

Abstract

A kinase-targeting cell-based high-throughput screen (HTS) against Trypanosoma brucei was recently reported, and this screening set included the Published Kinase Inhibitor Set (PKIS). From the PKIS was identified 53 compounds with pEC50 ≥ 6. Utilizing the published data available for the PKIS, a statistical analysis of these active antiparasitic compounds was performed, allowing identification of a set of human kinases having inhibitors that show a high likelihood for blocking T. brucei cellular proliferation in vitro. This observation was confirmed by testing other established inhibitors of these human kinases and by mining past screening campaigns at GlaxoSmithKline. Overall, although the parasite targets of action are not known, inhibitors of this set of human kinases displayed an enhanced hit rate relative to a random kinase-targeting HTS campaign, suggesting that repurposing efforts should focus primarily on inhibitors of these specific human kinases. We therefore term this statistical analysis-driven approach “preferred lead repurposing”.
Keywords: preferred lead repurposing, Trypanosoma brucei, Trypanosoma cruzi, Leishmania major, Plasmodium falciparum, Published Kinase Inhibitor Set
Neglected tropical diseases (NTDs) are a collection of infectious diseases that contribute to a significant level of morbidity and mortality, particularly in the developing world. For example, human African trypanosomiasis (HAT) is caused by the insect-borne protozoan pathogen Trypanosoma brucei, and the resulting infection results in 1.6 million disability-adjusted life years.1 Although the disease burden caused by these NTDs is high, there is little financial incentive to engage in the costly process for the discovery and development of drugs, which leaves a current pharmacopeia of mixed efficacy, safety, and convenience. To wit, one of the front-line treatments for HAT is melarsoprol, which is a brain-penetrant organoarsenic agent that itself has at least a 5% mortality rate.2
As a result, we and others have been advancing efforts to repurpose classes of investigational drug agents as new leads for HAT and other NTDs. Termed “target repurposing”3 or “piggy-back”4 drug discovery, these efforts entail assessment of established inhibitors of human enzymes that are homologous to essential parasite enzymes and pursuit of these and related analogues as a launching point for new antiparasitic agents. We have found that kinase inhibitors represent a particularly fertile area for application of target repurposing,5−9 and our pursuit of this family of inhibitors led to a high-throughput kinase-targeted inhibitor screen against T. brucei. In that 42444 compound screening campaign we identified 797 potent (pEC50 ≥ 6) and selective (>100×) inhibitors of the bloodstream form of the parasite.10
Included in the high-throughput screening (HTS) set was the Published Kinase Inhibitor Set (PKIS) released by GlaxoSmithKline (GSK), which consists of a set of 369 kinase inhibitor compounds with inhibitory data available against 224 human kinases at two concentrations (0.1 and 1 μM), providing information regarding each compound’s human kinase selectivity.11 Publicly available in screening plates upon request from GSK, this inhibitor set has been utilized by a number of research groups outside GSK, culminating in a number of papers describing exploration of a wide range of programs of relevance to human health.12
During the analysis of our HTS results, we noted that there were particular families of kinase inhibitors present in the PKIS collection that showed high potency against T. brucei, with good-to-excellent selectivity over host cells (HepG2, see Table S1 in the Supporting Information). This observation led us to ask whether a more rigorous analysis of the PKIS screening hits might uncover more information about which human kinases may share inhibitor sensitivity similar to that of T. brucei cells in vitro.
Although target repurposing approaches are most often launched by a bioinformatic analysis that matches human-to-parasite based on target protein sequence similarity, we hypothesized that an analysis of the PKIS may lead to an ability to match on the basis of similarity of inhibitor sensitivity. Such an analysis could allow a more focused approach to repurposing established kinase inhibitor chemotypes, even in the absence of information about specific parasite target inhibition. We felt that this approach could be valuable given previous work that demonstrated that repurposing chemical matter from human homologues can be successful and be causing their effect, at least in part, by inhibition of the parasite homologue, such as for Aurora kinase,13 or GSK3β.14 However, because much is unknown about the function of many T. brucei kinases, multiple targets may be contributing to the antiproliferative effects of a given kinase inhibitor, as was observed for DDD34425, a potent inhibitor of T. brucei PK50.14 Furthermore, although T. brucei does not express canonical protein tyrosine kinases,15 human tyrosine kinase inhibitors have previously been successfully repurposed to launch lead discovery programs.7,16
In light of these observations, we wished to utilize the unique juxtaposition of T. brucei and human kinase activity data to identify whether there was a significant probability that inhibitors of particular human kinases were more active against T. brucei when compared to a wider set of kinase inhibitors and whether these might provide a good starting point to launch new lead discovery efforts, even in the absence of parasite target information.
