

Abstract
Ethyl acetate and dichloromethane extract of Dillenia suffruticosa (EADS and DCMDS, respectively) can be a potential anticancer agent. The effects of EADS and DCMDS on the growth of HeLa cervical cancer cells and the expression of apoptotic-related proteins had been investigated in vitro. Cytotoxicity of the extracts toward the cells was determined by 5-diphenyltetrazolium bromide assay, the effects on cell cycle progression and the mode of cell death were analyzed by flow cytometry technique, while the effects on apoptotic-related genes and proteins were evaluated by quantitative real-time polymerase chain reaction, and Western blot and enzyme-linked immunosorbent assay, respectively. Treatment with DCMDS inhibited (P < 0.05) proliferation and induced apoptosis in HeLa cells. The expression of cyclin B1 was downregulated that led to G2/M arrest in the cells after treatment with DCMDA. In summary, DCMDS induced apoptosis in HeLa cells via endoplasmic reticulum stress-induced apoptotic pathway and dysregulation of mitochondria. The data suggest the potential application of DCMDS in the treatment of cervical cancer.
Keywords: Apoptosis, cell cycle arrest, cervical cancer, Dillenia suffruticosa, endoplasmic reticulum stress
INTRODUCTION
Cervical cancer ranks as the third most common cause of cancer-related mortality in women.[1] Several studies have reported that 99% of human papillomavirus (HPV) infections are associated with cervical cancer[2,3] with 50–70% and 7–20% of the cases are caused by HPV 16 and HPV 18, respectively.[4,5,6] As such, several epidemiological studies have documented the success of prophylactic HPV vaccination program as a preventive measure for cervical cancer but not solely to treat it. Indeed, a significant reduction in the incidence and mortality of cervical cancer does not only rely on screening program or vaccination. Thus, the need to discover a potent and more effective therapy is vital.
Currently, cisplatin is used as a gold standard drug in the management of advanced cervical cancer. Despite the effectiveness, the adverse effects of cisplatin such as hematologic toxicity, neutropenia, anemia, nausea, diarrhea, and emesis have become a major concern.[7,8,9,10] In addition, cisplatin treatment often results in the development of chemoresistance, leading to therapeutic failure.[9,11] Hence, attentions have been focusing in identifying new agents particularly those present in natural products which have been reported to have a long history for treatment of cancer.
Dillenia suffruticosa (Griffith ex Hook. f. and Thomson) Martelli or Wormia suffruticosa is indigenous to Malay Archipelago. This plant is traditionally used to treat cancerous growth[12] and fever, for wound healing[13] and to relief rheumatism.[14] D. suffruticosa extract also possesses antifungal[15] and antibacterial properties,[16] and antiviral properties against dengue type 2 virus.[17] From our previous study, ethyl acetate (EADS) and dichloromethane (DCMDS) extract of the root of D. suffruticosa exhibited strong cytotoxicity toward breast cancer (MCF-7 and MDA-321) and cervical cancer (HeLa) cell line due to induction of apoptosis and cell cycle arrest.[18,19,20,21] However, the actual mechanism underlying apoptosis induced by EADS and DCMDS in HeLa cells was unknown. Hence, this study evaluated the effects of the extracts on cell cycle progression and the expression of intrinsic and extrinsic apoptotic-related genes and proteins in HeLa cells.
RESULTS AND DISCUSSION
Cytotoxicity of ethyl acetate and dichloromethane extract of Dillenia suffruticosa via cell cycle arrest and induction of apoptosis
Ethyl acetate extract of D. suffruticosa exerted low toxicity toward HeLa cells at 24 h whereby the IC50 was undetectable even at the highest concentration employed (>100 μg/mL). On the other hand, a good drug-response curve was obtained for DCMDS with IC50 of 32.98 ± 1.68 μg/mL. DCMDS was most cytotoxic at 72 h (IC50 = 11.95 ± 2.07 μg/mL). Based on that, only DCMDS was used for further investigation. Both EADS and DCMDS showed lower IC50 values toward the normal cells 3T3 and Vero exhibited compared to HeLa [Table 1].
