

Abstract
Study Objective:
The adolescent brain may be uniquely affected by acute sleep deprivation (ASD) and chronic sleep restriction (CSR), but direct evidence is lacking. We used electron microscopy to examine how ASD and CSR affect pyramidal neurons in the frontal cortex of adolescent mice, focusing on mitochondria, endosomes, and lysosomes that together perform most basic cellular functions, from nutrient intake to prevention of cellular stress.
Methods:
Adolescent (1-mo-old) mice slept (S) or were sleep deprived (ASD, with novel objects and running wheels) during the first 6–8 h of the light period, chronically sleep restricted (CSR) for > 4 days (using novel objects, running wheels, social interaction, forced locomotion, caffeinated water), or allowed to recover sleep (RS) for ∼32 h after CSR. Ultrastructural analysis of 350 pyramidal neurons was performed (S = 82; ASD = 86; CSR = 103; RS = 79; 4 to 5 mice/group).
Results:
Several ultrastructural parameters differed in S versus ASD, S versus CSR, CSR versus RS, and S versus RS, although the different methods used to enforce wake may have contributed to some of the differences between short and long sleep loss. Differences included larger cytoplasmic area occupied by mitochondria in CSR versus S, and higher number of secondary lysosomes in CSR versus S and RS. We also found that sleep loss may unmask interindividual differences not obvious during baseline sleep. Moreover, using a combination of 11 ultrastructural parameters, we could predict in up to 80% of cases whether sleep or wake occurred at the single cell level.
Conclusions:
Ultrastructural analysis may be a powerful tool to identify which cellular organelles, and thus which cellular functions, are most affected by sleep and sleep loss.
Citation:
de Vivo L, Nelson AB, Bellesi M, Noguti J, Tononi G, Cirelli C. Loss of sleep affects the ultrastructure of pyramidal neurons in the adolescent mouse frontal cortex. SLEEP 2016;39(4):861–874.
Keywords: chronic sleep restriction, electron microscopy, interindividual variability, lysosomes, mitochondria
Significance.
To our knowledge, this is the first study that used electron microscopy to measure the effects of acute sleep deprivation and chronic sleep restriction on the adolescent brain. Sleep loss is common, and it is assumed that its negative effects are more devastating in the young brain. Yet, direct evidence is lacking. In frontal cortex of adolescent mice, we found that after chronic sleep restriction pyramidal neurons show signs of lysosomal activation and deposition of lipofuscin granules. We also found that chronic sleep loss may unmask inter-individual anatomical differences not obvious during baseline. Finally, using a combination of ultrastructural parameters we could predict, at the single cell level, whether a mouse has been awake or asleep, and for how long.
INTRODUCTION
Sleep loss impairs many cognitive functions, from attention and learning ability to verbal fluency and inhibitory control,1,2 but the underlying mechanisms are poorly understood. Animal studies have identified many molecular, biochemical, and electrophysiological correlates of sleep deprivation, including the cortical induction of stress response genes,3,4 the build-up of extracellular glutamate5 and adenosine,6 as well as the occurrence of microsleeps, short sleep events that only last a few seconds but may have disastrous consequences for performance.7 Most experiments so far have focused on acute total sleep deprivation, but there is increasing interest in the effects of chronic sleep restriction (CSR), a more common condition in modern society. Studies in humans have shown that CSR causes cumulative deficits in performance and exacerbates the cognitive effects of acute total sleep loss,8,9 but very few animal studies have investigated the cellular consequences of CSR.10 It is also generally assumed that the negative consequences of acute sleep loss and CSR would be more marked during sensitive periods such as adolescence, when the brain is undergoing substantial remodeling, but direct evidence is lacking.
Molecular studies have suggested that several cellular functions in the brain may be sensitive to sleep and wake, with neurons preferentially carrying out certain functions in one of the two behavioral states. Thus, protein and lipid synthesis and membrane formation may be favored by sleep, energy production and synaptic growth may occur mainly during wake, and extended wake may lead to signs of cellular stress.3,11–13 One approach to understand why sleep loss is detrimental to the brain is to examine its effects on the ultrastructure of brain cells, and specifically on the organelles that mediate cellular functions that are sensitive to sleep or lack of sleep. This analysis requires the high spatial resolution only afforded by electron microscopy (EM). EM, however, has rarely been used in sleep studies and never, to our knowledge, to assess the effects of CSR on the adolescent brain. New findings in rodents also suggest that sleep may not be as global as previously thought, with groups of neurons being “offline” when the rest of the brain is awake.14,15 During this phenomenon of “local” sleep, a few neurons during wake are experiencing short down states resembling those normally observed during nonrapid eye movement (NREM) sleep, perhaps in response to excessive neuronal activity and/or plasticity.16 Yet, the evidence for local sleep comes from extracellular recordings that lack single cell resolution, and the exact “cost” of activity and plasticity for brain cells remains unknown.
The primary goal of this study was to use EM to assess the effects of acute sleep deprivation (ASD) and chronic sleep restriction (CSR) on the ultrastructure of cortical pyramidal neurons in the frontal cortex of adolescent mice, and to do so we compared each wake condition to the same sleep group (S). In the cell bodies of these glutamatergic cells, which represent ∼80% of all cortical neurons, we measured mitochondria and various components of the endocytotic pathway–early endosomes (EE), multivesicular bodies (MVBs), and lysosomes– which together carry out most basic cellular functions, from nutrient intake and membrane turnover to energy production and prevention of metabolic and oxidative stress.17,18
METHODS
Experimental Groups
All animal procedures followed the National Institutes of Health's Guide for the Care and Use of Laboratory Animals. Facilities were reviewed and approved by the Institutional Animal Care and Use Committee of the University of Wisconsin-Madison, and were inspected and accredited by AAALAC (Association for Assessment and Accreditation of Laboratory Animal Care). Adolescent (∼1 mo old) Yellow Fluorescent Protein line H mice (YFP-H mice, Jackson Laboratory, Bar Harbor, ME) were used because the sleep pattern in this strain is well characterized, including during adolescence.19 Moreover, the current study is part of a larger effort to measure sleep/wake dependent changes in cortical synapses at the ultrastructural level, and previous studies in our laboratory have used confocal and two-photon in vivo microscopy in YFP-H mice to characterize sleep/wake-dependent20 and development-related21 changes in spine density. Mice were housed in environmentally controlled recording chambers with free access to food and water (12 h:12 h light-dark cycle; lights on at 08:00). They were divided in four experimental groups (n = 17, 8 females): sleeping (S) mice had access to a running wheel and novel objects during the dark phase, and then could sleep during the first 6–8 h of the light phase before sacrifice (the presence of novel objects did not affect sleep duration at night, which was 4.6 ± 0.15 h, mean ± standard deviation); ASD mice also were spontaneously awake most of the night, and then were kept awake for the first 6–8 h of the day by giving them access to a running wheel and novel objects; CSR mice were sleep restricted for > 4 days (102 h; see next paragraphs); recovery sleep (RS) mice were allowed to rest undisturbed for ∼32 h after 4 days (96 h) of CSR. Sleep and wake behavior was assessed by direct visual observation and using motion detection monitored with infrared video cameras, and quantified at 1-sec time resolution. All mice were sacrificed between 14:40 and 18:00 to maintain the time of brain collection within the same 3-h circadian window for all experimental groups. RS mice underwent 96 h of CSR, not 102 h as did the CSR mice, to allow sleep ad libitum for almost two complete light periods after CSR while maintaining the time of sacrifice in the late afternoon.
Chronic Sleep Restriction
CSR started at postnatal day 26 (P26, day 1) at lights on and ended after the first 6 h of the light phase on day 5 (P30). During the light period animals were kept awake using exposure to novel objects, a running wheel, social interactions with their siblings, and by changing cages and bedding. Mice were given caffeinated water at 0.1 mg/mL (∼15 mg/kg). Previous studies showed that this concentration results in plasma levels of caffeine in mice similar to those in the plasma of most coffee drinkers.22 Administration of caffeinated water ceased during RS in the RS group. During CSR room lights were also turned off for brief periods in the first 6 h of the light period (∼20 min/h for a total of 2 h) to provide an additional stimulus to stay awake. At night, mice were transferred for several hours a night to a conveyer over water apparatus23 that runs continually at slow speed, allowing the animals to sleep for brief periods (< 8 sec) before being forced to move or fall into the water. In a pilot study, seven mice at P21 were implanted for chronic EEG recordings under isoflurane anesthesia (1–2% in 100% O2), as detailed in the study by Nelson et al.19 Mice then underwent CSR for 4 days (from P26 to P29), during which electroencephalography (EEG) and motion detection were recorded continuously, followed by 2 days of recovery sleep. Nonrapid eye movement (NREM) sleep, rapid eye movement (REM) sleep, and wake were scored by visual inspection of 4-sec epochs (SleepSign; Kissei Comtec, Nagano, Japan) according to standard criteria. Wake was characterized by low voltage, high-frequency electroencephalography (EEG) pattern and phasic electromyographic activity. NREM sleep was characterized by the occurrence of high amplitude slow waves and low tonic muscle activity. During REM sleep, EEG pattern was similar to that during wake, but only heart beats and occasional twitches were evident in the electromyography signal.