To accomplish this, we systematically analyzed the PKIS data to find statistical associations between human kinase inhibition and T. brucei inhibition. Before that, the 369 compounds included in the PKIS were assessed for completeness of data: despite meeting the primary HTS cutoff, four compounds had incomplete EC50 data against T. brucei cells, and/or HepG2 cellular selectivity data. Thus, the overall compound set was reduced to 365.
Compounds were binned on the basis of their T. brucei pEC50 (Table 1). For the human kinase inhibitory analysis, each compound was assigned a score on the basis of whether the compound showed ≥70% inhibition (scored 1) or <70% inhibition (scored 0) at 0.1 μM. The total number of inhibiting compounds was calculated for each kinase. In general, these calculations gave us the number of compounds with a T. brucei pEC50 in a particular range (e.g., pEC50 ≥ 6) that displayed an inhibitory activity against the selected human kinase higher than or equal to the selected cutoff of 70% inhibition at 0.1 μM. This process was repeated for higher cutoff values of kinase inhibition (≥80 and ≥90% inhibition at 0.1 μM) and for several T. brucei pEC50 ranges. A total of 2016 2×2 contingency tables were constructed. Fold enrichments were calculated, and a chi-squared contingency table test was performed to evaluate the statistical significance of the enrichments.17,18 A contingency table was considered significant when its chi-squared test p value was <0.01.
Table 1. Inhibitors Binned on the Basis of T. brucei Proliferation Inhibition.
| pEC50 range | no. of compounds |
|---|---|
| <4 | 253 |
| ≥4 | 112 |
| ≥6 | 53 |
| ≥7 | 13 |
The contingency tables for each kinase were then sorted by their fold enrichment (see Methods for definition). In the interest of using the same cutoff value as used in the original T. brucei HTS, we elected to utilize the pEC50 ≥ 6 cutoff to denote “active” parasite proliferation inhibitors, which provided a large enough sample to provide a statistically valid analysis. The highest-scored kinases (by fold enrichment) and their respective p values are reported in Table 2.
Table 2. Highly Scored Human Kinases for Active T. brucei Inhibitors Defined as pEC50 ≥ 6, Grouped by Human Kinase Percent Inhibition Cutoffsa.
| 70% inhibition cutoff |
80% inhibition cutoff |
90% inhibition cutoff |
||||||
|---|---|---|---|---|---|---|---|---|
| kinase | family | Nb (active vs T. brucei) | enrichment | p value | enrichment | p value | enrichment | p value |
| DYRK1B | CMGC | 6 (6) | 41.20 | 6.06 × 10–8 | 35.32 | 1.19 × 10–6 | ||
| DYRK1A | CMGC | 6 (6) | 41.20 | 6.06 × 10–8 | 23.54 | 3.92 × 10–4 | ||
| ARK5 | CAMK | 4 (4) | 29.43 | 2.24 × 10–5 | ||||
| CDK1/cyclinB | CMGC | 3 (3) | 23.54 | 3.92 × 10–4 | ||||
| CDK3/cyclinE | CMGC | 3 (3) | 23.54 | 3.92 × 10–4 | ||||
| HIPK1 | CMGC | 6 (5) | 17.66 | 1.07 × 10–5 | 17.66 | 1.07 × 10–5 | ||
| CDK5/p35 | CMGC | 2 (2) | 17.66 | 6.17 × 10–3 | ||||
| NEK9 | other | 2 (2) | 17.66 | 6.17 × 10–3 | ||||
| HIPK4 | CMGC | 2 (2) | 17.66 | 6.17 × 10–3 | ||||
| LCK | TK | 2 (2) | 17.66 | 6.17 × 10–3 | ||||
| GSK3α | CMGC | 13 (9) | 11.77 | 4.26 × 10–8 | 11.77 | 5.89 × 10–5 | ||
| CLK2 | CMGC | 7 (5) | 11.77 | 5.89 × 10–5 | 11.77 | 5.89 × 10–5 | 17.66 | 1.07 × 10–5 |
| TSSK1 | CAMK | 7 (5) | 11.77 | 5.89 × 10–5 | 11.77 | 5.89 × 10–5 | 17.66 | 6.17 × 10–3 |
