

Abstract
Study Objectives:
Kleine-Levin syndrome (KLS) is a rare disorder of relapsing sleepiness. The hypothesis was that the syndrome is related to a change in the vigilance peptide orexin A.
Methods:
From 2002 to 2013, 57 patients with relapsing hypersomnolence were clinically assessed in a referral academic center in Beijing, China, and 44 (28 males and 16 females; mean age 18.3 ± 8.9 y (mean ± standard deviation, range 9–57 y) were determined to have clinical and behavioral criteria consistent with KLS. Cerebrospinal fluid orexin A levels and diurnal blood pressure were measured in relapse versus remission in a subgroup of patients.
Results:
Presenting symptoms included relapsing or remitting excessive sleepiness–associated parallel complaints of cognitive changes (82%), eating disorders (84%); depression (45%); irritability (36%); hypersexuality (18%); and compulsions (11%). Episodes were 8.2 ± 3.3 days in duration. In relapse, diurnal values for blood pressure and heart rate were lower (P < 0.001). In a subgroup (n = 34), cerebrospinal fluid orexin A levels were ∼31% lower in a relapse versus remission (215.7 ± 81.5 versus 319.2 ± 95.92 pg/ml, P < 0.001); in three patients a pattern of lower levels during subsequent relapses was documented.
Conclusions:
There are lower orexin A levels in the symptomatic phase than in remission and a fall and rise in blood pressure and heart rate, suggesting a role for orexin dysregulation in KLS pathophysiology.
Citation:
Wang JY, Han F, Dong SX, Li J, An P, Zhang XZ, Chang Y, Zhao L, Zhang XL, Liu YN, Yan H, Li QH, Hu Y, Lv CJ, Gao ZC, Strohl KP. Cerebrospinal fluid orexin A levels and autonomic function in Kleine-Levin syndrome. SLEEP 2016;39(4):855–860.
Keywords: CSF, hypersomnolence, orexin, syndrome
Significance.
Kleine Levin Syndrome is a rare disorder of recurrent hypersomnia, accompanied by autonomic and behavioral symptoms. This investigation connects the relapsing hypersomnia to cardiovascular downregulation and to reductions in CSF orexin A. Such biological markers may be clues to the mechanisms for the presentation and suggests orexinergic dysfunction as a target for therapy
INTRODUCTION
Kleine-Levin syndrome (KLS) is a rare disorder of episodic hypersomnia (∼1–1.5/million persons). It is distinguished from other disorders of excessive sleepiness by recovery and, in Western clinical reports, ancillary features of cognitive impairment, apathy and/or disinhibition, hyperphagia, hypersexuality, derealization, and dysautonomia.1 Symptoms remit over days to weeks; relapses occur; and this cyclic pattern is a defining feature. The course of KLS usually ends in clinical remission. Clinical acumen is needed for recognition and diagnosis.
Functional imaging identifies thalamic hypoperfusion,2,3 thalamic activation,4 and/or widespread hypermetabolism.5 Some features are present in remission as well as relapse3; for instance, thalamic-pontine connectivity appears altered in and out of the disease state, with reversible deficits present in the dorsal pons.6 Confounding variables in the interpretation of imaging studies is the dynamic entry and variable length of both remission and recovery.1
Narcolepsy with cataplexy is also a disorder of hypersomnolence characterized by a pathologic loss of orexin containing neurons in the hypothalamus with chronically and consistently very low cerebrospinal fluid (CSF) orexin levels.1,7 In KLS reports mentioning orexin levels, a twofold reduction in CSF orexin A (also known as hypocretin 1 levels in a patient with Prader-Willi syndrome meeting clinical criteria for KLS8 and another reported a low range level in a 14-y-old girl with syndromic KLS.9 Other reports are mixed. Katz and Roper10 reported CFS markers in two siblings in and out of episode and reported a low value in one; Knudsen et al.11 reported values for one patient in the normal range; and Bourgin et al.12 in their review incidentally reported normal levels in six patients and intermediate in two patients. These records suggest that orexin levels may vary but are not the very low values observed in narcolepsy with cataplexy.
Variations in orexin levels occur above the pathologic range. Animal models indicate that CNS orexin levels are lower but not absent in induced depression.13 Hypothalamic orexin levels vary among common mouse strains, increased by the substitution of a single chromosome.14 In animal models of orexin deficiency and in human narcolepsy, blood pressure is generally lower.15 Indeed, lower blood pressures and heart rate (HR) are reported in the hypersomnia phase in KLS patients.16 These observations form the rationale for systematic collection of CSF orexin levels and autonomic function over the course of KLS relapse.
As a referral center for hypersomnolence in Beijing China, we encountered over time a number of KLS patients,17 and were exploring an orexin hypothesis. We found lower orexin levels, and a lower blood pressure and HR, associated with the symptomatic phase of the syndrome.
METHODS
Human Subjects
Patients were recruited at the Sleep Center at People's Hospital, Beijing University. In 2004, the institutional review board of Beijing University approved the retrospective collection of clinical information from 2002, and prospective collection of all clinical information, CSF samples, and measurements of continuous blood pressure and HR. Written informed consent was obtained from all subjects or parents (if the child was a minor). At any time the parents or child could selectively decline sleep testing, the lumbar puncture and/or blood pressure/ pulse measures, or withdraw.