Table 1.
Cytotoxicity of EADS and DCMDS towards human cell lines as determined by MTT assay. Results are presented as mean±SD of triplicate of three independent experiments. *Significant different from control at P<0.05. α and β are significantly different
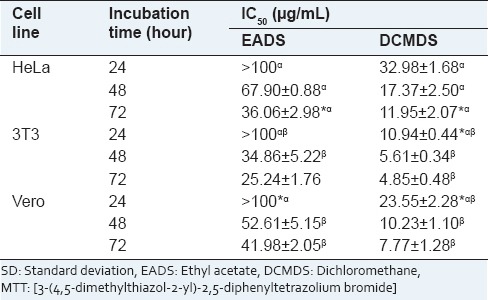
In this experiment, the human cervical cancer cells HeLa demonstrated different response toward EADS and DCMDS. The cytotoxic effects of both extracts were found to be in a time- and dose-dependent manner. The response to the cellular stress depends on many factors such as the cell type, duration, the number and intensity of the new stimuli.[22] This response in turn initiates a number of complex signaling cascade pathways to maintain the cell homeostasis.[23] DCMDS was more cytotoxic than EADS. Cytotoxicity of an extract is very much depending on its phytochemical components. EADS contains saponins, triterpenes, and tannins/polyphenolic while sterols were detected in DCMDS.[21] Plant sterols have been reported to alter the signaling pathways that prevent cell proliferation.[24] Even though EADS and DCMDS were more cytotoxic to the normal cells 3T3 and Vero compared to HeLa, the toxicity of the extracts to the normal cells in vivo might be different. This is due to the fact that an in vitro system is just evaluating a single cell type, organ, and isolated cells of other organs whereby no interaction and communication between the organs occurs.[25,26,27] In addition, the natural product has been claimed to exert minimum side effects compared with available synthetic agents to treat cervical cancer.[28]
DNA content analysis shows that there was a significant increase in the G2/M peak of HeLa cells treated with DCMDS (12.5 µg/mL and 25 µg/mL) at 24 h incubation as compared to the control [Figure 1]. The number of cells at G0/G1 and S phase decreased significantly at 24 h (P < 0.05). Decreased percentage of cells indicates that they were not arrested at that particular phase. At 50 µg/mL, DCMDS caused cell cycle arrest at G2/M after 48 and 72 h (P < 0.05) [Figure 1]. The protein analysis shows that cyclin B1 that is involved in the progression of cells from G2 phase to mitosis in cell cycle control was downregulated [Figure 2]. It shows that DCMDS arrested HeLa cells at G2/M. Thus, cell cycle arrest is one of the anticancer properties of the extract.[29]
Figure 1.

Effect of dichloromethane extract of Dillenia suffruticosa (DCMDS) on the cycle of HeLa cells. DCMDS induced arrest at the G2/M phase. Results are the mean ± standard deviation of triplicate of the experiment. *Significant as compared to the control
Figure 2.