| LRRK2 | TKL | 7 (5) | 11.77 | 5.89 × 10–5 | 9.81 | 7.05 × 10–4 | ||
| ROS | TK | 7 (5) | 11.77 | 5.89 × 10–5 | ||||
| GSK3β | CMGC | 12 (8) | 10.59 | 5.65 × 10–7 | 9.81 | 7.05 × 10–4 | ||
| LTK | TK | 9 (6) | 10.30 | 2.04 × 10–5 | 7.35 | 2.22 × 10–3 | 9.81 | 7.05 × 10–4 |
| CDK2/cyclinE | CMGC | 9 (6) | 10.30 | 2.04 × 10–5 | 17.66 | 6.17 × 10–3 | ||
| TTK | other | 6 (4) | 9.81 | 7.05 × 10–4 | ||||
| IRR | TK | 8 (5) | 8.83 | 2.29 × 10–4 | 7.84 | 7.14 × 10–3 | 7.84 | 7.14 × 10–3 |
| CDK2/cyclinA | CMGC | 13 (8) | 8.83 | 2.23 × 10–6 | 8.24 | 7.37 × 10–5 | ||
| IGF1R | TK | 10 (6) | 8.24 | 7.37 × 10–5 | 8.24 | 7.37 × 10–5 | 10.30 | 2.04 × 10–5 |
| ALK | TK | 10 (6) | 8.24 | 7.37 × 10–5 | 7.06 | 6.95 × 10–4 | ||
| LRRK2-G2019S | TKL | 5 (3) | 7.84 | 7.14 × 10–3 | 11.77 | 2.13 × 10–3 | ||
| PYK2 | TK | 9 (5) | 7.06 | 6.95 × 10–4 | ||||
| INSR | TK | 11 (6) | 6.86 | 2.18 × 10–4 | 6.86 | 2.18 × 10–4 | 7.06 | 6.95 × 10–4 |
| PLK1–002 | other | 16 (8) | 5.88 | 5.37 × 10–5 | 8.83 | 2.23 × 10–6 | 11.77 | 1.61 × 10–6 |
| ABL-Q252H | TK | 5 (3) | 7.84 | 7.14 × 10–3 | ||||
| ABL-M351T | TK | 3 (2) | 7.84 | 7.14 × 10–3 | ||||
Kinases with missing data do not display a statistically significant enrichment at the respective percent inhibition cutoff.
N, number of inhibitors from the PKIS that show ≥70% inhibition of the preferred kinase at 0.1 μM concentration; the number in parentheses represents the number of these inhibitors with T. brucei pEC50 ≥ 6.
On the basis of these results, we note that the most highly scored kinases belong to the human CMGC kinase family, which includes DYRK, CDK, GSK-3, and HIPK. Lower fold enrichment values have been found for tyrosine kinases (TK) (LCK, ROS, LTK, IGF1R, ALK, PYK2, INSR, and ABL mutant variants) and tyrosine-kinase-like kinases (TKL) (LRRK2 and its mutant variant LRRK2-G2019S). A graphical representation of the kinase enrichment scores, grouped by kinase family, is reported in Figure 1.
Figure 1.

Fold enrichment of human kinases, grouped by family, using three human kinase percent inhibition cutoffs (70, 80, and 90%). Kinases missing histogram bars do not display a statistically significant enrichment at the respective percent inhibition cutoff.
The translation of these observations to prospective application would be desirable. Thus, on the basis of the statistical analysis performed, we selected from commercial vendors 26 established inhibitors of the “preferred” human kinases (Tables S2 and S3, Supporting Information). To assess the structural similarity of the commercial inhibitors with the PKIS compounds, the Tanimoto coefficients between all of the former were calculated against all of the latter. We wished to ensure that any enhancement to the hit rate would not be the result of testing close analogues of the original PKIS compounds. The similarity assessment results are shown in Figure 2 in the form of box plots that show the range of structural similarity between the purchased compounds and PKIS inhibitors, grouped by the preferred kinase. (These data are also found in Table S4 of the Supporting Information.)
Figure 2.

Box plots showing Tanimoto scores of the purchased kinase inhibitors shown in Table 3 to members of the PKIS that inhibit the same primary human target. The primary human target is defined as the highest percent inhibition at 0.1 μM drug concentration.