Fifty-seven patients with recurrent hypersomnia were identified from 2002 to early 2013, with collection ending in June 2013, for this analysis. Thirteen patients were excluded because sleepiness episodes were < 2 days, or there was the presence of a primary neurological disorder including head trauma, or primary psychiatric disorders, or because symptoms were related to menstruation or pregnancy. The remaining 44 participants had history of episodic sleepiness lasting > 2 days (hypersomnia) consistent with KLS.18 All patients were seen at least twice after initial assessment and clinical information was collected over at least one sleepiness/remission episode. Many had several episodes with assessments and sometimes recovery through the end of the study period.
A common, detailed sleep history and neurological examination were obtained at presentation, and confirmed by one or more of the coauthors. Behavioral symptoms were assessed by internists with sleep expertise, without specificity for psychiatric dimensions. Brain computed tomography (CT) and/or magnetic resonance imaging (MRI) scan, and HLADQB106:02 testing were performed. Polysomnography (PSG) and/or multiple sleep latency test (MSLT) was performed in 36 participants: 15 both in remission and relapse, 16 in remission only, and 5 in relapse only. In eight participants, no studies were performed because of lack of consent and/or financial resources.
CSF by lumbar puncture was collected between 10:00 and 13:00 in those who consented to the procedure. De-identified CSF samples were frozen immediately and stored at −80°C for batch analysis. Samples were from 18 patients both in and out of hypersomnia phases, with 14 being collected from disease to recovery and 4 collected between recovery and disease. There were seven patients with samples collected only in the symptomatic phase, and eight patients only in remission. Three patients provided consent for cerebrospinal fluid samples measurement over two episodes, and one within the same episode at three time points: hypersomnia, partial remission (insomnia phase), and recovery.
Measurements of Orexin A
Hypocretin-1 (orexin A) levels were determined using a 125I radioimmunoassay kit (Phoenix Pharmaceuticals, Belmont, CA),19 for which there are known reference values. Samples were measured in duplicate and results averaged. The lower limit of detection is ∼20 pg/mL; the analytic imprecision is ∼5%. Measures in remission and relapse were performed in the same assay.
24-Hour Blood Pressure Measurement
Blood pressure and HR were measured over 24 h using ambulatory blood pressure (Model #90217-18Q; Spacelabs Health-care, Snoqualmie, WA) in (relapse) and out (remission) of hypersomnia episodes in 24 patients. The other 20 patients were not recorded because of equipment unavailability or family constraints.
Data Analysis
Group data are presented as mean and standard deviation (SD). For the primary analyses, differences were tested between clinical illness and remission. Comparisons in the same individual used paired t-tests. Comparisons of subjects with values only in illness or only in remission were made by nonpaired t-tests. The comparison of daytime or nighttime blood pressure levels between remission and relapse were analyzed by a general linear model, using univariate analysis, and by analyses of several points together as a sum for point-to-point comparison. Diurnal values were examined by a cosinor analysis fitting the general cosine function, CS(t) = 1M + A cos (rut + o) to the family of datasets, where CS(t) is the blood pressure or HR value at time t. We report mesor (midline value = M) and the amplitude (A).20 Significance levels are provided in the text or legend. Statistical tests were carried out using SPSS 13.0 software (IBM, Armonk, NY).
RESULTS
During this time period there were 44 patients (28 males and 16 females; mean age 18.3 ± 8.9 y, range 9–57 y at the time of presentation) who met criteria for a condition of hypersomnia lasting > 2 days with a later remission. The age of onset was estimated by the majority (88%) as in their second decade of life. There was no significant difference in the age of onset for males (14.3 ± 4.4 y; range 9–34 y) or females (14.3 ± 7.9 y; range 9–44 y), with average values similar to international experience.1 The average body mass index (BMI; 20.2 ± 2.9 kg/m2) was within the normal range of age-matched healthy Chinese controls (18.5 to 24 kg/m2). CT and/or MRI scans were not informative, showing no anatomic abnormalities. Thirty-three percent carried the human leukocyte antigen HLADQB106:02, similar to rates in a Chinese healthy population.17 PSG (n = 36) was unremarkable. MSLT revealed a sleep latency (SL) of 15.2 ± 3.3 min (range 8.2–20) in remission (n = 31), and 11.2 ± 5.0 min (range 1.7–20) in relapse (n = 20). In 15 patients with MSLT in both remission and relapse, a longer SL (15.3 ± 3.4 min versus 10.4 ± 5.4 min, P = 0.009) was found in remission versus relapse. Fifteen patients (75%) with an MSLT in relapse were pathologically sleepy (≤ 8 min mean sleep onset). No patient in remission had SL in a pathologic range. Four of the 31 patients in remission and 4 of the 20 (all children) in relapse had sleep onset rapid eye movement periods (SOREMs) ≥ 2. None met criteria for narcolepsy with SL ≤ 8 and SOREMs ≥ 2 on the MSLT.
