid A increased levels of the Notch1 whole receptor (Figure 5B). This was further analyzed by immunostainings which demonstrate again that Archazolid A abrogated NICD levels whereas the Notch1 whole receptor was increased (Figure 5D). Stainings for the lysosomal marker LAMP1 revealed large lysosomes that contained increased amounts of Notch1 in Archazolid A treated cells (Figure 5E). This suggested that Archazolid A inhibits Notch1 signaling in a way different from GSI: by capturing the Notch1 whole receptor in lysosomes and therefore inhibiting Notch1 cleavage and NICD generation at endolysosomal membranes.
Figure 5. Archazolid A inhibits Notch1 signaling.
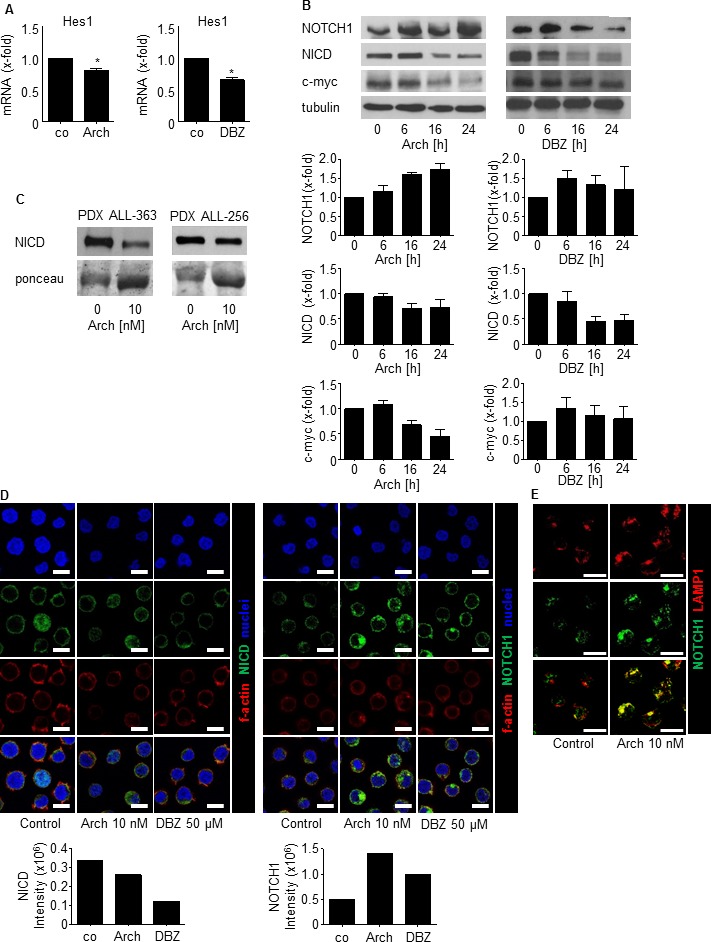
A. Hes1 mRNA expression of Jurkat cells treated with Archazolid A (Arch, 10 nM, 24h) or DBZ (50 μM, 24h) is shown. Archazolid A: paired t-test, *p = 0.0341, n = 3. DBZ: paired t-test, *p = 0.0090, n = 3. B. Immunoblots from Jurkat cells treated with Archazolid A (Arch, 10 nM, left panel) or DBZ (10 μM, right panel) for the indicated times and probed with antibodies for Notch, NICD, and c-myc are shown. Immunoblots for β-tubulin indicate equal loading. Bar graphs display quantitative evaluations of immunoblots for Notch1, NICD, and c-myc. n = 3. C. Immunoblots from PDX cells treated with Archazolid A (10 nM, 24h) and probed with antibodies for NICD are shown. Ponceau staining is used as loading control. D. Immunostainings from Jurkat cells treated with Archazolid A (Arch, 10 nM, 24h) or DBZ (50 μM, 24h) for NICD (green, left panels) and Notch1 (green, right panels) are shown. n = 3. Scale bar 10 μm. Bar graphs display quantitative evaluations of NICD and Notch1 intensities. E. Immunostainings from Jurkat cells treated with Archazolid A (Arch, 10 nM, 24h) for LAMP1 (red) and Notch1 (green) are shown. Merged pictures indicate colocalization of LAMP1 and Notch1 (yellow). n = 3. Scale bar 10 μm.
Figure 10. Archazolid A-induced apoptosis in PDX is in line with decreased levels of the anti-apoptotic protein survivin.
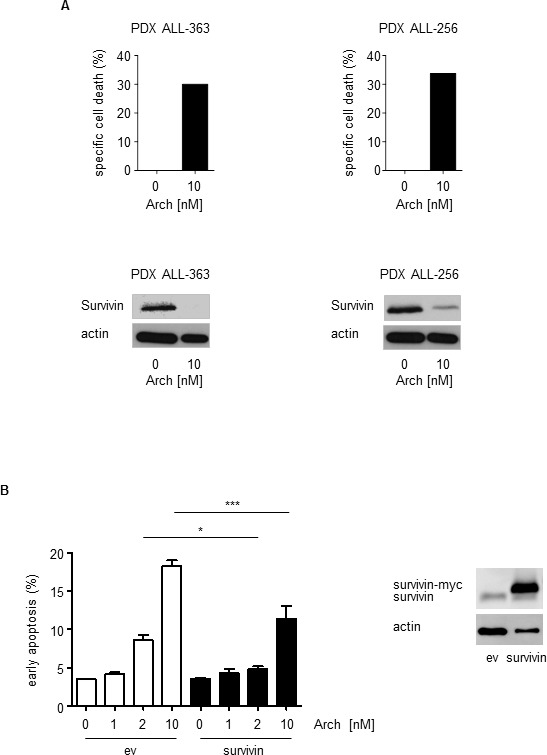
A. Upper panels show apoptosis rates (specific cell death) determined by PI exclusion staining of PDX leukemia samples treated with Archazolid A (Arch, 10 nM, 48 h). Lower panels display immunoblots from PDX cells from the same patients treated with Archazolid A (Arch, 10 nM, 24 h) and probed with antibodies for survivin. Immunoblots for actin indicate equal loading. B. Archazolid A mediated cell death is partially rescued by survivin overexpression. The graph shows early apoptosis (AnnexinV-positive and PI-negative cells) determined by AnnexinV/PI staining of Jurkat cells overexpressing either empty vector (ev) or survivin and treated with/without Archazolid A (Arch) at indicated concentrations for 48 h. One-Way ANOVA, Tukey, *p ≤ 0.05, ***p ≤ 0.001, n = 3. Immunoblots show overexpression of empty vector (ev) and survivin 24h after transfection; actin indicates equal loading.
However, NICD overexpression could not rescue from Archazolid A-mediated cell death (Figure 6A), suggesting no causal relationship between the Notch1 pathway and Archazolid A-induced leukemic cell death. Nevertheless, NICD overexpression induced Notch1 downstream target gene expression (Figure 6B) and rescued DBZ-induced inhibition of proliferation (Figure 6C), proving that NICD overexpression was functional. In order to better understand the inter-connection between V-ATPase and Notch1, we analyzed whether Notch inhibition affects levels of V-ATPase c-subunit. V-ATPase c-subunit expression was not affected by DBZ treatment (Figure 6D). This set of data indicated that Notch1 signaling inhibition is not the major relevant downstream signaling of Archazolid A in leukemic cell death induction.
Figure 6. NICD cannot rescue Archazolid A mediated induction of apoptosis.