Confident in the structural diversity of these purchased compounds over the original PKIS hits, we assessed them using the same T. brucei assay that was performed in the HTS campaign. The results of these screening experiments are shown in Table 3; 13 compounds showed a growth inhibition pEC50 ≥ 6. This result represents an overall 50% hit rate (13/26) (compared to 4.2% for the overall HTS, which was performed on a random set of kinase-targeting inhibitors (1797 compounds with pEC50 ≥ 6, of 42444)). The T. brucei hit rate of the PKIS collection was 14.3% (53/369).
Table 3. Inhibitors of Preferred Human Kinase Purchased and Tested against Protozoan Pathogens and Host Cells.
| T. brucei |
Leishmania |
T. cruzi |
P. falciparum |
host
cell tox |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| no. | compd | compd name | human kinase (pIC50) | pEC50 (within 46%) | Promast pEC50 | Amast pEC50 | pEC50 (μM) (within 21%) | D6 pEC50 | W2 pEC50 | C235 pEC50 | 3T3 pTC50 | HepG2 pTC50 |
| 1 | NEU-838 | crizotinib | ALK (7.6) |
6.4 | 6.2 | 5.8 | 5.1 | 6.1 | 7.4 | 7.0 | 4.8 | 5.4 |
| 2 | NEU-844 | TAE684 | ALK (8.5) |
7.0 | 6.5 | 5.9 | 5.3 | 6.3 | 6.4 | 6.3 | 4.6 | 5.3 |
| 3 | NEU-893 | GSK1838705A | ALK (9.3) |
5.7 | <4.6 | <4.8 | 5.0 | 5.6 | 5.8 | 5.8 | 4.8 | 5.1 |
| 4 | NEU-967 | NVP-AEW5414 | IGF-1R (6.8) |
5.7 | 7.0 | 5.4 | 5.4 | 5.9 | 6.0 | 5.9 | 4.8 | 5.5 |
| 5 | NEU-968 | BMS-2652465 | CDK1-cyclinB (8.2) | 6.3 | 6.6 | <4.8 | <4.3 | 4.8 | 5.0 | 4.9 | <4 | <4.4 |
| 6 | NEU-969 | PHA-8481256 | CDK2-cyclinA (7.3) | 7.6 | 6.5 | 5.3 | 7.4 | 5.2 | 5.1 | 5.2 | <4 | 5.0 |
| 7 | NEU-970 | GSK1904529A7 | IGF-1R (7.6) |
5.9 | <4.9 | 5.5 | <4.3 | 6.8 | 7.0 | 6.9 | <4 | <4.4 |
| 8 | NEU-971 | JNJ-77066218 | CDK2-cyclinE (8.5) | 6.5 | <4.6 | <4.8 | <4.3 | 5.7 | 6.0 | 5.9 | <4 | 5.2 |
| 9 | NEU-973 | ON-019109 | PLK1 (8.0) |
<5.5 | <4.6 | <4.8 | <4.3 | <4.7 | <4.7 | <4.7 | <4 | <4.4 |
| 10 | NEU-974 | CH542480210 | ALK (8.7) |
<5.5 | 6.9 | <4.8 | <4.3 | 5.4 | <4.7 | <4.7 | <4 | <4.4 |
| 11 | NEU-975 | dinaciclib | CDK2 (9.0) |
7.4 | 5.8 | 5.6 | 7.0 | 5.9 | 6.1 | 6.5 | <4 | <4.4 |
| P35/CDK5 (9.0) | ||||||||||||
| 12 | NEU-976 | linsitinib | IGF1R (7.5) |
<5.5 | 5.1 | <4.8 | 5.0 | 6.3 | 5.7 | 6.3 | <4 | 4.5 |
| 13 | NEU-977 | roscovitine 13 | P35/CDK5 (9.0) |
<5.5 | 5.2 | <4.8 | <4.3 | 6.1 | 5.7 | 5.7 | <4 | 4.5 |
| 14 | NEU-978 | CHIR-9801414 | GSK3α/β (9.2) |
<5.5 | 5.2 | <4.8 | <4.3 | <4.7 | <4.7 | <4.7 | <4 | 4.8 |
| 15 | NEU-979 | volasertib | PLK1 (9.1) |
<5.5 | 5.8 | 5.1 | 4.9 | 6.8 | 6.8 | 6.8 | 4.6 | 5.5 |
| 16 | NEU-980 | BI 253616 | PLK1 (9.1) |
6.2 | 5.4 | 5.1 | 4.7 | 7.7 | 8.0 | 7.9 | 4.5 | 5.6 |
| 17 | NEU-982 | AT751918 | p35/CDK5 (7.9) |
6.5 | <4.6 | <4.8 | 5.9 | 5.9 | 6.1 | 6.5 | <4 | <4.4 |
| 18 | NEU-984 | SNS-03219, 20 | CDK9/cyclinT (8.4) |