Tissue Preparation for Electron Microscopy
Under deep anesthesia (3% isoflurane), mice were perfused intracardially with a solution of 0.05 M phosphate buffered saline (PBS) followed by 2.5% glutaraldehyde and 4% paraformaldehyde dissolved in 0.1M sodium cacodylate buffer (41°C and pH 7.4). Brains were removed and kept in the same fixative overnight at 4°C. Tissue slices (120 μm) were cut on a vibratome and kept in a cryoprotectant solution until the day of processing. Sections were rinsed in cacodylate buffer, and incubated for 1 h on ice with a solution of 1.5% potassium ferrocyanide/2% osmium tetroxide. Then, they were exposed to a solution of 1% thiocarbohydrazide for 20 min at room temperature. Sections were placed in 2% osmium tetroxide for 30 min, and incubated overnight with 1% uranyl acetate at 4°C. The following day, the tissue was stained with a solution of lead aspartate for 30 min at 60°C (pH 5.5), and dehydrated using ice-cold solutions of freshly prepared 35%, 50%, 75%, 80%, 90%, 95%, and 100% ethanol. Sections were placed in propylene oxide for 10 min and then impregnated with 25%, 50%, 75% Durcupan ACM resin (Electron Microscopy Science, Hatfield PA) mixed with propylene oxide (2 h each). Tissue was placed in 100% Durcupan overnight and then into fresh Durcupan for 2 h. Sections were flat embedded with ACLAR embedding film (Electron Microscopy Science) and placed in a 60°C oven for 48–72 h. After polymerization, small squares of resin embedded tissue (1 mm2) from frontal cortex (1.85 mm anterior to bregma, 1.5 mm lateral) were excised under a stereomicroscope and glued on the tip of the metal pin. The block of tissue was trimmed and coated with silver paint to minimize specimen charging during imaging.
Image Acquisition
Images were obtained using a ΣIGMA VP field emission scanning electron microscope (Carl Zeiss NTS Ltd, Thornwood, NY) equipped with a backscattered electron detector. Images were acquired using an aperture of 30 μm, high vacuum, acceleration voltage of 1.2 kV; image resolution (xy plane) was between 1 and 3 nm. Pyramidal neurons were sampled in layer II of frontal cortex using established ultrastructural features of the perikaryon to distinguish them from non-pyramidal neurons.24–26 Specifically, neurons included in the analysis had a triangular shape with one apex directed toward layer I and often tapering into an apical dendrite, a large nucleus with 0 or 1 small indentation along one side, a gradual demarcation between cell body and basal dendrites, a widespread endoplasmic reticulum. Pyramidal neurons were often observed in small groups, with adjacent perikarya in direct apposition and containing a multilaminar subsurface complex.24 For each neuron, one section that included a large nucleus fully surrounded by cytoplasm was selected for analysis and used to count mitochondria, EEs, MVBs, and lysosomes and to measure their area. Segmentation was done manually by three people who were blind to experimental condition. All data were tested for accuracy and consistency by the same experienced tracer (LdV). Quantitative analysis was carried out with open source software Fiji.27
Regression Analysis
Raw measurements of density and percentage for each cell were pooled and standardized (z-score) in MATLAB (Math-Works, Natick MA) before applying binary logistic regression analysis. One dependent binomial variable (i. S = 1, ASD = 0; ii. S = 1, CSR = 0; iii. S = 1, RS = 0; iv. ASD = 1, CSR = 0; v. RS = 1, CSR = 0) and 11 continuous independent variables, i.e., ultrastructural parameters that showed changes across groups, were used to build each regression model. A backward stepwise procedure was used to remove nonsignificant predictors from the final model.
Statistical Analysis
Nonparametric statistics was used to compare the ultrastructural features from different experimental conditions. Kruskal-Wallis (KW) analysis of variance was used to compare more than two groups. Wilcoxon rank sum test (W) was used as a post hoc test, with α value of significance set at 0.05. The statistical variance component analysis was performed separating, in each experimental group, the between-mice variance σ 2bm from the within-mice variance σ2wm by means of a linear mixed model in R. All mixed models were estimated with a restricted maximum likelihood method. To quantify traitlike interindividual variability we assessed the ratio of between mice variance σ2bm to the total variance σ2bm + σ2wm (Rσ2bm). To test the hypothesis Rσ2bm = 0, a nonparametric bootstrapping procedure was utilized. We fitted a null model without the random subject effect, and then bootstrapped observations were generated by resampling the estimated residuals from this model. Statistical significance of the Rσ2bm values was assessed by comparing the estimated within-subject variance in the full model, to the within-subject variance in the null model. If the within-subject variance was significantly lower in the full model than the null model, then there was evidence that the Rσ2bm > 0.
RESULTS
We studied four groups of adolescent mice (∼1 m old) that were either allowed to sleep during the first 6–8 h of the light period (S mice), sleep deprived during this time (ASD mice), chronically sleep restricted for more than 4 days (102 h, CSR mice), or allowed to recover sleep ad libitum for ∼32 h after 4 days of CSR (RS mice) (Figure 1A). As detailed in the Methods, all mice were sacrificed within the same 3-h window in the afternoon. To maximize the chances of seeing significant ultrastructural changes, we required both ASD and CSR to be highly effective in enforcing wake. Crucially, we also wanted these methods to enforce wake in a realistic manner. Thus, the ASD protocol consisted in 6 to 8 “intense” hours of continuous wake with enhanced locomotion and exploration, whereas the CSR protocol used a combination of enhanced locomotion and exploration, as well as social interactions, caffeine and short changes in light schedule to produce a consistently severe sleep loss for 4 days. Of note, the use of additional strategies to enforce wake in the CSR groups was inevitable, because in pilot experiments we found that the exposure to novel objects and the voluntary use of a running wheel keep mice awake only for a few hours.
Figure 1.

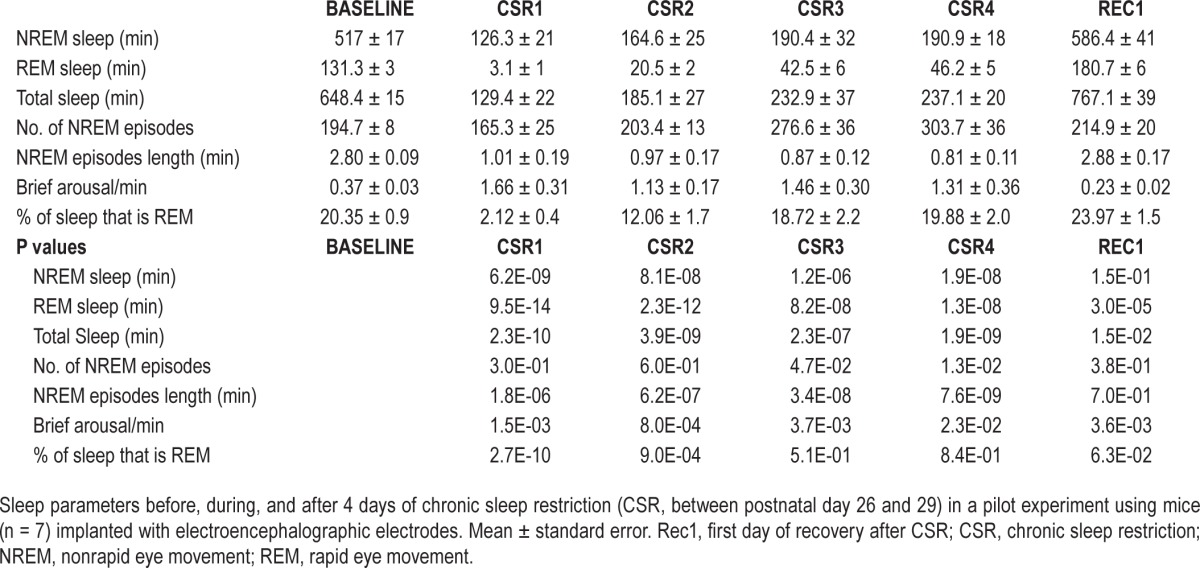
(A) Experimental groups used in the electron microscopy (EM) study. The arrow indicates the time of brain collection, which in all groups occurred between ∼15:00 and 18:00. Recovery sleep (RS) mice were sacrificed at postnatal day P31, to allow sleep ad libitum for ∼32 h after the end of chronic sleep restriction (CSR). The comparisons shown to the right are those discussed in the main text. Figures S1–S4 (supplemental material) also include the comparison ASD vs. CSR. (B) Changes in sleep duration during and after 4 days of CSR (between P26 and P29) in a pilot experiment using mice (n = 7) implanted with electroencephalography (EEG) electrodes. Baseline values of nonrapid eye movement (NREM) and rapid eye movement (REM) sleep duration at P26 were 517 ± 17 min and 131.3 ± 3 min (mean ± standard error) respectively, consistent with published values.19
To first validate the method to enforce CSR, seven mice were implanted with EEG electrodes and sleep restricted for 4 days (between P26 and P29) by exposure to novel objects, social interaction, access to a running wheel, caffeinated water, and forced locomotion. This paradigm decreased total sleep time across the 4 days by ∼70% (first day 80%; last day 63%), corresponding to a decline of 68% in NREM sleep and 79% in REM sleep relative to an undisturbed baseline day (Figure 1B). The sleep obtained during CSR was also more fragmented than during baseline, with shorter NREM sleep bouts and more frequent brief arousals during NREM sleep (Figure 1B, Table 1). As expected, a rebound in sleep time was present in RS mice during the first 24 h after CSR, with increases above baseline in total sleep (+18%), NREM sleep (+14%), and REM sleep (+39%) (Figure 1B, Table 1).
Table 1.
Sleep electroencephalographic analysis of chronic sleep restriction.
For the EM experiments, mice were not implanted with EEG electrodes to avoid possible damage to the cortex. Instead, they were constantly monitored by direct visual inspection and with infrared cameras, and overall motor activity was quantified and used to estimate sleep/wake amounts. We previously demonstrated that although this method cannot distinguish NREM sleep from REM sleep, it consistently estimates total sleep time with ∼93% accuracy.20 As expected, S and ASD mice spent most of their last 8 h asleep (∼80%) or awake (> 95%), respectively. Estimated sleep and wake amounts in CSR and RS mice were consistent with those found in the pilot EEG experiments, with a ∼70% decrease in overall sleep duration during CSR and a sleep rebound during recovery.
