

Radiation therapy has been used for more than 100 years to treat human disease with some of the first applications to cancers in the 1890s. Radiotherapy delivers ionizing radiation to target tissues leading to fragmentation and ionization of biomolecules, water, oxygen and other chemicals that generate reactive oxygen species (ROS). Direct and indirect ROS-mediated damage to DNA, membranes, proteins and other cellular components lead to cell death. Cancer tissues are more sensitive to this damage. The exact mechanism and mode of cell death, and the basis for selective cancer toxicity are still areas of active investigation.
Besides radiation therapy, cancer is also treated with immunotherapy, relying on provocation of the adaptive immune system to attack cancer cells. In immunotherapy, both the cellular and humoral adaptive immune response can target and eliminate abnormal cells based on antigenic differences between tumor and host. Immunotherapy activates natural killer cells or cytotoxic T cells that are specific for the tumor and attack the cancer. Taking advantage of the humoral immune response, multiple antibody treatments are available that bind to cancer antigens and activate complement-mediated cytotoxicity via the classical pathway to kill the tumor cell. This is one aspect of tumor immunity where the complement system is thought to be essential. The antibody defines the aberrant cell and the complement system lyses the tumor cell via the membrane attack complex, C5b-9. In the recent report by Surace et al. (1) a novel interaction between radiotherapy and complement is described with a different sequence of events: Radiation results in necrotic tumor cells and initiation of transient complement activation in the local tumor environment to generate anaphylatoxins, C3a and C5a, which then stimulate adaptive immunity to contribute to tumor cell killing. Surace et al. define C3a and C5a as complement components that are critical for recruitment of the cellular immune response and tumor cell killing following radiotherapy.
To further appreciate this finding, let’s step back and think about the complement system in general. Now this system is often viewed in medical training in the same vein as the coagulation cascade: a series of numbers and letters to memorize. However, bear in mind that this is an ancient evolutionarily conserved defense system present in horseshoe crabs and sea sponges, so it has proved its worth. In the human, deficiencies in complement system components increase the likelihood of immune complex disease and serious bacterial infections. However, if complement activation is excessive or normal control mechanisms are overwhelmed, complement-mediated pathology may occur. Autoimmune diseases are a prime example of antigen-antibody-mediated complement activation leading to pathology, i.e., glomerulonephritis, lupus, etc.
Some major principles of complement system should be mentioned: (I) the system is always operating at a slow steady state level of activation that can be pushed into amplification with a wide variety of stimuli; (II) the pathways produce many different biologically active products even if the pathway does not continue to lysis of the bacteria or tumor cell; (III) the system is intended to act locally and systemic activation indicates escape from local control. As seen in Figure 1, the complement system can be activated via 3 major pathways: the classical, the mannose binding lectin, and the alternative pathway. Each of these pathways converges on a central event—cleavage of circulating C3 into C3b and C3a. While C3a is released, C3b covalently binds to macromolecules or cells or is degraded into cleavage products such as iC3b, C3c and C3d. Bound C3b (termed C3 deposition), but not the C3 degradation products, participate in cleavage of C5. Both C3b and degradation products may amplify the humoral immune response or opsonize altered cells for phagocytosis. C3b contributes to formation of a molecular complex that then cleaves the C5 molecule resulting in generation of C5a and C5b. C3a and C5a are termed anaphylatoxins and are approximately 9,000 kD fluid phase molecules that interact with seven transmembrane G protein coupled receptors: C3aR, C5a1R (CD88) and C5a2R (2). C5b with complement components C6-9 forms a membrane attack complex creating a pore in the membrane, lysing the target cell when insufficient complement regulatory proteins are present. The inciting stimuli that initiates complement activation following radiotherapy is unclear from the study of Surace et al. (1), though they have provided some evidence that necrotic cells are involved or associated with the complement activation. What is clear is that the fluid phase activation products C3a and C5a are critically important for the radiotherapy effect in the mouse model. Radiotherapy induces local transient activation of complement, at least through C3 and C5. The generation of C3a and/or C5a and their receptor interactions are critically important for the maturation and activation of the adaptive immune response for a positive therapeutic effect of radiation therapy.
Figure 1.
