

Abstract
Background
We previously reported that talimogene laherparepvec, an oncolytic herpes virus encoding granulocyte-macrophage colony-stimulating factor (GM-CSF), resulted in an objective response rate of 26 % in patients with advanced melanoma in a phase II clinical trial. The response of individual lesions, however, was not reported. Since talimogene laherparepvec is thought to mediate anti-tumor activity through both direct tumor cytolysis and induction of systemic tumor-specific immunity, we sought to determine the independent response rate in virus-injected and non-injected lesions.
Methods
Fifty patients with stage IIIC or IV melanoma were treated with talimogene laherparepvec in a multi-institutional single-arm open-label phase II clinical trial. In this study patients were treated until a complete response was achieved, all accessible tumors disappeared, clinically significant disease progression, or unacceptable toxicity. This report is a post hoc analysis of the systemic effects of talimogene laherparepvec in injected lesions and two types of uninjected lesions—non-visceral lesions and visceral lesions.
Results
Eleven of 23 patients (47.8 %) had a ≥ 30 % reduction in the total burden of uninjected non-visceral lesions, and 2 of 12 patients (16.7 %) had a ≥ 30 % reduction in the total burden of visceral lesions. Among 128 evaluable lesions directly injected with talimogene laherparepvec, 86 (67.2 %) decreased in size by ≥ 30 % and 59 (46.1 %) completely resolved. Of 146 uninjected non-visceral lesions, 60 (41.1 %) decreased in size by ≥ 30 %, the majority of which (44 [30.1 %]) completely resolved. Of 32 visceral lesions, 4 (12.5 %) decreased in size by ≥ 30 %, and 3 (9.4 %) completely resolved. The median time to lesion response was shortest for lesions that were directly injected (18.4 weeks), followed by uninjected non-visceral lesions (23.1 weeks) and visceral lesions (51.3 weeks), consistent with initiation of a delayed regional and systemic anti-tumor immune response to talimogene laherparepvec.
Conclusions
These results support a regional and systemic effect of talimogene laherparepvec immunotherapy in patients with advanced melanoma.
Keywords: Herpes virus, Immunotherapy, Melanoma, Oncolytic virus, Talimogene laherparepvec, T-VEC
Background
The therapeutic landscape for the treatment of metastatic melanoma has been changing dramatically over the last few years driven by progress in the clinical application of targeted therapy and tumor immunotherapy [1]. Immunotherapy has gained considerable attention, in part because of high response rates with some monotherapy and combination therapy regimens (such as immune checkpoint inhibitors), evidence of overall survival benefit in randomized clinical trials, and the durability often obtained with immunotherapy agents [2–4]. There is now emerging evidence that lymphocyte-predominant tumors, characterized with high numbers of effector T cells (and a weighted effector T cell/regulatory T cell ratio), may be more susceptible to immunotherapy and strategies to increase the lymphocytic infiltration to tumors are a high priority for improving clinical responses to immunotherapy.
Talimogene laherparepvec, an oncolytic herpes simplex virus type 1 (HSV-1) [5], was gene modified to elicit increased selectivity and rescue replication in tumors as well as improved tumor antigen presentation. It contains deletions of the neurovirulence factors ICP34.5 and a factor that blocks peptide loading onto the major histocompatibility complex (MHC) called ICP47. These changes reduce the pathogenicity of the virus. Talimogene laherparepvec also contains an insertion of the human granulocyte-macrophage colony-stimulating factor (GM-CSF) gene sequence at the deleted ICP34.5 coding sequence sites to enhance systemic immune response [6]. We previously reported that in a multi-institutional, single-arm, open-label, phase II clinical trial, intralesional injection of talimogene laherparepvec resulted in an ORR of 26 % in patients with stage IIIC and stage IV melanoma. The majority of responding patients had a durable remission in both injected and uninjected non-visceral lesions (with MART-specific CD8+ T cells observed in both subsets) [7] including visceral sites for 7–31 months, indicating a possible systemic effect [8]. Recently, a randomized phase III clinical trial confirmed this objective response rate and demonstrated an especially high durable response rate for patients with stage IIIB/C and IV M1a disease [9]. This trial led to the U.S. Food and Drug Administration (FDA) approval of talimogene laherparepvec for local treatment of unresectable cutaneous, subcutaneous, and nodal lesions in patients with melanoma recurrent after initial surgery. Talimogene laherparepvec has not been shown to improve overall survival in patients with visceral metastases [10]. Talimogene laherparepvec has been associated with a tolerable safety profile and provides a new strategy for directly killing tumor cells, promoting local lymphocyte infiltration, and use in combination regimens to improve clinical responses to tumor immunotherapy. The ability of talimogene laherparepvec to mediate systemic clinical anti-tumor activity compared to exerting a more local effect on lymphocyte responses has been previously described [8] but not fully quantified.
This report is a post hoc analysis from a phase II study in melanoma of potential systemic effects of talimogene laherparepvec based on an analysis of individual virus-injected lesions and two types of uninjected lesions—non-visceral lesions and visceral lesions. We sought to determine the independent response rate as well as median time to lesion response in these different types of lesions. The results provide insight into the level of systemic activity possible with talimogene laherparepvec in patients with melanoma and provide support for combination studies in which talimogene laherparepvec-induced anti-tumor immunity can be enhanced with other agents known to expand or inhibit suppression of antigen-specific T cells.