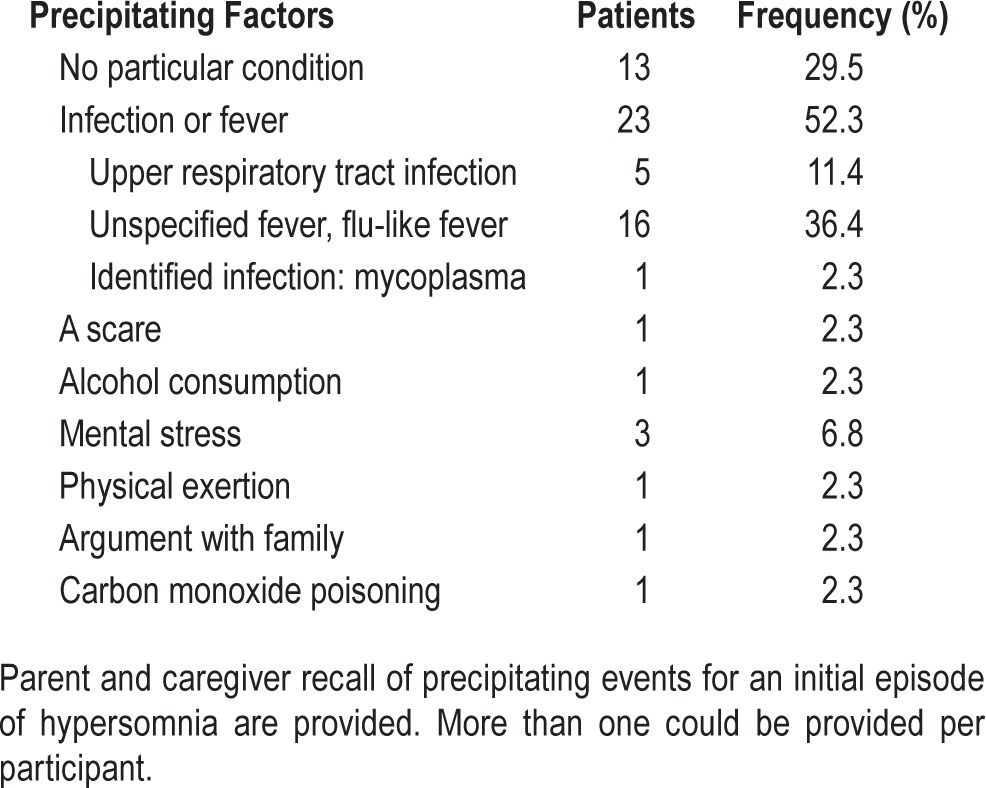
Sleep attacks, sleep drunkenness, prolonged sleep, sleeping during meals, etc. (hypersomnia) had developed acutely over days to weeks to the point of affecting family, social, and personal life and resulted in a patient or family seeking medical advice. The family had consulted other health care providers often (94%), and patients had undergone at least one remission. Recall by parents or the patient of any inciting events for the initial illness are presented in Table 1. Often with relapse there was no precipitant identified; with remissions there were occasional reports of difficulty in sleeping (insomnia, n = 4) giving way to “normal” behavior over several days.
Table 1.
Precipitating factors in 44 patients with Kleine-Levin syndrome.
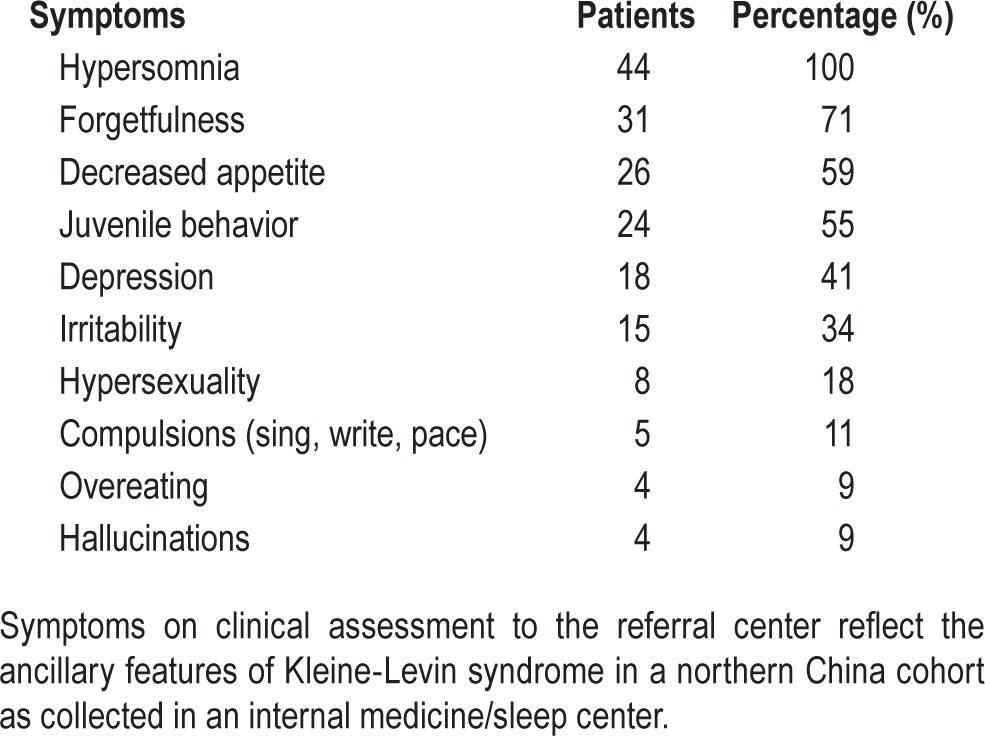
Twenty-nine patients had experienced five or more episodes, and only two patients were assessed after only two cycles. The average episode length was 8.2 ± 3.3 days (overall range: 3 to 30 days), with 20 (68%) between 7 and 10 days. Shortest lengths were an average of 4.6 ± 2.1 days and the longest, 13.7 ± 8 days. One patient reported an episode as short as 3 days, and two patients reported an episode lasting > 16 days. In the hypersomnolent phase there were elicited symptoms of cognitive changes, eating disorders, depressive features, irritability, hypersexuality, and compulsions (Table 2). In 19 patients there have been no episodes for 1 y or more, and 12 have been asymptomatic for 4 y or more.