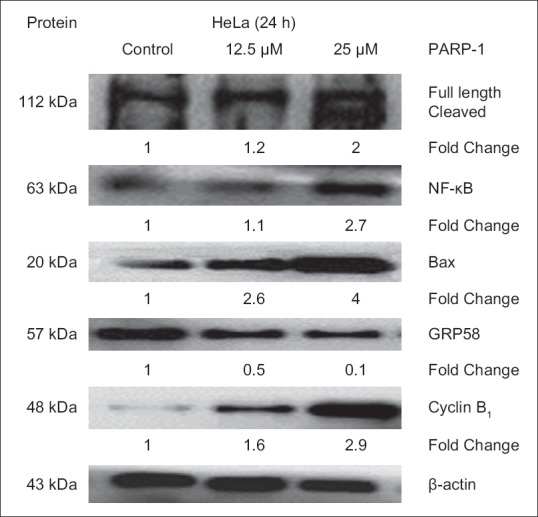
Expression level of the apoptotic-related proteins in HeLa cells determined by Western Blot analysis. β-actin acts as a loading control. Protein bands were quantified by densitometric analysis. Relative expression was calculated by the optical density ratio of protein to β-actin and normalized against the control. PARP-1: Poly (adenosine diphosphate-ribose) polymerase
Based on the Annexin V/FITC analysis, the DCMDS-treated cells (12.5 µg/mL) with high apoptotic index were obviously noted after 48 h, with significant increase in the early (Q2) and late apoptotic (Q3) cells. The number increased with increase in the concentration of the extract. The number of viable cells (Q1) in the treatment of 25 µg/mL DCMDS was very much lower (P < 0.05) as compared to the control cells at 72 h [Figure 3]. The percentage of necrotic cells was considerably low compared with apoptotic cells (P < 0.05). The effects of the treatment were dependent on dose and time. Exposure to a high concentration (50 µg/mL) of DCMDS further increased the rate of apoptosis and suppressed HeLa cells proliferation. A recombinant phosphatidylserine-binding protein, Annexin V interacts with phosphatidyl serine residues (ligand for phagocytes on the surface of the apoptotic cell)[30] and can be used for the detection of apoptosis.[31,32] Low number of viable cells and high percentage of cells at early and late apoptosis following treatment with DCMDS proved that the extract caused the cells to die primarily through apoptosis, not necrosis. It is of the advantage of apoptosis-inducing agent that it will prevent damage to the surrounding normal healthy cells or inflammation.
Figure 3.

Flow cytometric analysis of the mode of cell death induced by dichloromethane extract of Dillenia suffruticosa (DCMDS) toward HeLa cells. Number of apoptotic cells increased following exposure to increasing concentration of DCMDS. Q1 represents Annexin V (−)/PI (+) (necrotic cells); Q2 represents Annexin V (+)/PI (+) (late apoptotic and dead cells); Q3 represents Annexin V (+)/PI (−) (viable and early apoptotic cells); Q4 represents Annexin V (−)/PI (−) (healthy and viable cells)
Expression of the apoptotic genes and proteins in HeLa cells following treatment with dichloromethane extract of Dillenia suffruticosa
Effects of dichloromethane extract of Dillenia suffruticosa on the activity of caspases
Following exposure to DCMDS (25 ug/mL), the expression of caspase-3, -8, -9, and -12 in HeLa cells was up-regulated at 24 h [Figure 4]. It indicates that caspase-3, -8, -9, and -12 were activated in HeLa cells treated with DCMDS.
Figure 4.

Effect of dichloromethane extract of Dillenia suffruticosa on the activity of caspase-3, -8, and -9 (a) and caspase-12 (b). Caspase activity was enhanced after increasing the dose to 25 μg/ml. Results are presented as mean ± standard deviation; n = 3. *Significant different from the respective control at P < 0.05
Involvement of poly (adenosine diphosphate-ribose) polymerase-1 and NF-κB in dichloromethane extract of Dillenia suffruticosa-induced apoptosis
The expression level of poly (adenosine diphosphate-ribose) polymerase-1 (PARP-1) and NF-κB was positively correlated with the rate of DCMDS-induced apoptosis [Figures 2 and 5]. Increasing the concentration of DCMDS resulted in increasing level of PARP-1 cleavage activity and the expression level of NF-κB [Figures 2 and 5].
Figure 5.
