

Abstract
Vascular endothelial cell growth factor (VEGF) is increased in diabetic macular edema. Compound 49b, a novel β-adrenergic receptor agonist, is protective in a type 1 diabetic rat model. We questioned whether Compound 49b could decrease VEGF levels, suggesting that Compound 49b may be effective against edema. Two-month diabetic rats received topical Compound 49b for 7 days only and/or insulin-like growth factor binding protein 3 (IGFBP-3) siRNA. We also measured endothelial nitric oxide synthase (eNOS) and protein kinase C (PKC)ζ and PKCδ phosphorylation. Retinal endothelial cells (RECs) cultured in high glucose were treated with Compound 49b and IGFBP-3 siRNA for evaluation of the same signaling pathways. Compound 49b significantly decreased VEGF through increased IGFBP-3 in the diabetic retina. Compound 49b also reduced eNOS, PKCζ and PKCδ phosphorylation in the diabetic retina and REC. Compound 49b regulated a number of proteins involved in REC barrier properties.
Keywords: Beta-adrenergic receptor, diabetic retinopathy, VEGF
Introduction
Diabetic retinopathy is a leading cause of blindness, with rates significantly increasing worldwide. Diabetic macular edema and proliferative retinopathy are two complications commonly associated with blindness. Current therapies for diabetic macular edema include monthly intravitreal injections of anti-vascular endothelial cell growth factor (VEGF) and/or steroid injections or laser photocoagulation (Agarwal et al., 2015; Googe et al., 2011). While it is clear that VEGF is increased due to hypoxia (Das et al., 2015), other potential signaling pathways may offer additional therapeutic options.
One pathway that others have suggested that interacts with VEGF to regulate endothelial cell barrier properties is insulin-like growth factor binding protein 3 (IGFBP-3) (Kielczewski et al., 2011). Kielczewski et al. (2009) found that IGFBP-3 regulated VEGF and barrier properties through modulation of sphingomyelinase levels. We have also observed that IGFBP-3 can regulate VEGF in retinal endothelial cells (RECs) cultured in high glucose (Zhang et al., 2015). In our work, IGFBP-3 regulated eNOS and PKCζ to decrease VEGF levels (Zhang et al., 2015). Our finding of IGFBP-3 regulation of eNOS is in agreement with work by Jarajapu et al. (2012) done in human microvascular endothelial cells in vitro and in rat posterior cerebral arteries ex vivo.
In addition to IGFBP-3 regulation of endothelial cell barrier properties, various protein kinase C (PKC) isoforms have been reported to regulate blood–retinal barrier break-down in diabetes (Frey & Antonetti, 2011). Work in RECs and mice demonstrated that inhibition of PKCδ led to improved blood retinal barrier properties (Kim et al., 2010). Similarly, others reported that TNFα can activate PKCδ to increase bovine REC permeability (Aveleira et al., 2010). Since TNFα is increased in the diabetic retina, it is possible that TNFα modulation of PKCδ may be involved in retinal barrier dysfunction (Koizumi et al., 2003; Zhang et al., 2013). We have reported that IGFBP-3 can decrease TNFα levels in the retina (Zhang & Steinle, 2014), thus it is possible that IGFBP-3 may regulate PKCδ. In addition to PKCδ, work also suggests that PKCζ may be involved in retinal barrier changes in response to diabetes. Work in diabetic mice and RECs in high glucose demonstrated that inhibition of PKCζ could reduce VEGF-induced hyperpermeability (Song et al., 2014). Similar work by Titchenell et al. (2012) also found that inhibition of atypical PKC (aPKC, PKCζ & ι/λ) prevented VEGF-induced barrier dysfunction. While data show that PKC isoforms can regulate VEGF, additional studies demonstrate that VEGF can activate PKC to regulate occludin, a key barrier protein which regulates paracellular permeability (Harhaj et al., 2006). Thus, there appears to be a reciprocal relationship between VEGF and some PKC isoforms.
We have reported that Compound 49b, a novel β-adrenergic receptor agonist, can regulate IGFBP-3 (Zhang et al., 2012). Since others have shown that IGFBP-3 can regulate VEGF to alter endothelial cell barrier properties, we tested the hypothesis that Compound 49b would increase IGFBP-3 in the retina of diabetic rats and in RECs, leading to reduced VEGF levels through inhibition of PKCδ and PKCζ.
Methods
Rats
All animal work was approved by the Institutional Animal Care and Use Committee at the University of Tennessee Health Science Center Protocol #1992. Male Lewis rats were purchased from Charles River Laboratories. One group of rats was injected with citrate buffer alone, while the remaining five groups of rats received one injection of streptozotocin (60 mg/kg, IP) to induce diabetes. Blood glucose was checked on all rats with diabetes defined as blood glucose >250 mg/dl. Animals were diabetic for 2 months prior to treatments. Body weight and blood glucose are provided in Table 1.