Table 2.
Presenting symptoms in 44 patients with Kleine-Levin syndrome.
CSF values for orexin A collected in 33 patients are shown in Figure 1. In the 18 patients in whom CSF was collected over both phases, values for CSF orexin were 31% lower in relapse than in remission (319.2 ± 95.9 versus 215.7 ± 81.5 pg/ mL, P < 0.001). Those patients with CSF samples only in one or the other clinical state showed a similar trend (329.4 ± 69.3 versus 128.7 ± 47.0 pg/mL, P < 0.001). There were only three patients with values recorded below a proposed “lower limit” of normal, i.e., ≤ 110 pg/mL.1 CSF orexin A were collected over two episodes in three patients (Table 3). Although variations in difference were noted, in each the level of orexin was lower in relapse than in remission. In measurements of one patient in and following relapse, orexin levels during relapse were 138 pg/mL, toward recovery (when insomnia was noted) 279.5 pg/mL, and in remission 304 pg/mL.
Figure 1.
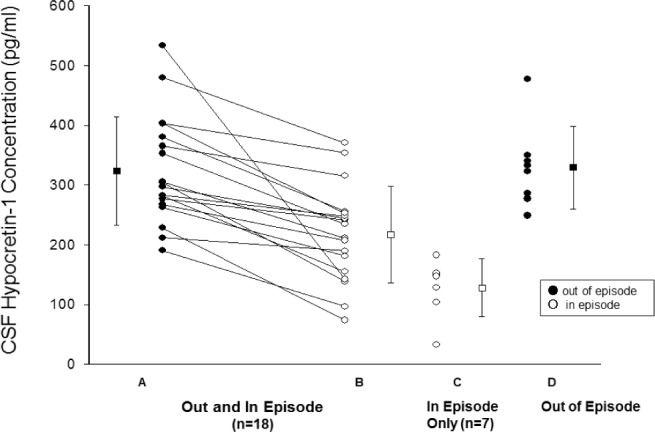
Concentration values for individuals for Orexin A (hypocretin-1) are shown for those values out (closed circles) and in (open circles) of the episode; patients values are connected by a line. On the right are values in different patients measured either in or out of the hypersomia episode. Individual values are shown as circles, and group values presented as mean ± standard deviation by a dash and whiskers. The literature lower limit of normal for CSF orexin is 110 pg/mL. Differences are significant (P < 0.001 P for: values in and out of episode in the same patient (A vs B, n = 18), paired t-test, and between values for groups of patients in (C, n = 7) or out of episode (D, n = 8), non-paired t-test.
Table 3.
CSF orexin-A levels over two episodes of relapse and remission of hypersomnolence in three participants.

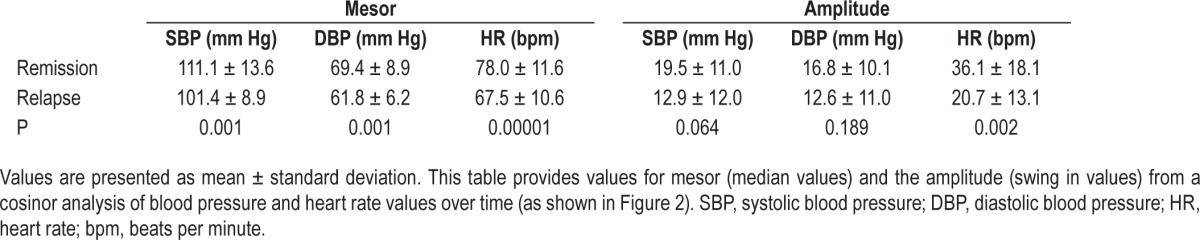
Blood pressure and HR were measured during a clinical phase and in remission in 24 patients (F/M = 9/15) (Figure 2). The daytime (06:00–22:00), nocturnal time (22:00–6:00), and 24-h levels of systolic blood pressure, diastolic blood pressure, and HR were all lower in the symptomatic state than that in remission (all: P < 0.01). Within this group, 18 had CSF orexin A levels measured before or after the diurnal recordings, and all exhibited values of blood pressure and HR in the disease state lower than in remission. Values calculated over the 24-h day (Table 4) found the mesor (mean values) for cardiovascular tone were different (lower) in the clinical phase; only in HR was the amplitude swing over the 24 h different between episodes. Peak, trough, and fit were higher in remission than in the hypersomnia episode (data not shown).