Methods
Study design
The full details of the overall design and methods for the phase II clinical trial have been previously reported [8]. Briefly, patients received up to 8 doses of talimogene laherparepvec over a 15-week period. Talimogene laherparepvec was administered at an initial dose of 106 plaque-forming units (PFU)/mL and injected into 1 or more skin or subcutaneous lesions (up to 4 mL total). Subsequent doses began 3 weeks after the first dose and consisted of talimogene laherparepvec at 108 PFU/mL (up to 4 mL total) every 2 weeks. The volume of talimogene laherparepvec delivered to each lesion depended on the size of the lesion measured on the day of virus administration. In this study, at least 1 lesion was to be left uninjected to assess systemic response. Uninjected lesions were to be at least 5 cm from the nearest injected lesion. Tumor responses were derived based on modified Response Evaluation Criteria in Solid Tumors (RECIST) version 1.0.
If indications of biological activity were observed after the initial 8 doses (stable disease or better, inflammatory response in an uninjected non-visceral lesion, and/or injection site reaction) and patients did not have evidence of clinically symptomatic disease progression or unacceptable toxicity, treatment could be continued for an additional 16 doses unless the investigator determined that another therapy was appropriate.
The protocol was approved by the site investigational review boards (see Acknowledgements) and by the U.S. FDA under an investigational new drug application. All patients provided written informed consent.
Response analysis in injected and uninjected lesions
To evaluate the systemic effect of talimogene laherparepvec (ie, beyond local effects in injected lesions), the following endpoints were evaluated by lesion type: patient incidence of overall lesion-type response, incidence of lesion response, maximum decrease in tumor burden (individual lesion and overall by lesion type) and the time to individual lesion response. Analyses of systemic effects were based on baseline or new measurable lesions from patients in a pre-planned safety analysis population. Evaluable lesions were defined as those with measurements recorded at ≥ 2 visits (with a measurable lesion size noted in the earliest assessment). All analyses were based on investigator assessment of lesion measurements and lesion injection status.
Statistical analysis
For this study descriptive statistics were used. Measurable lesion characteristics were displayed by lesion type (injected, uninjected non-visceral, and visceral), and the patient incidences of measurable lesion locations were tabulated by lesion type. Overall lesion-type burden was calculated as the sum of the longest diameters of all lesions of the same type at a study visit. The patient incidence of overall lesion response was reported as the proportion of patients with a ≥ 30 % decrease in overall lesion burden by lesion type. Graphical presentations of the maximum decrease in overall lesion-type burden were produced using waterfall plots. The maximum decrease in the size of individual lesions (based on the longest diameter) was categorized (>0 %, ≥ 30 %, 100 %) and presented by lesion type. The incidence of lesion response was reported as the proportion of lesions with a ≥ 30 % decrease in size. Graphical presentations of the maximum decrease in baseline or new measurable lesions were produced using waterfall plots.
The time to lesion response by lesion type was analyzed using the Kaplan-Meier method for all evaluable lesions of the same lesion type. Kaplan-Meier estimates of event quartiles and the corresponding 95 % confidence intervals, when estimable, were based on a sign test [11]. Time to lesion response was evaluated from the date of the baseline measurement to the date of first response (for new lesions, the first appearance of the lesion was considered the baseline measurement).
Results
Measurable lesion characteristics
Evaluable patients for the lesion-level analyses (patients with at least 1 lesion with measurements at ≥ 2 visits) and overall lesion-type burden analyses (patients with ≥ 2 visits with measurement of all lesions of the same type) are summarized in Table 1. Of the 50 patients who received talimogene laherparepvec injected into a lesion in the phase II clinical trial, 48 patients (96.0 %) also had at least 1 uninjected lesion (Table 1). This included 26 patients (52.0 %) with only uninjected non-visceral lesions at baseline and 22 patients (44.0 %) with at least 1 visceral lesion at baseline. Of 400 baseline or new lesions, 306 were evaluable (i.e., had measurements at ≥ 2 visits), including 146 (47.7 %) uninjected non-visceral lesions and 32 (10.5 %) visceral lesions). The most common anatomic locations of uninjected, non-visceral lesions were lymph nodes, cutaneous metastases, or subcutaneous nodules. Visceral lesions were most commonly located in the lungs and liver (Table 2).
Table 1.
Patient characteristics of measurable lesion type in talimogene laherparepvec study
| Any Measurable Lesion | Evaluablea Lesion | Evaluable for overall lesion- type burdenb | |
|---|---|---|---|
| n (%) | n (%) | n (%) | |
| Any | 50 (100) | 47 (100) | 37 (100) |
| At least 1 baseline uninjected lesion | 48 (96.0) | 41 (87.2) | 35 (94.6) |
| Baseline uninjected, non-visceral only | 26 (52.0) | 23 (48.9) | 23 (62.2) |
| At least 1 visceral lesion at baseline | 22 (44.0) | 15 (31.9) | 12 (32.4) |
Denominators are the numbers of patients
aEvaluable indicates at least 2 assessments with valid measurements per investigator
bEvaluable indicates at least 2 visits with non-missing overall lesion-type burden for each respective patient subgroup, or overall tumor burden for “Any”
All patients received at least one dose of talimogene laherparepvec
Table 2.