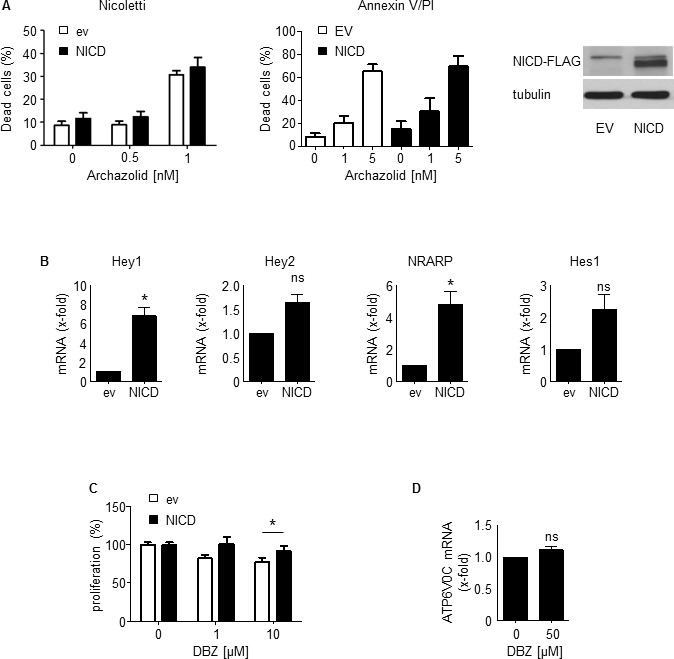
A. Apoptosis rate determined by Nicoletti assay (left bar graph) and by Annexin V/PI staining (right bar graph) of Jurkat cells overexpressing either empty vector or NICD after treatment with Archazolid A (48h) is shown. Immunoblots show NICD overexpression. n = 3. B. Increased expression of Notch downstream targets Hey1 (paired t-test, *p = 0.0197), Hey2 (paired t-test, p = 0.0576), NRARP (paired t-test, *p = 0.0407), Hes1 (paired t-test, p = 0.1117) after NICD overexpression (24h) is shown. n = 3. C. Proliferation of Jurkat cells overexpressing either empty vector or NICD after treatment with DBZ at indicated concentrations for 72h is shown. t-test, *p = 0.0209, n = 3. D. Expression of V-ATPase subunit c (ATP6V0C) of Jurkat cells treated with DBZ (50 μM, 24h) is shown. Non-significant (ns), paired t-test, p = 0.1886.
Inhibition of y-secretase impairs growth but does not induce death of leukemic cell lines
DBZ-mediated γ-secretase inhibition reduced proliferation of leukemic cell lines Jurkat (EC50 15.5 μM) and CEM (EC50 12.7 μM) (Figure 7A, 7B) as well as clonogenic growth (Figure 7C, 7D). However, DBZ neither induced death of leukemic cell lines (Figure 7E-7G), nor of human leukemic PDX samples (Figure 7H). This set of data suggests that the Notch1 pathway is not essential for the effects of Archazolid A on leukemic cell death.
Figure 7. y-secretase inhibition by DBZ inhibits growth but does not induce leukemic cell death.
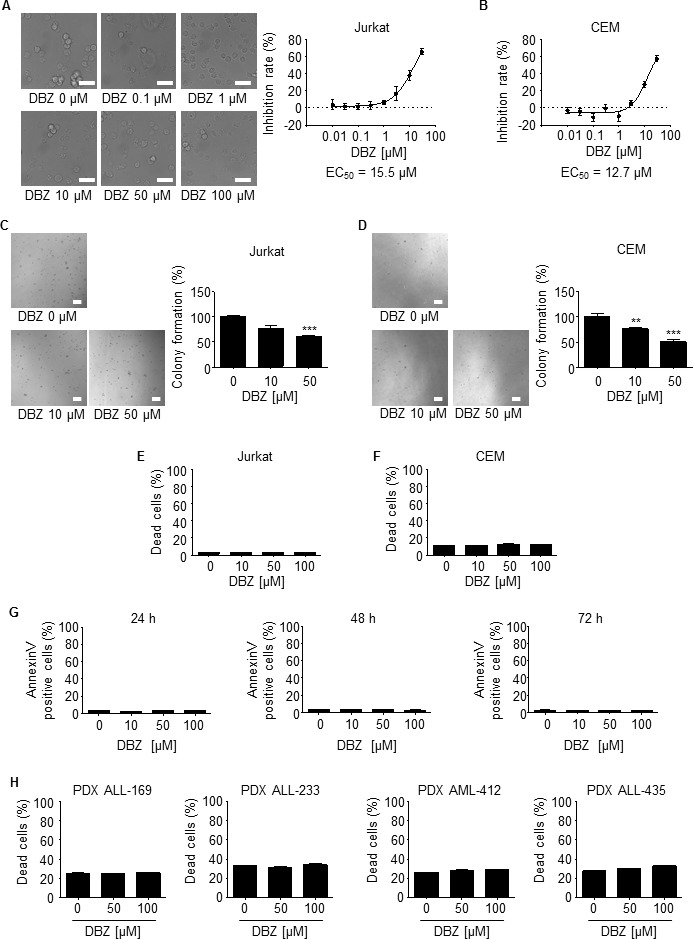
A., B. DBZ inhibits proliferation of leukemic cells. Inhibition rates of proliferation of Jurkat A. and CEM cells B. after treatment with DBZ for 72h at indicated concentrations are shown. EC50 is indicated. n = 3. Scale bar 50 μm. C., D. Colony formation of Jurkat C. and CEM cells D. after treatments with DBZ at indicated concentrations is shown. Scale bar 100 μm. One-Way ANOVA, Tukey, **p ≤ 0.01, ***p ≤ 0.001, n = 3. E., F. Apoptosis rate determined by Nicoletti assay of Jurkat E. and CEM cells F. after treatments with DBZ at indicated concentrations for 72h is shown. n = 3. G. Apoptosis rate determined by Annexin V/PI staining of Jurkat cells after treatments with DBZ at indicated concentrations for 24h, 48h, and 72h is shown. 24h and 48h: n = 2. 72h: n = 3. H. Apoptosis rate determined by PI exclusion of PDX leukemic cells with/without treatments with DBZ (48h) at indicated concentrations is shown.
Archazolid A decreases survivin by inducing cell cycle arrest and interfering with the iron metabolism in leukemic cells
In search for other pathways of Archazolid A responsible for cell death, we analyzed the anti-apoptotic protein survivin, a crucial regulator of cell death in leukemic cells [41-43]. In fact, Archazolid A decreased survivin protein levels (Figure 8A, 8B and Supplementary Figure 1) which was not due to changed survivin mRNA (Figure 8C). In contrast, DBZ showed no significant effect on survivin (Figure 8A-8C), again providing evidence that Archazolid A-mediated anti-leukemic effects are not based on Notch1 inhibition. Because survivin expression strongly depends on the cell cycle [41-43], subsequently, cell cycle analysis was performed. In fact, cells without Archazolid A treatment pass S-phase (8h) to reach (16h) and accumulate (20h, 24h) in G2-phase. In contrast, Archazolid A treated cells accumulate in S-phase (8h, 16h, 20h) but do not traverse to G2-phase. This suggests that Archazolid A treated cells die during S-phase which is confirmed by cell death analysis showing that Archazolid A-induced S-phase arrest is in parallel with apoptosis induction (Figure 8D).
Figure 8. Archazolid A decreases the anti-apoptotic protein survivin and interferes with the cell cycle in leukemic cells.