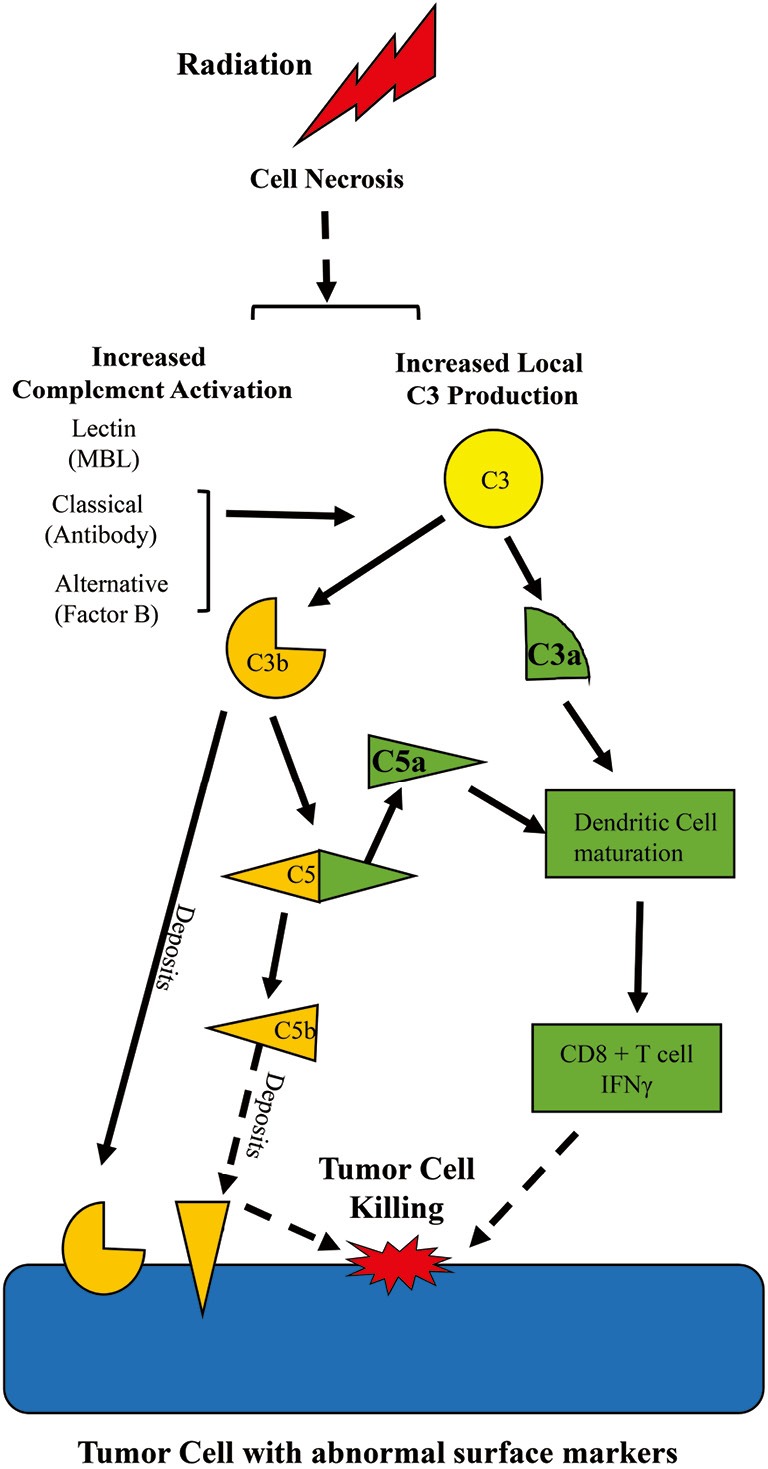
Radiation-induced complement activation initiates adaptive immune response and tumor cell death. The study of Surace et al. (1) demonstrates that radiotherapy induces cell necrosis, increases local production of C3 within the tumor and a transient local increase in complement activation with C3a and C5a generation. Increased complement activation results in C3 deposition on tumor cells. C3a and/or C5a are critically important for stimulation of dendritic cell maturation and interferon production by cytotoxic CD8+ T cells leading to a beneficial clinical response and tumor cell killing. Orange symbols represent activation of the innate immune response while green symbols represent activation of the adaptive immune response. Solid arrows represent data within the Surace et al. manuscript, while dashed arrows represent implied actions consistent with literature and existing data.