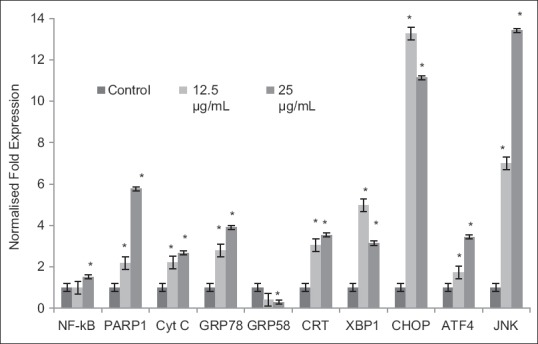
Level of mRNA expression of the selected genes in HeLa cells as determined by quantitative real-time polymerase chain reaction analysis. The level of mRNA expression correlates with the dosage of dichloromethane extract of Dillenia suffruticosa. Expression of all the genes was up-regulated except for GRP58. Fold changes of CCAAT/enhancer binding protein homologous protein and Jun NH2-terminal kinase mRNA were higher than others. Results are presented as mean ± standard deviation; n = 3. *Significant different from the respective control at P < 0.05
Involvement of p53, Bcl-2, and Bax in dichloromethane extract of Dillenia suffruticosa -induced apoptosis
The expression of p53 was lower in the control compared to the treated HeLa cells. The expression of p53 protein significantly increased at 12.5 µg/mL and 25 µg/mL of DCMDS compared with the control (P < 0.05) after 24 and 48 h. In contrast, the expression of anti-apoptotic Bcl-2 significantly decreased following treatment with DCMDS at 48 h [Figure 6]. The expression of Bax increased [Figure 2] p53-mediated apoptosis eliminated both types of cells with damaged DNA or an aberrant cell cycle.[33] Based on the data, it is evident that the expression level of p53 increased with decreased expression of Bcl-2 following treatment with DCMDS. It is known that p53 regulates a set of anti-apoptotic genes including members of the Bcl-2 family. Bax and IGF-BP3 are the genes that are regulated by p53 which could influence the decision to proceed to the apoptotic pathway or not.[34] On the other hand, Bcl-2 is a proto-oncogene that protects permanent cells from apoptosis.[35] Bax binds to Bcl-2 and antagonizes its ability to block apoptosis. Bcl-2 protects against diverse cytotoxic entities namely ultraviolet irradiation and cytotoxic drugs.[36,37] It is then postulated that downregulation of Bcl-2 expression causes HeLa cells to lose of their protective mechanism and promote apoptosis, as a passive process of activation of p53 by DCMDS.[38]
Figure 6.

Effects of treatment with dichloromethane extract of Dillenia suffruticosa (DCMDS) on the level of p53 and Bcl-2 in HeLa cells. Upregulation of p53 was noted after DCMDS treatment which was accompanied by downregulation of Bcl-2. Results are presented as mean ± standard deviation; n = 3. *Significant different from respective control at P < 0.01
Dichloromethane extract of Dillenia suffruticosa induced apoptosis via endoplasmic reticulum stress pathway
To gain more insights into the molecular events of DCMDS-induced apoptosis in the endoplasmic reticulum (ER), gene expression analysis was performed. DCMDS was found to upregulate the expression of a key indicator of ER stress responses, glucose-regulated protein 78/binding immunoglobulin protein (GRP78/BiP) and its downstream transcription factor CCAAT/enhancer binding protein (C/EBP)-homologous protein or growth and DNA damage (GADD)-inducible transcription factor 153 (CHOP/GADD153). From the quantitative real-time polymerase chain reaction (PCR) analysis, GRP78 mRNA level increased up to 4 folds in HeLa cells after treatment with DCMDS, while CHOP mRNA level increased dramatically up to 13 folds [Figure 6]. Activating transcription factor 4 (ATF4) level increased, which was accompanied by an increase in the X-box binding protein 1 (XBP1) mRNA splicing. The spliced form of the XBP-1 mRNA was detected at low concentration (12.5 µg/mL) of DCMDS treatment but decreased at higher concentration (25 µg/mL). Furthermore, calreticulin (CRT) mRNA was also up-regulated. However, ER chaperone GRP58 was downregulated as the concentration of DCMDS increased [Figures 2 and 6].