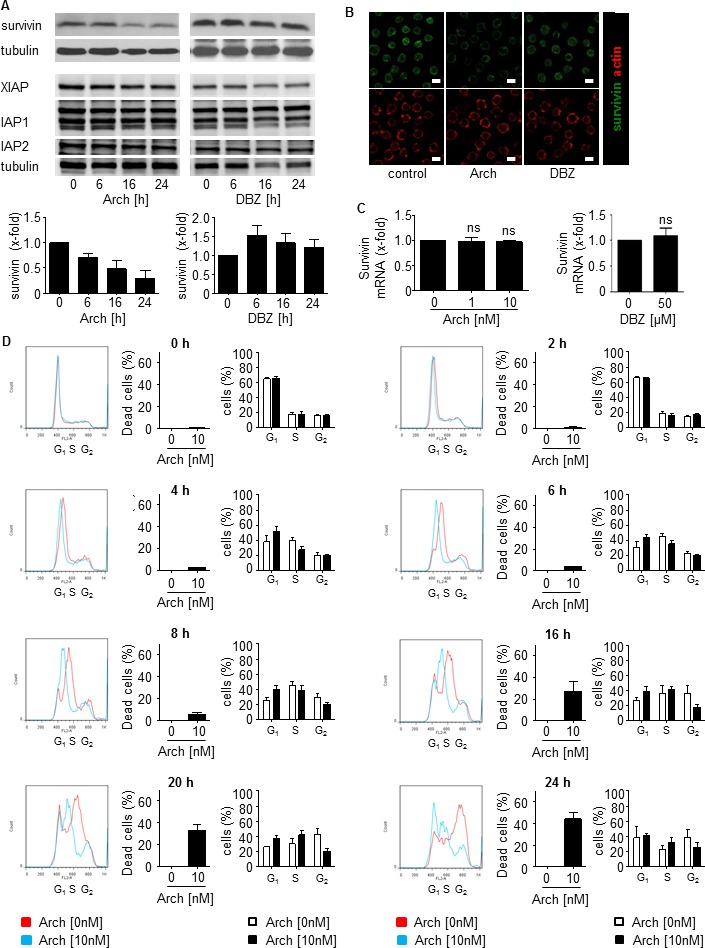
A. Archazolid A decreases the anti-apoptotic protein survivin. Immunoblots from Jurkat cells treated with Archazolid A (Arch, 10 nM, left panel) or DBZ (50 μM, right panel) for the indicated times and probed with antibodies for survivin, XIAP, IAP1, and IAP2 are shown. Immunoblots for tubulin indicate equal loading. n = 3. Bar graphs display the quantitative evaluation of survivin expression. B. Immunostainings for survivin (green) and actin (red) after treatment with/without Archazolid A (Arch, 10 nM, 24h) and DBZ (50 μM, 24h) is shown. Scale bar 10 μm. C. Archazolid A (Arch) and DBZ do not interfere with survivin mRNA expression. Survivin mRNA levels from Jurkat cells treated with Archazolid A (1 and 10 nM) and DBZ (50 μM) for 24h are shown. Not significant (ns), Archazolid A: One-Way ANOVA, DBZ: paired t-test, n = 3. D. Archazolid A (Arch) induces S-phase cell cycle arrest of Jurkat cells. Cell cyle analysis and apoptosis measurement after aphidicolin synchronization (24h) and subsequent treatment with Archazolid A for indicated times is shown. Control cells (untreated, Archazolid A 0 nM) are indicated in red, Archazolid A (Arch, 10 nM) treated cells are indicated in blue. One representative out of three independent experiments is shown.
In line with recent studies from our group [15, 44] that elucidated that interference of Archazolid with the iron metabolism leads to S-phase cell cycle arrest in breast cancer, Archazolid A stabilized Hif1α in leukemic cells (Figure 9A-9C) which was abrogated by simultaneous treatment with iron citrate (Figure 9C). In contrast, Notch inhibition by DBZ did not affect Hif1α (Figure 9D). As shown previously [44], Archazolid A-mediated cell death was partially rescued by iron citrate (Figure 9E). Because Hif1α stabilization is generally pro-proliferative, but also anti-proliferative and cell death inducing properties have been described [45-48], we analyzed the effects of Hif1α induction on cell death and survivin expression in leukemic cells. Induction of Hif1α by deferoxamine (DFO) induced leukemic cell death and decreased survivin, which was even enhanced by concomitant Archazolid A treatment (Figure 9F, 9G). This set of data suggests that interference of Archazolid A with the iron metabolism contributes to cell death in leukemic cells.
Figure 9. Archazolid A interferes with the iron metabolism in leukemic cells.
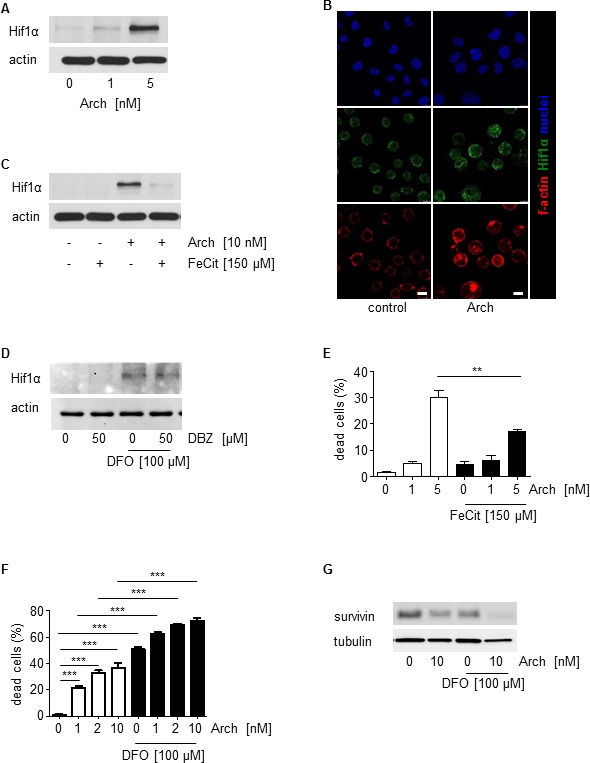
A. Archazolid A increases Hif1α. Immunoblots show Hif1α levels of Jurkat cells with/without Archazolid A (Arch) treatment at indicated concentrations for 24h. Actin indicates equal loading. B. Immunostainings for Hif1α (green) and f-actin (red) after treatment with/without Archazolid A (Arch, 10 nM, 24h) is shown. Nuclei are labeled with Hoechst33342 (blue). Scale bar 7.5 μm. C. Archazolid A mediated Hif1α increase is abrogated by iron citrate. Immunoblots show Hif1α levels of Jurkat cells with/without Archazolid A (Arch) and iron citrate (FeCit) treatment at indicated concentrations for 24h. Actin indicates equal loading. n = 3. D. Inhibition of Notch by DBZ does not influence Hif1α. Immunoblots of Jurkat cells treated with DBZ and deferoxamine (DFO) at indicated concentrations for 24h for Hif1α and actin (loading control) are shown. E. Archazolid A mediated cell death is partially rescued by iron citrate. The graph shows cell death of Jurkat cells treated with/without Archazolid A (Arch) and iron citrate (FeCit) at indicated concentrations for 48 h. Mann Whitney test, **p = 0.0022, n = 3. F. DFO induces cell death in Jurkat cells and is enhanced by Archazolid A. Nicoletti assay of cells treated with/without Archazolid A (Arch) and DFO at indicated concentrations for 48 h is shown. One-Way ANOVA, Tukey's post test, ***p ≤ 0.001, n = 3. G. Survivin is decreased by DFO which is enhanced by Archazolid A. Immunoblots for survivin and tubulin (loading control) from cells treated with/without DFO (100 μM) and Archazolid A (Arch, 10 nM) for 48h are shown; n = 3.
Finally, coincident with apoptosis induction, survivin levels were decreased by Archazolid A treatment of PDX human patient samples as shown for the PDX samples PDX ALL-363 and PDX ALL-256 (Figure 10A, Figure 4C). Moreover, survivin overexpression partially rescued Archazolid A induced cell death in leukemic cells (Figure 10B). This set of data suggests decreased survivin as relevant mechanism of Archazolid A to induce leukemic cell death.
DISCUSSION
Our study demonstrates that V-ATPase inhibition by Archazolid A exerts anti-leukemic effects, suggesting V-ATPase inhibition as potential option for T-ALL treatment.
During recent years, V-ATPase has emerged as interesting target for cancer therapy. Amongst others, including various studies of our group, it has been shown that V-ATPase is implicated in cancer cell metastasis [16, 49], invasion [50, 51], tumor cell death [15, 52], anoikis resistance [17], cellular stress response [15], breast cancer trastuzumab resistance [34], and regulation of the secretion profile of cancer and cancer-associated cells [18, 53]. Only few reports point to a function of V-ATPase in leukemia or hematopoietic cells in general. In detail, it was shown that proton pump inhibitors induce apoptosis of human B-cells [54] and lysosome disruption has been associated with anti-leukemic effects in acute myeloid leukemia (AML) [55]. Moreover, V-ATPase was suggested to regulate leukemic cell adhesion [56] and V-ATPase inhibition by Bafilomycin A reduced leukemic cell growth [35].