6.9 | <4.6 | <4.8 | < 4.3 | 5.3 | 5.5 | 5.7 | <4 | <4.4 |
| 19 | NEU-985 | AZD543821 | CDK2/cyclinE (8.2) |
7.5 | 6.4 | 5.6 | 5.8 | 5.9 | 6.0 | 5.9 | <4 | 4.9 |
| 20 | NEU-986 | SB 41528622 | GSK-3α (7.1) |
6.1 | <4.6 | <4.8 | <4.3 | 4.8 | 4.6 | 4.6 | 4.2 | <4.4 |
| 21 | NEU-987 | flavopiridol | CDK1/2/4/6 (7.4) |
6.8 | 6.4 | 5.5 | 4.9 | 5.7 | 5.7 | 5.8 | < 4 | < 4.4 |
| 22 | NEU-989 | TWS11925 | GSK-3β (7.5) |
6.0 | 4.7 | <4.8 | <4.3 | 5.0 | 4.7 | 5.1 | 4.4 | <4.4 |
| 23 | NEU-990 | SB 21676322 | GSK-3α (7.5) |
<5.5 | 5.6 | <4.8 | <4.3 | <4.7 | <4.7 | <4.7 | <4 | <4.4 |
| 24 | NEU-991 | tideglusib | GSK-3β (7.2) |
<5.5 | 6.5 | <4.8 | 4.7 | 4.6 | 4.6 | 4.7 | 4.1 | <4.4 |
| 25 | NEU-1007 | PHA-79388727 | p25/CDK5 (8.3) |
5.7 | <4.6 | <4.8 | <4.3 | 5.7 | 5.6 | 5.6 | <4 | <4.4 |
| 26 | NEU-1049 | harmine | DYRK1B (7.6) |
<5.5 | 5.9 | 5.9 | 4.8 | <4.7 | <4.7 | <4.7 | <4 | <5.1 |
For example, Figure 3 shows the progression from GSK1173862A, an “active” ALK inhibitor in the PKIS, to four purchased ALK inhibitors. We note that two of the purchased inhibitors of human ALK (crizotinib and NVP-TAE684) are potent T. brucei proliferation inhibitors, despite their structural uniqueness compared to the original PKIS hit chemotype.
Figure 3.

Progression of a representative inhibitor class from a preferred human kinase to other ALK chemotypes with T. brucei proliferation activity.
We wished to test the significance of the observed enrichments using a wider set of investigational inhibitors of “preferred” kinases. We performed a retrospective analysis of HTS hit compounds that had previously emerged from internal GSK kinase inhibitor discovery programs. The expectation would be that if we were to select compounds from the HTS that were originally designed for one of the “preferred” human kinases, we should see an enhancement of hit rate against T. brucei when compared to inhibitors that were originally designed for other human kinases. The GSK database was searched to find screening hits active against the preferred kinases. After the removal of any PKIS compounds and their analogues included in this data set, nine human kinase screens were selected where at least 100 molecules were shared with the T. brucei screen. Contingency tables were built by comparing “actives/inactives” of T. brucei versus “actives/inactives” of the tested kinase, where “actives” are defined as having ≥80% inhibition in single-concentration assays. Chi-squared contingency table tests were performed and the corresponding p values determined as described above. Of these kinases analyzed in this way (ALK, ARK5, CDK2/cyclinA, GSK3β, DYRK1A, IGF1R, LCK, LRRK2, and PLK1), all showed conclusive evidence of significance in the chi-square test (p < 0.01 and enrichments > 1; Figure 4).
Figure 4.