Figure 2.
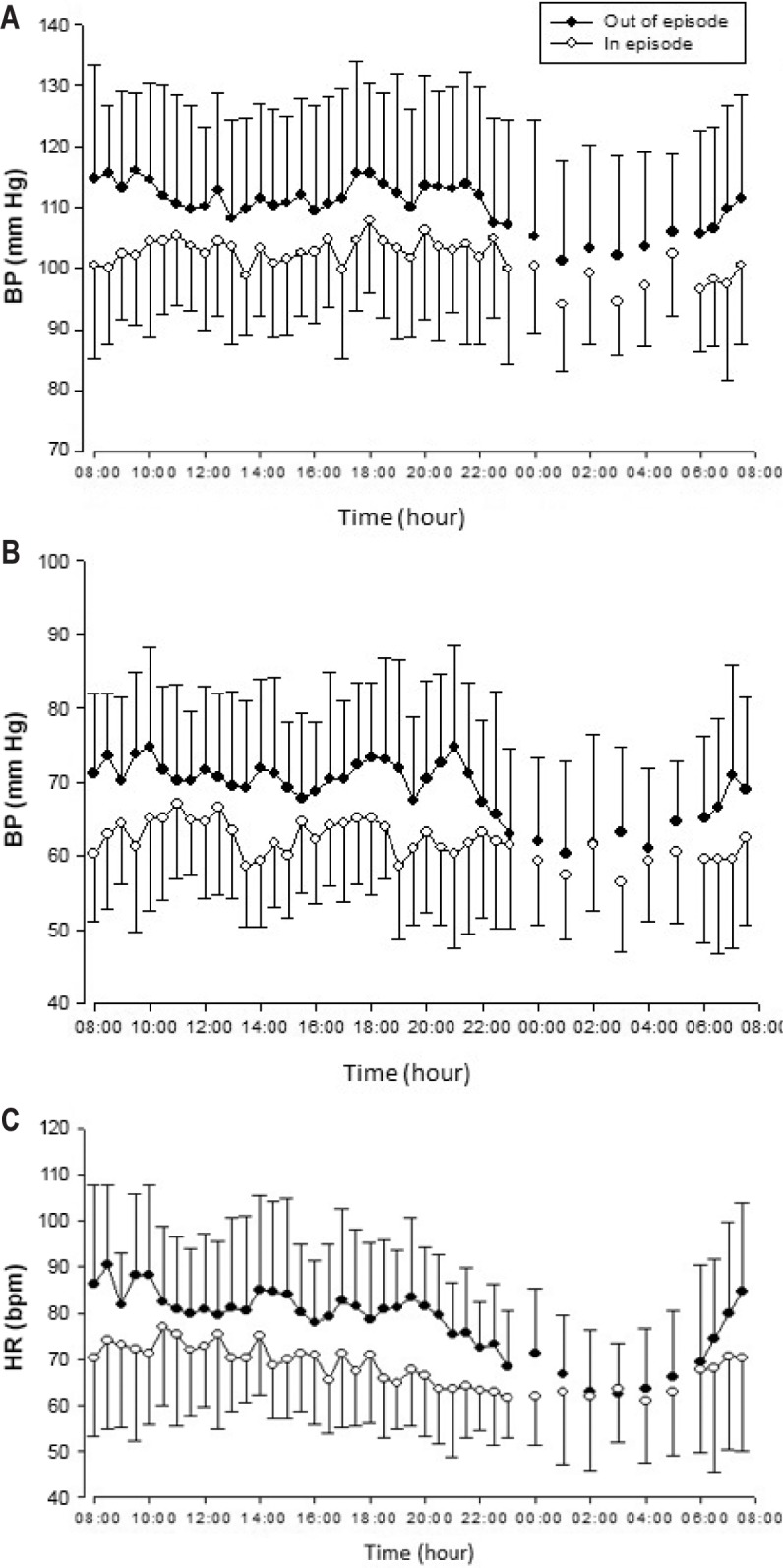
Three 24-h profiles of systolic (A) and diastolic (B) blood pressure and heart rate (C) represent collection of ambulatory values in 24 patients in and out of the episodic clinical state. Data are presented as the mean ± standard deviation. Significance levels are presented in the text and the circadian analysis presented in Table 4. BP, blood pressure; HR, heart rate; bpm, beats per minute.
Table 4.
Diurnal fitting of diurnal blood pressure and heart rate (cosinor summary).