In search for the signaling pathway responsible for the anti-leukemic effects of Archazolid A-mediated V-ATPase inhibition, first the Notch pathway was analyzed as it is one of the most prominent oncogenic pathways in T-ALL [57]. Targeted therapies addressing the Notch pathway have been proposed as auspicious options for T-ALL treatment. Despite promising in vitro results and initial clinical effectiveness of GSIs that block Notch activatory cleavage, this benefit only in some studies translates into improved overall survival [58-60]. In fact, most human T-ALL cell lines are resistant to GSIs that fail to induce leukemic cell death [57]. Therefore, to inhibit Notch signaling with a mechanism different from GSIs might be a promising approach. Our study shows that V-ATPase reduces Notch1 signaling by capturing the Notch whole receptor in the endolysosomal compartment and inhibiting its activatory cleavage. This is in line with reports demonstrating that impaired endolysosomal function by V-ATPase inhibition blocks γ-secretase mediated Notch activatory cleavage at the endolysosomal membrane [11, 35, 37, 38]. However, our rescue experiments revealed that leukemic cell death by Archazolid A was not caused by inhibition of Notch signaling. Moreover, although our results revealed that GSI treatment reduced leukemic cell growth, cell death was not induced. Thus, Archazolid A-mediated induction of leukemic cell death was not based on Notch1 pathway inhibition.
Our work suggests the anti-apoptotic protein survivin as target addressed by Archazolid A to induce leukemic cell death. Survivin exerts multiple cellular functions: it participates in the regulation of cell division, apoptosis, stress response, migration, and metastasis. Whereas it is nearly absent in normal tissues, it is overexpressed in most human tumors, including hematopoietic malignancies [42, 43, 61-63]. Survivin expression has been associated with leukemia progression and poor clinical outcome [64-66]. In consequence, survivin is addressed in cancer therapy by molecular antagonists like antisense oligonucleotides, siRNA, or hammerhead ribozymes, as well as small molecules or cancer immunotherapeutics [67]. Inhibition of survivin has shown clinical benefits and chemosensitizing effects in leukemia [42, 43, 68-70]. By describing potent anti-leukemic effects of Archazolid A, our study probably expands the class of small molecule survivin antagonists.
Survivin expression strongly depends on the cell cycle, i.e. survivin is upregulated and stabilized during G2-phase [41]. Besides regulation at the transcriptional level, changes in protein stability essentially contribute to survivin expression during the cell cycle: proteasome-dependent destruction of survivin has been shown in interphase cells whereas at metaphase, mitotic phosphorylation of survivin by Cdk1 has been associated with increased protein stability [71]. In fact, in line with recent studies from our group addressing V-ATPase in cancer [15, 44], our results presented here suggest that Archazolid decreased survivin at the protein level by inducing S-phase cell cycle arrest in leukemic cells which probably was due to its interference with the iron metabolism.
In line with previous studies from our group showing that Archazolid B led to cellular stress response and induction of Hif1α [15, 44], our present study suggests that the induction of Hif1α contributes to Archazolid A induced leukemic cell death. Although the stabilization of Hif1α during hypoxia is generally pro-proliferative as stabilization of Hif1α influences the survival of tumor cells, Hif1α has also tumor-inhibiting properties. In detail, Hif1α overexpression inhibits cell proliferation by expressing cell cycle inhibitors like p53, p21, and p27 and promotes apoptosis by inducing pro-apoptotic molecules such as p53, Nip3, and Noxa [45-48].
A recent study from our group demonstrated that inhibition of V-ATPase by Archazolid B led to iron depletion by disruption of transferrin receptor recycling which subsequently reduced ribonucleotide reductase (RNR) activity, induced S-phase cell cycle arrest and finally caused cell death [44]. Interestingly, iron overload due to inefficient erythropoiesis or blood transfusion represents a major problem in patients suffering from myelodysplastic syndromes (MDS), clonal disorders with ineffective hematopoiesis and an increased risk of transformation into acute myeloid leukemia. Iron overload is associated with osteopenia and osteoporosis in these patients [72, 73]. In consequence, V-ATPase inhibition in hematopoietic diseases might be an interesting object for further studies.
To conclude, our study provides evidence for V-ATPase inhibition as new alternative strategy and Archazolid A as interesting new compound for T-ALL therapy.
MATERIALS AND METHODS
Cells
Cell lines: Human leukemia Jurkat T cells (J16, S-Jurkat) were kindly provided by P.H. Krammer and H. Walczak, Heidelberg, Germany. S-Jurkat cells were cultured in RPMI 1640 (PAN Biotech, Aidenbach, Germany) containing 10% FCS (PAA Laboratories, Cölbe, Germany) and 1% pyruvate (Merck, Darmstadt, Germany). CCRF-CEM cells were kindly provided by Dr. Joachim Arend (Mainz, Germany) and cultured in RPMI 1640 containing 10% FCS.
Reagents
The γ-secretase inhibitor DBZ was purchased from Merck Millipore. The pan-caspase inhibitor QVD-OPh (551476) was purchased from Calbiochem. Deferoxamine (DFO) was purchased from Sigma.
Patient-derived xenograft (PDX) leukemic cells
The xenograft mouse model, the transplantation of patient's primary tumor cells, and the cell amplification have been described previously [39, 40]. In detail, primary leukemic cells were obtained from diagnostic bone marrow aspiration or peripheral blood sampling before onset of treatment. Xenografts were established by injecting 107 fresh or frozen/thawed primary leukemic cells into NSG mice (NOD SCID gamma mice, i.e. mice with dysfunctional gamma chain of the IL-2R receptor) by tail vein injection. Development of leukemia was monitored by repetitive blood sampling in mice and by determining human leukemic cells in flow cytometry upon staining of murine CD45 and human CD38. At clear leukemic engraftment or by latest after 25 weeks, mice were sacrificed and human patient-derived xenograft (PDX) leukemic cells were isolated either from bone marrow or from spleens of mice using Ficoll purification. Phenotypic markers and genetic aberrations in PDX cells compared to primary leukemic tumor cells remained mainly stable as shown before [25]. For stimulation and during experiments, cells were maintained in RPMI 1640 medium containing 20% FCS and 1% glutamine. Freshly isolated PDX leukemic cells were stimulated as indicated in vitro.
Primary non-tumor human peripheral blood mononuclear cells (PBMCs)
Isolation of primary non-tumor human PBMCs was performed using the “Ficoll-Paque PLUS” kit from GE Healthcare according to the manufacturer's instructions. Briefly, EDTA (1.5mg/ml) blood (2 ml) was mixed with balanced salt solution (2 ml), added on top of Ficoll-Paque PLUS (3ml) in a 15ml centrifuge tube, and centrifuged (400×g, 40min, RT). Afterwards, PBMCs were carefully withdrawn from the interface between the upper plasma layer and the lower Ficoll-Paque PLUS layer, washed twice by 3 volumes of balanced salt solution, centifuged (100×g, 10 min, RT) and resuspended in culture medium (RPMI 1640+20%FCS+1%Glutamine). PBMCs were directly used for the respective assays.
For FACS analysis, gating of lymphocytes was performed and only these cells were included in the evaluation.
Tansfection of cells
Cell transfection was performed by using the Amaxxa system with the cell line nucleofector kit V (Lonza, VCA-1003) according to the manufacturer's instructions. 1×106 cells were electroporated (program X-001). NICD plasmid was from addgene (20183). Survivin plasmid was from Sino Biologicals (pCMV3-BIRC5-myc, HG10356-CM G09AU4M62). For respective assays, cells were used 24h (NICD) or 48h (survivin) after transfection.
Proliferation
CellTiter-Blue® assay was performed according manufacturer's instructions. Briefly, cells were seeded (96 well plates, 3×103 cells/well) for 24 h, treated with Archazolid A or DBZ at indicated concentrations and incubated for 70 h. CTB solution (20μl) was added and cells were incubated for 2h. Fluorescence was measured with a Tecan reader (Maennedorf, Austria).
PDX viability
PDX leukemic cells were seeded (96 well plates, 1×105 cells/well) and the cells were treated with Archazolid A at indicated concentrations for 72h. Cell Titer Blue assay was performed according manufacturer's instructions as described before at “proliferation”. For calculating viability, the value from the time of plating was subtracted.