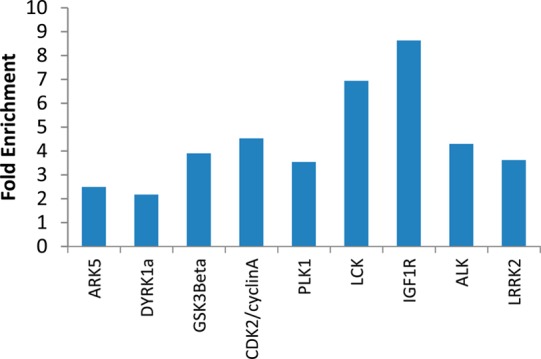
Fold enrichment of hit rate in T. brucei for actives in different preferred kinases utilizing the 80% human kinase inhibition cutoff. Data from single-concentration assays were retrieved (at least 100 compounds shared between T. brucei screen and kinase screen).
In addition, we checked that “non-preferred” kinases from our analysis did not show statistical association with T. brucei inhibition by using non-PKIS compounds in retrospective GSK screens (Figure S4 in the Supporting Information).
These results, obtained with compounds structurally different from the PKIS and from the commercial set of inhibitors, further demonstrate that these particular human kinases are “preferred” in terms of their susceptibility to inhibitors that also show a high probability of anti-T. brucei activity.
Finally, we were interested to know whether these “preferred” T. brucei proliferation inhibitors would have a similar activity against other protozoan parasites. We reasoned that this would be possible because the three related kinetoplastid parasites, T. brucei, T. cruzi, and Leishmania major, have highly similar kinomes.15 Thus, in the interest of trying to ascertain cross-parasite relevance of these “preferred” kinase inhibitors, we also assessed these compounds against T. cruzi intracellular amastigotes (Tulahuen strain), as well as against L. major promastigotes and intracellular amastigotes. In addition, we tested these inhibitors against the malaria-causing protozoan Plasmodium falciparum (D6 strain). Little activity was observed, indicating that these inhibitors are apparently “preferred“ only as T. brucei growth inhibitors (see Table 3 and Figure S2 in the Supporting Information).
In summary, by testing the PKIS against T. brucei cells, we were able to identify kinases having inhibitors that are predisposed to be active against T. brucei cells. By performing a prospective analysis using structurally unique, commercially available inhibitors of these preferred kinases, and a retrospective analysis of historical GSK kinase HTS data, we confirmed that, in fact, inhibitors of these kinases are excellent starting points for launching new T. brucei drug discovery efforts. In contrast to using a human/parasite target homology-based approach for compound selection (also known as target repurposing), the utilization of such a “preferred” kinase analysis represents a method for a ligand-centric hit identification process that we term “preferred lead repurposing”. Although we have presented evidence of such an approach working for hit identification for T. brucei drug discovery, the hits identified are not necessarily promising hits for other protozoan parasites. In due course, as PKIS screening results are reported for other pathogens such as T. cruzi, L. major, or P. falciparum, we look forward to identification of “preferred kinases” that can help focus lead repurposing efforts for these pathogens, as well. Finally, we note that several of the potent and nontoxic T. brucei proliferation inhibitors that we have identified in this work are compounds that have advanced into human clinical trials for a variety of indications. Encouraged by this, we have launched a program to further evaluate some of these compounds for direct repurposing against HAT and have initiated systematic medicinal chemistry programs for others to optimize potency and properties to be consistent with the targeted profile for new HAT therapeutics.
We note that some compounds may not be well suited for antiparasitic agents by virtue of their potent activity against human kinases. However, given that some of these kinase inhibitors are tolerated in humans for extended dosing regimens and that antiparasitic therapies would likely require a much shorter treatment period, we posit that in some cases exquisite selectivity for T. brucei cells versus over host kinases may not be required. On the other hand, it would seem prudent to minimize such host kinase activities, which would be the goal of further medicinal chemistry optimization. We propose that focusing on inhibitors of “preferred” human kinases provides a strong starting point for such optimization programs.
Methods
Biological Assays
The T. brucei and HepG2 cell assays were performed as previously described,10 as were the P. falciparum, L. major, and T. cruzi assays.8 The HepG2 cells were sourced ethically, and their research use was in accord with the terms of the informed consent.
Statistical Analyses
A 2×2 contingency table was constructed, crossing T. brucei “active/inactive” with human kinase “active/inactive” (see Supporting Information, Figure S1). A total number of 2016 contingency tables were generated (224 kinases × 3 kinase % inhibition cutoffs × 3 pEC50 cutoffs), which considered 81760 T. brucei activit–-human kinase activity combinations (365 × 224).