Importantly, the role of complement activation in inducing tumor specific immunity following radiotherapy is not limited to a single type of cancer. The study examined mouse models of melanoma and colon cancer as well as human skin cancer and found a transient, but clearly effective, C3a- and C5a-dependent stimulation of cellular infiltration in each. The role of C3a and C5a was evaluated using mice deficient in the specific receptors, C3aR and C5a1R (CD88) as well as blockade using antibody or small molecule antagonists of the G-protein coupled receptors. Importantly, dexamethasone treatment down-regulated the anaphylatoxin receptors and radiotherapy efficacy, suggesting a need to consider the potential loss of treatment efficacy when using dexamethasone to reduce radiation toxicity.
The location and distribution of activated C3 may also be important. Surace shows accumulation of C3 along blood vessels following 20 Gy. Vascular injury, and damage to endothelial cells in particular, has been implicated as especially important in the effects of high dose irradiation such as used by Surace (3,4). Irradiated endothelial cells increase acid sphingomyelinase activity generating ceramide that is critical to endothelial cell injury and subsequent anti-tumor activity of high dose irradiation. Ceramide transport proteins have been implicated in complement activation (5). Together, these observations may describe a pathway of endothelial cell injury, ceramide activation and complement targeting leading to vascular injury, shutting down nutrient supplies to cancer and ultimately tumor cell death.
The traditional immune response to tumor cells includes antigen presenting cell activation of cytotoxic CD8+ T cells to produce γ-interferon. Activation of C3 by radiotherapy up-regulated C3a and C5a receptors on dendritic cells and maturation of these critical antigen presenting cells, induced γ-interferon production by cytotoxic T cells, and prevented the regulatory T cell infiltrate. Previous studies demonstrated that C3 is required for in vitro dendritic cell maturation and pro-inflammatory γ-interferon CD4 and CD8 response, as well as in mouse and human graft vs. host disease (6-8). Importantly, therapeutic blockade of C3 activation with Compstatin inhibits the pro-inflammatory Th1 response without altering the regulatory T cell response (9). Additional studies demonstrated that the absence or down regulation of complement regulatory protein, Crry, induced interferon production in a mouse bladder cancer model (10). The study by Surace et al. (1) did not examine expression of complement regulatory proteins on the tumor cells. Future studies will require investigation of complement regulator expression on the tumor stroma, immune cells within the tumor and tumor cells themselves.
Direct and indirect cancer cell damage from radiotherapy can increase tumor immunogenicity. Indeed, cell disruption and death following radiotherapy have been described as an in situ personal vaccination against cancer (11). T cell stimulation and activation against cancer may lead to tumor responses outside the irradiated field. Such events, where one site of metastasis is irradiated and both targeted and other sites of disease respond, is termed an abscopal effect (11). Abscopal effects are relatively rare but have been described, particularly in melanoma. Despite the ability of radiotherapy to enhance an anti-cancer immune reaction, systemic anti-cancer immune activity is seen only rarely. One explanation for the limited immune response is the difficulty in determining foreign cancer cells as distinct from self. Immune checkpoint activities dampen T cell activation when antigens presented to T cells are weak or potentially self-related. The recent emergence of immune checkpoint inhibitors may offer exciting new and effective combinations of immunotherapy and radiotherapy (12). Treatments to enhance the anticancer complement response may accentuate T cell activation. Removing T cell inhibition with immune checkpoint inhibitors may provide the necessary combination treatment to unleash a truly effective anti-cancer immune response.
All complement components can be produced in the liver. However, studies have also demonstrated that many components, and C3 in particular, are produced by immune cells locally, particularly in ischemic environments (13). The Surace et al. manuscript demonstrated that not only immune cells but also tumor cells themselves produce C3. This is similar to findings described in gastric tumors (14). Initial investigations into the specific pathway of complement activation by Surace et al. indicated involvement of both the classical and alternative pathway with no evidence of lectin pathway activation. The inciting stimuli for complement activation have yet to be defined, but the data suggest that necrotic cells may be involved.