Apoptosis and cell cycle
Apoptosis and cell cycle analysis were performed according to the Nicoletti method [74]. In brief, cells were seeded (24 well plates, 0.5×104 cells/well for 48 h, 2.5×104 cells/well for 72 h) and treated with Archazolid A or DBZ at indicated concentrations. For DNA staining, cells were permeabilized and stained by resuspension in hypotonic fluorochrome solution (HFS) containing propidium iodide (PI, 50 μg/ml) and incubated at 4°C overnight. Subsequently, flow cytometry was performed on a FACSCalibur (Becton Dickinson, Heidelberg, Germany). The sub-G1 peak accounts for apoptotic cells and was determined according to Nicoletti [74]. Cell cycle was analyzed by using FlowJo 7.6 (Tree Star Unc., Ashland, USA).
PI exclusion
Cells were seeded (96 well plates, 1×105 cells/well and treated with Archazolid A or DBZ at indicated concentrations. Subsequently, cells were incubated with PI (5 μg/ml, 5 min) that was added to the cell suspension before analysis by flow cytometry. Subsequently, flow cytometry was performed on a FACSCalibur (Becton Dickinson, Heidelberg, Germany) using the Fl-2 laser.
As cells were not permeabilized, PI staining was exclusive for dead cells, whereas viable cells were not stained by PI. For quantification, PI positive dead cells can be seen in dot plots at the left and in histograms at the right and are shown in red; PI negative viable cells can be seen in dot plots at the right and in histograms at the left and are shown in green (Figure 4A).
Annexin V/PI staining
Annexin V/PI staining was performed by using the Annexin V-FITC Apoptosis Detection Kit (eBioscience Dx, BMS500FI/300CE) according to the manufacturer's protocol. Briefly, cells were treated as indicated, collected, centrifuged (600g, 4°C, 10min), washed with PBS, centrifuged again (600g, 4°C, 10min) and resuspended in binding buffer. 5μl AnnexinV-FITC were added to 195μl of cell suspension, cells were mixed and incubated at RT for 10 min (cover from light), centrifuged (600g, 4°C, 10min), washed with 200μl Binding Buffer and resuspended in Binding Buffer. 10μl PI (20μg/ml) was added to 190 μl of cell suspension and FACS analysis was performed using Fl-2 and Fl-3 lasers. AnnexinV positive cells have been considered as apoptotic. AnnexinV positive and PI negative cells have been considered as early apoptotic.
For primary PDX leukemic cells, the specific apoptosis was calculated according to the following formula: % specific apoptosis = 100 × (% total apoptosis of treated cells - % total apoptosis of untreated cells) / (100 - % total apoptosis untreated cells) [75].
For primary non-tumor human PBMCs, gating of lymphocytes was performed and only these cells were included in the evaluation.
Colony formation
Cells were seeded (6 well plates, 5×105 cells/well) and treated with Archazolid A or DBZ at indicated concentrations for 24h. Cells were freshly plated (96 well plates, 5×103 cells/well) in RPMI 1640 Medium containing 40% FCS, 0.52% Methylcellulose, and 1% Sodium Pyruvate and incubated for 11 d. Images from each well were taken with a Zeiss 510 Meta Confocal Microscope. Colonies were counted by using Image J with the cell counter plugin.
Immunoblotting
Immunoblotting was performed as described [76]. The following primary antibodies were used: actin (MAB1501 Millipore), BCL-XL (2762 Cell Signaling), BNIP3 (ab10433 Abcam), Caspase 3 (sc-7148 Santa Cruz), Caspase 9 (9506 Cell Signaling), Hif1α (610958 BD), IAP-1 (4952 Cell Signaling), IAP-2 (3130 Cell Signaling), NICD (4147 Cell Signaling), Notch1 (4380 Cell Signaling), c-Myc (sc-788 Santa Cruz), survivin (2803 Cell Signaling), β-tubulin (2164 Cell Signaling), XIAP (610717 BD).
RT-PCR
mRNA was isolated using the Qiagen RNeasy Mini Kit. For reverse transcription the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems) was used. RT-PCR was performed with the 7300 Real Time PCR System.
For RT-PCR of Notch downstream targets, the following Taqman gene expression assays were used: Hes1 Hs00172878, HEY1 Hs00232618, HEY2 Hs00232622, NRARP Hs01104102 (Life Technologies Corporation, Carlsbad, CA, USA). GAPDH was used as housekeeper.
For RT-PCR for V-ATPase subunits, SYBR® Green PCR Master Mix (4309155, Life Technologies) was used. A list with the primer sequences for all V-ATPase subunits is included in Supplementary Table 1.
Confocal microscopy
For Immunostaining with antibodies, cells were treated as indicated, collected, resuspended and seeded on chrome alum-gelatin (0.05% CrK(SO4)2·12H2O, 0.4% gelatine in H2O) coated coverslips (30 min). Cells were fixed (4% PFA, 10mins), permablized (0.2% Triton X 100 in PBS, 5min), blocked (0.2% BSA in PBS, 1 h), incubated with primary antibodies (0.2% BSA in PBS, 1h) and subsequently secondary antibodies (0.2% BSA in PBS, 1h) and mounted. The following primary antibodies were used: EEA1 (sc-6415, Santa Cruz), LAMP1 (H4A3, Developmental Studies Hybridoma Bank), NICD (4147 Cell Signalling), Notch1 (4308, Cell Signaling). Alexa Fluor conjugated secondary antibodies (Molecular Probes) were used.
For LysoTracker experiment, cells were treated as indicated, collected, and stained with LysoTracker (Molecular Probes) for 45min. After Hoechst33342 staining (5 μg/ml, 5min), confocal microscopy was performed with a Zeiss LSM 510 META confocal microscope.
For the evaluation of the size of the endolysosomal compartment, EEA1 staining was performed. The size of the endolysosomal compartment was analyzed by calculating the EEA1-positive area per cell by using ImageJ. Approximately 20 cells per cell line were randomly selected for the evaluation.
The quantification of the intensities of Notch1 and NICD staining was performed by using Image J. The total intensity of the whole image was divided by the number of cells. Cells on the border of the images were excluded.
Statistic evaluation
All experiments were performed at least 3 times in duplicates/triplicates. Results are expressed as mean value ± SEM. One-way ANOVA/Tukey and individual t-tests were conducted using GraphPad Prism (version 5.04, GraphPad Software, Inc.). P values less than 0.05 were considered as significant.
SUPPLEMENTARY MATERIAL FIGURE AND TABLE
Acknowledgments
The authors thank Kerstin Loske and Jana Peliskova for their help with the experiments.
Footnotes
FUNDING
S.Z. was supported by the Chinese Scholarship Council (CSC). A.M.V. received financial support by the German Research Foundation (DFG, FOR 1406, Vo 376-14/15)
CONFLICTS OF INTEREST
The authors declare no conflict of interest.