DISCUSSION
In this relatively large Northern Chinese cohort with the rare syndrome of KLS, we found characteristic episodic sleepiness associated with a reduction in CSF orexin A levels. Furthermore, CSF levels will fall a second time upon a second hypersomnia episode. We further show that symptoms and lower levels are associated with objective changes in autonomic functions, consistent with known actions of the peptide.
The 1.8:1 ratio of males to females and childhood and adolescent age of onset of this cohort is consistent with the existing literature.1 Unlike Western samples, however, the BMI was within the normal population range. In addition, there was a relatively low prevalence of reporting of hypersexuality and hyperphagia and greater reporting of a decreased appetite.1 Such differences from Western reports may result from cultural factors, effects of genetic background, or publication bias, but these clinical features, like hypersomnia, remit with recovery.
The current report indicates that blood pressure and HR are reduced in relapse. A lower blood pressure and a non-dipping pattern is observed in narcolepsy, compared to healthy controls,21 and in narcolepsy orexin deficiency was the primary predictor of a blunted HR response to arousals in both non-rapid eye movement and rapid eye movement sleep.22 In animal models there are similar trends of decreasing and increasing autonomic tone with decreased and increased peptide levels or actions.23,24 In the current study, there was reduced diurnal blood pressure and reduced circadian mesor during episodes, but circadian peak and trough were not affected. A potential confounder in a relationship between reduced CSF orexin levels and cardiovascular tone in human disorders of hypersomnolence would be the effects of sleep length and inactivity, which might also result in a lowering of HR and blood pressure. More detailed activity profiles in these disorders might be informative.
A chronically low CSF orexin level is the signature feature of narcolepsy with cataplexy,11 and of all reports there is only one case of spontaneous clinical remission.7 In the current study, orexin levels were almost always above the limit considered diagnostic for narcolepsy (Figure 1). This subtle change could explain the inconsistency of stereotypic narcoleptic-like laboratory findings in KLS patients, and some reports of a “normal” CSF orexin. The etiology of narcolepsy with cataplexy involves an interaction between an environmental trigger and a genetically susceptible host, resulting in symptoms and a loss of orexigenic neurons in the hypothalamus.7 In KLS, there are no such susceptibility markers. Nevertheless, an association with an environmental trigger is still possible, as suggested by a recent report of KLS associated with mild upper respiratory tract infections in a population-based study from Taiwan.25
Orexin measurements in other hypersomnolence syndromes do not show as unique an association as it does here in KLS. For instance, orexin CSF levels may be normal in patients with Parkinson disease and hypersomnia, not explained by another disorder.26 In patients with Guillain-Barré syndrome and sleepiness, one-third can have undetectable orexin levels, another one-third had moderately reduced levels, and one-third had levels in the normal range.27 Recently, an interaction between beta-amyloid and orexin CSF levels was observed distinguishing symptoms in patients with Alzheimer disease, and higher orexin levels were associated with agitation.28 In non-KLS disorders other peptides are suspected to be relevant to the symptom of sleepiness/alertness. Histamine, a somnogenic peptide, while in normal range in sleep apnea,14,29 may be low in narcolepsy without the presence of a very low orexin level.30 Another marker, lipocalin-type prostaglandin D synthase (LPGDS), a brain enzyme that determines the level of prostaglandin D2, a substance with endogenous somnogenic effects, has been studied in narcolepsy but reports are nonconclusive: low values in one report,31 and increased values in another.32 Although we report in KLS a consistent decrease and increase in CSF orexin levels with relapse and remission, we cannot exclude the actions of other peptides or neurotransmitters.
This large cohort is restricted geographically, and different from Western Hemisphere cohorts in terms of ethnic background and/or ecological circumstance in which the illness develops. The landscape of clinical presentations in this rare disease was assessed in a single center, with somewhat different results from those described in Western cohorts. Within this cohort, those consenting to CSF levels and/or blood pressure monitoring did not appear clinically different from the whole group; nevertheless, a hidden bias might have led participation in or consent for CSF results. In addition, we performed assessments using sleep questionnaires, without psychiatric assessments that might tease out cognitive features such as derealization, apathy, etc., in and out of the hypersomnia episode.4 As in other rare disorders, there is a need for an international, multidisciplinary effort to develop standard assessments for studies of pathogenesis.
The current findings in KLS do not indicate an etiology, but are translationally relevant by connecting relapsing hypersomnia to cardiovascular downregulation and reductions in CSF orexin A. Because KLS is rare, its treatment is currently empiric.7 The case reports of success with lithium, modafinil, clarithromycin, acetazolamide, etc. presumably are not acting directly on the orexinergic system, and success could be explained as much by spontaneous recovery as by physician action.33–36
DISCLOSURE STATEMENT
This was not an industry supported study. This work was supported by research grants from the Ministry of Science and Technology (2015CB856405, 2014DFA31500), NSFC (81420108002) and the Ministry of Education (20120001110028) in China. Dr. Strohl is supported in part by the VA Research Service and has consulted for Inspire Medical and Seven Dreamers. The other authors have indicated no financial conflicts of interest.