Surace et al. employed two different strategies to assess the critical importance of C3a and C5a in the effectiveness of radiation therapy. One was the use of mice deficient in C3aR, C5a1R or both. This sound strategy assesses the importance of the receptor throughout development in determining if radiation therapy will be effective. The authors also blocked C5a1R with specific antibodies and C3aR with a small molecule antagonist. These approaches add important evidence that the lack of C3a and C5a receptors during development in knockout animals is not the critical event. However, use of inhibitors is always limited by the specificity of the treatment. For example, the C3aR antagonist SB290157 also exhibits agonist activity and may stimulate C3aR and mobilize neutrophils in intestinal ischemia reperfusion injury (15,16). Literature reported doses of SB290157 range from 2–30 mg/kg with minimal data indicating that these doses are C3aR specific and lack off target effects. In fact in the rat model of placental ischemia induced hypertension, 5 mg/kg SB290157 attenuated increased circulating C3a, an unexpected finding for a simple antagonist (15). Limited solubility of SB290157 in aqueous solutions results in experimental delivery as a suspension in saline or PBS.
Although focusing on tumors implanted into mice, Surace et al. also demonstrates complement activation in a limited number of human skin tumors. For further translation to human, both complement and dexamethasone studies should be expanded to include more relevant tumor models that include mutations found in human melanomas, such as the BrafV600E mutation which occurs in over 50% of human melanomas (17,18). As radiotherapy today frequently includes multiple doses of radiation, it will be critical to determine if multiple doses produce similar results and if radiation dosing in mice translates to dosing in humans. Radiotherapy produces inflammation in normal tissue adjacent to target cancer tissues that may lead to side effects such as nausea, emesis, brain edema, pneumonitis and other toxicities. Dexamethasone is frequently used to address these and other side effects. Based on the findings of Surace et al., dexamethasone inhibits radiation induced C3 activation and more importantly allows tumor growth. These intriguing data suggest the use of dexamethasone to protect normal tissues from radiotherapy may also protect tumor tissues. Dexamethasone was recently found to decrease short-term pain flares after radiotherapy for bone metastases without decreasing long term (6 weeks) pain control, suggesting dexamethasone may not uniformly produce tumor protection (19). Very few studies have addressed the possibility of cancer protection resulting from dexamethasone effects on tumor microenvironment, but given the widespread use of dexamethasone with radiotherapy and the findings of Surace, a closer examination of tumor protection with dexamethasone may be worthwhile.
Future studies are needed to understand the specific, and at times opposing, effects of C3a and C5a as well as the role of the second C5a receptor, known as C5a2R (C5L2). The exact role for C5a2R in the overall actions of C5a is unresolved (2). While Surace et al. examined tumor growth and dendritic cell maturation in mice deficient for either C3aR or C5a1R, the radiotherapy-induced T cell response and dexamethasone studies were performed in wild-type and double knock out (C3aR−/−C5a1R−/−) mice. Thus, future studies should examine the role of each specific anaphylatoxin and the additional receptor, C5a2R. This is particularly important as C3a and C5a have opposing actions in multiple diseases including asthma, lupus, and septic shock where C5a is pro-inflammatory and C3a is anti-inflammatory (20). The C3a-C5a dichotomy may also be due to possible interactions of either C5a or C3a with C5a2R. This soluble receptor may be a decoy receptor that interacts with C5a1R to negatively regulate the response of one or both anaphylatoxins (21-24). Delineating the important mediators and receptors may provide a mechanism to explain the excess toxicity in patients with lupus receiving radiotherapy (25).
Therapeutic intervention in the complement system creates a challenge because this system plays an essential role in host defense. The antibody to C5, eculizumab, has successfully treated paroxysmal nocturnal hemoglobinuria and atypical hemolytic uremic syndrome. Both of these diseases involve uncontrolled complement activation due to improperly functioning endogenous complement regulators. Blocking C5 activation prevents formation of the membrane attack complex and complement-mediated cell damage. Eculizumab also minimizes C5a production presenting a distinct disadvantage during radiation therapy unless preserving C3a action is sufficient. Inhibiting C5 activation also increases susceptibility of individuals to meningococcal infection so that monitoring and vaccination are essential. The therapeutic ratio in radiotherapy compares the dose of radiation that kills tumor cells with the dose that causes complications in normal tissue. Inhibiting complement activation may be an opportunity to enhance the therapeutic ratio of radiotherapy. Targeting complement inhibitors to normal tissues but not adjacent cancer tissues may contain normal tissue injury and maximize radiotherapy-induced tumor cell damage. For example, inhaled inhibitors for patients receiving radiotherapy to the esophagus or breast may provide lung protection without compromising target effects.