REFERENCES
- 1.Van Vlierberghe P, Ferrando A. The molecular basis of T cell acute lymphoblastic leukemia. J Clin Invest. 2012;122:3398–3406. doi: 10.1172/JCI61269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Koch U, Radtke F. Notch in T-ALL: new players in a complex disease. Trends Immunol. 2011;32:434–442. doi: 10.1016/j.it.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 3.Pancewicz J, Nicot C. Current views on the role of Notch signaling and the pathogenesis of human leukemia. BMC Cancer. 2011;11:502. doi: 10.1186/1471-2407-11-502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.De Falco F, Sabatini R, Del Papa B, Falzetti F, Di Ianni M, Sportoletti P, Baldoni S, Screpanti I, Marconi P, Rosati E. Notch signaling sustains the expression of Mcl-1 and the activity of eIF4E to promote cell survival in CLL. Oncotarget. 2015;6:16559–16572. doi: 10.18632/oncotarget.4116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosati E, Sabatini R, Rampino G, Tabilio A, Di Ianni M, Fettucciari K, Bartoli A, Coaccioli S, Screpanti I, Marconi P. Constitutively activated Notch signaling is involved in survival and apoptosis resistance of B-CLL cells. Blood. 2009;113:856–865. doi: 10.1182/blood-2008-02-139725. [DOI] [PubMed] [Google Scholar]
- 6.Rosati E, Sabatini R, De Falco F, Del Papa B, Falzetti F, Di Ianni M, Cavalli L, Fettucciari K, Bartoli A, Screpanti I, Marconi P. gamma-Secretase inhibitor I induces apoptosis in chronic lymphocytic leukemia cells by proteasome inhibition, endoplasmic reticulum stress increase and notch down-regulation. Int J Cancer. 2013;132:1940–1953. doi: 10.1002/ijc.27863. [DOI] [PubMed] [Google Scholar]
- 7.Tian C, Yu Y, Jia Y, Zhu L, Zhang Y. HES1 activation suppresses proliferation of leukemia cells in acute myeloid leukemia. Annals of hematology. 2015;94:1477–83. doi: 10.1007/s00277-015-2413-0. [DOI] [PubMed] [Google Scholar]
- 8.Kannan S, Sutphin RM, Hall MG, Golfman LS, Fang W, Nolo RM, Akers LJ, Hammitt RA, McMurray JS, Kornblau SM, Melnick AM, Figueroa ME, Zweidler-McKay PA. Notch activation inhibits AML growth and survival: a potential therapeutic approach. J Exp Med. 2013;210:321–337. doi: 10.1084/jem.20121527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kato T, Sakata-Yanagimoto M, Nishikii H, Ueno M, Miyake Y, Yokoyama Y, Asabe Y, Kamada Y, Muto H, Obara N, Suzukawa K, Hasegawa Y, Kitabayashi I, Uchida K, Hirao A, Yagita H, et al. Hes1 suppresses acute myeloid leukemia development through FLT3 repression. Leukemia. 2015;29:576–585. doi: 10.1038/leu.2014.281. [DOI] [PubMed] [Google Scholar]
- 10.Vaccari T, Lu H, Kanwar R, Fortini ME, Bilder D. Endosomal entry regulates Notch receptor activation in Drosophila melanogaster. J Cell Biol. 2008;180:755–762. doi: 10.1083/jcb.200708127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vaccari T, Duchi S, Cortese K, Tacchetti C, Bilder D. The vacuolar ATPase is required for physiological as well as pathological activation of the Notch receptor. Development. 2010;137:1825–1832. doi: 10.1242/dev.045484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nishi T, Forgac M. The vacuolar (H+)-ATPases—nature's most versatile proton pumps. Nat Rev Mol Cell Biol. 2002;3:94–103. doi: 10.1038/nrm729. [DOI] [PubMed] [Google Scholar]
- 13.Forgac M. Vacuolar ATPases: rotary proton pumps in physiology and pathophysiology. Nat Rev Mol Cell Biol. 2007;8:917–929. doi: 10.1038/nrm2272. [DOI] [PubMed] [Google Scholar]
- 14.Cotter K, Capecci J, Sennoune S, Huss M, Maier M, Martinez-Zaguilan R, Forgac M. Activity of plasma membrane V-ATPases is critical for the invasion of MDA-MB231 breast cancer cells. J Biol Chem. 2015;290:3680–3692. doi: 10.1074/jbc.M114.611210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.von Schwarzenberg K, Wiedmann RM, Oak P, Schulz S, Zischka H, Wanner G, Efferth T, Trauner D, Vollmar AM. Mode of cell death induction by pharmacological vacuolar H+-ATPase (V-ATPase) inhibition. J Biol Chem. 2013;288:1385–1396. doi: 10.1074/jbc.M112.412007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wiedmann RM, von Schwarzenberg K, Palamidessi A, Schreiner L, Kubisch R, Liebl J, Schempp C, Trauner D, Vereb G, Zahler S, Wagner E, Muller R, Scita G, Vollmar AM. The V-ATPase-inhibitor archazolid abrogates tumor metastasis via inhibition of endocytic activation of the Rho-GTPase Rac1. Cancer Res. 2012;72:5976–5987. doi: 10.1158/0008-5472.CAN-12-1772. [DOI] [PubMed] [Google Scholar]
- 17.Schempp CM, von Schwarzenberg K, Schreiner L, Kubisch R, Muller R, Wagner E, Vollmar AM. V-ATPase inhibition regulates anoikis resistance and metastasis of cancer cells. Mol Cancer Ther. 2014;13:926–937. doi: 10.1158/1535-7163.MCT-13-0484. [DOI] [PubMed] [Google Scholar]
- 18.Kubisch R, Frohlich T, Arnold GJ, Schreiner L, von Schwarzenberg K, Roidl A, Vollmar AM, Wagner E. V-ATPase inhibition by archazolid leads to lysosomal dysfunction resulting in impaired cathepsin B activation in vivo. Int J Cancer. 2014;134:2478–2488. doi: 10.1002/ijc.28562. [DOI] [PubMed] [Google Scholar]
- 19.Chung C, Mader CC, Schmitz JC, Atladottir J, Fitchev P, Cornwell ML, Koleske AJ, Crawford SE, Gorelick F. The vacuolar-ATPase modulates matrix metalloproteinase isoforms in human pancreatic cancer. Laboratory investigation; a journal of technical methods and pathology. 2011;91:732–743. doi: 10.1038/labinvest.2011.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nishisho T, Hata K, Nakanishi M, Morita Y, Sun-Wada GH, Wada Y, Yasui N, Yoneda T. The a3 isoform vacuolar type H(+)-ATPase promotes distant metastasis in the mouse B16 melanoma cells. Mol Cancer Res. 2011;9:845–855. doi: 10.1158/1541-7786.MCR-10-0449. [DOI] [PubMed] [Google Scholar]
- 21.Michel V, Licon-Munoz Y, Trujillo K, Bisoffi M, Parra KJ. Inhibitors of vacuolar ATPase proton pumps inhibit human prostate cancer cell invasion and prostate-specific antigen expression and secretion. Int J Cancer. 2013;132:E1–10. doi: 10.1002/ijc.27811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Avnet S, Di Pompo G, Lemma S, Salerno M, Perut F, Bonuccelli G, Granchi D, Zini N, Baldini N. V-ATPase is a candidate therapeutic target for Ewing sarcoma. Biochimica et biophysica acta. 2013;1832:1105–1116. doi: 10.1016/j.bbadis.2013.04.003. [DOI] [PubMed] [Google Scholar]
- 23.Perut F, Avnet S, Fotia C, Baglio SR, Salerno M, Hosogi S, Kusuzaki K, Baldini N. V-ATPase as an effective therapeutic target for sarcomas. Exp Cell Res. 2014;320:21–32. [PubMed] [Google Scholar]
- 24.Di Cristofori A, Ferrero S, Bertolini I, Gaudioso G, Russo MV, Berno V, Vanini M, Locatelli M, Zavanone M, Rampini P, Vaccari T, Caroli M, Vaira V. The vacuolar H+ ATPase is a novel therapeutic target for glioblastoma. Oncotarget. 2015;6:17514–31. doi: 10.18632/oncotarget.4239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hamm R, Zeino M, Frewert S, Efferth T. Up-regulation of cholesterol associated genes as novel resistance mechanism in glioblastoma cells in response to archazolid B. Toxicology and applied pharmacology. 2014;281:78–86. doi: 10.1016/j.taap.2014.08.033. [DOI] [PubMed] [Google Scholar]
- 26.Huss M, Wieczorek H. Inhibitors of V-ATPases: old and new players. J Exp Biol. 2009;212(Pt 3):341–346. doi: 10.1242/jeb.024067. [DOI] [PubMed] [Google Scholar]