Endoplasmic reticulum is an organelle that serves many functions including calcium homeostasis, cellular protein quality control and the transport of synthesized proteins to the Golgi apparatus.[39] ER stress happens when ER function is disrupted that subsequently causes accumulation of unfolded proteins in the ER. It triggers the unfolded protein response (UPR) to reduce the excessive protein loading which signals temporary inhibition of protein translation, degradation of misfolded proteins, and the induction of molecular chaperones and folding enzymes to increase the ER capacity of protein folding and degradation.[40]
In our study, DCMDS caused upregulation of GRP78/BiP and its downstream transcription factor CHOP or growth-arrest and DNA-damage-inducible gene 153 (GADD153) (CHOP/GADD153) [Figure 5]. CHOP, c-Jun NH2-terminal kinase (JNK), and caspases have been implicated in mediating apoptotic signals in response to ER stress.[41] During prolonged ER stress, CHOP is one of the most highly up-regulated genes.[42] All three UPR signaling pathways (PKR-like ER kinase [PERK], inositol-requiring transmembrane kinase [IRE1], and ATF6) are involved in inducing CHOP transcription, although the PERK pathway is essential.[43] Conversely, overexpression of CHOP promotes apoptosis in response to ER stress caused by thapsigargin and increases cellular reactive oxygen species, which most likely contribute to ER stress-associated cell death.[44] In response to ER stress, IRE1 alpha has been found to recruit the adaptor protein TRAF2 to the ER membrane. This recruitment is regulated by c-Jun NH2-terminal inhibitory kinase, which has been reported to interact with both IRE1α and TRAF2. The IRE1α/TRAF2 complex then recruits apoptosis signal-regulating kinase 1 (ASK1), causing activation of ASK1 and the downstream JNK pathway. JNK phosphorylates Bcl-2 and BH3-only protein, initiating mitochondria-mediated apoptosis. Together, these results suggest that the ER stress sends signals to the mitochondria via the regulation of Bcl-2 proteins by both CHOP and JNK.[41] Activation of JNK pathway could lead to FasL induction and cell death. Activation of FasL followed by caspase-8 can initiate the extrinsic pathway.[45] In this study, NF-κB was also activated, promoting DCMDS-mediated induction of Fas or FasL and thus increasing the level of apoptosis in HeLa cells. NF-κB acts as a pro-apoptotic factor in this context. This indicates that the extrinsic pathway is involved in DCMDS-induced apoptosis.
The ER stress-induced apoptosis also involves caspases. In mice, procaspase-12 is localized on the cytoplasmic side of the ER and is cleaved and activated specifically by ER stress. Calpains, a family of calcium-dependent cysteine proteases, have been shown to play a role in caspase-12 activation[46] and calpain-deficient MEFs have reduced ER stress-induced caspase-12 activation and are resistant to ER stress-associated apoptosis.[47]
GRP58 might play a role in DCMDS-induced ER stress-induced apoptosis. Following treatment with DCMDS, the rate of apoptosis increased with downregulation of GRP58 but upregulation of other ER stress proteins. Our findings were in concordance with previous reports that upregulation of GRP78 will cause downregulation of (GRP58).[48] The expression level of ER chaperone GRP58 reduced in a dose-dependent manner [Figures 2 and 5]. Overexpression of GRP58 is associated with the ER stress due to disruption of the calcium homeostasis,[49] hypoxia and virus or bacterial infection. In human ovarian cancer,[50] prostate cancer,[51] breast cancer, lung cancer, uterine cancer, and stomach cancer,[52] GRP58 level was enhanced when compared with normal tissues of similar origin. However, downregulation of GRP58 expression has also been documented in gastric cancer,[53] esophageal cancer,[54] cervical carcinoma[55] and renal cell carcinoma.[56] A few studies showed the association of GRP58 with the inhibition of cancer cell proliferation. Knock-down of GRP58 was associated with the inhibition of breast cancer cell proliferation[57] and melanoma cells.[58] Downregulation of GRP58 was associated with poor prognosis of early stage cervical cancer.[59] A recent study showed that this stress-responsive protein which is activated on glucose deprivation modulates invasiveness of cervical cancer.[60] In their investigation, knockdown of GRP58 in HeLa cells led to decreased cell invasiveness and inhibition of lung metastasis in a xenograft mouse model.