Traditional approaches to enhance the therapeutic ratio of radiotherapy include greater precision in delivery or use of higher doses. Indeed, high precision delivery is now technically possible for almost all sites using intensity modulated radiation therapy (IMRT), image guided radiation therapy (IGRT) and even proton irradiation. Further advances in precise delivery are limited more by our ability to identify microscopic tumor cells rather than depositing dose precisely in tissue. Dose escalation also appears to have limitations as well. As pointed out by Yamoah et al. (26) recently, several dose escalation studies have failed to show improved benefit. Therefore, improving the efficacy of radiotherapy depends on a more complete understanding of radiation effects on cancer and normal tissue and exploiting these differences for therapeutic gain. The insight provided by Surace et al. opens many new possibilities to selectively augment the action of radiation against cancer.
Acknowledgements
Funding: This work was supported in part by NIH HL109843 (JF Regal, SD Fleming), NIH AI107005 (SD Fleming) and GM103418 (SD Fleming).
Footnotes
Provenance: This is a Guest Editorial commissioned by Section Editor Hongcheng Zhu, MD, PhD (Department of Radiation Oncology, The First Affiliated Hospital of Nanjing Medical University, Nanjing, China).
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Surace L, Lysenko V, Fontana AO, et al. Complement is a central mediator of radiotherapy-induced tumor-specific immunity and clinical response. Immunity 2015;42:767-77. 10.1016/j.immuni.2015.03.009 [DOI] [PubMed] [Google Scholar]
- 2.Klos A, Wende E, Wareham KJ, et al. International Union of Basic and Clinical Pharmacology. [corrected]. LXXXVII. Complement peptide C5a, C4a, and C3a receptors. Pharmacol Rev 2013;65:500-43. 10.1124/pr.111.005223 [DOI] [PubMed] [Google Scholar]
- 3.Garcia-Barros M, Paris F, Cordon-Cardo C, et al. Tumor response to radiotherapy regulated by endothelial cell apoptosis. Science 2003;300:1155-9. 10.1126/science.1082504 [DOI] [PubMed] [Google Scholar]
- 4.Rao SS, Thompson C, Cheng J, et al. Axitinib sensitization of high Single Dose Radiotherapy. Radiother Oncol 2014;111:88-93. 10.1016/j.radonc.2014.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bode GH, Losen M, Buurman WA, et al. Complement activation by ceramide transporter proteins. J Immunol 2014;192:1154-61. 10.4049/jimmunol.1301673 [DOI] [PubMed] [Google Scholar]
- 6.Baruah P, Simpson E, Dumitriu IE, et al. Mice lacking C1q or C3 show accelerated rejection of minor H disparate skin grafts and resistance to induction of tolerance. Eur J Immunol 2010;40:1758-67. 10.1002/eji.200940158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ghannam A, Fauquert JL, Thomas C, et al. Human complement C3 deficiency: Th1 induction requires T cell-derived complement C3a and CD46 activation. Mol Immunol 2014;58:98-107. 10.1016/j.molimm.2013.11.010 [DOI] [PubMed] [Google Scholar]
- 8.Ghannam A, Pernollet M, Fauquert JL, et al. Human C3 deficiency associated with impairments in dendritic cell differentiation, memory B cells, and regulatory T cells. J Immunol 2008;181:5158-66. 10.4049/jimmunol.181.7.5158 [DOI] [PubMed] [Google Scholar]