Table 1.
Mean body weight, blood glucose and intraocular pressure (IOP) in control (Ctrl) and diabetic (STZ) rats left untreated or treated with 1mM Compound 49b alone or in combination of IGFBP-3 siRNA (Bp3siRNA) or scrambled siRNA (ScsiRNA) for 2 months and 7 days.
| Groups | Number | Body weight (g) | Glucose (mg/dl) | IOP (mmHg) |
|---|---|---|---|---|
| Ctrl | 15 | 458±18.6 | 86.7±8.2 | 14.6±2.8 |
| STZ | 10 | 236±32.5* | 487.3±102.2* | 13.8±4.3 |
| STZ+49b | 10 | 238±29.8* | 523.5±70.8* | 13.5±2.8 |
| STZ+BP3siRNA | 9 | 232±24.3* | 511±52.1* | 14.1±2.6 |
| STZ+BP3siRNA+49b | 10 | 229±21.5* | 501.5±58.2* | 16.3±3.1 |
| STZ+ScsiRNA | 8 | 233±25.2* | 491±92.7* | 14.5±3.3 |
p<0.05 versus control (Ctrl).
Injection
Rat IGFBP-3 siRNA and non-silencing CONTROL siRNA were obtained from Dharmacon siDESIGN center (Fisher Scientific, Pittsburgh, PA). Lyophilized siRNAs were reconstituted, condensed with Enhancer R and mixed with TransMessenger™ Transfection Reagent (Qiagen, Valencia, CA), at a final concentration of 0.5 μg/μl. For the administration of siRNA, 1 μg of siRNA was injected using a 10-μl Hamilton® microsyringe into the vitreous of each eye once every other day for six days. Some eyes were also treated with 1 mg/kg Compound 49b (4 ul volume) daily for 7 days after siRNA injection. Electroretinogram (ERG) analyses were performed on each eye prior to treatment initiation and prior to sacrifice and isolation of retina.
Compound 49b treatment
Compound 49b is a β1/β2-adrenergic receptor agonist (Figure 1). Compound 49b is dissolved in saline and administered as an eye drop at a 1 mg/kg dose. It is administered topically in 4 ul to each eye at the same time each day, as we have done previously (Zhang et al., 2012).
Figure 1.

Compound 49b is a β1/β2-adrenergic receptor agonist. Figure shows the chemical structure of Compound 49b.
Electroretinogram
Prior to treatments (2 months diabetes) and prior to sacrifice (7 days of treatment) for biochemical analyses, animals were subjected to ERG analyses to evaluate the changes in the electrical activity of the retina as we have done previously (Jiang et al., 2013; Zhang et al., 2012). After dark adaptation overnight, ERG responses were recorded from both eyes using platinum wire corneal electrodes, forehead reference electrode and ground electrode in the tail. Pupils were fully dilated using 1% tropicamide solution (Alcon, Ft. Worth, TX). Methylcellulose (Celluvise; Allergan, Irvine, CA) drops were applied to maintain a good electrical connection, while body temperature was maintained at 37 °C using a water-based heating pad. ERG waveforms were recorded with a bandwidth of 0.3–500 Hz and sampled at 2 kHz by a digital acquisition system and were analyzed using a custom-built program, which allowed a measurement of a-wave, b-wave and oscillatory potential from all animals (MatLab, Mathworks, Natick, MA). Statistics were done on the mean±SD amplitudes of the a- and b-wave of each treatment group.
Intraocular pressure (IOP) was measured monthly using a tonometer (TonoLab, Colonial Medical Supply, Franconia, NH). Briefly, the tip of the probe of the tonometer was placed at the cornea of the eye. During measurements, the tip of the probe hits the cornea six times and gave the IOP reading of that eye. This procedure was carried out for both the eyes as we have done previously (Zhang et al., 2012). IOP levels are presented in Table 1.
Retinal endothelial cells (RECs)
Primary human RECs were acquired from Cell System Corporation (CSC, Kirkland, WA). Cells were grown in M131 medium containing microvascular growth supplements (Invitrogen, Carlsbad, CA) (MVGS), 10 μg/ml gentamycin and 0.25 μg/ml amphotericin B. In the high glucose condition, cells were transferred to high glucose (25mM) (Cell Systems) medium, supplemented with MVGS and antibiotics for 3 days. Only primary cells within passage 6 were used. Cells were quiesced by incubating in high or normal glucose medium without MVGS for 24 h prior to all experiments. For the work with siRNA, ON-TARGETplus SMARTpool human IGFBP-3 siRNA (Dharmacon, Inc., Fisher Scientific, Pittsburgh, PA) was used at a final concentration of 20nM using RNAiMAX transfection reagent according to the manufacturer’s instructions. For control of siRNA experiments, non-targeting siRNA #1 (Dharmacon) was used as a nonspecific control. RECs were transfected with siRNA at a final concentration of 20nM using RNAiMAX transfection reagent according to the manufacturer’s instructions. The cells were used for experiments 24 h after transfection.
Western blotting
Whole retinal lysates and REC lysates were placed into lysis buffer containing protease and phosphatase inhibitors. The lysates were kept on ice for 30 min and cleared by centrifugation at 12,000 rpm for 20 min at 4 °C. Equal amounts of protein from the lysates were separated on the precast tris-glycine gels (Invitrogen, Carlsbad, CA) and blotted onto a nitrocellulose membrane. The blots were blocked with TBST (10mM Tris-HCl buffer, pH 8.0, 150mM NaCl, 0.1% Tween 20) containing 5% (w/v) bovine serum albumin and then incubated with respective primary antibodies. Primary antibodies used were anti-phospho-PKCζ, PKCζ, PKCδ, phospho-PKCδ, phospho-eNOS and eNOS (Cell Signaling, Danvers, MA). IGFBP-3, VEGF and β-actin antibodies were purchased from Santa Cruz Technology (Santa Cruz, CA). Membranes were then washed with TBST and incubated with horseradish peroxidase labeled secondary antibodies. The antigen–antibody complexes were detected using West Pico Chemilluminescence reagent. Mean densitometry of the bands were assessed using Kodak Image Station 4000MM software (Carestream, Rochester, NY).
Statistics
All the experiments were repeated a minimum of three times. Data are presented as mean±standard error of mean. Data was analyzed with a Kruskal–Wallis test, followed by Dunn’s test with p values<0.05 considered statistically significant. One representative blot is shown for Western blot data.
Results
Compound 49b improved b-wave and oscillatory potential amplitudes through increasing IGFBP-3 levels
Rats were placed into six groups, with five groups receiving streptozotocin to induce diabetes. Electroretinograms were done at 2 months of diabetes, and again 7 days after Compound 49b and IGFBP-3 siRNA treatment. Figure 2 shows that Compound 49b was highly effective in improving a- and b-wave amplitudes in the diabetic animals (Figure 2, left and middle panels), which agrees well with our previously published work (Zhang et al., 2012). Compound 49b was not able to improve b-wave amplitudes and oscillatory potentials when IGFBP-3 was knocked down with siRNA, suggesting that Compound 49b regulates retinal function through restoration of IGFBP-3 levels in diabetes.
Figure 2.

Compound 49b improves a-wave and b-wave amplitudes, as well as oscillatory potentials through increasing IGFBP-3 levels. Electroretinogram measurements were made at 2 months of diabetes and after 7 days of treatments in control (Ctrl), diabetic (Diab), diabetic+Compound 49b (diab+49b), diabetic+IGFBP-3 siRNA (Diab+Bp3 siRNA), diabetic+IGFBP-3 siRNA+Compound 49b (Diab+BP3 siRNA+49b) and diabetic+scrambled siRNA (Diab+scsiRNA). *p<0.05 versus control, #p<0.05 versus diabetic, $p<0.05 versus Diab+49b. N=5–6 for all groups. For a-wave, b-wave and oscillatory potentials before measurements, all groups are significantly different than the control.
IGFBP-3 regulated phosphorylation of eNOS in diabetic rats
Diabetes significantly reduced IGFBP-3 levels both in rats (Figure 3A) and REC (Figure 3C), which agrees with published data (Zhang et al., 2012). The goal of this work was to determine whether Compound 49b requires increased IGFBP-3 to regulate downstream signaling pathways related to endothelial cell permeability. Compound 49b significantly increased IGFBP-3 in both rats and REC. Diabetes significantly increased the phosphorylation of eNOS, which was inhibited by Compound 49b in both rats (Figure 3B) and cells (Figure 3D). Compound 49b actions on eNOS required IGFBP-3, as knockdown of IGFBP-3 with siRNA was significantly different than Compound 49b treatment alone.
Figure 3.

Compound 49b regulated IGFBP-3 and eNOS inversely in the diabetic rat retina and in REC cultured in high glucose. Figures A, B show IGFBP-3 and eNOS levels in control (Ctrl), diabetic (Diab), diabetic+Compound 49b (diab+49b), diabetic+IGFBP-3 siRNA (Diab+Bp3 siRNA), diabetic+IGFBP-3 siRNA+Compound 49b (Diab+BP3 siRNA+49b) and diabetic+scrambled siRNA (Diab+scsiRNA) rat retina. Figure A demonstrates that IGFBP-3 siRNA was effective in reducing IGFBP-3 levels in the diabetic rat retina. Figure B demonstrates that Compound 49b can decrease eNOS levels in the diabetic rat retina through increased IGFBP-3 levels. Figures C and D represent measurements of IGFBP-3 (C) and eNOS (D) in REC cultured in normal (NG) or high glucose (HG) and high glucose and treated with Compound 49b (HG+49b), high glucose, IGFBP-3 siRNA and Compound 49b (HG+Bp3 siRNA+49b) and high glucose with scrambled siRNA+49b (HG +Sc +49b). *p<0.05 versus NG or control, #p<0.05 versus HG or diabetes, $p<0.05 versus HG+49b or Diab+49b. N=5–6 for rat retina and N=4 for cell work.
Compound 49b regulates PKCδ and PKCζ in diabetes through IGFBP-3 levels
Others have suggested that IGFBP-3 may be involved in retinal barrier changes (Jarajapu et al., 2012). We have data that eNOS interacts with PKCζ in REC (Zhang et al., in press). In this work, our goal was to investigate whether Compound 49b could increase IGFBP-3, leading to altered PKCδ and PKCζ phosphorylation. Figure 4 demonstrates that diabetes increased both PKCδ and PKCζ phosphorylation in rat retina (Figure 4A and B) and in REC cultured in high glucose (Figure 4C and D). Compound 49b significantly reduced both PKC isoforms investigated. When Compound 49b was combined with IGFBP-3 siRNA treatment, Compound 49b was less effective in reducing PKC phosphorylation in both animal and cell samples, suggesting that IGFBP-3 is required for Compound 49b to regulate these PKC isoforms.
Figure 4.

Inhibition of PKCδ and PKCζ by Compound 49b is mediated by IGFBP-3. Figures A, B show phosphorylation of PKCδ (A) and PKCζ(B) in control (Ctrl), diabetic (Diab), diabetic+Compound 49b (Diab+49b), diabetic+IGFBP-3 siRNA (Diab+Bp3 siRNA), diabetic+IGFBP-3 siRNA+Compound 49b (Diab+BP3 siRNA+49b) and diabetic+scrambled siRNA (Diab+scsiRNA) rat retina. Figure A demonstrates that IGFBP-3 siRNA was effective in reducing PKCδ phosphorylation. Figure B demonstrates that Compound 49b can decrease phosphorylation of PKCζ in the diabetic rat retina through increased IGFBP-3 levels. Figures C and D represent measurements of PKCδ (C) and PKCζ (D) in REC cultured in normal (NG) or high glucose (HG) and high glucose and treated with Compound 49b (HG +49b), high glucose, IGFBP-3 siRNA and Compound 49b (HG+Bp3 siRNA+49b) and high glucose with scrambled siRNA+49b (HG+Sc+49b). *p<0.05 versus NG or control, #p<0.05 versus HG or diabetes, $p<0.05 versus HG+49b or Diab+49b. N=5–6 for rat retina and N=4 for cell work.
VEGF is reduced by Compound 49b actions on IGFBP-3
Diabetes significantly increased VEGF levels in both animals and cells, as has been reported extensively in the literature (Das et al., 2015). Since one of the common therapeutics for diabetic macular edema is anti-VEGF injections, it is important to understand the regulation of VEGF in the diabetic retina. In this work, we demonstrated that β-adrenergic receptor stimulation via Compound 49b treatment significantly reduced VEGF levels (Figure 5A and B). However, when Compound 49b was added in conjunction with IGFBP-3 siRNA, Compound 49b was not as effective in reducing VEGF levels, suggesting that β-adrenergic receptors regulated VEGF through IGFBP-3 actions (Figure 6).
Figure 5.

Compound 49b significantly reduced VEGF in the diabetic rat retina and in REC cultured in high glucose. Figure A shows VEGF levels in control (Ctrl), diabetic (Diab), diabetic+Compound 49b (Diab+49b), diabetic+IGFBP-3 siRNA (Diab+Bp3 siRNA), diabetic+IGFBP-3 siRNA+Compound 49b (Diab+BP3 siRNA+49b) and diabetic+scrambled siRNA (Diab+scsiRNA) rat retina. Figure A demonstrates that Compound 49b significantly reduces VEGF levels through increasing IGFBP-3 levels. Figure B represent measurements of VEGF in REC cultured in normal (NG) or high glucose (HG) and high glucose and treated with Compound 49b (HG+49b), high glucose, IGFBP-3 siRNA and Compound 49b (HG+Bp3 siRNA+49b) and high glucose with scrambled siRNA+49b (HG+Sc+49b). High glucose conditions significantly increase VEGF levels, which are reduced by Compound 49b. *p<0.05 versus NG or control, #p<0.05 versus HG or diabetes, $p<0.05 versus HG+49b or Diab+49b. N=5–6 for rat retina and N=4 for cell work.
Figure 6.
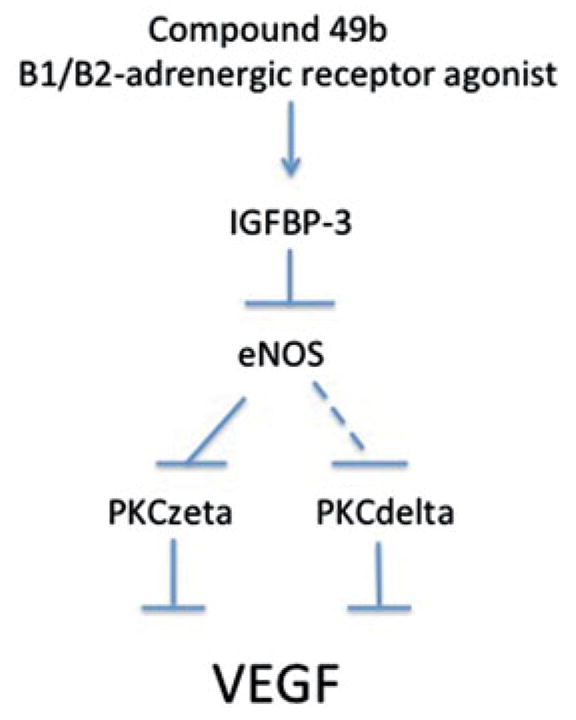
Compound 49b regulates VEGF through IGFBP-3 actions. Figure is a schematic of the proposed pathway by which Compound 49b may regulate VEGF.
Discussion
The goal of this work was to investigate whether our novel β-adrenergic receptor agonist, Compound 49b, could reduce VEGF levels in an acute model of type 1 diabetes. Additionally, our goal was to determine whether Compound 49b’s actions to increase IGFBP-3 were required for the reduction in VEGF levels. The ability of Compound 49b to reduce VEGF was somewhat in question, as others have reported that β-adrenergic receptor antagonists are effective in the oxygen-induced retinopathy model (Dal Monte et al., 2015), which is highly dependent on VEGF changes. However, work by this same group demonstrated that isoproterenol, a β-adrenergic receptor agonist, could reduce VEGF levels in mice exposed to excessive oxygen levels (Dal Monte et al., 2012). Additionally, others have also reported that norepinephrine can increase VEGF levels and angiogenesis in cancer cells through hypoxia-inducible factor-1α actions (Park et al., 2011). This data on norepinephrine actions agrees with work on mouse retinal explants that demonstrated that β3-adrenergic receptor agonists could increase VEGF (Dal Monte et al., 2013). However, others have suggested that β-adrenergic receptor antagonists might potentiate VEGF-induced angiogenesis in normoxia (Stati et al., 2014), suggesting that the oxygen levels of the tissue in question may affect the response to VEGF. Thus, it was unclear whether Compound 49b, a β1/β2-adrenergic receptor agonist, would decrease VEGF. Data in Figure 5 demonstrate that Compound 49b can significantly decrease VEGF in the diabetic rat retina and in REC cultured under high glucose conditions. Additionally, the data clearly demonstrate that Compound 49b requires IGFBP-3 to reduce VEGF. This is in agreement with work by Jarajapu et al. (2012) that showed that IGFBP-3 regulated VEGF.
Since the data demonstrated that Compound 49b regulated VEGF through increased IGFBP-3 levels, we wanted to determine the cellular pathways by which Compound 49b/IGFBP-3 reduced VEGF levels in the diabetic retina and REC. In a paper by Zhang et al. (2015), we demonstrated that IGFBP-3 regulated VEGF through decreased eNOS and PKCζ levels. The finding of IGFBP-3 regulation of eNOS follows previous work by Jarajapu et al. (2012). Thus, we sought to determine if Compound 49b-induced increases in IGFBP-3 was required for IGFBP-3 regulation of eNOS and PKC. We found that diabetes significantly increased phosphorylation of eNOS, which was reduced following topical Compound 49b through increased IGFBP-3 levels. This is in contrast to work in embryonic stem cells, where others found that β-adrenergic receptor antagonists decreased eNOS levels (Sharifpanah et al., 2014). Additionally, some have found that IGFBP-3 can increase eNOS levels in other vascular models (Kielczewski et al., 2009); however, these actions of IGFBP-3 were to protect the vasculature. The differences in our findings likely lie in the models investigated. Others have reported a strong correlation between eNOS levels and VEGF in tumor growth. When VEGF levels were increased, eNOS levels were also significantly increased (Barbieri et al., 2012), which agrees with our findings of a correlation between eNOS and VEGF.
In addition to eNOS, some groups have demonstrated that PKC isoforms are involved in VEGF regulation and maintenance of the blood retinal barrier (Geraldes & King, 2010). While it is unclear if PKCδ and PKCζ regulate eNOS to alter VEGF signaling or vice versa, it is clear that eNOS, PKCδ and PKCζ are involved in the regulation of VEGF and endothelial cell barrier integrity (Wang et al., 2010). Our data demonstrate that Compound 49b requires IGFBP-3 to decrease both PKCδ and PKCζ (Figure 4). Work in the Goto Kakizaki type 2 diabetic rat model demonstrated that PKCζ is involved in diabetic barrier integrity; however, they focused on the epithelium barrier between the retinal pigmented epithelial cells (Omri et al., 2011, 2013). Similar to our findings, Song et al. (2014) found that inhibition of PKCζ decreased VEGF levels in the streptozotocin-induced type 1 diabetic rat model, as well as in REC. In addition to PKCζ, inhibition of PKCδ also decreased VEGF-induced increases in endothelial cell permeability (Kim et al., 2010). Additional studies demonstrated that PKCδ activates eNOS to reduce atherosclerosis through altered platelet aggregation in postmenopausal women with type 2 diabetes (Munoz et al., 2012). Taken together, these data suggest that eNOS, PKCζ and PKCδ can interact to regulate VEGF levels and endothelial cell permeability. Our data extend these findings to add that β-adrenergic receptors regulate these pathways through IGFBP-3 actions.
These findings on Compound 49b further our previous work demonstrating that Compound 49b improved ERG amplitudes in type 1 diabetic rats, as well as prevented retinal thinning and cell loss in the ganglion cell layer (Zhang et al., 2012). Our findings of neuronal and vascular changes in the type 1 diabetic rat are in agreement with findings recently reported in a review article describing the interactions between the neural and vascular changes that occur in the retina in response to diabetes (Simo & Hernandez, 2014). Thus, in addition to improving the functional, neuronal and vascular changes in diabetic retinopathy, these studies suggest that Compound 49b can also regulate VEGF, a key protein in vascular permeability.
Conclusions
In conclusion, our data support existing literature to suggest that IGFBP-3 is a key regulator of VEGF in the retina. Since IGFBP-3 is reduced in the streptozotocin-induced type 1 diabetic rat model (Zhang et al., 2012), reduction of VEGF would require any novel drug to increase IGFBP-3. We have already demonstrated that Compound 49b, a β-adrenergic receptor agonist, successfully increases IGFBP-3 to reduce apoptosis in the retina. We also confirmed that Compound 49b can improve retinal ERG amplitudes through increasing IGFBP-3 levels, as Compound 49b was not as effective when IGFBP-3 siRNA was used. This study demonstrated that Compound 49b significantly reduced VEGF levels through IGFBP-3-mediated modulation of eNOS, PKCζ and PKCδ pathways in both diabetic rat retina and REC cultured in high glucose conditions.
Acknowledgments
Declaration of interest
This work was supported by the National Eye Institute (R01EY022045 to JJS), P30EY04068 (PI:Hazlett) and an Unrestricted Grant to the Department of Ophthalmology from Research to Prevent Blindness (PI:Jurzch, Kresge Eye Institute).
References
- Agarwal A, Sarwar S, Sepah YJ, Nguyen QD. What have we learnt about the management of diabetic macular edema in the antivascular endothelial growth factor and corticosteroid era? Curr Opin Ophthalmol. 2015;26:177–183. doi: 10.1097/ICU.0000000000000152. [DOI] [PubMed] [Google Scholar]
- Aveleira CA, Lin CM, Abcouwer SF, Ambrosio AF, Antonetti DA. TNF-alpha signals through PKCzeta/NF-kappaB to alter the tight junction complex and increase retinal endothelial cell permeability. Diabetes. 2010;59:2872–2882. doi: 10.2337/db09-1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbieri A, Palma G, Rosati A, Giudice A, Falco A, Petrillo A, Petrillo M, et al. Role of endothelial nitric oxide synthase (eNOS) in chronic stress-promoted tumour growth. J Cell Mol Med. 2012;16:920–926. doi: 10.1111/j.1582-4934.2011.01375.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dal Monte M, Cammalleri M, Mattei E, Filippi L, Bagnoli P. Protective effects of beta1/2 adrenergic receptor deletion in a model of oxygen-induced retinopathy. Invest Ophthalmol Vis Sci. 2015;56:59–73. doi: 10.1167/iovs.14-15263. [DOI] [PubMed] [Google Scholar]
- Dal Monte M, Filippi L, Bagnoli P. Beta3-adrenergic receptors modulate vascular endothelial growth factor release in response to hypoxia through the nitric oxide pathway in mouse retinal explants. Naunyn Schmiedebergs Arch Pharmacol. 2013;386:269–278. doi: 10.1007/s00210-012-0828-x. [DOI] [PubMed] [Google Scholar]
- Dal Monte M, Martini D, Latina V, Pavan B, Filippi L, Bagnoli P. Beta-adrenoreceptor agonism influences retinal responses to hypoxia in a model of retinopathy of prematurity. Invest Ophthalmol Vis Sci. 2012;53:2181–2192. doi: 10.1167/iovs.11-9408. [DOI] [PubMed] [Google Scholar]
- Das A, Stroud S, Mehta A, Rangasamy S. New treatments for diabetic retinopathy. Diabetes Obes Metab. 2015;17:219–230. doi: 10.1111/dom.12384. [DOI] [PubMed] [Google Scholar]
- Frey T, Antonetti DA. Alterations to the blood–retinal barrier in diabetes: Cytokines and reactive oxygen species. Antioxid Redox Signal. 2011;15:1271–1284. doi: 10.1089/ars.2011.3906. [DOI] [PubMed] [Google Scholar]
- Geraldes P, King GL. Activation of protein kinase C isoforms and its impact on diabetic complications. Circ Res. 2010;106:1319–1331. doi: 10.1161/CIRCRESAHA.110.217117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Googe J, Brucker AJ, Bressler NM, Qin H, Aiello LP, Antoszyk A, Beck RW, et al. Randomized trial evaluating short-term effects of intravitreal ranibizumab or triamcinolone acetonide on macular edema after focal/grid laser for diabetic macular edema in eyes also receiving panretinal photocoagulation. Retina. 2011;31:1009–1027. doi: 10.1097/IAE.0b013e318217d739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harhaj NS, Felinski EA, Wolpert EB, Sundstrom JM, Gardner TW, Antonetti DA. VEGF activation of protein kinase C stimulates occludin phosphorylation and contributes to endothelial permeability. Invest Ophthalmol Vis Sci. 2006;47:5106–5115. doi: 10.1167/iovs.06-0322. [DOI] [PubMed] [Google Scholar]
- Jarajapu YP, Cai J, Yan Y, Li Calzi S, Kielczewski JL, Hu P, Shaw LC, et al. Protection of blood retinal barrier and systemic vasculature by insulin-like growth factor binding protein-3. PLoS One. 2012;7:e39398. doi: 10.1371/journal.pone.0039398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Zhang Q, Liu L, Tang J, Kern TS, Steinle JJ. Beta2-adrenergic receptor knockout mice exhibit A diabetic retinopathy phenotype. PLoS One. 2013;8:e70555. doi: 10.1371/journal.pone.0070555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielczewski JL, Jarajapu Y, McFarland EL, Cai J, Afzal A, Li Calzi S, Chang KH, et al. Insulin-like growth factor binding protein-3 mediates vascular repair by enhancing nitric oxide generation. Circ Res. 2009;105:897–905. doi: 10.1161/CIRCRESAHA.109.199059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielczewski JL, Li Calzi S, Shaw LC, Cai J, Qi X, Ruan Q, Wu L, et al. Free insulin-like growth factor binding protein-3 (IGFBP-3) reduces retinal vascular permeability in association with a reduction of acid sphingomyelinase (ASMase) Invest Ophthalmol Vis Sci. 2011;52:8278–8286. doi: 10.1167/iovs.11-8167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Kim JH, Jun HO, Yu YS, Kim KW. Inhibition of protein kinase C delta attenuates blood–retinal barrier breakdown in diabetic retinopathy. Am J Pathol. 2010;176:1517–1524. doi: 10.2353/ajpath.2010.090398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koizumi K, Poulaki V, Doehmen S, Welsandt G, Radetzky S, Lappas A, Kociok N, et al. Contribution of TNF-alpha to leukocyte adhesion, vascular leakage, and apoptotic cell death in endotoxininduced uveitis in vivo. Invest Ophthalmol Vis Sci. 2003;44:2184–2191. doi: 10.1167/iovs.02-0589. [DOI] [PubMed] [Google Scholar]