For EM analyses we focused on layer II of the frontal cortex for several reasons. In both humans and rodents, the slow wave activity (SWA) of NREM sleep is largest in frontal areas. SWA reflects the propensity for sleep, as its levels increase with wake duration and decline during sleep. SWA is also a marker of sleep intensity, because arousal thresholds during sleep are higher when SWA is higher.28 Thus, frontal cortex may accumulate the largest need for sleep, may be most sensitive to the effects of sleep loss, and/or the changes in neural activity that underlie the occurrence of slow waves may be most pronounced in this area. We focused specifically on layer II because sleep need increases not only with the duration of wake but also with its “intensity,” i.e. depending on the extent of wake-related learning and synaptic plasticity, and supragranular layers are known to be highly plastic.29–31
A total of 350 pyramidal neurons were analyzed in 17 mice, ∼20 neurons/mouse, including 82 in S (n = 4 mice), 86 in ASD (n = 4), 103 in CSR (n = 5), and 79 in RS (n = 4) conditions. In the cytoplasm of each cell body we counted mitochondria, EEs, MVBs, and lysosomes and measured their area (one section/ cell). In all analyses we compared each wake condition (ASD or CSR) to the same S group. RS mice were compared to CSR and to S groups, to test the extent to which RS could reverse the effects of CSR (Figure 1A).
Mitochondrial Changes after Chronic Sleep Loss
More than 3,000 mitochondria were measured in each experimental group (S = 3,532; ASD = 3,817; CSR = 4,390; RS = 3,620). Their density did not change across groups (KW, P = 0.31), but in CSR and RS there was a shift of the mitochondrial population toward bigger sizes (KW, P < 0.0001; W: S versus CSR, P < 0.0001, S versus ASD, P = 0.82, CSR versus RS, P = 0.31, S versus RS, P = 0.0034). The shift occurred preferentially in mitochondria of small (< 0.05 μm2, KW, P = 0.0033, S versus CSR, P = 0.0033, CSR versus RS, P = 0.0019, RS versus S, P = 0.78) and medium size (0.051–0.25 μm2, KW, P = 0.034, S versus CSR, P = 0.039 CSR versus RS, P = 0.788, RS versus S, P = 0.027; Figure 2A), which together accounted for ∼95% of all mitochondria, with no change in large mitochondria (> 0.25 μm2, KW P = 0.45). Moreover, extra-large mitochondria, defined as those with area ≥ 1 μm2 (Figure 2B), were present in a minority of cells in ASD (maximum 0.99 μm2), CSR (maximum 1.13 μm2), and RS (maximum 1.16 μm2) but never observed in S (maximum 0.78 μm2). These extra-large mitochondria did not show abnormal features such as loss of matrix density or decrease in cristae number. In fact, obvious mitochondrial abnormalities such those associated with cellular stress (e.g., donut or blob shapes32) or neurodegeneration (e.g., pale and patchy matrix, few cristae, and discontinuous outer membrane in Parkinson disease and Alzheimer disease33) were not seen in any experimental group. The increased mitochondrial size in CSR and RS led to a higher percentage of cytoplasm occupied by mitochondria in these two groups relative to S (CSR +14%, RS +10.4%; KW, P = 0.003, W: S versus CSR, P = 0.0013, S versus RS, P = 0.0016; CSR versus RS, P = 0.89, S versus ASD, P = 0.06, Figure 2C). Finally, a small minority (0–10%) of mitochondria had an hourglass shape (Figure 2D). Although present in all experimental groups, hourglass mitochondria represented a smaller proportion of the entire mitochondrial population in S than in all other groups (KW, P = 0.011, W: S versus ASD, P = 0.048, S versus CSR, P = 0.0015, CSR versus RS, P = 0.51, S versus RS, P = 0.011).
Figure 2.
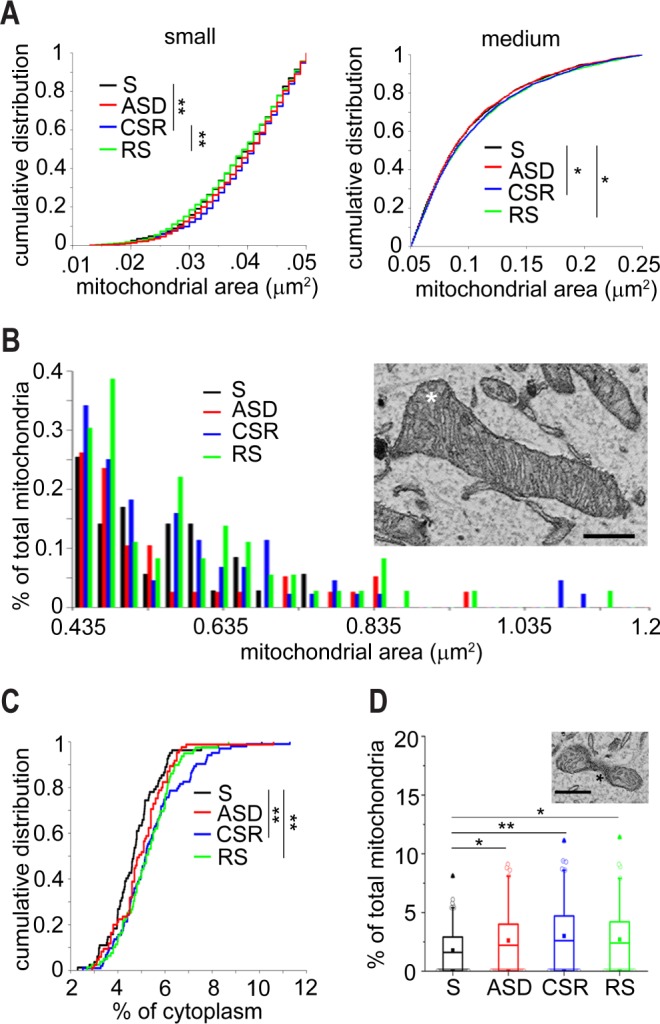
Mitochondrial changes in response to sleep loss. (A) Cumulative distribution of size for small (left) and medium (right) mitochondria. (B) Distribution of extra-large mitochondria in the four experimental conditions. The micrograph shows an example of an extra large mitochondrion (asterisk), scale bar = 0.5 μm. (C) Cumulative distribution of the percentage of cytoplasm occupied by mitochondria in S (3,532 mitochondria), ASD (3,817 mitochondria), CSR (4,390 mitochondria), and RS (3,620 mitochondria). (D) Percentage of mitochondria with an hourglass shape. The micrograph shows an example of an hourglass mitochondrion (asterisk). CSR, chronic sleep restriction; RS, recovery sleep; S, sleep; ASD, acute sleep deprivation. Scale bar = 0.5 μm. *P < 0.05, **P < 0. 01.
In summary, we found three mitochondrial parameters that changed due to sleep and wake. CSR differed from S in all three: more cytoplasmic area occupied by mitochondria, presence of very large mitochondria, and greater proportion of hourglass mitochondria. All three parameters were also significantly different in RS relative to S, whereas the last two differed between ASD and S.
Effects of Sleep and Wake on the Early Endocytic Pathway
The endocytic recycling pathway starts at the plasma membrane and ends in lysosomes. Between these two extremes lies a continuum of intermediate endosomal compartments, a dynamic population of organelles that exchange their content while also undergoing gradual maturation.17,34,35 EEs are considered the first step of the endocytic pathway, where most molecules are sorted to be either quickly recycled back to the plasma membrane or directed to lysosomes for degradation and slow recycling.18,36 MVBs represent a second sorting station, originating both from the maturation of EEs and from the exchange of material with other endosomal compartments.17 We defined EEs as spherical membrane delimited compartments with a clear lumen and containing amorphous material or 1 internal vesicle (Figure 3A, left), and MVBs as organ-elles characterized by a single outer membrane that enclosed more than one vesicle (Figure 3A, right). Because endosomal compartments differ for content, morphology, molecular composition, and internal pH, our classification is probably a simplification of a more complex pathway.
Figure 3.

Effects of sleep and wake on the endosomal compartments. (A) Left panel: example of an early endosome (EE); right panel: example of a multivesicular body (MVB). (B) Box plot representing the density of EEs across experimental groups (99 in S, 113 in ASD, 67 in CSR, and 117 in RS). (C) Cumulative distribution of the percentage of cytoplasm occupied by EEs across groups. (D) Density of EEs (white bars) and MVBs (black bars) across groups. (E) Left: schematic illustration of invagination and back-fusion events: MVB biogenesis and formation of internal vesicles occurs by invagination of MVB's outer membrane and incorporation of receptors and lipids coming from the plasma membrane; during back-fusion, internal vesicles fuse with the MVB's outer membrane and then bud off to bring recycled material to the plasma membrane. Right: micrographs with examples of invagination and back-fusion in MVBs and EEs. (F) Percentage of total endosomal compartments undergoing either invagination or back-fusion across all conditions. CSR, chronic sleep restriction; RS, recovery sleep; S, sleep; ASD, acute sleep deprivation. Scale bar for all micrographs = 0.5 μm. Bars indicate mean ± standard deviation. *P < 0.05, **P < 0.01, ***P < 0.001.
We found that the size of EEs was similar across all conditions, ranging from 0.024 μm2 to 0.53 μm2, but their density was lower in CSR than in S and RS (KW, P = 0.0001, W: CSR versus S, P = 0.010, CSR versus RS, P = 0.0001; Figure 3B). As a result, the percentage of cytoplasm occupied by EEs was lower in CSR than in S and RS (KW, P = 0.0004; W: CSR versus S, P = 0.0054, CSR versus RS, P = 0.0002; Figure 3C).
We found no significant difference in MVBs across conditions, neither in size or density nor in the percentage of occupied cytoplasm. What changed across groups, however, was the relative proportion between EEs and MVBs, which in S mice favored MVBs (Figure 3D). This imbalance was lost in ASD, where EEs and MVBs were present in equal percentage (MW, P = 1) and augmented in CSR, where MVBs largely outnumbered EEs (MW two-tailed test: S, P = 0.03, CSR, P = 0.016) and accounted for almost 70% of all endosomal compartments.