Anatomic location of measurable uninjected lesions in talimogene laherparepvec study
| Any | Evaluablea | |
|---|---|---|
| n (%) | n (%) | |
| Any uninjected non-visceral, number of patients (%) | 49 (100) | 42 (100) |
| Any non-visceral, number of patients (%) | 48 (98.0) | 39 (92.9) |
| Head/Neck, Front, number of lesions (%) | 2 (4.1) | 1 (2.4) |
| Head/Neck, Back | 1 (2.0) | 1 (2.4) |
| Head/Neck, Right | 4 (8.2) | 4 (9.5) |
| Head/Neck, Left | 4 (8.2) | 4 (9.5) |
| Trunk, Front | 13 (26.5) | 10 (23.8) |
| Trunk, Back | 6 (12.2) | 5 (11.9) |
| Lower Limb, Right | 5 (10.2) | 5 (11.9) |
| Lower Limb, Left | 8 (16.3) | 8 (19.0) |
| Upper Limb, Right | 4 (8.2) | 3 (7.1) |
| Upper Limb, Left | 2 (4.1) | 1 (2.4) |
| Right Hand, Palm | 1 (2.0) | 1 (2.4) |
| Right Hand, Back | 1 (2.0) | 1 (2.4) |
| Groin | 2 (4.1) | 2 (4.8) |
| Lymph node, specify | 21 (42.9) | 16 (38.1) |
| Other | 16 (32.7) | 8 (19.0) |
| Any visceral, number of patients (%) | 23 (46.9) | 15 (35.7) |
| Eye, number of lesions (%) | 1 (2.0) | 0 (0.0) |
| Brain | 3 (6.1) | 0 (0.0) |
| Lung | 17 (34.7) | 12 (28.6) |
| Gastrointestinal Tract | 3 (6.1) | 1 (2.4) |
| Kidney | 3 (6.1) | 0 (0.0) |
| Adrenal | 4 (8.2) | 1 (2.4) |
| Liver | 8 (16.3) | 4 (9.5) |
| Pancreas | 3 (6.1) | 2 (4.8) |
| Spleen | 4 (8.2) | 0 (0.0) |
Denominator is the total number of patients. Patients may have multiple lesions
aEvaluable indicates at least 2 assessments with valid measurements
All patients received at least one dose of talimogene laherparepvec
Patient incidence of overall lesion-type response
Of the 23 evaluable patients with baseline uninjected non-visceral disease, 11 patients (47.8 %) had a ≥ 30 % reduction in the total burden of uninjected non-visceral lesions (Table 3). Of the 12 evaluable patients with visceral disease, 2 patients (16.7 %) had a ≥ 30 % reduction in the total burden of visceral lesions. In general, the observed response rate per investigator was consistent with the rate of overall lesion-type response (derived from the tumor burden change within a lesion subset; see Table 3). The overall tumor response rate for patients with only uninjected non-visceral lesions was 43.5 % per investigator and 47.8 % based on the change in the total patient burden of uninjected non-visceral lesions. The overall tumor response rate for patients with visceral-only lesions was 16.7 %, both per investigator and based on the change in the total patient burden of visceral lesions.
Table 3.
Summary of talimogene laherparepvec responses by lesion-type
| Overall tumor response | Overall lesion-type response | |||
|---|---|---|---|---|
| Any | OR | CR | Yes | |
| n (%) | n (%) | n (%) | n (%) | |
| Any | 37 (100) | 14 (37.8) | 8 (21.6) | 15 (40.5) |
| At least 1 baseline uninjected lesion | 35 (100) | 12 (34.3) | 6 (17.1) | 13 (37.1) |
| Baseline uninjected non-visceral only | 23 (100) | 10 (43.5) | 5 (21.7) | 11 (47.8) |
| At least 1 visceral lesion at baseline | 12 (100) | 2 (16.7) | 1 (8.3) | 2 (16.7) |
Denominator is the total number of patients in corresponding patient lesion-type subgroup
Objective response (OR), and complete response (CR) as per investigator-reported responses. Overall lesion response was reported as the proportion of patients with a ≥ 30 % decrease in overall lesion burden. OR was evaluated by modified RECIST.
Overall lesion type response: max decrease ≥ 30 % in overall lesion-type burden from baseline in patient lesion-type subgroup and in total tumor burden for “Any”
All patients received at least one dose of talimogene laherparepvec
Incidence of lesion response
The waterfall plot of the maximum decrease of individual lesions is shown in Fig. 1a. Among 128 evaluable lesions directly injected with talimogene laherparepvec, 86 (67.2 %) decreased in size by ≥ 30 %, and 59 (46.1 %) completely resolved (Fig. 1a). Responses were also observed in baseline and new uninjected lesions, including both non-visceral and visceral lesions as shown in the waterfall plot for baseline and new measurable lesions that appeared during the course of study treatment (Fig. 1b). Of 146 uninjected non-visceral lesions, 60 (41.1 %) decreased in size by ≥ 30 %, the majority of which (44 [30.1 %]) completely resolved. The 44 uninjected non-visceral, non-visceral baseline or new lesions that completely resolved were in 6 of 24 patients with a baseline or new uninjected non-visceral lesion that had a CR. Of 32 visceral lesions, 4 (12.5 %) decreased in size by ≥ 30 %, the majority of which (3 [9.4 %]) completely resolved (Fig. 1c). The 3 baseline or new visceral lesions that completely resolved were in 1 of 12 patients with baseline or new visceral lesions that had a CR.
Fig. 1.