- Munoz YC, Gomez GI, Moreno M, Solis CL, Valladares LE, Velarde V. Dehydroepiandrosterone prevents the aggregation of platelets obtained from postmenopausal women with type 2 diabetes mellitus through the activation of the PKC/eNOS/NO pathway. Horm Metab Res. 2012;44:625–631. doi: 10.1055/s-0032-1309056. [DOI] [PubMed] [Google Scholar]
- Omri S, Behar-Cohen F, de Kozak Y, Sennlaub F, Verissimo LM, Jonet L, Savoldelli M, et al. Microglia/macrophages migrate through retinal epithelium barrier by a transcellular route in diabetic retinopathy: Role of PKCzeta in the Goto Kakizaki rat model. Am J Pathol. 2011;179:942–953. doi: 10.1016/j.ajpath.2011.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omri S, Behar-Cohen F, Rothschild PR, Gelize E, Jonet L, Jeanny JC, Omri B, Crisanti P. PKCzeta mediates breakdown of outer blood–retinal barriers in diabetic retinopathy. PLoS One. 2013;8:e81600. doi: 10.1371/journal.pone.0081600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SY, Kang JH, Jeong KJ, Lee J, Han JW, Choi WS, Kim YK, et al. Norepinephrine induces VEGF expression and angiogenesis by a hypoxia-inducible factor-1alpha protein-dependent mechanism. Int J Cancer. 2011;128:2306–2316. doi: 10.1002/ijc.25589. [DOI] [PubMed] [Google Scholar]
- Sharifpanah F, Saliu F, Bekhite MM, Wartenberg M, Sauer H. Beta-adrenergic receptor antagonists inhibit vasculogenesis of embryonic stem cells by downregulation of nitric oxide generation and interference with VEGF signalling. Cell Tissue Res. 2014;358:443–452. doi: 10.1007/s00441-014-1976-8. [DOI] [PubMed] [Google Scholar]
- Simo R, Hernandez C European Consortium for the Early Treatment of Diabetic R. Neurodegeneration in the diabetic eye: New insights and therapeutic perspectives. Trends Endocrinol Metab. 2014;25:23–33. doi: 10.1016/j.tem.2013.09.005. [DOI] [PubMed] [Google Scholar]
- Song HB, Jun HO, Kim JH, Yu YS, Kim KW, Kim JH. Suppression of protein kinase C-zeta attenuates vascular leakage via prevention of tight junction protein decrease in diabetic retinopathy. Biochem Biophys Res Commun. 2014;444:63–68. doi: 10.1016/j.bbrc.2014.01.002. [DOI] [PubMed] [Google Scholar]
- Stati T, Musumeci M, Maccari S, Massimi A, Corritore E, Strimpakos G, Pelosi E, et al. Beta-blockers promote angiogenesis in the mouse aortic ring assay. J Cardiovasc Pharmacol. 2014;64:21–27. doi: 10.1097/FJC.0000000000000085. [DOI] [PubMed] [Google Scholar]
- Titchenell PM, Lin CM, Keil JM, Sundstrom JM, Smith CD, Antonetti DA. Novel atypical PKC inhibitors prevent vascular endothelial growth factor-induced blood-retinal barrier dysfunction. Biochem J. 2012;446:455–467. doi: 10.1042/BJ20111961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Wu B, Sun Y, Xu T, Zhang X, Zhou M, Jiang W. Translocation of protein kinase C isoforms is involved in propofol-induced endothelial nitric oxide synthase activation. Br J Anaesth. 2010;104:606–612. doi: 10.1093/bja/aeq064. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Guy K, Pagadala J, Jiang Y, Walker RJ, Liu L, Soderland C, et al. Compound 49b prevents diabetes-induced apoptosis through increased IGFBP-3 levels. Invest Ophthalmol Vis Sci. 2012;53:3004–3013. doi: 10.1167/iovs.11-8779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Jiang Y, Miller MJ, Peng B, Liu L, Soderland C, Tang J, et al. IGFBP-3 and TNF-alpha regulate retinal endothelial cell apoptosis. Invest Ophthalmol Vis Sci. 2013;54:5376–5384. doi: 10.1167/iovs.13-12497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Jiang Y, Steinle JJ. IGFBP-3 reduces eNOS and PKCzeta phosphorylation, leading to lowered VEGF levels. Mol Vis. 2015;21:604–611. [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Steinle JJ. IGFBP-3 inhibits TNF-alpha production and TNFR-2 signaling to protect against retinal endothelial cell apoptosis. Microvasc Res. 2014;95:76–81. doi: 10.1016/j.mvr.2014.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]