ACKNOWLEDGMENTS
The authors thank Dr. L. Lin from Stanford University for the help with the measurement of CSF orexin. We also thank Drs. Mignot and Dauvilliers for advice and input in the conduct and presentation of the study. Author contributions: Conception, design, and supervision of this study resulted from core, collaborative effort by Drs. F. Han, J.Y. Wang, X. Song, X.S. Dong, and K.P. Strohl; recruitment, collection, and verification of clinical data over the 11-y period of the study were performed by J Li, P. An, X.Z. Zhang, L. Zhao, X.L. Zhang, Y. N. Liu, H. Yan, Q.H. Li, Y. Hu, Z.C. Gao; statistical and model analyses were performed by Y. Chang and J. Lv; drafting of the manuscript was by Drs. Han, Wang, Dong, and Strohl; all authors reviewed and approved the final submission.
REFERENCES
- 1.Arnulf I, Rico TJ, Mignot E. Diagnosis, disease course, and management of patients with Kleine-Levin syndrome. Lancet Neurol. 2012;11:918–28. doi: 10.1016/S1474-4422(12)70187-4. [DOI] [PubMed] [Google Scholar]
- 2.Huang YS, Guilleminault C, Kao PF, Liu FY. SPECT findings in the Kleine-Levin syndrome. Sleep. 2005;28:955–60. doi: 10.1093/sleep/28.8.955. [DOI] [PubMed] [Google Scholar]
- 3.Kas A, Lavault S, Habert MO, Arnulf I. Feeling unreal: a functional imaging study in patients with Kleine-Levin syndrome. Brain. 2014;137:2077–87. doi: 10.1093/brain/awu112. [DOI] [PubMed] [Google Scholar]
- 4.Engstrom M, Karlsson T, Landtblom AM. Thalamic activation in the Kleine-Levin syndrome. Sleep. 2014;37:379–86. doi: 10.5665/sleep.3420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dauvilliers Y, Bayard S, Lopez R, Comte F, Zanca M, Peigneux P. Widespread hypermetabolism in symptomatic and asymptomatic episodes in Kleine-Levin syndrome. PloS One. 2014;9:e93813. doi: 10.1371/journal.pone.0093813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Engstrom M, Karlsson T, Landtblom AM. Reduced thalamic and pontine connectivity in kleine-levin syndrome. Front Neurol. 2014;5:42. doi: 10.3389/fneur.2014.00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mignot EJ. A practical guide to the therapy of narcolepsy and hypersomnia syndromes. Neurotherapeutics. 2012;9:739–52. doi: 10.1007/s13311-012-0150-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dauvilliers Y, Baumann CR, Carlander B, et al. CSF hypocretin-1 levels in narcolepsy, Kleine-Levin syndrome, and other hypersomnias and neurological conditions. J Neurol Neurosurg Psychiatry. 2003;74:1667–73. doi: 10.1136/jnnp.74.12.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Podesta C, Ferreras M, Mozzi M, Bassetti C, Dauvilliers Y, Billiard M. Kleine-Levin syndrome in a 14-year-old girl: CSF hypocretin-1 measurements. Sleep Med. 2006;7:649–51. doi: 10.1016/j.sleep.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 10.Katz JD, Ropper AH. Familial Kleine-Levin syndrome: two siblings with unusually long hypersomnic spells. Arch Neurol. 2002;59:1959–61. doi: 10.1001/archneur.59.12.1959. [DOI] [PubMed] [Google Scholar]
- 11.Knudsen S, Jennum PJ, Alving J, Sheikh SP, Gammeltoft S. Validation of the ICSD-2 criteria for CSF hypocretin-1 measurements in the diagnosis of narcolepsy in the Danish population. Sleep. 2010;33:169–76. doi: 10.1093/sleep/33.2.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bourgin P, Zeitzer JM, Mignot E. CSF hypocretin-1 assessment in sleep and neurological disorders. Lancet Neurol. 2008;7:649–62. doi: 10.1016/S1474-4422(08)70140-6. [DOI] [PubMed] [Google Scholar]
- 13.Feng P, Hu Y, Li D, et al. The effect of clomipramine on wake/sleep and orexinergic expression in rats. J Psychopharmacol. 2009;23:559–66. doi: 10.1177/0269881108089606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feng P, Hu Y, Vurbic D, Akladious A, Strohl KP. Chromosome 1 replacement increases brain orexins and antidepressive measures without increasing locomotor activity. J Psychiatr Res. 2014;59:140–7. doi: 10.1016/j.jpsychires.2014.08.008. [DOI] [PubMed] [Google Scholar]
- 15.Kukkonen JP, Leonard CS. Orexin/hypocretin receptor signalling cascades. Br J Pharmacol. 2014;171:314–31. doi: 10.1111/bph.12324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hegarty A, Merriam AE. Autonomic events in Kleine-Levin syndrome. Am J Psychiatry. 1990;147:951–2. doi: 10.1176/ajp.147.7.951. [DOI] [PubMed] [Google Scholar]