With time, MVBs enlarge, increase the number of internal vesicles, and become more electron dense. Formation of MVBs internal vesicles is thought to occur through invagination of the endosomal limiting membrane and endocytosis of soluble cell components from the cytoplasm.17 The cargo of MVBs vesicles can then be degraded inside lysosomes, undergo back-fusion, and be recycled to the plasma membrane, or be released extracellularly in intact vesicles. We observed both invagination and back-fusion in EEs and MVBs (Figure 3E). These events occurred at similar rates in S and RS, whereas after ASD and CSR back-fusion events became at least twice as frequent as invaginations (MW: ASD, P = 0.028, CSR, P = 0.0079; Figure 3F). Although the fate of back-fusing vesicles needs to be established, this result suggests that wake increases the need to quickly recycle cell components, perhaps receptors and membrane lipids.
Evidence for Past Lysosomal Activation after Chronic Sleep Loss
Lysosomes receive and degrade macromolecules from the endocytic and autophagic membrane-trafficking pathways and are divided in primary and secondary lysosomes based on their appearance. Primary lysosomes were defined here as membrane delimited organelles with homogenous electron-dense lumen24 (Figure 4A). They were often observed close to the cisternae of the endoplasmic reticulum, MVBs, or the Golgi apparatus, from which they originate, but never in direct contact with the plasma or nuclear membrane. They had different sizes (range 0.014–0.637 μm2) and shapes, the most common being ovoidal, but sometimes protrusions and invagi-nations were evident. In ASD mice, primary lysosomes were significantly larger than in S (38% increase; KW, P < 0.0001, W: ASD versus S, P < 0.0001; Figure 4B), but also significantly less numerous (KW, P = 0.0012, W: ASD versus S, P = 0.0024; Figure 4C). Overall, the percentage of cell cytoplasm occupied by primary lysosomes was similar across all conditions (0.48%, KW, P = 0.21).
Figure 4.

Lysosomal changes due to sleep loss. (A) Micrographs of a primary lysosome (asterisks). (B) Cumulative distribution of the size of primary lysosomes. (C) Box plot showing the density of primary lysosomes across groups. Secondary lysosomes were 117 in S, 83 in ASD, 222 in CSR, and 77 in RS. (D) Micrograph of secondary lysosomes (asterisk). (E) Cumulative distribution of the size of secondary lysosomes. (F) Box plot showing the density of secondary lysosomes across groups. (G) Example of a lipofuscin-like granule. (H) Average density of lipofuscin-like granules in each animal. CSR, chronic sleep restriction; RS, recover sleep; S, sleep; ASD, acute sleep deprivation. Scale bars = 0.5 μm. *P < 0.05, **P < 0.01, ***P < 0.001.
Secondary lysosomes are believed to derive from primary lysosomes after invagination of a cytosolic substrate,37 after the fusion of primary lysosomes with late endosomes or autophagosomes,38 or through the fusion of endosomes with vesicles coming from the trans-Golgi network and containing hydrolytic enzymes necessary for substrate digestion.35 Secondary lysosomes are considered markers of past lysosomal activation and were defined here as membrane delimited organelles with heterogeneous dark lumen containing whorls of membranes and granules of different size and density (Figure 4D).24 Their shape was often irregular, and their size ranged between 0.022 μm2 and 1.81 μm2. ASD and RS were associated with larger secondary lysosomes than S and CSR (25% increase in size; KW, P < 0.0001; W: ASD versus S, P = 0.0008, RS versus S, P = 0.0004, RS versus CSR, P < 0.0001; Figure 4E), but the density of secondary lysosomes was greater in CSR than in S and RS (KW, P < 0.0001; W: CSR versus S, P = 0.0003, CSR versus RS, P < 0.0001; Figure 4F). As a result, the percentage of cell cytoplasm occupied by secondary lysosomes was greater in CSR than in S and RS (W: CSR versus S, P = 0.029, CSR versus RS, P = 0.021). Autophagosomes, defined as double-membrane vesicles that sequester part of the cytoplasm and its organelles to fuse with lysosomes, were also observed in many neurons (167 total), but their density was similar across conditions (0.005 autophagosomes/μm2).
The production of reactive oxygen species is the consequence of normal oxidative metabolism in mitochondria. Cells are equipped with many antioxidant systems that help reduce macromolecular damage, but cannot completely prevent it, resulting in damaged proteins and lipids that become biological waste. Lipofuscins—thought to derive from the incomplete digestion of mitochondrial protein and lipid components—deposit gradually in cells and enlarge to occupy increasing portions of the lysosomal compartment.39 In proliferating cells, lipofuscins are diluted efficiently through cell division, but in postmitotic cells, like neurons, they accumulate progressively with time. Across all 17 animals we counted 499 lysosomes but only 33 granules that had the appearance of lipofuscins (Figure 4G), consistent with the fact that the latter are usually rare in young mice. The size of these granules did not change across groups. However, half of these granules were found in CSR (16 granules total, 5 of 5 mice), whereas only six granules were found in S (2 of 4 mice), five granules in ASD (4 of 4 mice), and six granules in RS (2 of 4 mice) (Figure 4H), suggesting increased waste production with chronic sleep loss, consistent with increased metabolic activity and/or cellular stress.
Sleep Loss Unmasks Interindividual Variability
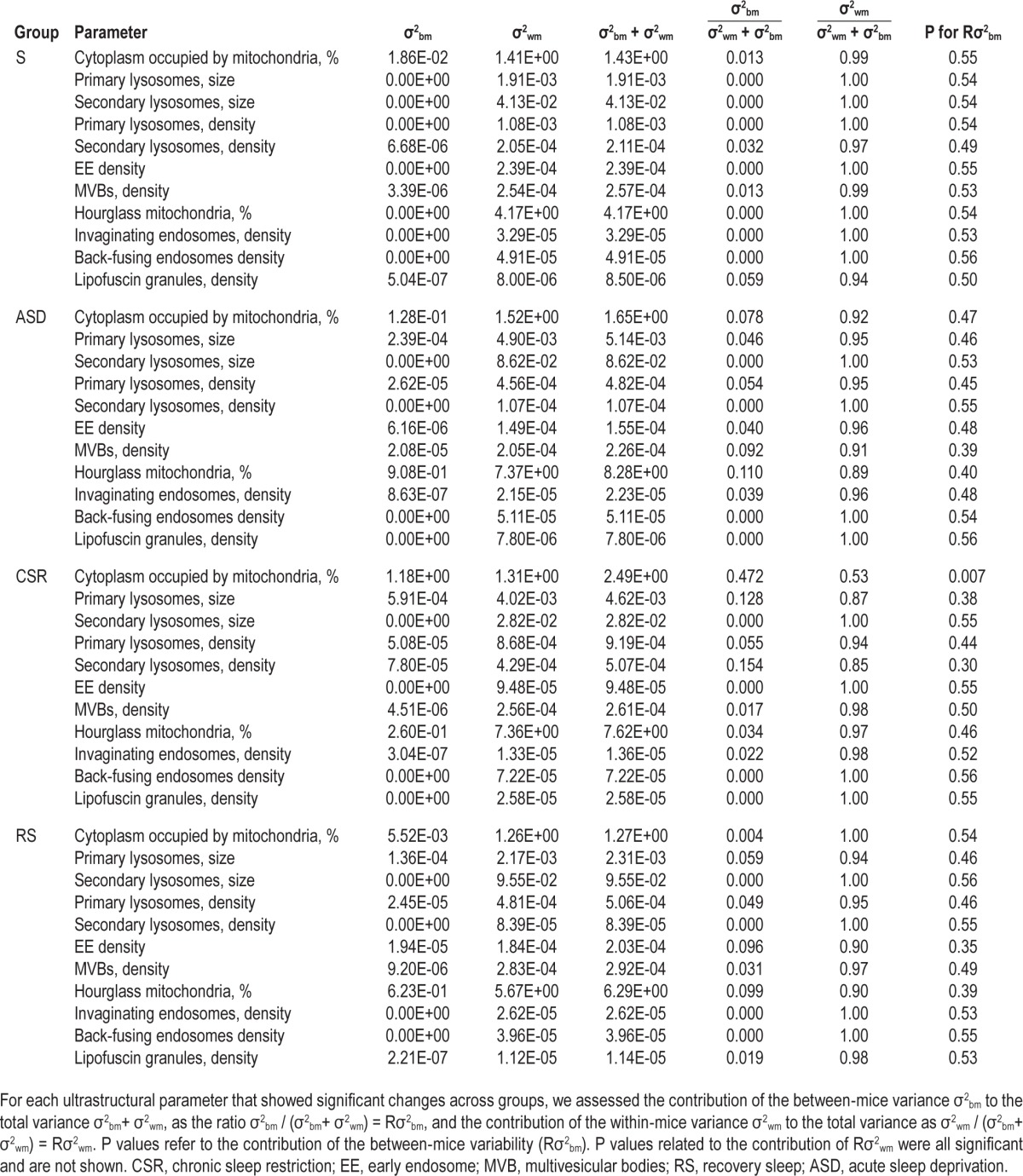
All ultrastructural parameters measured in the current study showed some variability among cells of the same experimental group. Because all analyses were done after pooling cells within each group, this variability could reflect differences among cells of the same mouse (within), and/or among different animals (between). To distinguish between these two possibilities we performed a variance component analysis, separating the between-mice variance σ2bm from the within-mice variance σ2wm in the data. For each ultrastructural parameter that showed a significant difference according to the previous separate analyses we measured the contribution of σ2bm or σ2wm to the total variance by calculating the ratio σ2bm / σ2bm + σ2wm, (called Rσ2bm), and the ratio σ2wm / σ2bm + σ2wm, (called Rσ2wm) (Table 2). Within-mice variance was significantly different from zero for all parameters and its contribution to the total variance was high in all groups but not significantly different among them, even if tended to be lower in ASD, CSR, and RS than in S. Between-mice variance, by contrast, in almost all cases was significantly lower than within-mice variance and not significantly different from zero, resulting in low contribution of the between-mice variance to the total variance. The only exception was the percentage of cytoplasm occupied by mitochondria in the CSR group, whose between-mice variance was significantly above zero, resulting in a Rσ2bm of ∼0.5. Together, these results indicate that much of the variability observed inside each group originated from the differences among the cells within each mouse, which was similar across groups. Of note, the Rσ2bm was low but showed a clear trend to increase with the extent of sleep loss. Indeed, the mean Rσ2bm within each group, calculated by averaging the ratios for all 11 parameters, was lowest in S (0.0106), higher in ASD (0.0417), highest in CSR (0.0803), and declined in RS to a level intermediate between S and ASD (0.0325). Thus, our data suggest that sleep loss can unmask inter-individual differences that are not obvious under baseline conditions of sleep.