From these tables, two conditional probabilities have been calculated as reported below:
Finally, fold enrichment has been calculated as ratio of the two conditional probabilities:
If the number of compounds falling in quadrant A (Tb+|Kinase+), that is, active against the selected kinase and active against T. brucei, is higher than the number of compounds falling in quadrant C (Tb–|Kinase+), that is, active against the selected kinase but inactive against T. brucei, the fold enrichment will approach values higher than 1, whereas in the opposite cases the fold enrichment for that particular human kinase will be lower than 1. We elected to utilize the T. brucei pEC50 ≥ 6 cutoff, which provided a large enough sample to provide a statistically valid analysis, thus reducing the number of contingency tables to 672 (224 kinases × 3 kinase % inh cutoffs × 1 pEC50 cutoff). p values (chi-squared test) were calculated using R version 2.15.2 (2012-10-26).
Inhibitor Procurement
Compounds were manually selected from Selleck Chemicals on the basis of their activity against “preferred” kinases (Table S3); compounds were used as received.
Similarity Calculations
To get an idea of the structural similarity of the commercial inhibitors tested and the PKIS compounds, the Tanimoto coefficient was calculated for each inhibitor and each PKIS compound by using the RDKit cheminformatic toolkit19 and encoding the structures by means of topological fingerprints. The results were represented as box plots using the R program.
Acknowledgments
We acknowledge funding from The Tres Cantos Open Lab Foundation (TC-007) and the National Institutes of Health (R01AI082577, R01AI114685).
Glossary
Abbreviations
- PKIS
Published Kinase Inhibitor Set
- NTD
neglected tropical disease
- HAT
human African trypanosomiasis
- GSK
GlaxoSmithKline
- HTS
high-throughput screening
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsinfecdis.5b00136.
Additional figures and data tables are compiled, including the full HTS results of the PKIS compounds tested against T. brucei cells, and all of the screening data of the purchased compounds against all of the parasites tested (PDF)
Author Contributions
Contributed to experimental designs: M.P.P., M.N., E.A.. Performed statistical, computational, and cheminformatic analyses: E.A., H.X., G.C. Performed biological assays: R.G.-D., C.C.-O., M.B., P.M., J.E., N.E.R., P.J.L., S.E.L.. Wrote the manuscript: E.A., S.X., G.C., M.P.P. Contributed to data analyses: P.M., A.R., R.J.S.
All of the compound structures and assay data included in this work are also publicly available at www.collaborativedrug.com.
The authors declare no competing financial interest.
Supplementary Material
References
- The Global Burden of Disease: 2012 Update; http://www.who.int/entity/healthinfo/global_burden_disease (accessed Jan 23, 2012).
- Kuepfer I.; Schmid C.; Allan M.; Edielu A.; Haary E. P.; Kakembo A.; Kibona S.; Blum J.; Burri C. (2012) Safety and efficacy of the 10-day melarsoprol schedule for the treatment of second stage rhodesiense sleeping sickness. PLoS Neglected Trop. Dis. 6, e1695. 10.1371/journal.pntd.0001695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollastri M. P.; Campbell R. K. (2011) Target repurposing for neglected diseases. Future Med. Chem. 3, 1307–1315. 10.4155/fmc.11.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nallan L.; Bauer K. D.; Bendale P.; Rivas K.; Yokoyama K.; Horney C. P.; Pendyala P. R.; Floyd D.; Lombardo L. J.; Williams D. K.; Hamilton A.; Sebti S.; Windsor W. T.; Weber P. C.; Buckner F. S.; Chakrabarti D.; Gelb M. H.; Van Voorhis W. C. (2005) Protein farnesyltransferase inhibitors exhibit potent antimalarial activity. J. Med. Chem. 48, 3704–3713. 10.1021/jm0491039. [DOI] [PubMed] [Google Scholar]