- 17.Han F, Lin L, Li J, et al. Presentations of primary hypersomnia in Chinese children. Sleep. 2011;34:627–32. doi: 10.1093/sleep/34.5.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.American Academy of Sleep Medicine. Westchester, IL: American Academy of Sleep Medicine; 2005. International classification of sleep disorders, 2nd edition: diagnostic and coding manual. [Google Scholar]
- 19.Taheri S, Mignot E. The genetics of sleep disorders. Lancet Neurol. 2002;1:242–50. doi: 10.1016/s1474-4422(02)00103-5. [DOI] [PubMed] [Google Scholar]
- 20.Cahan C, Decker MJ, Arnold JL, Goldwasser E, Strohl KP. Erythropoietin levels with treatment of obstructive sleep apnea. J Appl Physiol. 1995;79:1278–85. doi: 10.1152/jappl.1995.79.4.1278. [DOI] [PubMed] [Google Scholar]
- 21.Andlauer O, Moore HT, Hong SC, et al. Predictors of hypocretin (orexin) deficiency in narcolepsy without cataplexy. Sleep. 2012;35:1247–55F. doi: 10.5665/sleep.2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sorensen GL, Knudsen S, Petersen ER, et al. Attenuated heart rate response is associated with hypocretin deficiency in patients with narcolepsy. Sleep. 2013;36:91–8. doi: 10.5665/sleep.2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fronczek R, Thijs RD. Autonomic alterations in narcolepsy-contrasting results in mice and men. Sleep. 2013;36:9–10. doi: 10.5665/sleep.2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shirasaka T, Takasaki M, Kannan H. Cardiovascular effects of leptin and orexins. Am J Physiol Regul Integr Comp Physiol. 2003;284:R639–51. doi: 10.1152/ajpregu.00359.2002. [DOI] [PubMed] [Google Scholar]
- 25.Huang YS, Guilleminault C, Lin KL, Hwang FM, Liu FY, Kung YP. Relationship between Kleine-Levin syndrome and upper respiratory infection in Taiwan. Sleep. 2012;35:123–9. doi: 10.5665/sleep.1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Overeem S, van Hilten JJ, Ripley B, Mignot E, Nishino S, Lammers GJ. Normal hypocretin-1 levels in Parkinson's disease patients with excessive daytime sleepiness. Neurology. 2002;58:498–9. doi: 10.1212/wnl.58.3.498. [DOI] [PubMed] [Google Scholar]
- 27.Nishino S, Kanbayashi T, Fujiki N, et al. CSF hypocretin levels in Guillain-Barre syndrome and other inflammatory neuropathies. Neurology. 2003;61:823–5. doi: 10.1212/01.wnl.0000081049.14098.50. [DOI] [PubMed] [Google Scholar]
- 28.Dauvilliers YA, Lehmann S, Jaussent I, Gabelle A. Hypocretin and brain beta-amyloid peptide interactions in cognitive disorders and narcolepsy. Front Aging Neurosci. 2014;6:119. doi: 10.3389/fnagi.2014.00119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kanbayashi T, Kodama T, Kondo H, et al. CSF histamine contents in narcolepsy, idiopathic hypersomnia and obstructive sleep apnea syndrome. Sleep. 2009;32:181–7. doi: 10.1093/sleep/32.2.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nishino S, Sakurai E, Nevsimalova S, et al. Decreased CSF histamine in narcolepsy with and without low CSF hypocretin-1 in comparison to healthy controls. Sleep. 2009;32:175–80. doi: 10.1093/sleep/32.2.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bassetti CL, Hersberger M, Baumann CR. CSF prostaglandin D synthase is reduced in excessive daytime sleepiness. J Neurol. 2006;253:1030–3. doi: 10.1007/s00415-006-0153-8. [DOI] [PubMed] [Google Scholar]
- 32.Jordan W, Tumani H, Cohrs S, et al. Narcolepsy increased L-PGDS (beta-trace) levels correlate with excessive daytime sleepiness but not with cataplexy. J Neurol. 2005;252:1372–8. doi: 10.1007/s00415-005-0870-4. [DOI] [PubMed] [Google Scholar]
- 33.Yao CC, Lin Y, Liu HC, Lee CS. Effects of various drug therapies on Kleine-Levin syndrome: a case report. Gen Hosp Psychiatry. 2013;35:102 e7–9. doi: 10.1016/j.genhosppsych.2012.03.010. [DOI] [PubMed] [Google Scholar]
- 34.Rezvanian E, Watson NF. Kleine-levin syndrome treated with clarithromycin. Jj Clin Sleep Med. 2013;9:1211–2. doi: 10.5664/jcsm.3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zink AN, Perez-Leighton CE, Kotz CM. The orexin neuropeptide system: physical activity and hypothalamic function throughout the aging process. Front Syst Neurosci. 2014;8:211. doi: 10.3389/fnsys.2014.00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oliveira MM, Conti C, Prado GF. Pharmacological treatment for Kleine-Levin syndrome. Cochrane Database Syst Rev. 2013;8:CD006685. doi: 10.1002/14651858.CD006685.pub3. [DOI] [PubMed] [Google Scholar]