- 9.Ma Q, Li D, Carreño R, et al. Complement component C3 mediates Th1/Th17 polarization in human T-cell activation and cutaneous GVHD. Bone Marrow Transplant 2014;49:972-6. 10.1038/bmt.2014.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Varela JC, Imai M, Atkinson C, et al. Modulation of protective T cell immunity by complement inhibitor expression on tumor cells. Cancer Res 2008;68:6734-42. 10.1158/0008-5472.CAN-08-0502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Formenti SC, Demaria S. Combining radiotherapy and cancer immunotherapy: a paradigm shift. J Natl Cancer Inst 2013;105:256-65. 10.1093/jnci/djs629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Esposito A, Criscitiello C, Curigliano G. Immune checkpoint inhibitors with radiotherapy and locoregional treatment: synergism and potential clinical implications. Curr Opin Oncol 2015;27:445-51. 10.1097/CCO.0000000000000225 [DOI] [PubMed] [Google Scholar]
- 13.Pope MR, Hoffman SM, Tomlinson S, et al. Complement regulates TLR4-mediated inflammatory responses during intestinal ischemia reperfusion. Mol Immunol 2010;48:356-64. 10.1016/j.molimm.2010.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kitano E, Kitamura H. Synthesis of the third component of complement (C3) by human gastric cancer-derived cell lines. Clin Exp Immunol 1993;94:273-8. 10.1111/j.1365-2249.1993.tb03443.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lillegard KE, Loeks-Johnson AC, Opacich JW, et al. Differential effects of complement activation products c3a and c5a on cardiovascular function in hypertensive pregnant rats. J Pharmacol Exp Ther 2014;351:344-51. 10.1124/jpet.114.218123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu MC, Brennan FH, Lynch JP, et al. The receptor for complement component C3a mediates protection from intestinal ischemia-reperfusion injuries by inhibiting neutrophil mobilization. Proc Natl Acad Sci U S A 2013;110:9439-44. 10.1073/pnas.1218815110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature 2002;417:949-54. 10.1038/nature00766 [DOI] [PubMed] [Google Scholar]
- 18.Chin L. The genetics of malignant melanoma: lessons from mouse and man. Nat Rev Cancer 2003;3:559-70. 10.1038/nrc1145 [DOI] [PubMed] [Google Scholar]
- 19.Chow E, Meyer RM, Ding K, et al. Dexamethasone in the prophylaxis of radiation-induced pain flare after palliative radiotherapy for bone metastases: a double-blind, randomised placebo-controlled, phase 3 trial. Lancet Oncol 2015;16:1463-72. 10.1016/S1470-2045(15)00199-0 [DOI] [PubMed] [Google Scholar]
- 20.Coulthard LG, Woodruff TM. Is the complement activation product C3a a proinflammatory molecule? Re-evaluating the evidence and the myth. J Immunol 2015;194:3542-8. 10.4049/jimmunol.1403068 [DOI] [PubMed] [Google Scholar]
- 21.Bamberg CE, Mackay CR, Lee H, et al. The C5a receptor (C5aR) C5L2 is a modulator of C5aR-mediated signal transduction. J Biol Chem 2010;285:7633-44. 10.1074/jbc.M109.092106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen NJ, Mirtsos C, Suh D, et al. C5L2 is critical for the biological activities of the anaphylatoxins C5a and C3a. Nature 2007;446:203-7. 10.1038/nature05559 [DOI] [PubMed] [Google Scholar]
- 23.Rittirsch D, Flierl MA, Nadeau BA, et al. Functional roles for C5a receptors in sepsis. Nat Med 2008;14:551-7. 10.1038/nm1753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scola AM, Johswich KO, Morgan BP, et al. The human complement fragment receptor, C5L2, is a recycling decoy receptor. Mol Immunol 2009;46:1149-62. 10.1016/j.molimm.2008.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pinn ME, Gold DG, Petersen IA, et al. Systemic lupus erythematosus, radiotherapy, and the risk of acute and chronic toxicity: the Mayo Clinic Experience. Int J Radiat Oncol Biol Phys 2008;71:498-506. 10.1016/j.ijrobp.2007.10.014 [DOI] [PubMed] [Google Scholar]
- 26.Yamoah K, Showalter TN, Ohri N. Radiation Therapy Intensification for Solid Tumors: A Systematic Review of Randomized Trials. Int J Radiat Oncol Biol Phys 2015;93:737-45. 10.1016/j.ijrobp.2015.07.2284 [DOI] [PMC free article] [PubMed] [Google Scholar]