- 27.Bockelmann S, Menche D, Rudolph S, Bender T, Grond S, von Zezschwitz P, Muench SP, Wieczorek H, Huss M. Archazolid A binds to the equatorial region of the c-ring of the vacuolar H+-ATPase. J Biol Chem. 2010;285:38304–38314. doi: 10.1074/jbc.M110.137539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huss M, Sasse F, Kunze B, Jansen R, Steinmetz H, Ingenhorst G, Zeeck A, Wieczorek H. Archazolid and apicularen: novel specific V-ATPase inhibitors. BMC biochemistry. 2005;6:13. doi: 10.1186/1471-2091-6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huss M, Vitavska O, Albertmelcher A, Bockelmann S, Nardmann C, Tabke K, Tiburcy F, Wieczorek H. Vacuolar H(+)-ATPases: intra- and intermolecular interactions. Eur J Cell Biol. 2011;90:688–695. doi: 10.1016/j.ejcb.2011.04.009. [DOI] [PubMed] [Google Scholar]
- 30.Sasse F, Steinmetz H, Hofle G, Reichenbach H. Archazolids, new cytotoxic macrolactones from Archangium gephyra (Myxobacteria). Production, isolation, physico-chemical and biological properties. The Journal of antibiotics. 2003;56:520–525. doi: 10.7164/antibiotics.56.520. [DOI] [PubMed] [Google Scholar]
- 31.Menche D, Hassfeld J, Li J, Rudolph S. Total synthesis of archazolid A. Journal of the American Chemical Society. 2007;129:6100–6101. doi: 10.1021/ja071461o. [DOI] [PubMed] [Google Scholar]
- 32.Roethle PA, Chen IT, Trauner D. Total synthesis of (−)-archazolid B. Journal of the American Chemical Society. 2007;129:8960–8961. doi: 10.1021/ja0733033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rath S, Liebl J, Furst R, Vollmar AM, Zahler S. Regulation of endothelial signaling and migration by v-ATPase. Angiogenesis. 2014;17:587–601. doi: 10.1007/s10456-013-9408-z. [DOI] [PubMed] [Google Scholar]
- 34.von Schwarzenberg K, Lajtos T, Simon L, Muller R, Vereb G, Vollmar AM. V-ATPase inhibition overcomes trastuzumab resistance in breast cancer. Mol Oncol. 2014;8:9–19. doi: 10.1016/j.molonc.2013.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kobia F, Duchi S, Deflorian G, Vaccari T. Pharmacologic inhibition of vacuolar H+ ATPase reduces physiologic and oncogenic Notch signaling. Mol Oncol. 2014;8:207–220. doi: 10.1016/j.molonc.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Perez-Sayans M, Somoza-Martin JM, Barros-Angueira F, Rey JM, Garcia-Garcia A. V-ATPase inhibitors and implication in cancer treatment. Cancer Treat Rev. 2009;35:707–713. doi: 10.1016/j.ctrv.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 37.Yan Y, Denef N, Schupbach T. The vacuolar proton pump, V-ATPase, is required for notch signaling and endosomal trafficking in Drosophila. Dev Cell. 2009;17:387–402. doi: 10.1016/j.devcel.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Valapala M, Hose S, Gongora C, Dong L, Wawrousek EF, Samuel Zigler J, Jr, Sinha D. Impaired endolysosomal function disrupts Notch signalling in optic nerve astrocytes. Nat Commun. 2013;4:1629. doi: 10.1038/ncomms2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Terziyska N, Castro Alves C, Groiss V, Schneider K, Farkasova K, Ogris M, Wagner E, Ehrhardt H, Brentjens RJ, zur Stadt U, Horstmann M, Quintanilla-Martinez L, Jeremias I. In vivo imaging enables high resolution preclinical trials on patients‘ leukemia cells growing in mice. PLoS One. 2012;7:e52798. doi: 10.1371/journal.pone.0052798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vick B, Rothenberg M, Sandhofer N, Carlet M, Finkenzeller C, Krupka C, Grunert M, Trumpp A, Corbacioglu S, Ebinger M, Andre MC, Hiddemann W, Schneider S, Subklewe M, Metzeler KH, Spiekermann K, et al. An advanced preclinical mouse model for acute myeloid leukemia using patients‘ cells of various genetic subgroups and in vivo bioluminescence imaging. PLoS One. 2015;10:e0120925. doi: 10.1371/journal.pone.0120925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Altieri DC. Survivin, cancer networks and pathway-directed drug discovery. Nat Rev Cancer. 2008;8:61–70. doi: 10.1038/nrc2293. [DOI] [PubMed] [Google Scholar]
- 42.Park E, Gang EJ, Hsieh YT, Schaefer P, Chae S, Klemm L, Huantes S, Loh M, Conway EM, Kang ES, Hoe Koo H, Hofmann WK, Heisterkamp N, Pelus L, Keerthivasan G, Crispino J, et al. Targeting survivin overcomes drug resistance in acute lymphoblastic leukemia. Blood. 2011;118:2191–2199. doi: 10.1182/blood-2011-04-351239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Purroy N, Abrisqueta P, Carabia J, Carpio C, Calpe E, Palacio C, Castellvi J, Crespo M, Bosch F. Targeting the proliferative and chemoresistant compartment in chronic lymphocytic leukemia by inhibiting survivin protein. Leukemia. 2014;28(10):1993–2004. doi: 10.1038/leu.2014.96. [DOI] [PubMed] [Google Scholar]
- 44.Schneider LS, von Schwarzenberg K, Lehr T, Ulrich M, Kubisch-Dohmen R, Liebl J, Trauner D, Menche D, Vollmar AM. Vacuolar-ATPase Inhibition Blocks Iron Metabolism to Mediate Therapeutic Effects in Breast Cancer. Cancer Res. 2015;75:2863–2874. doi: 10.1158/0008-5472.CAN-14-2097. [DOI] [PubMed] [Google Scholar]
- 45.Greijer AE, van der Wall E. The role of hypoxia inducible factor 1 (HIF-1) in hypoxia induced apoptosis. Journal of clinical pathology. 2004;57:1009–1014. doi: 10.1136/jcp.2003.015032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Koshiji M, Huang LE. Dynamic balancing of the dual nature of HIF-1alpha for cell survival. Cell Cycle. 2004;3:853–854. doi: 10.4161/cc.3.7.990. [DOI] [PubMed] [Google Scholar]
- 47.Sermeus A, Michiels C. Reciprocal influence of the p53 and the hypoxic pathways. Cell death & disease. 2011;2:e164. doi: 10.1038/cddis.2011.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fridman JS, Lowe SW. Control of apoptosis by p53. Oncogene. 2003;22:9030–9040. doi: 10.1038/sj.onc.1207116. [DOI] [PubMed] [Google Scholar]
- 49.Sennoune SR, Bermudez LE, Lees JC, Hirsch J, Filleur S, Martinez-Zaguilan R. Vacuolar H+-ATPase is down-regulated by the angiogenesis-inhibitory pigment epithelium-derived factor in metastatic prostate cancer cells. Cell Mol Biol (Noisy-le-grand) 2014;60:45–52. [PubMed] [Google Scholar]
- 50.Hinton A, Sennoune SR, Bond S, Fang M, Reuveni M, Sahagian GG, Jay D, Martinez-Zaguilan R, Forgac M. Function of a subunit isoforms of the V-ATPase in pH homeostasis and in vitro invasion of MDA-MB231 human breast cancer cells. J Biol Chem. 2009;284:16400–16408. doi: 10.1074/jbc.M901201200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fan SH, Wang YY, Lu J, Zheng YL, Wu DM, Zhang ZF, Shan Q, Hu B, Li MQ, Cheng W. CERS2 Suppresses Tumor Cell Invasion and Is Associated with Decreased V-ATPase and MMP-2/MMP-9 activities in Breast Cancer. J Cell Biochem. 2015;116:502–13. doi: 10.1002/jcb.24978. [DOI] [PubMed] [Google Scholar]
- 52.Graham RM, Thompson JW, Webster KA. Inhibition of the vacuolar ATPase induces Bnip3-dependent death of cancer cells and a reduction in tumor burden and metastasis. Oncotarget. 2014;5:1162–1173. doi: 10.18632/oncotarget.1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Scherer O, Steinmetz H, Kaether C, Weinigel C, Barz D, Kleinert H, Menche D, Muller R, Pergola C, Werz O. Targeting V-ATPase in primary human monocytes by archazolid potently represses the classical secretion of cytokines due to accumulation at the endoplasmic reticulum. Biochem Pharmacol. 2014;91:490–500. doi: 10.1016/j.bcp.2014.07.028. [DOI] [PubMed] [Google Scholar]
- 54.De Milito A, Iessi E, Logozzi M, Lozupone F, Spada M, Marino ML, Federici C, Perdicchio M, Matarrese P, Lugini L, Nilsson A, Fais S. Proton pump inhibitors induce apoptosis of human B-cell tumors through a caspase-independent mechanism involving reactive oxygen species. Cancer Res. 2007;67:5408–5417. doi: 10.1158/0008-5472.CAN-06-4095. [DOI] [PubMed] [Google Scholar]
- 55.Sukhai MA, Prabha S, Hurren R, Rutledge AC, Lee AY, Sriskanthadevan S, Sun H, Wang X, Skrtic M, Seneviratne A, Cusimano M, Jhas B, Gronda M, MacLean N, Cho EE, Spagnuolo PA, et al. Lysosomal disruption preferentially targets acute myeloid leukemia cells and progenitors. J Clin Invest. 2013;123:315–328. doi: 10.1172/JCI64180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Funayama K, Murai F, Shimane M, Nomura H, Asano S. Adhesion-induced drug resistance in leukemia stem cells. Pharmacology. 2010;86:79–84. doi: 10.1159/000305344. [DOI] [PubMed] [Google Scholar]
- 57.O'Neil J, Grim J, Strack P, Rao S, Tibbitts D, Winter C, Hardwick J, Welcker M, Meijerink JP, Pieters R, Draetta G, Sears R, Clurman BE, Look AT. FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to gamma-secretase inhibitors. J Exp Med. 2007;204:1813–1824. doi: 10.1084/jem.20070876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Breit S, Stanulla M, Flohr T, Schrappe M, Ludwig WD, Tolle G, Happich M, Muckenthaler MU, Kulozik AE. Activating NOTCH1 mutations predict favorable early treatment response and long-term outcome in childhood precursor T-cell lymphoblastic leukemia. Blood. 2006;108:1151–1157. doi: 10.1182/blood-2005-12-4956. [DOI] [PubMed] [Google Scholar]
- 59.van Grotel M, Meijerink JP, Beverloo HB, Langerak AW, Buys-Gladdines JG, Schneider P, Poulsen TS, den Boer ML, Horstmann M, Kamps WA, Veerman AJ, van Wering ER, van Noesel MM, Pieters R. The outcome of molecular-cytogenetic subgroups in pediatric T-cell acute lymphoblastic leukemia: a retrospective study of patients treated according to DCOG or COALL protocols. Haematologica. 2006;91:1212–1221. [PubMed] [Google Scholar]
- 60.Mansour MR, Sulis ML, Duke V, Foroni L, Jenkinson S, Koo K, Allen CG, Gale RE, Buck G, Richards S, Paietta E, Rowe JM, Tallman MS, Goldstone AH, Ferrando AA, Linch DC. Prognostic implications of NOTCH1 and FBXW7 mutations in adults with T-cell acute lymphoblastic leukemia treated on the MRC UKALLXII/ECOG E2993 protocol. J Clin Oncol. 2009;27:4352–4356. doi: 10.1200/JCO.2009.22.0996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Carter BZ, Milella M, Altieri DC, Andreeff M. Cytokine-regulated expression of survivin in myeloid leukemia. Blood. 2001;97:2784–2790. doi: 10.1182/blood.v97.9.2784. [DOI] [PubMed] [Google Scholar]
- 62.Carter BZ, Qiu Y, Huang X, Diao L, Zhang N, Coombes KR, Mak DH, Konopleva M, Cortes J, Kantarjian HM, Mills GB, Andreeff M, Kornblau SM. Survivin is highly expressed in CD34(+)38(−) leukemic stem/progenitor cells and predicts poor clinical outcomes in AML. Blood. 2012;120:173–180. doi: 10.1182/blood-2012-02-409888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fukuda S, Hoggatt J, Singh P, Abe M, Speth JM, Hu P, Conway EM, Nucifora G, Yamaguchi S, Pelus LM. Survivin modulates genes with divergent molecular functions and regulates proliferation of hematopoietic stem cells through Evi-1. Leukemia. 2015;29:433–40. doi: 10.1038/leu.2014.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Smolewski P, Robak T. Inhibitors of apoptosis proteins (IAPs) as potential molecular targets for therapy of hematological malignancies. Curr Mol Med. 2011;11:633–649. doi: 10.2174/156652411797536723. [DOI] [PubMed] [Google Scholar]
- 65.Glodkowska-Mrowka E, Solarska I, Mrowka P, Bajorek K, Niesiobedzka-Krezel J, Seferynska I, Borg K, Stoklosa T. Differential expression of BIRC family genes in chronic myeloid leukaemia—BIRC3 and BIRC8 as potential new candidates to identify disease progression. Br J Haematol. 2014;164:740–742. doi: 10.1111/bjh.12663. [DOI] [PubMed] [Google Scholar]
- 66.Moore AS, Alonzo TA, Gerbing RB, Lange BJ, Heerema NA, Franklin J, Raimondi SC, Hirsch BA, Gamis AS, Meshinchi S. BIRC5 (survivin) splice variant expression correlates with refractory disease and poor outcome in pediatric acute myeloid leukemia: a report from the Children's Oncology Group. Pediatr Blood Cancer. 2014;61:647–652. doi: 10.1002/pbc.24822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Altieri DC. Targeting survivin in cancer. Cancer Lett. 2013;332:225–228. doi: 10.1016/j.canlet.2012.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Raetz EA, Morrison D, Romanos-Sirakis E, Gaynon P, Sposto R, Bhojwani D, Bostrom BC, Brown P, Eckroth E, Cassar J, Malvar J, Buchbinder A, Carroll WL. A phase I study of EZN-3042, a novel survivin messenger ribonucleic acid (mRNA) antagonist, administered in combination with chemotherapy in children with relapsed acute lymphoblastic leukemia (ALL): a report from the therapeutic advances in childhood leukemia and lymphoma (TACL) consortium. J Pediatr Hematol Oncol. 2014;36:458–463. doi: 10.1097/MPH.0b013e3182a8f58f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen J, Pise-Masison CA, Shih JH, Morris JC, Janik JE, Conlon KC, Keating A, Waldmann TA. Markedly additive antitumor activity with the combination of a selective survivin suppressant YM155 and alemtuzumab in adult T-cell leukemia. Blood. 2013;121:2029–2037. doi: 10.1182/blood-2012-05-427773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Andersen MH, Svane IM, Becker JC, Straten PT. The universal character of the tumor-associated antigen survivin. Clin Cancer Res. 2007;13:5991–5994. doi: 10.1158/1078-0432.CCR-07-0686. [DOI] [PubMed] [Google Scholar]
- 71.Altieri DC. Validating survivin as a cancer therapeutic target. Nat Rev Cancer. 2003;3:46–54. doi: 10.1038/nrc968. [DOI] [PubMed] [Google Scholar]
- 72.Bulycheva E, Rauner M, Medyouf H, Theurl I, Bornhauser M, Hofbauer LC, Platzbecker U. Myelodysplasia is in the niche-novel concepts and emerging therapies. Leukemia. 2015;29:259–68. doi: 10.1038/leu.2014.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Breccia M, Alimena G. Efficacy and safety of deferasirox in myelodysplastic syndromes. Ann Hematol. 2013;92:863–870. doi: 10.1007/s00277-013-1703-7. [DOI] [PubMed] [Google Scholar]
- 74.Nicoletti I, Migliorati G, Pagliacci MC, Grignani F, Riccardi C. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J Immunol Methods. 1991;139:271–279. doi: 10.1016/0022-1759(91)90198-o. [DOI] [PubMed] [Google Scholar]
- 75.Muscolini M, Cianfrocca R, Sajeva A, Mozzetti S, Ferrandina G, Costanzo A, Tuosto L. Trichostatin A up-regulates p73 and induces Bax-dependent apoptosis in cisplatin-resistant ovarian cancer cells. Mol Cancer Ther. 2008;7:1410–1419. doi: 10.1158/1535-7163.MCT-08-0299. [DOI] [PubMed] [Google Scholar]
- 76.Liebl J, Zhang S, Moser M, Agalarov Y, Demir CS, Hager B, Bibb JA, Adams RH, Kiefer F, Miura N, Petrova TV, Vollmar AM, Zahler S. Cdk5 controls lymphatic vessel development and function by phosphorylation of Foxc2. Nat Commun. 2015;6:7274. doi: 10.1038/ncomms8274. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.