Table 2.
Results of variance component analysis performed by separating the between-mice variance σ2bm from the within-mice variance σ2wm by mean of a linear mixed model.
A Combination of Ultrastructural Parameters Can Predict Sleep and Wake at the Cellular Level
We found that in the adolescent frontal cortex, sleep loss is associated with ultrastructural changes in mitochondria, endosomes, and lysosomes of pyramidal neurons. Looking at individual parameters and at each organelle separately, however, may not fully capture the extent of these changes. We thus considered all 11 parameters that showed a significant difference according to the previous separate analyses, and that could reasonably be assumed to be independent of each other. These parameters included the percentage of cytoplasm occupied by mitochondria; the percentage of hourglass mitochondria; density and size of primary and secondary lysosomes; density of EEs, MVBs, and lipofuscin granules; and density of total endosomal compartments undergoing invagination or back-fusion events. Through multivariate logistic regression analysis we tested how accurately one could assign each cell to the right experimental group using these 11 parameters. In the first predictive model, cells belonging to S mice were pooled and compared to cells belonging to ASD mice, obtaining a set of probabilities representing the likelihood of each cell of being correctly classified as S or ASD (S = 1, ASD = 0). Based on the probabilities generated by the logistic regression, each cell was then classified as S or ASD (cutoff probability = 0.5). The same regression analysis was then applied to compare S and CSR cells (S = 1, CSR = 0), S and RS cells (S = 1, RS = 0), and CSR and RS cells (RS = 1, CSR = 0). The probability distributions showed that, from an ultrastructural standpoint, the population of S cells is significantly different from the other three populations (ASD, CSR, RS), and that CSR cells also differ from RS cells (Figure 5A–5D; W, P < 0.0001). The percentage of correct cell classification was approximately 70%, ranging from 59% to 80% depending on the specific comparison (Table 3). Applying the regression function on smaller datasets of cells randomly selected from each group (29 cells from 4 S mice, 27 cells from 4 ASD mice, 37 cells from 5 CSR, 27 cells from 4 RS mice) gave similar results, although the probability of a correct classification was smaller than when using the full datasets (on average S versus ASD, 61%; S versus CSR, 60%; S versus RS, 66%; RS versus CSR, 72%).
Figure 5.
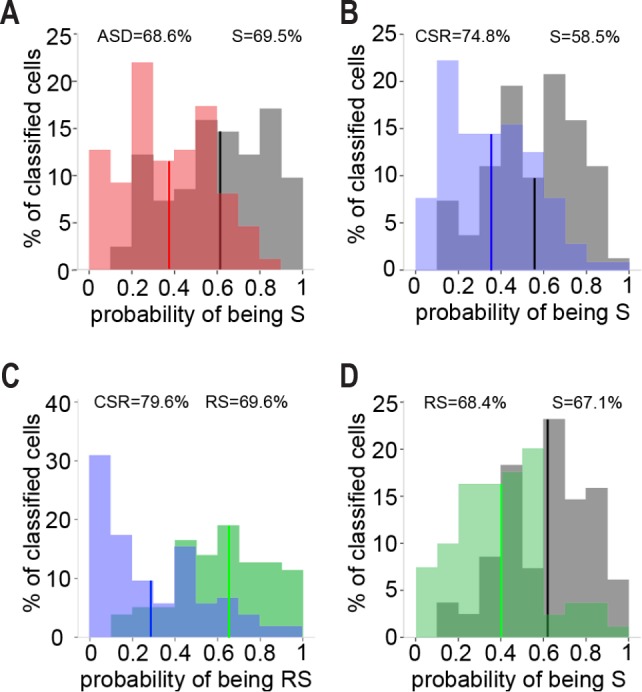
Correct cell classification using 11 ultrastructural parameters. Histograms of cell probabilities calculated by binary logistic regression analysis using 11 ultrastructural measures at single cell level. Percentages on top of each graph indicate correct cell classification in the respective experimental group. Vertical lines mark average probability. (A) Average probability of being S (S = 1, ASD = 0) for S and ASD populations of cells: 0.61 in S, 0.37 in ASD. (B) Average probability of being S (S = 1, CSR = 0) for S and CSR cells: 0.56 in S, 0.35 in CSR. (C) Average probability of being RS (RS = 1, CSR = 0) for RS and CSR cells: 0.62 in RS, 0.29 in CSR. (D) Average probability of being S (S = 1, RS = 0) for S and RS cells: 0.62 in S, 0.42 in RS. CSR, chronic sleep restriction; RS, recovery sleep; S, sleep; ASD, acute sleep deprivation.
Table 3.
Cell classification.
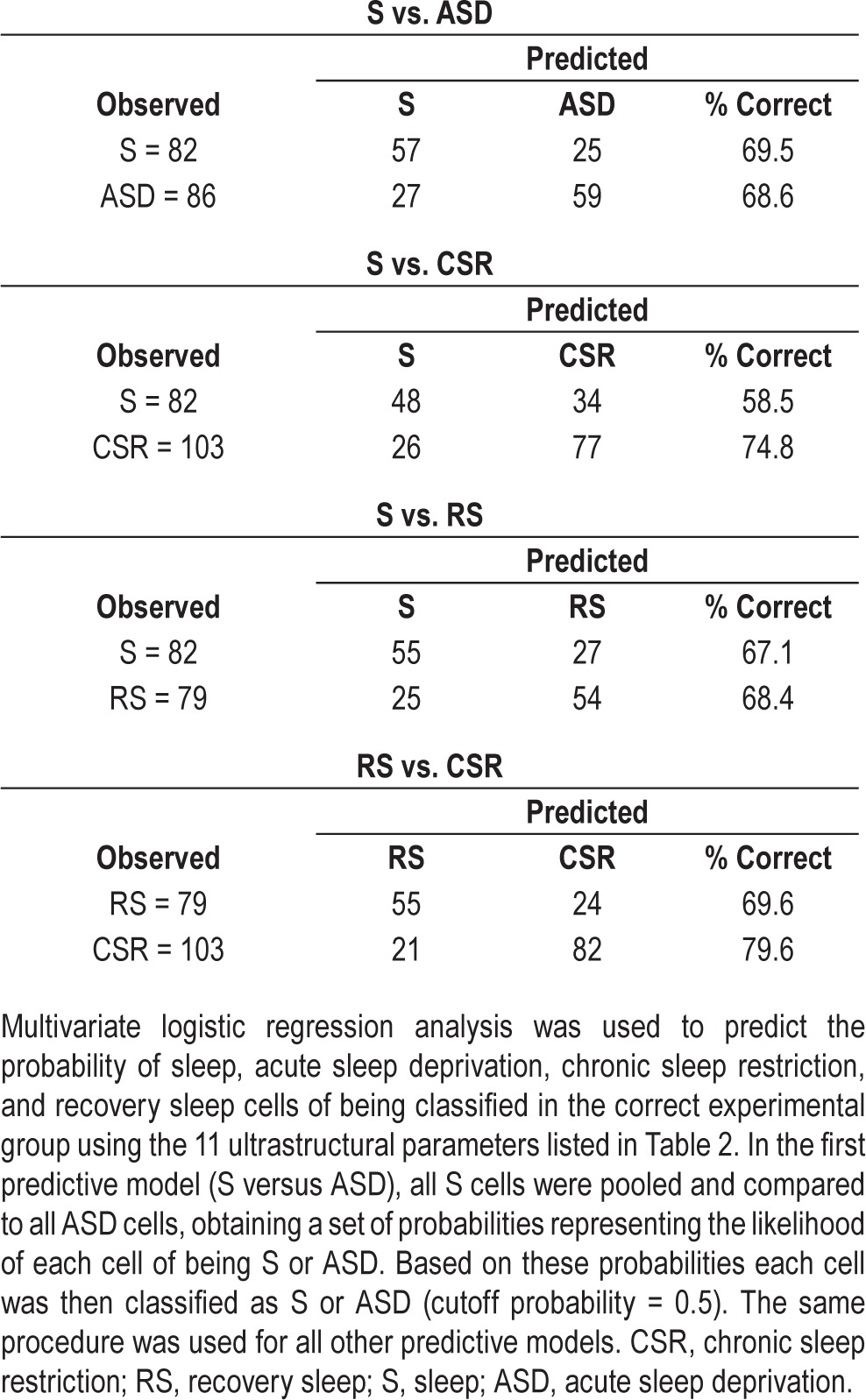
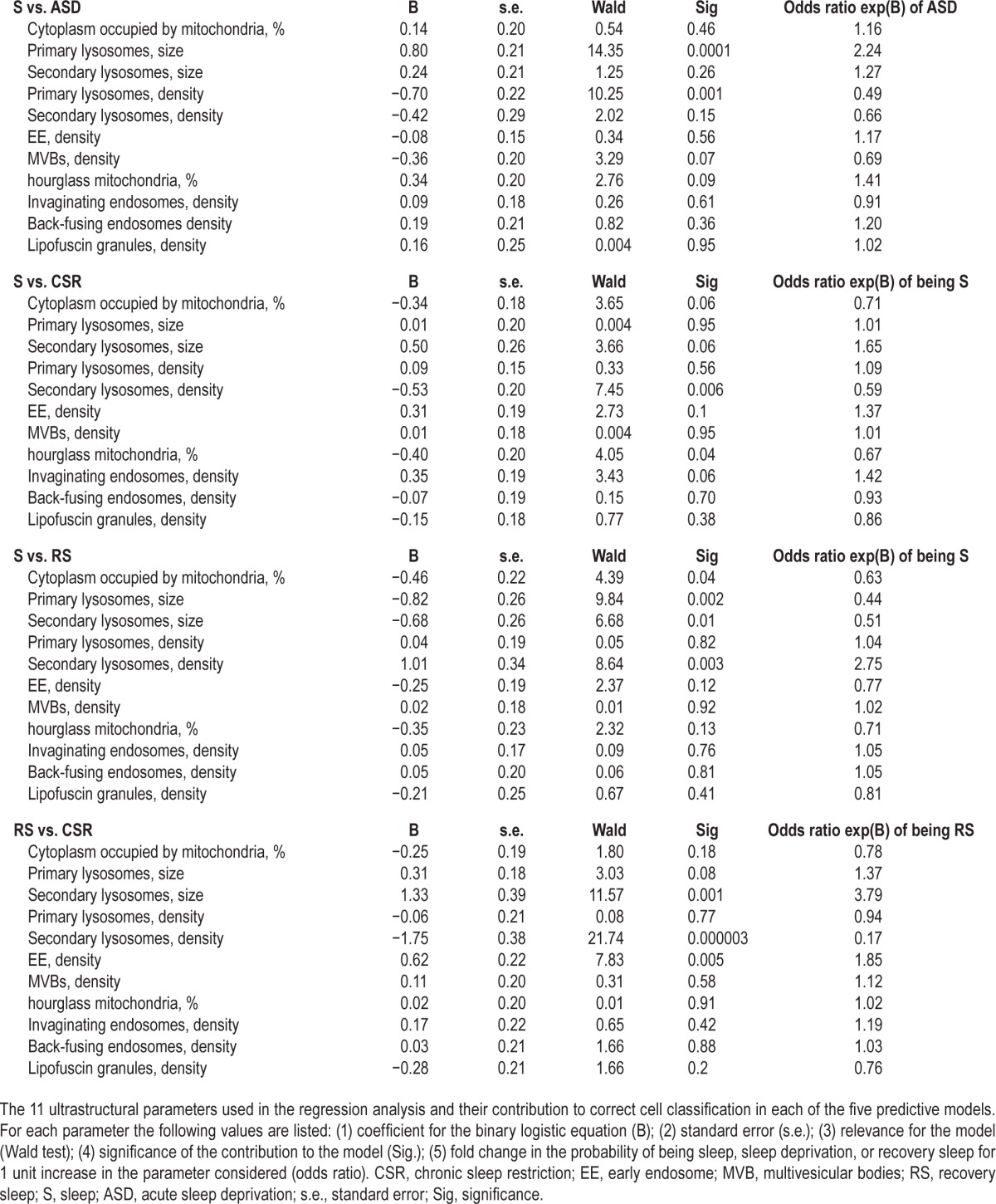
All parameters contributed to some extent to the correct cell classification, but their relative weight varied depending on the specific comparison (Table 4). Specifically, size and density of primary lysosomes were the most significant predictors to discriminate between S and ASD cells, while density of secondary lysosomes and the percentage of hourglass mitochondria mostly contributed to the distinction between S and CSR cells. Moreover, size of primary lysosomes and density of secondary lysosomes counted the most for the difference between S and RS. Finally, density and size of secondary lysosomes were the most informative features for the comparison between CSR and RS cells.
Table 4.
Regression features.
DISCUSSION
We describe for the first time quantitative ultrastructural differences in the cerebral cortex of adolescent mice after sleep, acute total sleep deprivation, and chronic sleep restriction, although the use of additional strategies to enforce wake in CSR mice relative to ASD mice, including caffeine and social interactions, may have contributed to some of the differences between short and long sleep loss. We know of very few published reports on the effects of sleep and wake at the ultrastructural level. Two preliminary EM studies, published only as abstracts, found no consistent changes in axonal microtubule density40,41 and no qualitative changes in mitochondria and other organelles in the brain of adult rats subjected to 8 days of total sleep deprivation relative to yoked controls.42 A more recent ultrastructural study43 describes “reparative changes” in anterior limbic cortex of adult rats after 12–36 h of ASD, but the analysis was based on a combined index of “repair”, without quantitative measurements for each organelle. Moreover, whether this index had predictive power for correct cell classification was not tested.
We found several parameters that could distinguish neurons from mice that slept for 6–8 h relative to those of mice that were kept awake during that time. Relative to S mice, in neurons from ASD mice there were relatively more hourglass mitochondria (a possible sign of increased turnover), some large mitochondria appeared, the relative proportion between EEs and MVBs was more balanced, back-fusion events increased, primary lysosomes became larger but less frequent, and secondary lysosomes grew in size. The cytoplasmic area occupied by mitochondria also increased in ASD mice relative to S mice, but not significantly. Although we do not know whether similar changes may occur between sleep and spontaneous wake, these results indicate that ultrastructural analyses are clearly capable of detecting mitochondrial, endosomal, and lysosomal changes even after just a few hours of sleep loss.
Mitochondrial changes relative to sleep were already present after ASD but became only significant, or more so, after CSR. Because brief exposure to light at night and caffeine were used to enforce CSR but were not employed during ASD, we cannot rule out that these stimuli in themselves, independent of their wake-promoting effects, may have contributed to the larger changes seen after CSR. However, we do not know of any evidence that brief exposure to light affects the ultrastructure of cortical cells. Similarly, although caffeine per se can affect mitochondrial activity and morphology via activation of calcium release from the endoplasmic reticulum (ER), this effect has been documented in muscle cells whose sarcoplasmic reticulum is rich in caffeine-sensitive ryanodine receptors, but not in cortical cells. In fact, the cell bodies of cortical pyramidal neurons express mostly IP3 receptors, not ryanodine receptors,44 and layer II pyramidal neurons in the rat adolescent frontal and parietal cortex are insensitive to adenosine.45 Note also that only a minority of mitochondria (5–20%) have been shown to be in close contact with the ER,46 whereas the most robust change that we found—an overall shift toward larger mitochondria–occurred in most mitochondria. Still, we cannot rule out that caffeine may have affected layer II pyramidal neurons indirectly, by acting on other neuronal populations or glial cells, as shown after prolonged (25 days) caffeine treatment in adolescent rats.47
The shift toward larger mitochondria is likely a sign of activation, reflecting a greater need for energy during wake relative to sleep, independent of wake duration. Overall energy metabolism increases in cerebral cortex in wake relative to sleep, and so do extracellular levels of glutamate, lactate, and oxygen, whereas glucose levels increase with sleep.5,48–51 In rat cerebral cortex, as few as 3 h of total ASD increase mitochondrial gene expression52,53 and mitochondrial enzymatic activity.53,54 However, whether CSR increases brain mitochondrial function is unknown. A recent study found that sleep fragmentation reduces the brain uptake of 2-deoxyglucose,10 but this protocol of sleep disruption caused only small decreases in total sleep amount, whereas in our CRS mice sleep was cut by 70%. In another study, rats subjected to 11 to 12 days of total sleep deprivation showed no change in whole brain glucose consumption and some regional decreases, but these differences were measured relative to yoked controls that had CSR.55 Based on our morphological findings of much larger mitochondrial changes in CSR mice than in ASD mice, we predict that there should be an overall upregulation of cortical mitochondrial activity associated with CSR. The other mitochondrial changes that we found with sleep loss (extra-large and hourglass mitochondria) could also indicate activation or injury, although the latter seems unlikely because (1) extra-large mitochondria were very rare, and in a few cases already present after ASD; (2) extra-large mitochondria did not show any obvious ultrastructural abnormality; and (3) the percentage of hourglass mitochondria was higher also in ASD mice relative to S mice, and did not differ between ASD and CSR animals.
Of note, the mitochondrial changes induced by CSR started to revert (small mitochondria in RS are similar to S) but did not completely returned to “baseline” sleep levels after 32 h of sleep. Moreover, based on 11 ultrastructural parameters, we found that neurons from S and RS mice could be distinguished from each other with the same accuracy as S cells could be distinguished from ASD or CRS cells. In humans, CSR leads to performance deficits that may not be fully resolved after 1 or 2 days of recovery.56,57 Despite obvious species differences, our EM findings may thus represent the structural counterpart of this behavioral evidence. Together, anatomical and functional results suggest that even after relatively short CSR (1–2 w) the brain may need more than a few hours of sleep to recover.
Other changes that were common to both ASD and CSR were in the number of endosomal compartments showing back-fusion, which increased by 40% in both ASD and CSR mice relative to S mice. This result suggests that brain cells respond to extended wake with an increased turnover of cellular components, at least in part through a mechanism of recycling that involves EEs and MVBs.
CSR, however, was specifically associated with more secondary lysosomes and relatively more mature MVBs than EEs, changes that were absent after ASD. Secondary lysosomes reflect past lysosomal activation to eliminate damaged cellular components including mitochondria, and to prevent or reduce cellular stress.18 Even short ASD triggers a stress protective response in brain cells, the unfolded protein response mediated by the ER,4 which helps restore ER function by upregulating the expression of molecular chaperones such as BiP/HSPA5. Transcriptomic and proteomic studies have found other presumed signs of cellular stress in the cortex of sleep deprived rodents, including increased messenger RNAs and/or protein levels of heat shock proteins, hemoglobin alpha 1/2 and beta, and cytochrome C, whose release from mitochondria may lead to apoptosis.13,58 Some of these changes were already present after short ASD, became more significant after days of total ASD, and were also present in yoked controls subjected to several days of CSR.13,58 The ultrastructural signs of past lysosomal activation found here after CSR provide direct anatomical evidence that even a partial and relatively short period of sleep loss (> 4 days) may lead to significant cellular stress, at least during adolescence. The presence of lipofuscin granules in all CSR mice, even though in very few cells, is also consistent with this hypothesis. We do not know whether the lysosomal activation triggered by CSR has long-term, irreversible consequences, nor do we know whether it would occur under less severe conditions of CSR. However, the number of lipofuscin granules was low in RS mice, suggesting that what we detected, mostly in CSR, were not mature lipofuscins/ceroid, which are thought to be undegradable, but perhaps an intermediate, immature form.59,60 Evidence of reversibility of lipofuscin formation in vivo is limited, but was described in motor cortex of squirrel monkeys rehabilitated after protein malnutrition.61
We found that cellular signatures of sleep and wake exist and can be used to estimate the probability of a pyramidal neuron to belong to a S, ASD, CSR, or RS mouse. By using a combination of 11 parameters related to mitochondria, endosomes and lysosomes, cell classification was correct, on average, in 70% of cases, a number likely to significantly increase when additional cellular measures will be added. The current degree of accuracy is already remarkable considering that, due to the time-consuming nature of EM analysis, we sampled one single plane for each cell, and “only” ∼20 cells were analyzed in each mouse (80–100 cells/group). The limitations of our sampling technique may be partly responsible for the variability observed among the cells within the same mouse, but if so, this factor affected all mice and all groups to a similar extent because within-mice variance did not change significantly across groups. Independent of these limitations, the current finding that sleep and wake are associated with reliable ultrastructural changes suggests that they could serve as markers to determine whether sleep or wake have occurred at the level of single neurons, and to further our understanding of the phenomenon of local sleep16 and its underlying cellular mechanisms.
Importantly, we found that between-mice variance tended to increase in mice that underwent ASD or CSR relative to S, and did not return to baseline level in the RS group. This trend toward higher interindividual variability after sleep loss, although not significant, is remarkable given that each group included only four or five animals, and may reflect true biological heterogeneity unmasked by sleep loss. The percentage of cytoplasm occupied by mitochondria appeared to contribute the most to the increase in between-mice variance, suggesting that metabolic parameters may be especially useful to identify susceptibility to sleep loss at the individual level. In humans, there are stable, trait-like differences in the susceptibility to cognitive impairment caused by ASD or CSR.62,63 Neuro-imaging studies found that differences in the activation of frontoparietal regions during a working memory task at rest are associated with differences in the extent of the cognitive decline during ASD.64,65 Moreover, differences in the micro-structure of the white matter over broad areas,66 and more specifically in the axonal pathways connecting frontal and parietal areas,67 can predict interindividual differences in the resistance to ASD. To our knowledge, no study in humans has looked for possible differences in gray matter, and there are no studies in sleep deprived rodents focusing on interindividual differences and their underlying mechanisms. Our findings, however, suggest that this may be a promising avenue for future experiments.
DISCLOSURE STATEMENT
This was not an industry-supported study. Support provided by grants 1R01MH099231 and 1P01NS083514 to Drs. Cirelli and Tononi. Dr. Tononi has consulted for Philips Respironics and has been involved in a research study in humans supported by Philips Respironics. Dr. Tononi is also a consultant for the Allen Institute for Brain Research. The article submitted is not related to any of these relationships. The other authors have indicated no financial conflicts of interest.
ACKNOWLEDGMENTS
The authors thank Drs. William Marshall and Armand Mensen for help with variance component and regression analysis.
REFERENCES
- 1.Killgore WD. Effects of sleep deprivation on cognition. Prog Brain Res. 2010;185:105–29. doi: 10.1016/B978-0-444-53702-7.00007-5. [DOI] [PubMed] [Google Scholar]
- 2.Goel N, Basner M, Rao H, Dinges DF. Circadian rhythms, sleep deprivation, and human performance. Prog Mol Biol Transl Sci. 2013;119:155–90. doi: 10.1016/B978-0-12-396971-2.00007-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cirelli C, Gutierrez CM, Tononi G. Extensive and divergent effects of sleep and wakefulness on brain gene expression. Neuron. 2004;41:35–43. doi: 10.1016/s0896-6273(03)00814-6. [DOI] [PubMed] [Google Scholar]
- 4.Naidoo N, Giang W, Galante RJ, Pack AI. Sleep deprivation induces the unfolded protein response in mouse cerebral cortex. J Neurochem. 2005;92:1150–7. doi: 10.1111/j.1471-4159.2004.02952.x. [DOI] [PubMed] [Google Scholar]
- 5.Dash MB, Douglas CL, Vyazovskiy V, Cirelli C, Tononi G. Long-term homeostasis of extracellular glutamate in the rat cerebral cortex across sleep and waking states. J Neurosci. 2009;29:620–9. doi: 10.1523/JNEUROSCI.5486-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Porkka-Heiskanen T, Strecker RE, McCarley RW. Brain site-specificity of extracellular adenosine concentration changes during sleep deprivation and spontaneous sleep: an in vivo microdialysis study. Neuroscience. 2000;99:507–17. doi: 10.1016/s0306-4522(00)00220-7. [DOI] [PubMed] [Google Scholar]
- 7.Durmer JS, Dinges DF. Neurocognitive consequences of sleep deprivation. Semin Neurol. 2005;25:117–29. doi: 10.1055/s-2005-867080. [DOI] [PubMed] [Google Scholar]
- 8.Cohen DA, Wang W, Wyatt JK, et al. Uncovering residual effects of chronic sleep loss on human performance. Sci Transl Med. 2010;2:14ra13. doi: 10.1126/scitranslmed.3000458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anderson C, Sullivan JP, Flynn-Evans EE, Cade BE, Czeisler CA, Lockley SW. Deterioration of neurobehavioral performance in resident physicians during repeated exposure to extended duration work shifts. Sleep. 2012;35:1137–46. doi: 10.5665/sleep.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.He J, Hsuchou H, He Y, Kastin AJ, Wang Y, Pan W. Sleep restriction impairs blood-brain barrier function. J Neurosci. 2014;34:14697–706. doi: 10.1523/JNEUROSCI.2111-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mackiewicz M, Zimmerman JE, Shockley KR, Churchill GA, Pack AI. What are microarrays teaching us about sleep? Trends Mol Med. 2009;15:79–87. doi: 10.1016/j.molmed.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maret S, Dorsaz S, Gurcel L, et al. Homer1a is a core brain molecular correlate of sleep loss. Proc Natl Acad Sci U S A. 2007;104:20090–5. doi: 10.1073/pnas.0710131104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cirelli C, Pfister-Genskow M, McCarthy D, Woodbury R, Tononi G. Proteomic profiling of the rat cerebral cortex in sleep and waking. Arch Ital Biol. 2009;147:59–68. [PMC free article] [PubMed] [Google Scholar]
- 14.Vyazovskiy VV, Olcese U, Hanlon EC, Nir Y, Cirelli C, Tononi G. Local sleep in awake rats. Nature. 2011;472:443–7. doi: 10.1038/nature10009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rector DM, Topchiy IA, Carter KM, Rojas MJ. Local functional state differences between rat cortical columns. Brain Res. 2005;1047:45–55. doi: 10.1016/j.brainres.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 16.Tononi G, Cirelli C. Sleep and the price of plasticity: from synaptic and cellular homeostasis to memory consolidation and integration. Neuron. 2014;81:12–34. doi: 10.1016/j.neuron.2013.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scott CC, Vacca F, Gruenberg J. Endosome maturation, transport and functions. Semin Cell Dev Biol. 2014;31:2–10. doi: 10.1016/j.semcdb.2014.03.034. [DOI] [PubMed] [Google Scholar]
- 18.Damme M, Suntio T, Saftig P, Eskelinen EL. Autophagy in neuronal cells: general principles and physiological and pathological functions. Acta Neuropathol. 2015;129:337–62. doi: 10.1007/s00401-014-1361-4. [DOI] [PubMed] [Google Scholar]
- 19.Nelson AB, Faraguna U, Zoltan JT, Tononi G, Cirelli C. Sleep patterns and homeostatic mechanisms in adolescent mice. Brain Sci. 2013;3:318–43. doi: 10.3390/brainsci3010318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maret S, Faraguna U, Nelson A, Cirelli C, Tononi G. Sleep and wake modulate spine turnover in the adolescent mouse cortex. Nature Neurosci. 2011;14:1418–20. doi: 10.1038/nn.2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de Vivo L, Faraguna U, Nelson AB, et al. Developmental patterns of sleep slow wave activity and synaptic density in adolescent mice. Sleep. 2014;37:689–700. doi: 10.5665/sleep.3570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu YP, Nolan B, Lou YR, Peng QY, Wagner GC, Conney AH. Voluntary exercise together with oral caffeine markedly stimulates UVB light-induced apoptosis and decreases tissue fat in SKH-1 mice. Proc Natl Acad Sci U S A. 2007;104:12936–41. doi: 10.1073/pnas.0705839104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Newman SM, Paletz EM, Obermeyer WH, Benca RM. Sleep deprivation in pigeons and rats using motion detection. Sleep. 2009;32:1299–312. doi: 10.1093/sleep/32.10.1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peters A, Palay S, Webster H. Philadelphia, PA: WB Saunders; 1991. The fine structure of the nervous system: the neurons and supporting cells. [Google Scholar]
- 25.Jones EG, Powell TPS. An anatomical study of converging sensory pathways within the cerebral cortex of the monkey. Brain. 1970;93:793–820. doi: 10.1093/brain/93.4.793. [DOI] [PubMed] [Google Scholar]
- 26.Peters A, Kaiserman-Abramof IR. The small pyramidal neuron of the rat cerebral cortex. The perikaryon, dendrites and spines. Am J Anat. 1970;127:321–55. doi: 10.1002/aja.1001270402. [DOI] [PubMed] [Google Scholar]
- 27.Schindelin J, Arganda-Carreras I, Frise E, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–82. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vyazovskiy VV, Cirelli C, Tononi G. Electrophysiological correlates of sleep homeostasis in freely behaving rats. Prog Brain Res. 2011;193:17–38. doi: 10.1016/B978-0-444-53839-0.00002-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fox K. Anatomical pathways and molecular mechanisms for plasticity in the barrel cortex. Neuroscience. 2002;111:799–814. doi: 10.1016/s0306-4522(02)00027-1. [DOI] [PubMed] [Google Scholar]
- 30.Diamond ME, Huang W, Ebner FF. Laminar comparison of somatosensory cortical plasticity. Science. 1994;265:1885–8. doi: 10.1126/science.8091215. [DOI] [PubMed] [Google Scholar]
- 31.Jiang B, Trevino M, Kirkwood A. Sequential development of long-term potentiation and depression in different layers of the mouse visual cortex. J Neurosci. 2007;27:9648–52. doi: 10.1523/JNEUROSCI.2655-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ahmad T, Aggarwal K, Pattnaik B, et al. Computational classification of mitochondrial shapes reflects stress and redox state. Cell Death Dis. 2013;4:e461. doi: 10.1038/cddis.2012.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Trimmer PA, Swerdlow RH, Parks JK, et al. Abnormal mitochondrial morphology in sporadic Parkinson's and Alzheimer's disease cybrid cell lines. Exper Neurol. 2000;162:37–50. doi: 10.1006/exnr.2000.7333. [DOI] [PubMed] [Google Scholar]
- 34.Huotari J, Helenius A. Endosome maturation. EMBO J. 2011;30:3481–500. doi: 10.1038/emboj.2011.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saftig P, Klumperman J. Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat Rev Mol Cell Biol. 2009;10:623–35. doi: 10.1038/nrm2745. [DOI] [PubMed] [Google Scholar]
- 36.Maxfield FR, McGraw TE. Endocytic recycling. Nat Rev Mol Cell Biol. 2004;5:121–32. doi: 10.1038/nrm1315. [DOI] [PubMed] [Google Scholar]
- 37.Li WW, Li J, Bao JK. Microautophagy: lesser-known self-eating. Cell Mol Life Sci. 2012;69:1125–36. doi: 10.1007/s00018-011-0865-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Luzio JP, Pryor PR, Bright NA. Lysosomes: fusion and function. Nat Rev Mol Cell Biol. 2007;8:622–32. doi: 10.1038/nrm2217. [DOI] [PubMed] [Google Scholar]
- 39.Sulzer D, Mosharov E, Talloczy Z, Zucca FA, Simon JD, Zecca L. Neuronal pigmented autophagic vacuoles: lipofuscin, neuromelanin, and ceroid as macroautophagic responses during aging and disease. J Neurochem. 2008;106:24–36. doi: 10.1111/j.1471-4159.2008.05385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kiyashchenko L. Ultrastructural changes in cytoskeleton of rat CNS nerve fibers under sleep deprivation. J Sleep Res. 1994;3:128. [Google Scholar]
- 41.Feng PF, Bergmann BM, Rechtschaffen A. Effect of total sleep deprivation on microtubule density in rat brain. Sleep Res. 1995;24:443. [Google Scholar]
- 42.Feng PF, Bergmann BM, Rechtschaffen A. Effect of total sleep deprivation on neuronal ultrastructure in the rat. Sleep Res. 1996;25:466. [Google Scholar]
- 43.Abushov BM. Morphofunctional analysis of the effects of total sleep deprivation on the CNS in rats. Neurosci Behav Physiol. 2010;40:403–9. doi: 10.1007/s11055-010-9271-y. [DOI] [PubMed] [Google Scholar]
- 44.Sharp AH, McPherson PS, Dawson TM, Aoki C, Campbell KP, Snyder SH. Differential immunohistochemical localization of inositol 1,4,5-trisphosphate- and ryanodine-sensitive Ca2+ release channels in rat brain. J Neurosci. 1993;13:3051–63. doi: 10.1523/JNEUROSCI.13-07-03051.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van Aerde KI, Qi G, Feldmeyer D. Cell type-specific effects of adenosine on cortical neurons. Cereb Cortex. 2015;25:772–87. doi: 10.1093/cercor/bht274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rizzuto R, Pinton P, Carrington W, et al. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science. 1998;280:1763–6. doi: 10.1126/science.280.5370.1763. [DOI] [PubMed] [Google Scholar]
- 47.Ardais AP, Borges MF, Rocha AS, Sallaberry C, Cunha RA, Porciuncula LO. Caffeine triggers behavioral and neurochemical alterations in adolescent rats. Neuroscience. 2014;270:27–39. doi: 10.1016/j.neuroscience.2014.04.003. [DOI] [PubMed] [Google Scholar]
- 48.Dash MB, Bellesi M, Tononi G, Cirelli C. Sleep/wake dependent changes in cortical glucose concentrations. J Neurochem. 2013;124:79–89. doi: 10.1111/jnc.12063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dash MB, Tononi G, Cirelli C. Extracellular levels of lactate, but not oxygen, reflect sleep homeostasis in the rat cerebral cortex. Sleep. 2012;35:909–19. doi: 10.5665/sleep.1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Naylor E, Aillon DV, Barrett BS, et al. Lactate as a biomarker for sleep. Sleep. 2012;35:1209–22. doi: 10.5665/sleep.2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wisor JP, Rempe MJ, Schmidt MA, Moore ME, Clegern WC. Sleep slow-wave activity regulates cerebral glycolytic metabolism. Cereb Cortex. 2013;23:1978–87. doi: 10.1093/cercor/bhs189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cirelli C, Tononi G. Differences in gene expression between sleep and waking as revealed by mRNA differential display. Brain Res Mol Brain Res. 1998;56:293–305. doi: 10.1016/s0169-328x(98)00057-6. [DOI] [PubMed] [Google Scholar]
- 53.Nikonova EV, Vijayasarathy C, Zhang L, et al. Differences in activity of cytochrome C oxidase in brain between sleep and wakefulness. Sleep. 2005;28:21–7. doi: 10.1093/sleep/28.1.21. [DOI] [PubMed] [Google Scholar]
- 54.Nikonova EV, Naidoo N, Zhang L, et al. Changes in components of energy regulation in mouse cortex with increases in wakefulness. Sleep. 2010;33:889–900. doi: 10.1093/sleep/33.7.889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Everson CA, Smith CB, Sokoloff L. Effects of prolonged sleep deprivation on local rates of cerebral energy metabolism in freely moving rats. J Neurosci. 1994;14:6769–78. doi: 10.1523/JNEUROSCI.14-11-06769.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Belenky G, Wesensten NJ, Thorne DR, et al. Patterns of performance degradation and restoration during sleep restriction and subsequent recovery: a sleep dose-response study. J Sleep Res. 2003;12:1–12. doi: 10.1046/j.1365-2869.2003.00337.x. [DOI] [PubMed] [Google Scholar]
- 57.Banks S, Van Dongen HP, Maislin G, Dinges DF. Neurobehavioral dynamics following chronic sleep restriction: dose-response effects of one night for recovery. Sleep. 2010;33:1013–26. doi: 10.1093/sleep/33.8.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cirelli C, Faraguna U, Tononi G. Changes in brain gene expression after long-term sleep deprivation. J Neurochem. 2006;98:1632–45. doi: 10.1111/j.1471-4159.2006.04058.x. [DOI] [PubMed] [Google Scholar]
- 59.Terman A, Brunk UT. On the degradability and exocytosis of ceroid/lipofuscin in cultured rat cardiac myocytes. Mech Ageing Dev. 1998;100:145–56. doi: 10.1016/s0047-6374(97)00129-2. [DOI] [PubMed] [Google Scholar]
- 60.Terman A, Brunk UT. Lipofuscin: mechanisms of formation and increase with age. APMIS. 1998;106:265–76. doi: 10.1111/j.1699-0463.1998.tb01346.x. [DOI] [PubMed] [Google Scholar]
- 61.Manocha SL, Sharma SP. Reversibility of lipofuscin accumulation caused by protein malnutrition in the motor cortex of squirrel monkeys, Saimiri scireus. Acta Histochem. 1977;58:219–31. doi: 10.1016/S0065-1281(77)80132-3. [DOI] [PubMed] [Google Scholar]
- 62.Van Dongen HP, Baynard MD, Maislin G, Dinges DF. Systematic interindividual differences in neurobehavioral impairment from sleep loss: evidence of trait-like differential vulnerability. Sleep. 2004;27:423–33. [PubMed] [Google Scholar]
- 63.Rupp TL, Wesensten NJ, Balkin TJ. Trait-like vulnerability to total and partial sleep loss. Sleep. 2012;35:1163–72. doi: 10.5665/sleep.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mu Q, Mishory A, Johnson KA, et al. Decreased brain activation during a working memory task at rested baseline is associated with vulnerability to sleep deprivation. Sleep. 2005;28:433–46. doi: 10.1093/sleep/28.4.433. [DOI] [PubMed] [Google Scholar]
- 65.Chee MW, Chuah LY, Venkatraman V, Chan WY, Philip P, Dinges DF. Functional imaging of working memory following normal sleep and after 24 and 35 h of sleep deprivation: correlations of fronto-parietal activation with performance. NeuroImage. 2006;31:419–28. doi: 10.1016/j.neuroimage.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 66.Rocklage M, Williams V, Pacheco J, Schnyer DM. White matter differences predict cognitive vulnerability to sleep deprivation. Sleep. 2009;32:1100–3. doi: 10.1093/sleep/32.8.1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cui J, Tkachenko O, Gogel H, et al. Microstructure of frontoparietal connections predicts individual resistance to sleep deprivation. NeuroImage. 2015;106:123–33. doi: 10.1016/j.neuroimage.2014.11.035. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.