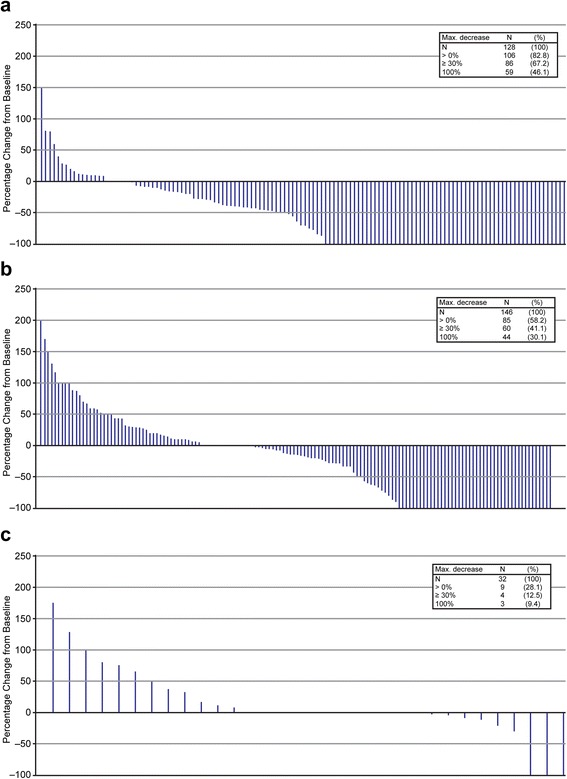
Maximum percent decrease in evaluable lesions: (a) Injected lesions, (b) Uninjected non-visceral lesions, and (c) Visceral lesions. Lesion measurements per investigator. Evaluable indicates at least 2 assessments with valid measurements. Uninjected lesion indicates baseline or new lesions never known to be injected. Safety analysis set consisted of the patients who received at least one dose of study therapy
Time to lesion response
The median time to lesion response (baseline or new lesions) is shown in Fig. 2 and was shortest for lesions that were directly injected (18.4 weeks) compared to all uninjected non-visceral lesions (29.1 weeks). On further analysis of uninjected lesions, uninjected non-visceral lesions responded at a median of 23.1 weeks compared to visceral lesions which responded at a mean of 51.3 weeks. These results are consistent with initiation of a delayed regional and systemic anti-tumor immune response to talimogene laherparepvec.
Fig. 2.
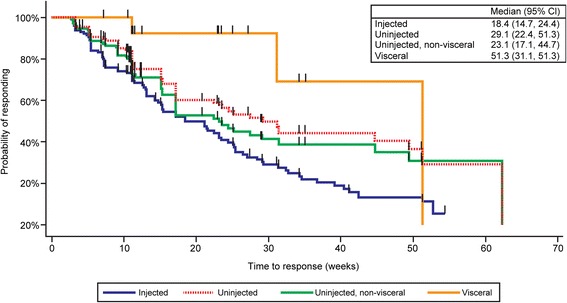
Lesion level time to response (Kaplan-Meier survival curves) for talimogene laherparepvec for injected, all uninjected, uninjected non-visceral, or visceral lesions in the phase II clinical trial of talimogene laherparepvec in patients with stage IIIC or IV melanoma. Safety analysis set consisted of patients who received at least one dose of study therapy
Discussion
Talimogene laherparepvec is an oncolytic herpes virus encoding GM-CSF and was designed to exert anti-tumor effects through both a direct oncolytic effect in injected lesions and through induction of systemic anti-tumor immunity. In a multi-institutional phase II clinical trial, an objective response rate of 26 % was reported and evidence of systemic anti-tumor immunity was also seen with MART-specific CD8+ T cells observed in both injected and uninjected lesions of responding patients [8]. In this post hoc analysis, we confirmed the total tumor burden reduction in both types of uninjected lesions—uninjected non-visceral and visceral lesions—as evidence of both regional and systemic immune response elicited by talimogene laherparepvec. However, the regional response rate was higher than systemic responses when analyzing both total tumor burden and individual tumor lesions. While this could relate to a slowly expanding T cell response primed close to the site of virus injection, it may also suggest that direct injection and oncolysis are important for maximizing therapeutic response of individual tumor lesions. Interestingly, the majority of lesions that had ≥ 30 % reduction in size went on to complete resolution, and this was seen across all types of lesions—injected, uninjected non-visceral, and visceral lesions. This supports the possibility that talimogene laherparepvec can successfully induce an effective systemic anti-tumor response with only a local, intratumoral, injection and immunotherapy mechanism as an important component of the mechanism of action for talimogene laherparepvec.
Numerous attempts to incite or modulate systemic immune responses via intralesional immunotherapy have been reported over the last 50 years. One of the earliest studies dates back to the 1970s when Dr. Donald Morton at the John Wayne Cancer Institute reported the effect of intralesional Bacillus Calmette-Guerin (BCG) in a cohort of patients with unresectable melanoma [12], although a subsequent randomized phase III trial failed to prove the systemic efficacy of this approach in 2004 [13]. Nevertheless, a number of phase I/II studies with intralesional cytokines (eg, GM-CSF, IL-2, interferons), viruses (eg, HSV-1, Coxsackievirus, Poxviruses), and plasmids (eg, Allovectin) have been conducted but their use has remained sporadic, and they have not progressed to become established treatments [14–17]. Talimogene laherparepvec was designed to increase both oncolytic effects through modulation of the HSV-1 vector for tumor-specific HSV-1 and to generate enhanced systemic response through deletion of the viral ICP47 gene and expression of GM-CSF [5]. The herpes ICP47 gene product blocks antigen presentation and normally prevents immune recognition of viral antigens. When this gene product is deleted, it is hypothesized that both viral-specific and pre-formed tumor-specific antigens are more likely to be presented by major histocompatibility complexes (MHC) and thus, generate T cell responses. GM-CSF expression within the local tumor microenvironment following injection of talimogene laherparepvec serves as a patient-specific, in situ, method of maturing dendritic cells and, hence priming local cytotoxic T cell responses. These results are consistent with other GM-CSF expressive immunotherapies [18–21] supporting systemic clinical effect. Further studies need to be done to better define whether viral- or tumor antigen-induced T cell repertoires are responsible for tumor rejection with talimogene laherparepvec and other oncolytic viruses.
In this study, we found a higher response rate of talimogene laherparepvec in the control of uninjected non-visceral lesions compared to the control of visceral lesions. This could relate to local spread of virus, which could mediate oncolytic effects on uninjected lesions, or it could be due to intrinsic differences in the therapeutic effectiveness of talimogene laherparepvec in various sites. Effector T cells are likely primed close to the injected tumors, likely in regional draining lymph nodes, and must be released into the systemic circulation, where they must survive and traffic to other sites of tumor growth. Expression of certain chemokine receptors and adhesion molecules on T cells are essential in this homing process. T cells primed in one location generally result in homing to a similar tissue compartment [22]. For example, native herpes simplex virus infection preferentially primes lymphocytes that express CCR4 and CCR10, chemokine receptors that promote trafficking to the skin [23]. In contrast, gut-tropic T cells express a high level of the chemokine receptor CCR9 and migrate preferentially to the lamina propria of the small intestine [24]. Dendritic cells (DCs) play an instructive role in efficient T cell priming and influence their function in a tissue-specific fashion by imparting variable chemokine receptor expression following priming [25]. DCs are also responsible for the imprinting of tissue-specific homing potential [26, 27]. Thus, DCs from the injected lesion may specifically prime effector T cells targeting tumors in similar locations, which could explain why uninjected non-visceral lesions were more susceptible to rejection after talimogene laherparepvec was injected into other skin-associated tumors. Further, the role of viral infection, local inflammation, and delivery of GM-CSF on the formation of resident T cells responding to tumor antigens is not fully understood. Resident memory CD8+ T cells (TRM) primed in the skin afford global protection within the skin, but do not circulate to other sites [28–30]. Their generation in this context, along with decreased induction of tissue-circulating effector memory T cells (TEM) or secondary lymphoid tissue-circulating central memory T cells (TCM), could serve as an explanation for the increased resolution of uninjected non-visceral lesions compared to visceral lesions.
Another possible explanation for the better response of uninjected non-visceral lesions may be that these lesions are simply closer to TVEC-injected lesions compared to visceral lesions and it may be easier and faster for activated T cells to migrate to these lesions due to the shorter distance. An alternative explanation involving distance from the injection lesion may be that uninjected non-visceral lesions are likewise infected during infection of nearby injected lesions. However, animal studies in which Newcastle Disease Virus was injected into one lesion failed to detect virus in contralateral lesions within the same tissue [31]. Further investigation is needed to better understand how tumors are rejected with talimogene laherparepvec treatment and the nature and activity of tumor-reactive T cells need to be more fully defined. Future clinical studies will examine the impact of direct injection of talimogene laherparepvec into visceral tumors and this may provide an opportunity to better understand if local injection can overcome the more limited visceral lesion response observed with injection of accessible skin and soft tissue lesions, which has characterized most oncolytic virus clinical trials to date. Another strategy in clinical development is to combine oncolytic viruses with other T cell promoting immunotherapy agents, most notable T cell and other immune checkpoint inhibitors. These agents may enhance the activation status of talimogene laherparepvec-primed T cell responses and would be expected to increase the therapeutic activity at both a local and systemic level.
Overall, we observed that in 12 out of 23 patients with evaluable visceral lesions, only 2 out of 12 patients had evaluable visceral lesion responses and only 4 out of 32 total evaluable visceral lesions had an objective response. In the pivotal phase III clinical trial, a higher response rate was observed in patients with unresectable stage IIIB/C and IV M1a disease, suggesting this population may be better suited for treatment with talimogene laherparepvec. A major limitation of our study was the relatively small sample size, which may have obscured the significance of the systemic response elicited by talimogene laherparepvec. An evaluation of lesion responses in patients treated on the much larger phase III clinical trial may help resolve this issue [9]. Other limitations of this study include the need to rely on investigator assessment, different methods of lesion measurement (e.g. clinical calipers for skin and soft tissue lesions vs. CT-guided imaging for visceral lesions), lack of immune response data and time bias since different lesion types were measured at different time points and immunotherapy may result in a delayed kinetics of therapeutic response. Nonetheless, these data provide some insight into the mechanism of talimogene laherparepvec activity and help identify appropriate patients for treatment with talimogene laherparepvec.
Conclusions
In conclusion, the ability of talimogene laherparepvec to initiate regional and systemic anti-tumor activity is supported by responses in both uninjected non-visceral and some visceral melanoma lesions. The time to lesion response was shortest for lesions that were directly injected, followed by uninjected non-visceral lesions and visceral lesions, consistent with initiation of a delayed systemic anti-tumor immune response following talimogene laherparepvec treatment. Given the recent FDA approval of talimogene laherparepvec for the treatment of melanoma, further studies are needed to better understand how talimogene laherparepvec mediates anti-tumor activity. New studies are planned to evaluate therapeutic responses through direct injection of visceral tumors and combinations with other T cell enhancing immunotherapy agents. These studies should provide new insight into the optimal patient selection for talimogene laherparepvec treatment and will likely guide further improvements in the therapeutic effectiveness of oncolytic viruses as a new class of drugs for the treatment of cancer.
Acknowledgements
We wish to acknowledge the patients and their families, friends, and caregivers as well as the site study staffs. We also acknowledge Mee Rhan Kim, PhD, from Amgen Inc. for writing assistance with this manuscript. Study procedures received approval from: Columbia University Institutional Review Board; North Memorial Health Care Institutional Review Board; UCSD Institutional Review Board; Colorado Multiple Institutional Review Board; UCLA Institutional Review Boards; Atlantic Health System Institutional Review Board; University of Texas Southwestern Institutional Review Board; and Ethics Committees of the Institute of Cancer Research and the Royal Marsden Hospital.
This work is original and has not been presented or published elsewhere.
Abbreviations
- BCG
Bacillus Calmette-Guerin
- CR
Complete response
- DCs
Dendritic cells
- FDA
Food and Drug Administration
- GM-CSF
Granulocyte-macrophage colony-stimulating factor
- HSV-1
Herpes simplex virus type 1
- IL-2
Interleukin-2
- MHC
Major histocompatibility complex
- ORR
Objective response rate
- PFU
Plaque-forming unit
- RECIST
Response Evaluation Criteria in Solid Tumors
- TRM
Resident memory CD8+ T cells
- TEM
Effector memory T cells
- TCM
Central memory T cells
Footnotes
Competing interests
HLK has been a member of advisory boards for Alkermes, Amgen Inc., EMD Serono, Merck, Prometheus and Sanofi and has participated in Speakers Bureau for Merck. RG has received research funding from Genentech, Novartis, BMS, Merck, Amgen Inc., and has been a consultant and/or advisory board member for Genentech, Novartis, BMS, and Amgen Inc. JG has received research support from Amgen, including for this study. EW has been a consultant to and/or been a member of advisory boards/speakers bureau for Amgen Inc. KH has received consultancy fees and travel support from Amgen Inc. MW is an employee of Amgen Inc. TA, TR, JN, AZ, and NNS have no financial competing interests. No non-financial competing interests exist for any of the authors.
Authors’ contributions
HLK, JN, AZ, and NNS participated in the study conduct, collected and analyzed data, collaborated in conception of this analysis, and helped to draft the manuscript. TA, TR, RG, JG, EW, and KH participated in the study conduct, collected and analyzed data, and helped to draft the manuscript. MW conducted biostatistical analysis of the data and helped to draft the manuscript. All authors approved the final version.
Contributor Information
Howard L. Kaufman, Phone: 732-235-6807, Email: howard.kaufman@rutgers.edu
Thomas Amatruda, Email: Thomas.Amatruda@USONCOLOGY.COM.
Tony Reid, Email: tonyreid@ucsd.edu.
Rene Gonzalez, Email: Rene.Gonzalez@ucdenver.edu.
John Glaspy, Email: JGlaspy@mednet.ucla.edu.
Eric Whitman, Email: Eric.Whitman@atlantichealth.org.
Kevin Harrington, Email: Kevin.Harrington@icr.ac.uk.
John Nemunaitis, Email: JNemunaitis@MaryCrowley.Org.
Andrew Zloza, Email: az255@cinj.rutgers.edu.
Michael Wolf, Email: wolfm@amgen.com.
Neil N. Senzer, Email: NSenzer@MaryCrowley.Org
References
- 1.Gerner RE, Moore GE, Dickey C. Combination chemotherapy in disseminated melanoma and other solid tumors in adults. Oncology. 1975;31:22–30. doi: 10.1159/000225002. [DOI] [PubMed] [Google Scholar]
- 2.Atkins MB, Lotze MT, Dutcher JP, Fisher RI, Weiss G, Margolin K, Abrams J, Sznol M, Parkinson D, Hawkins M, Paradise C, Kunkel L, Rosenberg SA. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol. 1999;17:2105–2116. doi: 10.1200/JCO.1999.17.7.2105. [DOI] [PubMed] [Google Scholar]
- 3.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJ, Lutzky J, Lorigan P, Vaubel JM, Linette GP, Hogg D, Ottensmeier CH, Lebbe C, Peschel C, Quirt I, Clark JI, Wolchok JD, Weber JS, Tian J, Yellin MJ, Nichol GM, Hoos A, Urba WJ. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Robert C, Ribas A, Wolchok JD, Hodi FS, Hamid O, Kefford R, Weber JS, Joshua AM, Hwu WJ, Gangadhar TC, Patnaik A, Dronca R, Zarour H, Joseph RW, Boasberg P, Chmielowski B, Mateus C, Postow MA, Gergich K, Elassaiss-Schaap J, Li XN, Iannone R, Ebbinghaus SW, Kang SP, Daud A. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet. 2014;384:1109–1117. doi: 10.1016/S0140-6736(14)60958-2. [DOI] [PubMed] [Google Scholar]
- 5.Hu JC, Coffin RS, Davis CJ, Graham NJ, Groves N, Guest PJ, Harrington KJ, James ND, Love CA, McNeish I, Medley LC, Michael A, Nutting CM, Pandha HS, Shorrock CA, Simpson J, Steiner J, Steven NM, Wright D, Coombes RC. A phase I study of OncoVEXGM-CSF, a second-generation oncolytic herpes simplex virus expressing granulocyte macrophage colony-stimulating factor. Clin Cancer Res. 2006;12:6737–6747. doi: 10.1158/1078-0432.CCR-06-0759. [DOI] [PubMed] [Google Scholar]
- 6.Liu BL, Robinson M, Han ZQ, Branston RH, English C, Reay P, McGrath Y, Thomas SK, Thornton M, Bullock P, Love CA, Coffin RS. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther. 2003;10:292–303. doi: 10.1038/sj.gt.3301885. [DOI] [PubMed] [Google Scholar]
- 7.Kaufman HL, Kim DW, DeRaffele G, Mitcham J, Coffin RS, Kim-Schulze S. Local and distant immunity induced by intralesional vaccination with an oncolytic herpes virus encoding GM-CSF in patients with stage IIIc and IV melanoma. Ann Surg Oncol. 2010;17:718–730. doi: 10.1245/s10434-009-0809-6. [DOI] [PubMed] [Google Scholar]
- 8.Senzer NN, Kaufman HL, Amatruda T, Nemunaitis M, Reid T, Daniels G, Gonzalez R, Glaspy J, Whitman E, Harrington K, Goldsweig H, Marshall T, Love C, Coffin R, Nemunaitis JJ. Phase II clinical trial of a granulocyte-macrophage colony-stimulating factor-encoding, second-generation oncolytic herpesvirus in patients with unresectable metastatic melanoma. J Clin Oncol. 2009;27:5763–5771. doi: 10.1200/JCO.2009.24.3675. [DOI] [PubMed] [Google Scholar]
- 9.Andtbacka RH, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, Delman KA, Spitler LE, Puzanov I, Agarwala SS, Milhem M, Cranmer L, Curti B, Lewis K, Ross M, Guthrie T, Linette GP, Daniels GA, Harrington K, Middleton MR, Miller WH, Jr, Zager JS, Ye Y, Yao B, Li A, Doleman S, VanderWalde A, Gansert J, Coffin RS. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol. 2015;33:2780–2788. doi: 10.1200/JCO.2014.58.3377. [DOI] [PubMed] [Google Scholar]
- 10.Imlygic™: (talimogene laherparepvec) prescribing information, Amgen Inc., Thousand Oaks, CA; 2015.
- 11.Brookmeyer R, Crowley J. A confidence-interval for the median survival-time. Biometrics. 1982;38:29–41. doi: 10.2307/2530286. [DOI] [Google Scholar]
- 12.Morton DL, Eilber FR, Holmes EC, Hunt JS, Ketcham AS, Silverstein MJ, Sparks FC. BCG immunotherapy of malignant melanoma: summary of a seven-year experience. Ann Surg. 1974;180:635–643. doi: 10.1097/00000658-197410000-00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Agarwala SS, Neuberg D, Park Y, Kirkwood JM. Mature results of a phase III randomized trial of bacillus Calmette-Guerin (BCG) versus observation and BCG plus dacarbazine versus BCG in the adjuvant therapy of American Joint Committee on Cancer Stage I-III melanoma (E1673): a trial of the Eastern Oncology Group. Cancer. 2004;100:1692–1698. doi: 10.1002/cncr.20166. [DOI] [PubMed] [Google Scholar]
- 14.Andtbacka RHI, Kaufman H, Daniels GA, Spitler LE, Lutzky J, Hallmeyer S, Whitman ED, Nemunaitis JJ, Zhou K, Karpathy R, Weisberg JI, Shafren D: CALM study: A phase II study of intratumoral coxsackievirus A21 in patients with stage IIIc and stage IV malignant melanoma. J Clin Oncol. 2013;31:abstract TPS3128.
- 15.Bedikian AY, Richards J, Kharkevitch D, Atkins MB, Whitman E, Gonzalez R. A phase 2 study of high-dose Allovectin-7 in patients with advanced metastatic melanoma. Melanoma Res. 2010;20:218–226. doi: 10.1097/CMR.0b013e3283390711. [DOI] [PubMed] [Google Scholar]
- 16.MacKie RM, Stewart B, Brown SM. Intralesional injection of herpes simplex virus 1716 in metastatic melanoma. Lancet. 2001;357:525–526. doi: 10.1016/S0140-6736(00)04048-4. [DOI] [PubMed] [Google Scholar]
- 17.Si Z, Hersey P, Coates AS. Clinical responses and lymphoid infiltrates in metastatic melanoma following treatment with intralesional GM-CSF. Melanoma Res. 1996;6:247–255. doi: 10.1097/00008390-199606000-00008. [DOI] [PubMed] [Google Scholar]
- 18.Olivares J, Kumar P, Yu Y, Maples PB, Senzer N, Bedell C, Barve M, Tong A, Pappen BO, Kuhn J, Magee M, Wallraven G, Nemunaitis J. Phase I trial of TGF-beta 2 antisense GM-CSF gene-modified autologous tumor cell (TAG) vaccine. Clin Cancer Res. 2011;17:183–192. doi: 10.1158/1078-0432.CCR-10-2195. [DOI] [PubMed] [Google Scholar]
- 19.Senzer N, Barve M, Kuhn J, Melnyk A, Beitsch P, Lazar M, Lifshitz S, Magee M, Oh J, Mill SW, Bedell C, Higgs C, Kumar P, Yu Y, Norvell F, Phalon C, Taquet N, Rao DD, Wang Z, Jay CM, Pappen BO, Wallraven G, Brunicardi FC, Shanahan DM, Maples PB, Nemunaitis J. Phase I trial of “bi-shRNAi(furin)/GMCSF DNA/autologous tumor cell” vaccine (FANG) in advanced cancer. Mol Ther. 2012;20:679–686. doi: 10.1038/mt.2011.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Soiffer R, Hodi FS, Haluska F, Jung K, Gillessen S, Singer S, Tanabe K, Duda R, Mentzer S, Jaklitsch M, Bueno R, Clift S, Hardy S, Neuberg D, Mulligan R, Webb I, Mihm M, Dranoff G. Vaccination with irradiated, autologous melanoma cells engineered to secrete granulocyte-macrophage colony-stimulating factor by adenoviral-mediated gene transfer augments antitumor immunity in patients with metastatic melanoma. J Clin Oncol. 2003;21:3343–3350. doi: 10.1200/JCO.2003.07.005. [DOI] [PubMed] [Google Scholar]
- 21.Soiffer R, Lynch T, Mihm M, Jung K, Rhuda C, Schmollinger JC, Hodi FS, Liebster L, Lam P, Mentzer S, Singer S, Tanabe KK, Cosimi AB, Duda R, Sober A, Bhan A, Daley J, Neuberg D, Parry G, Rokovich J, Richards L, Drayer J, Berns A, Clift S, Cohen LK, Mulligan RC, Dranoff G. Vaccination with irradiated autologous melanoma cells engineered to secrete human granulocyte-macrophage colony-stimulating factor generates potent antitumor immunity in patients with metastatic melanoma. Proc Natl Acad Sci U S A. 1998;95:13141–13146. doi: 10.1073/pnas.95.22.13141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mora JR, von Andrian UH. T-cell homing specificity and plasticity: new concepts and future challenges. Trends Immunol. 2006;27:235–243. doi: 10.1016/j.it.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 23.Koelle DM, Liu Z, McClurkan CM, Topp MS, Riddell SR, Pamer EG, Johnson AS, Wald A, Corey L. Expression of cutaneous lymphocyte-associated antigen by CD8(+) T cells specific for a skin-tropic virus. J Clin Invest. 2002;110:537–548. doi: 10.1172/JCI0215537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zabel BA, Agace WW, Campbell JJ, Heath HM, Parent D, Roberts AI, Ebert EC, Kassam N, Qin S, Zovko M, LaRosa GJ, Yang LL, Soler D, Butcher EC, Ponath PD, Parker CM, Andrew DP. Human G protein-coupled receptor GPR-9-6/CC chemokine receptor 9 is selectively expressed on intestinal homing T lymphocytes, mucosal lymphocytes, and thymocytes and is required for thymus-expressed chemokine-mediated chemotaxis. J Exp Med. 1999;190:1241–1256. doi: 10.1084/jem.190.9.1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sato A, Iwasaki A. Peyer’s patch dendritic cells as regulators of mucosal adaptive immunity. Cell Mol Life Sci. 2005;62:1333–1338. doi: 10.1007/s00018-005-5037-z. [DOI] [PubMed] [Google Scholar]
- 26.Mora JR, Bono MR, Manjunath N, Weninger W, Cavanagh LL, Rosemblatt M, Von Andrian UH. Selective imprinting of gut-homing T cells by Peyer’s patch dendritic cells. Nature. 2003;424:88–93. doi: 10.1038/nature01726. [DOI] [PubMed] [Google Scholar]
- 27.Mora JR, Cheng G, Picarella D, Briskin M, Buchanan N, von Andrian UH. Reciprocal and dynamic control of CD8 T cell homing by dendritic cells from skin- and gut-associated lymphoid tissues. J Exp Med. 2005;201:303–316. doi: 10.1084/jem.20041645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gebhardt T, Wakim LM, Eidsmo L, Reading PC, Heath WR, Carbone FR. Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat Immunol. 2009;10:524–530. doi: 10.1038/ni.1718. [DOI] [PubMed] [Google Scholar]
- 29.Gebhardt T, Whitney PG, Zaid A, Mackay LK, Brooks AG, Heath WR, Carbone FR, Mueller SN. Different patterns of peripheral migration by memory CD4+ and CD8+ T cells. Nature. 2011;477:216–219. doi: 10.1038/nature10339. [DOI] [PubMed] [Google Scholar]
- 30.Mueller SN, Gebhardt T, Carbone FR, Heath WR. Memory T cell subsets, migration patterns, and tissue residence. Annu Rev Immunol. 2013;31:137–161. doi: 10.1146/annurev-immunol-032712-095954. [DOI] [PubMed] [Google Scholar]
- 31.Zamarin D, Holmgaard RB, Subudhi SK, Park JS, Mansour M, Palese P, Merghoub T, Wolchok JD, Allison JP. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci Transl Med. 2014;6:226ra232. doi: 10.1126/scitranslmed.3008095. [DOI] [PMC free article] [PubMed] [Google Scholar]