- Diaz-Gonzalez R.; Kuhlmann F. M.; Galan-Rodriguez C.; Madeira da Silva L.; Saldivia M.; Karver C. E.; Rodriguez A.; Beverley S. M.; Navarro M.; Pollastri M. P. (2011) The susceptibility of trypanosomatid pathogens to PI3/mTOR kinase inhibitors affords a new opportunity for drug repurposing. PLoS Neglected Trop. Dis. 5, e1297. 10.1371/journal.pntd.0001297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochiana S. O.; Pandarinath V.; Wang Z.; Kapoor R.; Ondrechen M. J.; Ruben L.; Pollastri M. P. (2013) The human Aurora kinase inhibitor danusertib is a lead compound for anti-trypanosomal drug discovery via target repurposing. Eur. J. Med. Chem. 62, 777–784. 10.1016/j.ejmech.2012.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel G.; Karver C. E.; Behera R.; Guyett P. J.; Sullenberger C.; Edwards P.; Roncal N. E.; Mensa-Wilmot K.; Pollastri M. P. (2013) Kinase scaffold repurposing for neglected disease drug discovery: discovery of an efficacious, lapatinib-derived lead compound for trypanosomiasis. J. Med. Chem. 56, 3820–3832. 10.1021/jm400349k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel G.; Roncal N. E.; Lee P. J.; Leed S. E.; Erath J.; Rodriguez A.; Sciotti R. J.; Pollastri M. P. (2014) Repurposing human Aurora kinase inhibitors as leads for anti-protozoan drug discovery. MedChemComm 5, 655–658. 10.1039/c4md00045e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seixas J. D.; Luengo-Arratta S. A.; Diaz R.; Saldivia M.; Rojas-Barros D. I.; Manzano P.; Gonzalez S.; Berlanga M.; Smith T. K.; Navarro M.; Pollastri M. P. (2014) Establishment of a structure-activity relationship of the NVP-BEZ235 chemotype as a lead for African sleeping sickness. J. Med. Chem. 57, 4834–4848. 10.1021/jm500361r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz R.; Luengo-Arratta S. A.; Seixas J. D.; Amata E.; Devine W.; Cordon-Obras C.; Rojas-Barros D. I.; Jimenez E.; Ortega F.; Crouch S.; Colmenarejo G.; Fiandor J. M.; Martin J. J.; Berlanga M.; Gonzalez S.; Manzano P.; Navarro M.; Pollastri M. P. (2014) Identification and characterization of hundreds of potent and selective inhibitors of Trypanosoma brucei growth from a kinase-targeted library screening campaign. PLoS Neglected Trop. Dis. 8, e3253. 10.1371/journal.pntd.0003253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dranchak P.; MacArthur R.; Guha R.; Zuercher W. J.; Drewry D. H.; Auld D. S.; Inglese J. (2013) Profile of the GSK published protein kinase inhibitor set across ATP-dependent and-independent luciferases: implications for reporter-gene assays. PLoS One 8, e57888. 10.1371/journal.pone.0057888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drewry D. H.; Willson T. M.; Zuercher W. J. (2014) Seeding collaborations to advance kinase science with the GSK Published Kinase Inhibitor Set (PKIS). Curr. Top. Med. Chem. 14, 340–342. 10.2174/1568026613666131127160819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochiana S. O.; Pandarinath V.; Wang Z.; Kapoor R.; Ondrechen M. J.; Ruben L.; Pollastri M. P. (2013) The human Aurora kinase inhibitor danusertib is a lead compound for anti-trypanosomal drug discovery via target repurposing. Eur. J. Med. Chem. 62, 777–784. 10.1016/j.ejmech.2012.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbaniak M. D.; Mathieson T.; Bantscheff M.; Eberhard D.; Grimaldi R.; Miranda-Saavedra D.; Wyatt P.; Ferguson M. A. J.; Frearson J.; Drewes G. (2012) Chemical proteomic analysis reveals the drugability of the kinome of Trypanosoma brucei. ACS Chem. Biol. 7, 1858–1865. 10.1021/cb300326z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons M.; Worthey E. A.; Ward P. N.; Mottram J. C. (2005) Comparative analysis of the kinomes of three pathogenic trypanosomatids: Leishmania major, Trypanosoma brucei and Trypanosoma cruzi. BMC Genomics 6, 127. 10.1186/1471-2164-6-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behera R.; Thomas S. M.; Mensa-Wilmot K. (2014) New chemical scaffolds for human african trypanosomiasis lead discovery from a screen of tyrosine kinase inhibitor drugs. Antimicrob. Agents Chemother. 58, 2202–2210. 10.1128/AAC.01691-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patefield W. M. (1981) Algorithm AS159. An efficient method of generating r × c tables with given row and column totals. Appl. Stat. 30, 91–97. 10.2307/2346669. [DOI] [Google Scholar]
- Agresti A. (2007) An Introduction to Categorical Data Analysis, 2nd ed., Wiley, New York. [Google Scholar]
- RDKit; http://www.rdkit.org.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.