Some studies have linked ER stress with apoptosis mediated by the mitochondrial mechanisms involving the Bcl-2 family of proteins. During ER stress, Bax and Bak in the ER membrane undergo conformational changes.[61] This allows the release of calcium from the ER to the cytoplasm, which in turn activates m-Calpain. Subsequently, it cleaves and activates procaspase-12. Activated caspase-12 cleaves and activates procaspase-9, which in turn activates downstream caspases, including caspase-3.[62] In addition, calcium released from the ER is taken up by the mitochondria, causing mitochondrial inner membrane depolarization and caspase-8 activation. Cytochrome c is released into the cytoplasm to stimulate the assembly of the apoptosome (consisting of Apaf-1, cytochrome c, ATP, and procaspase-9), activation of procaspase-9, which in turn activates caspase-3, DNA fragmentation, and cell death.[63,64,65]
The expression of ER molecule, CRT was also up-regulated in DCMDS-treated HeLa cells. Within ER, CRT together with calnexin and GRP58 serve as a molecular chaperone to ensure proper folding of glycoproteins.[66] It is believed that treatment with DCMDS triggered the activation of the CRT exposure pathway by preapoptotic ER stress molecules and the phosphorylation of the eukaryotic translation initiation factor eIF2alpha by the kinase PERK, followed by caspase-8-mediated proteolysis, and activation of the pro-apoptotic proteins Bax and Bak. It is therefore concluded that DCMDS induced ER stress and caused cell death via apoptosis in HeLa cells.
EXPERIMENTAL
Preparation of ethyl acetate and dichloromethane extracts of Dillenia suffruticosa
The extracts were obtained using a sequential cold solvent extraction method and tested as described previously.[20]
Cell culture
The human cervical adenocarcinoma (HeLa), mouse fibroblast (3T3), and African green monkey kidney epithelial (Vero) cell lines (American Type and Culture Collection [ATCC], Rockville MD, USA) were grown in RPMI-1640 supplemented with 10% (v/v) of fetal bovine serum and 1% of penicillin-streptomycin (100 IU/mL of penicillin and 100 µg/mL of streptomycin) (PAA Laboratories, Linz, Austria). The cells were allowed to grow at 37°C with 5% CO2 atmosphere.
Determination of cytotoxicity of ethyl acetate and dichloromethane extract of Dillenia suffruticosa through induction of apoptosis and cell cycle arrest
Cytotoxicity was assessed using 3-4 5-dimethylthiazol- 2-yl]-2, 5-diphenyltetrazolium bromide assay[67] as described previously (Armania et al., 2013). The cells (0.7 × 105 cells/mL) were treated in a 96-well plate with different concentration of EADS and DCMDS (3.0–200 µg/mL) for 24, 48, and 72 h. The cell cycle distribution was analyzed using an FACScan flow cytometer (Becton Dickinson, Mountain View, CA, USA) 3. The population of cells in each phase and mode of cell death was determined using ModFit LT™ (Verity Software House, Topsham, Maine, USA) and FlowJo version 8.6 (Treestar Inc., San Carlos, CA), respectively.
Quantitative real-time polymerase chain reaction
HeLa cells were treated with DCMDS (12.5 µg/mL and 25 µg/mL) for 48 h. Untreated samples were included as controls. RNA was extracted using Total RNA Extraction Kit (Mini) cells according to the manufacturer's instructions (Real Biotech Corporation, USA) as described previously.[68] Highly purified salt-free primers were designed by Next Gene Scientific Sdn. Bhd. Malaysia and synthesized by AIT Biotech, Singapore: GRP78, F-5’-AGTTCTTCAATGGCAAGGAA-3’, R-5’ CAGTTCAATACCAAGTGTAAGG-3’ (NM_005347.4); GRP58, F-5’ CATGTACGTTGCTATCCAGGC-3’, R-5’-TGCTAAAGGAGAGAAGTTTG-3’ (NM_005313.4); CHOP, F-5’-ATGGCAGCTGAGTCATTGCCTTTC-3, R-’5’-AGAAGCAGGGTCAAGAGTGGTGAA-3’;[69] XBP, F-15’-GGAGTTAAGACAGCGCTTGGGGA-3’, R-5’-TGTTCTGGAGGGGTGACAACTGGG-3’;[70] ATF-4, F-5’-AACCGACAAAGACACCTTCG-3, R-’5’-ACCCATGAGGTTTGAAGTGC-3’;[71] CRT, F-5’-ACGAGCCAAGATTGATGACC-3, R-5’-CAGAAGCTCCACCACCAAAGAT-3’;[72] JNK, F-5’-GCCATTGATCACTGCTGCAC-3’, R-5’-GCGGGCGTCTAAAATTCTG-3’ (NM_139046); NF-κB, F-5’-TTCCACGATCACCAGGTAGG-3’, R-5’-TATCGAGTCGAGTACGCCAA-3’, (NM_001077493); PARP, F-5’-GTGTGGGACTTTTCCATCAAA-3’, R-5’-CCAGTGCCACACCGTTGAA-3’ (NM_001618); Cytochrome C, F-5’-TCCCCAGATGCCTTTGTT-3’, R-5’-TTCCATTCGCCCTTGTATTC-3’;[73] HPRT, F-5’-CTGCTACCACTACCTTCA-3’, R-5’-CTCCTTAATGTCACGCACGAT-3’ (NM_001101.3); β-actin, F-5’-CGAGATGTGATGAAGGAGATG-3’, R-5’-CCTGTTGACTGGTCATTACAA-3’ (NM_000194.2).
Polymerase chain reaction products were synthesized using IQ™ SYBR® Green Supermix (Bio-Rad, USA). Thermal cycling programs were performed using the three-step cycling protocol in low-profile 0.2 mL tube strips (Bio-Rad, Hercules, CA, USA) (Initial denaturation: 5 min; Denaturation: 95°C for 20 s; Annealing: 60°C for 30 s; Extension: 70°C for 5 s). Conditions for all PCRs were optimized in a gradient cycler (Mastercycler Gradient, Bio-Rad, USA) with regard to various annealing temperatures (50–60°C). All the reactions were performed in triplicate and a mixture without cDNA template (NTC) was used as the negative control. Data acquisition and analysis were performed using CFX Manager version 2.0 (Bio-Rad, USA).
Western blot analysis
Western blot analysis was performed as described previously.[74] The membrane was incubated overnight at 4°C with the primary antibody, rabbit anti-PARP, NF-κB, Bax, cyclin B1, mouse anti-GRP58 (Abcam, Cambridge, MA, USA), and anti-β-actin (Santa Cruz, USA) at a dilution of 1:2000. These steps were followed by incubation with horseradish peroxidise-conjugated secondary antibody, rabbit anti-mouse IgG (Santa Cruz, USA) for 60 min at 25°C on rocker (dilution 1:4000). All immunoblots were visualized by enhanced chemiluminescence plus Western blotting detection reagents (Abcam Corporation, USA). Densitometrical quantification of autoradiograms was analyzed by ImageJ software (version 1.41, National Institutes of Health, Bethesda, MD).
Enzyme-linked immunosorbent assay
Untreated and treated HeLa cells (12.5 µg/mL and 25 µg/mL) were scraped, washed twice with ice-cold PBS and lysed for 60 min at room temperature (27°C) in lysis buffer (1X) (Invitrogen, USA). The p53 and Bcl-2 protein concentration in the extract was estimated using the Human p53 Platinum enzyme-linked immunosorbent assay (ELISA) kit and the Human Bcl-2 Platinum ELISA kit, respectively (Bender MedSystems GmbH, Vienna, Austria). The protease activity of caspases-3, -8 and -9 was evaluated spectrophotometrically using Colorimetric Assay kit (Gene script, USA) and caspase-12 using the Human Caspase-12 ELISA kit (Cusabio Biotech, China). Experiments were performed in triplicate. The samples extinction values were determined using ELISA plate reader (Biotek, USA) at 450 nm.
Statistical analysis
Statistical analysis was performed with General Linear Model (Univariate), Duncan's multiples range test, and Dunnett's test using Statistical Package of Social Sciences (SPSS) for Window version 21.0 (SPSS Inc., Chicago, IL, USA). All the data are presented as mean ± standard deviation. A mean difference is considered significant when P < 0.05.
CONCLUSIONS
In short, this study demonstrates that EADS and DCMDS have the potential to be developed into an agent for the treatment of cervical cancer. DCMDS induced cellular stress in HeLa cells by activation of mitochondrial and ER stress signaling pathways that ultimately led to apoptosis. Thus, DCMDS may be a potential source of lead compounds for drug development.
Footnotes
Source of Support: This study was funded by Research University Grant Scheme of Universiti Putra Malaysia (04-01-09-0714RU) and Fundamental Research Grant Scheme (04-04-10-884FR)
Conflict of Interest: None declared.
REFERENCES
- 1.Ferlay J, Steliarova-Foucher E, Lortet-Tieulent J, Rosso S, Coebergh JW, Comber H, et al. Cancer incidence and mortality patterns in Europe: Estimates for 40 countries in 2012. Eur J Cancer. 2013;49:1374–403. doi: 10.1016/j.ejca.2012.12.027. [DOI] [PubMed] [Google Scholar]
- 2.Seoud M. Burden of human papillomavirus-related cervical disease in the extended middle East and north Africa-a comprehensive literature review. J Low Genit Tract Dis. 2012;16:106–20. doi: 10.1097/LGT.0b013e31823a0108. [DOI] [PubMed] [Google Scholar]
- 3.Stanley M. Pathology and epidemiology of HPV infection in females. Gynecol Oncol. 2010;117:S5–10. doi: 10.1016/j.ygyno.2010.01.024. [DOI] [PubMed] [Google Scholar]
- 4.Giuliano AR, Lazcano-Ponce E, Villa LL, Flores R, Salmeron J, Lee JH, et al. The human papillomavirus infection in men study: Human papillomavirus prevalence and type distribution among men residing in Brazil, Mexico, and the United States. Cancer Epidemiol Biomarkers Prev. 2008;17:2036–43. doi: 10.1158/1055-9965.EPI-08-0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Koutsky LA, Ault KA, Wheeler CM, Brown DR, Barr E, Alvarez FB, et al. A controlled trial of a human papillomavirus type 16 vaccine. N Engl J Med. 2002;347:1645–51. doi: 10.1056/NEJMoa020586. [DOI] [PubMed] [Google Scholar]
- 6.Muñoz N, Castellsagué X, de González AB, Gissmann L. Chapter 1: HPV in the etiology of human cancer. Vaccine. 2006;24(Suppl 3):S3/1–10. doi: 10.1016/j.vaccine.2006.05.115. [DOI] [PubMed] [Google Scholar]
- 7.Kong TW, Chang SJ, Paek J, Yoo SC, Yoon JH, Chang KH, et al. Comparison of concurrent chemoradiation therapy with weekly cisplatin versus monthly fluorouracil plus cisplatin in FIGO stage IIB-IVA cervical cancer. J Gynecol Oncol. 2012;23:235–41. doi: 10.3802/jgo.2012.23.4.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oike T, Ohno T, Noda SE, Sato H, Tamaki T, Kiyohara H, et al. Comparison of hematological toxicities between innovator and generic cisplatin formulations in cervical cancer patients treated with concurrent chemoradiotherapy. J Radiat Res. 2013;54:474–8. doi: 10.1093/jrr/rrs121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rose PG, Sill MW, McMeekin DS, Ahmed A, Salani R, Yamada SD, et al. A phase I study of concurrent weekly topotecan and cisplatin chemotherapy with whole pelvic radiation therapy in locally advanced cervical cancer: A Gynecologic Oncology Group study. