

Abstract
With the completion of the human genome sequence and genome sequence available for other vertebrate genomes, the task of manual annotation at the large genome scale has become a priority. Possibly even more important, is the requirement to curate and improve this annotation in the light of future data. For this to be possible, there is a need for tools to access and manage the annotation. Ensembl provides an excellent means for storing gene structures, genome features, and sequence, but it does not support the extra textual data necessary for manual annotation. We have extended Ensembl to create the Otter manual annotation system. This comprises a relational database schema for storing the manual annotation data, an application-programming interface (API) to access it, an extensible markup language (XML) format to allow transfer of the data, and a server to allow multiuser/multimachine access to the data. We have also written a data-adaptor plugin for the Apollo Browser/Editor to enable it to utilize an Otter server. The otter database is currently used by the Vertebrate Genome Annotation (VEGA) site (http://vega.sanger.ac.uk), which provides access to manually curated human chromosomes. Support is also being developed for using the AceDB annotation editor, FMap, via a perl wrapper called Lace. The Human and Vertebrate Annotation (HAVANA) group annotators at the Sanger center are using this to annotate human chromosomes 1 and 20.
The human genome sequence (The International Human Genome Sequencing Consortium 2001) was declared finished in April 2003. There are several other vertebrate genome sequences available (mouse, Waterston et al. 2002; rat, http://www.hgsc.bcm.tmc.edu/projects/rat), with more coming (chicken, http://genome.wustl.edu/projects/chicken/Chicken_Genome.pdf; dog, http://www.genome.gov/Pages/Research/Sequencing/SeqProposals/CanineSEQedited.pdf). To provide meaning to the sequence, functional regions, such as genes within it, must be located and annotated as well as other features of the genome that can be informative, such as CpG islands and poly(A) sites. One approach to this is automatic annotation such as that provided by Ensembl, which provides useful insight into gene position, structure, and function. The other approach is manual annotation and curation, in which expert annotators examine sequence-similarity results and automatic predictions, and create gene models from them. In theory, this should be the most reliable set of gene models. In practice, there have been problems with data consistency, attribution, and data communication. For the manual annotation data to be valuable, it must be of a consistent and high standard and be readily accessible and easy to curate. Such data will provide a vital data set for the post genomics era, where accurate gene models will be necessary for understanding such things as the functional consequences of variation, understanding disease, and providing reference genome annotation to aid in annotation of genomes with small amounts of organism-specific data.
The manual annotation effort of the human genome is of two types. The large genome centers, such as the Wellcome Trust Sanger Institute (http://www.sanger.ac.uk) and the Genome Sequencing Center at Washington University Medical School (http://genome.wustl.edu), are currently manually annotating whole chromosomes. There are also smaller research groups with a detailed knowledge of individual loci or genomic regions (such as the major histocompatibility complex) that will want to be able to add this information to existing generic annotation.
There are existing systems for storing and transferring annotation data. AceDB (Stein and Thierry-Mieg 1999) is a custom-built database system that is very flexible in the data it can represent. It also provides a graphical user interface (GUI) called FMap that enables manual annotation to be added. AceDB is used widely for storing human genome annotation (Stein and Thierry-Mieg 1999) and other organisms, as well as its initial purpose of annotating Caenorhabditis elegans (Harris et al. 2003). The GadFly database system (Mungall et al. 2002) is another system for storing annotation data, in which the genome-wide annotation of Drosophila is stored. CHADO (http://www.gmod.org) is a new annotation database/XML format that is being actively developed.
Accurate transferral of genome annotation between systems is a key aspect of maintaining and updating data from multiple sources. For instance, we need a way of transferring information from a database to a browser/genome editor and back again or between different editors. This involves representing the annotation in a flat file format in such a way that all of the data and their heirarchical relationships can be unambiguously represented. Flat file formats abound in bioinformatics, and we considered existing formats as to their suitability to avoid creating a new one. Two main criteria were used to assess the formats. Firstly, they should be able to represent all curated annotation produced so far, and secondly, the format should be unambiguous as to what data should go where. The first criterion is obviously essential, but the second one is equally so. If there are multiple people in different parts of the world creating and exchanging annotation, it must be obvious to everyone what piece of information goes where and what it represents. Failure to do this results in degradation and inconsistencies in a data set that has taken a lot of time and trouble to produce. In assessing the available formats, we first looked at General Feature Format (GFF) and Gene Transfer Format (GTF). These are two widely used flat file formats for transferring annotation data (http://www.sanger.ac.uk/Software/formats/GFF/GFF_Spec.shtml). The GFF format has a simple tab-delimited structure with the ability to incorporate a certain amount of hierarchical data, but we felt extending this to accommodate curated data would be pushing the format too far and would lose the simplicity that is one of its strengths. GTF is an extension of GFF and can represent gene structures well, but is limited in its ability to represent the extra textual and versioning information necessary for a curatable annotation system and thus was also rejected. The Distributed Annotation System (DAS) is a means of transferring and sharing annotation (Dowell et al. 2001). It provides an extensible markup language (XML) format and a client/server interface. Using XML allows people to represent richer data with its nested tag layout. The current implementation of DAS, however, aimed to keep the data simple, which, although a laudable aim, was thus limited in its ability to represent gene structure information. Additionally, it could not easily provide versioning or authorship information, and was therefore discarded. The only existing format that catered to most of our needs was GAME XML (http://www.fruitfly.org/annot/gamexml.dtd.txt). This was developed for the Drosophila melanogaster manual annotation and is a format for representing annotation data for transfer between sites and machines. Although this was the closest to satisfying our requirements, we were concerned that the format was still changing and also that some of the tags could be interpreted ambiguously, which could lead to data inconsistencies. Chado is a set of schema modules for building a model organism database. It aims to be flexible in the data it can represent. However, it was still in early development stages at the time we were assessing formats. Regrettably, we felt compelled to design a new, light-weight annotation format that would fulfill our requirements.
The decision to base the Otter system around a new format was not taken lightly. Our foremost consideration in designing the system was to preserve the consistency of the data. Past experience in importing external data both into Ensembl and into Otter has been that even though a data set purports to be in a certain format, there almost invariably exists a difference between the format definition and implementation. This has resulted in each different data source having its own custom-built parser to deal with slight variations in the data presented. This has even been the case in the GTF format, which has validators to test the integrity of the data. The need for custom-built parsers is not necessarily due to sloppiness on the part of the data authors or lack of documentation or tools on the part of the format authors, but rather part and parcel of the type of data we are dealing with. It has only been relatively recently that there has been consensus on how to define what constitutes a protein-coding gene. Even in that case, there are confusions about how to mark up multiple transcripts or where the coding region starts and in what coordinate system. Given that, in practice, code would have to be written for whatever format we chose, we made the decision to have our own tightly constrained format and provide parsers to and from other formats when and where necessary.
Deciding on a flat file format is only part of the solution, however. With the scale of the annotation task that currently exists, we wanted a system to enable storing and maintenance of genome-scale manual annotation data. The system also needed to be able to support annotation in multiple centers and allow transfer of the data between them. Also, we wanted a system with built-in versioning and history mechanisms to allow curation of the data over time as new information confirming or disproving the annotation becomes available. We call the system that has been developed the Otter manual-annotation system. The Otter system has been designed to be capable of storing annotation of the standard defined in the series of Human Annotation Workshops (HAWK) (http://www.sanger.ac.uk/HGP/havana/hawk.html). HAWK workshops have been attended by 15 different human sequence analysis groups and are intended to define a standard of annotation.
One important feature of the Otter system is that annotation is performed on genomic assemblies. This assembly information is maintained in the database and also in the transfer format. Annotation on individual Bacterial Artificial Chromosome (BAC) clones has been commonplace in the past, as it is time inefficient to wait until all clones have been sequenced before starting the annotation. Unfortunately, this substantially increases the complexity of the annotation task, due to genes spanning more than one clone. This leads to genes being annotated piecemeal and having to be joined together at a later date, which can lead to errors in gene naming and splice-site identification. There is also the problem of parts of annotation lying on overlaps between clones that may be lost when the clones are joined together into an assembly. Assembly differences are a common cause of errors when using annotation data. As a genome moves toward being finished, the bulk of the assembly may stay the same, but there will be a number of clones that will change, often around the centromere and around gaps. Updating assemblies if the data is stored in chromosomal coordinates can prove time consuming and error prone, as the change in length of one sequence at the start of a chromosome can lead to the updating of several million feature coordinates along the length of the rest of the chromosome. To avoid this, the data is still stored on assembly components that can be clones or whole-genome shotgun (WGS) contigs. This enables one clone in the assembly to change without having to change the rest of the data.
RESULTS AND DISCUSSION
The Otter annotation system consists of a relational database to store the annotation data, an XML format for transferring the data between machines, and a server to control access to the data and allow multiuser annotation.
The Database
The annotation data in Otter is stored in a MySQL (http://www.mysql.com) database. The schema is an extension of the core Ensembl schema for storing gene structures and the evidence for them (Stabenau et al. 2004). There are 21 extra tables that store the manual annotation data. Sixteen of these store the extra data required, which includes gene names, remarks on genes, transcripts and clones, author of annotations, synonyms for genes, and accessions of the evidence supporting transcript structures. The remaining extra tables provide support for stable id generation and the locking of clones.
Otter databases use the InnoDB table type of MySQL (available since version 3.23), which provides transaction support. This helps prevent incomplete data being stored back into the database, because only complete transactions are written back. In the case of Otter, a transaction is usually the storing of a complete modified region after an annotation session.
The schema is shown in the Supplemental data available online at www.genome.org. One thing to note is that the tables are joined through the stable ID rather than the internal IDs (such as transcript_id and gene_id) used for joining the core tables. This was a deliberate choice on our part to make it easier to port the Otter tables onto a non-Ensembl database.
The API
As the Otter database schema is based on the Ensembl schema, it was fitting to base the API to access the database around the core Ensembl API. As with the Ensembl core API, there is a set of datamodel classes that represent the annotation data (AnnotatedGene, Author, GeneRemark, etc.) and a set of data adaptors that contain the code to query the database and construct data-model-type objects. As the Ensembl API is object oriented, extending the core types is straightforward, so, for example, the Otter API extends the Ensembl Gene object to an AnnotatedGene object.
Figure 1 shows the extra classes and their relationships to the core Ensembl objects.
Figure 1.
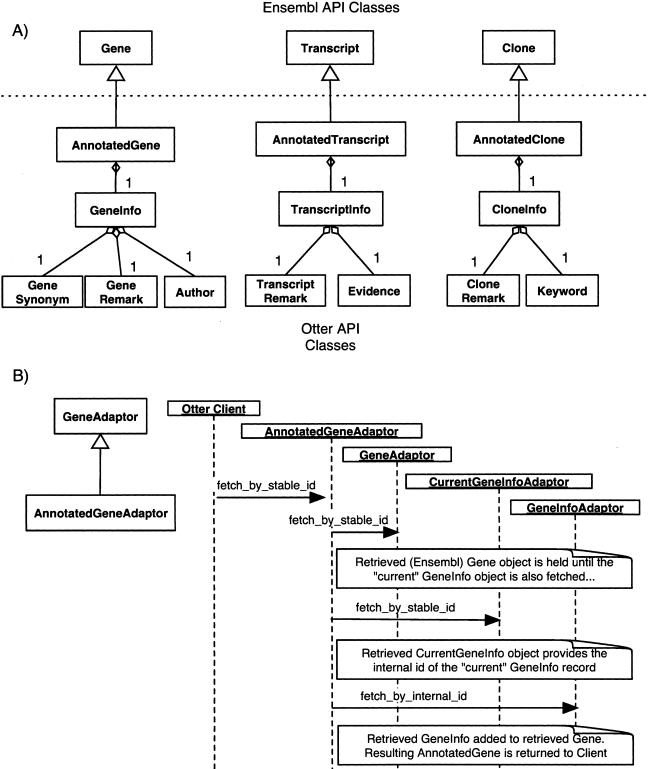
UML representations of the Otter datamodel. (A) The relationships between the Otter datamodel classes. The Gene, Transcript, and Clone classes at the top of the diagram are the Ensembl parent classes that are extended by Otter. The extensions contain member variables of types GeneInfo, TranscriptInfo, and CloneInfo, respectively, that hold the manual annotation information. (B) The sequence of calls necessary to fetch an AnnotatedGene from an Otter database with the AnnotatedGeneAdaptor.
A part of the API unique to Otter is the stable ID generation code (Ensembl has stable ids, but these are not generated within the core API). For Otter this is vital, because the stable ID is the unique key to join tables. Otter generates stable ids for each gene, transcript, exon, and translation added to the database. The stable id has a version associated with it as well as a creation timestamp and a modification timestamp. In this way, the Otter database is able to store multiple versions of the same annotation with the same stable ID but different versions, giving a history of how the annotation has changed. When the database is accessed through the API by default, only the most recent version of each annotation is returned.
The author of each annotation is also stored along with their e-mail address. This will be important if queries arise over particular annotations and the source of the annotation needs to be traced. To provide an insight into why each annotation was created, the Otter database also stores supporting evidence for each gene. This is stored at the transcript level, and it contains a list of the accessions of all database entries supporting the annotation and a type (EST, cDNA, Protein, Genomic, Other).
The decision to just store accessions for the sequences used as evidence for annotation is based on the fact that the existing Sanger annotation stored evidence in this way.
Scripts are provided to allow this transcript level evidence to be converted into the exon level evidence stored in the supporting_feature table of a standard Ensembl database.
Another part of the API unique to Otter is the Converter. This module handles conversion between XML or AceDB and Otter datamodel objects. This allows us to convert from XML into objects and then out to AceDB, for example. Alternatively, it makes it possible to import from AceDB format and store into the database.
There are two modules, the CloneLockBroker and AnnotationBroker, which provide other support functionality. The CloneLockBroker handles requests to set or release locks on a region of the genome stored in the database. These locks are required to stop multiple annotators editing the same region simultaneously. On the basis of experience of how annotators prefer to work, we do not support optimistic locking, because we consider that adding conflict resolution is probably an unnecessary extra level of complexity, given that currently individual annotators are assigned nonoverlapping regions to work on.
The AnnotationBroker handles the comparison between the currently stored and edited annotation for a region being written back. It produces a list of changed or added annotations that need storing in the database, and increments the version where necessary. It also handles adding stable IDs to new annotations that will not have them.
Gene merging and splitting are two common edits that annotators perform. When a gene is merged, the gene gets one of the stable IDs of the original genes. The transcripts retain their stable ids. When a gene is split, one of the genes retains the stable id of the original, and again, the transcript stable ids are retained. As the Otter system retains all previous versions of the annotation, it is possible to trace the history of any particular piece of annotation.
The XML Format
We designed the Otter XML format to represent the annotation stored in an Otter database, the genomic assembly on which the annotation is built, and optionally, the DNA sequence for the region. It does not represent the similarity hits and automatic predictions from which annotations are built (these must be retrieved from a separate source, such as an Ensembl database or a flat file, such as a GFF file). Limiting the XML to representing annotation was a deliberate decision on our part to try to remove the potential for misuse of general `feature' tags that can occur as extra types of annotation are added. To handle new types of annotation, we would prefer to add new tags with well-specified meanings.
Figure 2 shows all of the Otter XML tags. The Otter XML tags are described in detail at http://www.sanger.ac.uk/Users/jgrg/otter_xml.html. We provide a brief description here.
Figure 2.

The tags in the Otter XML hierarchy for representing genome annotation.
A section of Otter XML is delimited by an Otter tag pair. Within that are one or more sequence_set tags. These contain the assembly information and annotations. The assembly information (either contigs or WGS reads) is represented within sequence_fragment tags. These contain tags for the versioned accession (accession and version), identifier (id), and the assembly mapping information (assembly_start, assembly_end, fragment_ ori, fragment_offset). Also, as remarks and keywords can be added to clones, there are tags for these (remark and keyword) and their author (author and author_email).
Optionally, sequence can be included within DNA tags at the sequence_set level. This is optional, because more efficient methods exist for fetching sequence at many sites.
The gene annotation is a three-level hierarchy (genes contain transcripts that contain exons, and optionally, a translation). The XML mirrors this. There are locus tags that contain one or more transcript tags. These contain one or more exon tags (within an exon_set tag), and optionally, tags representing the translation location (translation_start and translation_end). All three of the main levels have stable_id tags. The translation stable ID is contained within a translation_stable_id tag. Within both locus and transcript tags, author and author_email tags represent the author of the annotation. At these two levels, remark tags contain remarks about the annotation and a name tag and contain its annotator assigned (official Human Genome Organisation (HUGO); Wain et al. 2002) name.
Other tags at the locus level contain the locus synonyms (synonym) and whether the locus is previously known or novel (known).
Other tags at the transcript level represent whether it is believed to represent a complete coding sequence (cds_start_not_ found, cds_end_not_found) and mRNA (mRNA_start_not_ found, mRNA_end_not_found) and its type (transcript_class). The evidence for each transcript is represented by a hierarchy of tags within the transcript evidence tags within an evidence_set tag. The evidence tag contains name and type tags.
To represent the exon location within the exon tags, there are start, end, strand, and frame tags.
This XML format has been agreed on by the G16 groups (T. Hubbard, pers. comm.) as a means for annotation transfer between groups.
The Server
One of the requirements for an annotation database back end is that multiple annotators will need to access the database simultaneously from different machines, potentially at different sites. To satisfy this requirement, we have written a set of Common Gateway Interface (CGI) scripts and a small perl-based Web server. We wrote our own Web server for two reasons. Firstly, we wanted to make installing the Otter system as simple as possible. Having our own server negates the need to compile and install a server such as Apache. Secondly, having a very simple Web server code makes debugging the requests much easier. If desired, Apache or any other Web server can be used in place of the provided one.
The CGI scripts and their roles are shown in Table 1. They provide a simple interface to the database. The data is transferred to and from the server in Otter XML format. Figure 3 shows the client / server interactions. If errors occur on the server side, they are returned as text error messages within Otter response tags.
Table 1.
CGI Scripts in the Otter Server
| get_region | Retrieve a genomic region from the server, optionally locking it for editing. Users are identified by name and e-mail address. |
| write_region | Write a region back to the server. The region must have been previously locked by the same user. This is a POST request. |
| unlock_region | Unlock a region without writing it. |
| get_sequence | Get the sequence for a region as a string. |
| datasets | List the datasets on the server. This request returns details of databases containing dna; name of assembly is required. |
Figure 3.
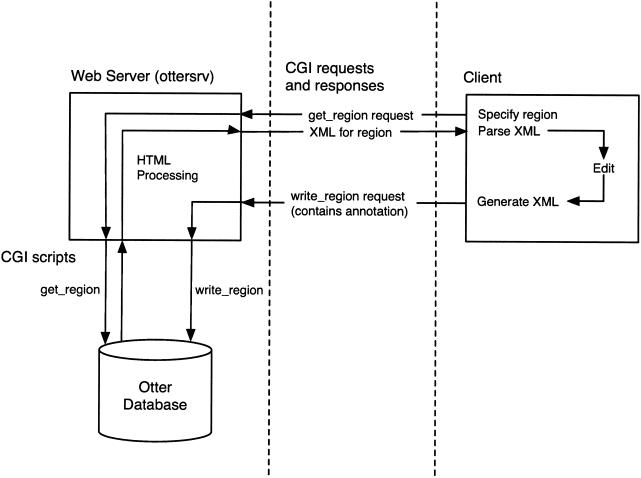
Otter server client interactions. The two main interactions between client (for example, Apollo), server, and database are shown. These are fetching a region from the server (get_region request) and writing it back (write_region request).
To illustrate the interactions between client and server, the requests involved in an annotation session are described here. First, the client sends a get_region request specifying a particular chromosomal region (say chromosome 1 1–2 Mb). Optionally, an assembly name can be specified. Details of the user name and host are also sent if the user wants to edit the data rather than just view it. For an edit session, the get_region CGI script creates locks in the database for the clones that make up the specified region using the user information supplied. It then generates an Otter XML representation of the annotation (if any) on the region from the database, which is returned to the client for display. If an error occurs, for example, if the database cannot be opened, an Otter response is sent indicating the nature of the error, which the client should display.
The user edits and creates new annotation in the client. When the user wants to save the new data back to the server (probably by selecting a save menu option), a write_region request is sent to the server. The client generates an XML representation of all of the annotation on the region that is included in the write_region request. The write_region CGI script extracts the annotation from the database into Otter objects, converts the XML received in the request into Otter objects, and then compares the two sets. Any changes are identified, and the modified data stored back to the database. A response is sent back to the client indicating that the store was successful. Until the server acknowledges successful storing of data from a write_region request, the client is responsible for maintaining a backup of the annotation session. When the server does successfully store the data, it is immediately available for others to access.
Technical problems, such as badly formatted XML, are checked for during the write_region request. However, the server does not perform checks on the biological quality of the annotation, such as checking that translations do not contain stop codons or that phase is consistent between exons when it stores the annotation. As the Otter database is an extended Ensembl database, scripts available for assessing Ensembl predictions can be applied to the manual annotation in the Otter database as a post-processing step.
The use of an XML format and a CGI server makes it relatively straightforward to add Otter support to various front-end GUIs. Adding support to Apollo (Lewis et al. 2002) is described below. Support for the AceDB front end FMap (via the lace/otter interface; Deloukas et al. 2001) is also complete.
Clients
Two annotation tools now have Otter client support. These are Apollo and Otter/Lace.
Apollo is a desktop graphical application that allows the straightforward browsing and annotation of large genomes. It accepts input from and writes to a variety of data sources including GAME, Ensembl, GFF, and Otter. It does this via a set of `pluggable' data adapters; in particular, it is capable of reading and writing annotations in Otter XML, either via http from an Otter server or directly to/from a file. Figure 4 shows the Ensembl/Otter data adapter's interface in Apollo. The annotations are usually read at the same time as analyses and sequence information are read from an Ensembl database. This illustrates that it is the clients' responsibility to fetch the analyses results for the region on which the annotations will be based from a separate datasource to the Otter server. Figure 5 shows the Apollo main window with data loaded. This provides a unified view of the computational results and manual annotations for a genome, and allows the user to create or modify annotations and write these back to an Otter server or data file.
Figure 4.
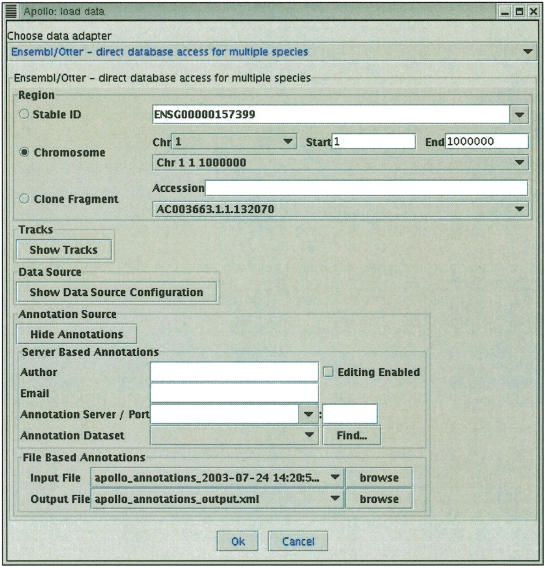
The interface for the data adapter used to connect Apollo to read predictions from an Ensembl database and annotations from an Otter-XML formatted file, or from an Otter server.
Figure 5.
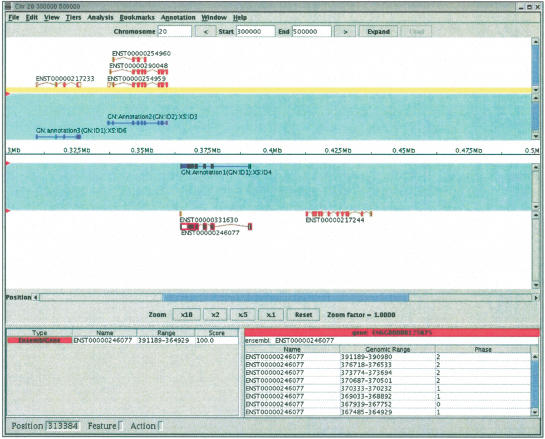
Ensembl gene predictions and otter annotations displayed in Apollo. The annotations added (in blue) to the gene predictions can be saved as Otter-format XML files, or written directly to the Otter server.
Otter/Lace is a perl wrapper round the AceDB annotation editor, FMap. The Human and Vertebrate Annotation (HAVANA) group annotators at the Sanger center are using this to annotate human chromosomes 1 and 20.
Format Conversion
There is already a large amount of data stored in various systems. We wanted to be able to import this data for the following various reasons:
VEGA site—present finished chromosome scale annotation with a consistent interface.
Comparison against automatic predictions, such as the Ensembl predictions.
We have code to convert from various formats to Otter objects, which can be written to a database. These include GTF, EMBL, GAME (via Apollo adaptor code), and AceDB. We also recognized the need to output the data in formats other than Otter XML and provide exporters to AceDB and GTF format.
VEGA and Other Databases
The VEGA database (http://vega.sanger.ac.uk) is a database repository of high-quality manual annotation that uses an Otter database as its back end. It contains current versions of the manual annotation of human chromosomes 6 (Mungall et al. 2003), 13 (A. Dunham, in prep.), 14 (Heilig et al. 2003), 20 (Deloukas et al. 2001), and 22 (Collins et al. 2003), Zebrafish annotation of 168 clones (Jekosch 2004) and the mouse region Del36H (http://mrcseq.har.mrc.ac.uk/chr13.html). The site runs off Ensembl Web code with extensions to display extra information from the Otter database.
Figure 6 shows an example ContigView page from the Ensembl Web site. The gene structures from the VEGA database are displayed on the Ensembl Web site in the “Vega transcript” track on ContigView. Not all of the annotation can be shown, as the assembly in VEGA can be different from that in Ensembl, in which case, only the annotation that can be cleanly transferred between the assemblies is shown.
Figure 6.
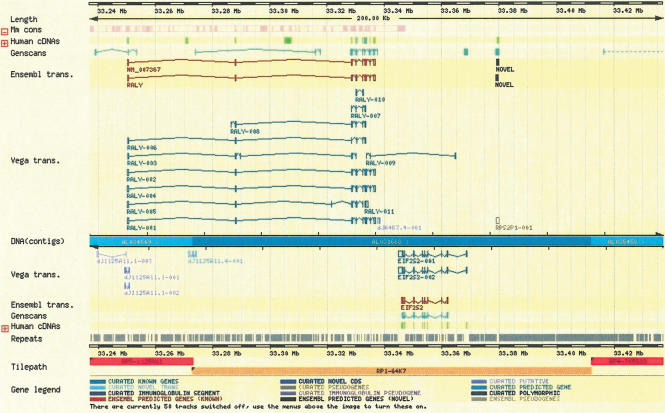
VEGA annotation displayed in ContigView. The Vega trans. track contains manual annotation transferred from the VEGA database into an Ensembl database.
Otter annotation databases have been created for other organisms such as Schizosaccharomyces pombe and Streptomyces coelicolor (K. James, pers. comm.).
Future Directions
Protein-coding genes have been the center of annotation efforts up to now. However, it is now recognized that there are many RNA genes (Eddy 2001) and other conserved noncoding regions that would benefit from manual annotation. These could be stored within the gene/transcript/exon model in an Otter database. However, it may prove to be better to create new tables, objects, and XML tags to store and transfer annotations for these.
The current transcript level supporting evidence is not ideal for determining exactly why particular annotations have been created. It will be fairly simple to add support for exon level supporting evidence to Otter. As annotators are not currently providing this, we have not supported it in the format.
To ease installation problems, we are planning to release an Otter distribution for MacOSX and i86 Linux containing all of the components (perl binary and modules, graphics libraries, jars) necessary to run an Otter server, the Apollo client, and an Ensembl Web site.
METHODS
The Otter API is written in Perl. It requires at least Perl version 5.6. The code is open source and is available from the ensembl cvs repository in the ensembl-otter module. Details of how to download the code are available at http://cvsweb.sanger.ac.uk. The Ensembl core code (ensembl-core module) must also be installed. Otter databases are stored in MySQL version 4.0, and MySQL must have been built to support InnoDB type tables.
Acknowledgments
We thank the users of our Web site and data sets and the developers on our mailing lists for much useful feedback and discussion. We particularly acknowledge the members of the annotation team at the Wellcome Trust Sanger Institute and the Apollo development team at Berkeley and Harvard. The Ensembl project is principally funded by the Wellcome Trust with additional funding from EMBL.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.1864804.
Footnotes
[Supplemental material is available online at www.genome.org.]
References
- Collins, J.E., Goward, M.E., Cole, C.G., Smink, L.J., Huckle, E.J., Knowles, S., Bye, J.M., Beare, D.M., and Dunham, I. 2003. Reevaluating human gene annotation: A second-generation analysis of chromosome 22. Genome Res. 13: 27-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deloukas, P., Matthews, L.H., Ashurst, J., Burton, J., Gilbert, J.G., Jones, M., Stavrides, G., Almeida, J.P., Babbage, A.K., Bagguley, C.L., et al. 2001. The DNA sequence and comparative analysis of human chromosome 20. Nature 414: 865-871. [DOI] [PubMed] [Google Scholar]
- Dowell, R.D., Jokerst, R.M., Day, A., Eddy, S.R., and Stein, L. 2001. The distributed annotation system. BMC Bioinformatics 2: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy, S.R. 2001. Non-coding RNA genes and the modern RNA world. Nat. Rev. Genet. 2: 919-929. [DOI] [PubMed] [Google Scholar]
- Harris, T., Lee, R., Schwarz, E., Bradnam, K., Lawson, D., Chen, W., Blasier, D., Kenny, E., Cunningham, F., Kishore, R., et al. 2003. WormBase: A cross-species database for comparative genomics. Nucleic Acids Res. 31: 133-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilig, R., Eckenberg, R., Petit, J.L., Fonknechten, N., Da Silva, C., Cattolico, L., Levy, M., Barbe, V., de Berardinis, V., Ureta-Vidal, A., et al. 2003. The DNA sequence and analysis of human chromosome 14. Nature 421: 601-607. [DOI] [PubMed] [Google Scholar]
- The International Human Genome Sequencing Consortium 2001. Initial sequencing and analysis of the human genome. Nature 409: 860-921. [DOI] [PubMed] [Google Scholar]
- Jekosch, K. 2004. The zebrafish genome project: Sequence analysis and annotation. In Methods in cell biology: Zebrafish, genetics, genomics and informatics (eds. H.W. Detrich et al.). Elsevier Academic Press, London, UK, San Diego, CA (in press). [DOI] [PubMed]
- Lewis, S.E., Searle, S.M.J., Harris, N., Gibson, M., Iyer, V., Ricter, J., Wiel, C., Bayraktaroglu, L., Birney, E., Crosby, M.A., et al. 2002. Apollo: A sequence annotation editor. Genome Biol. 3: RESEARCH0082. [DOI] [PMC free article] [PubMed]
- Mungall, C.J., Misra, S., Berman, B.P., Carlson, J., Frise, E., Harris, N., Marshall, B., Shu, S., Kaminker, J.S., Prochnik, S.E., et al. 2002. An integrated computational pipeline and database to support whole-genome sequence annotation. Genome Biol. 3: RESEARCH0081.1–0081.11. [DOI] [PMC free article] [PubMed]
- Mungall, A.J., Palmer, S.A., Sims, S.K., Edwards, C.A., Ashurst, J.L., Wilming, L., Jones, M.C., Horton, R., Hunt, S.E., Scott, C.E., et al. 2003. The DNA sequence and analysis of human chromosome 6. Nature 425: 805-811. [DOI] [PubMed] [Google Scholar]
- Stabenau, A., McVicker, G., Melsopp, C., Proctor, G., Clamp, M., and Birney, E. 2004. The Ensembl core software libraries. Genome Res. (this issue). [DOI] [PMC free article] [PubMed]
- Stein, L.D. and Thierry-Mieg, J. 1999. AceDB: A genome database management system. Comput. Sci. Eng. 1: 44-52. [Google Scholar]
- Wain, H.M., Lovering, R.C., Bruford, E.A., Lush, M.J., Wright, M.W., and Povey, S. 2002. Guidelines for human gene nomenclature. Genomics 79: 464-470. [DOI] [PubMed] [Google Scholar]
- Waterston, R.H., Lindblad-Toh, K., Birney, E., Rogers, J., Abril, J.F., Agarwal, P., Agarwala, R., Ainscough, R., Alexandersson, M., An, P., et al. 2002. Initial sequencing and comparative analysis of the mouse genome. Nature 420: 520-562. [DOI] [PubMed] [Google Scholar]
WEB SITE REFERENCES
- http://www.sanger.ac.uk/Software/formats/GFF/GFF_Spec.shtml; GFF format spec.
- http://www.mysql.com/; MySQL database.
- http://www.fruitfly.org/annot/gamexml.dtd.txt; GAME XML DTD.
- http://cvsweb.sanger.ac.uk; Public CVS repository for the Ensembl software.
- http://www.sanger.ac.uk; The Wellcome Trust Sanger Institute. [DOI] [PubMed]
- http://www.sanger.ac.uk/Users/jgrg/otter_xml.html; Description of Otter annotation exchange format.
- http://vega.sanger.ac.uk; The Vertebrate Genome Annotation (VEGA) Web site.
- http://www.hgsc.bcm.tmc.edu/projects/rat; Rat genome sequencing homepage.
- http://genome.wustl.edu/projects/chicken/Chicken_Genome.pdf; Chicken genome sequencing proposal.
- http://www.genome.gov/Pages/Research/Sequencing/SeqProposals/CanineSEQedited.pdf; Dog genome sequencing proposal.
- http://genome.wustl.edu; The Genome Sequencing Center at Washington University Medical School.
- http://www.sanger.ac.uk/HGP/havana/hawk.html; The HAWK manual annotation workshop.
- http://mrcseq.har.mrc.ac.uk/chr13.html; Del36H sequencing project proposal.
- http://www.gmod.org; Generic model organism database construction set.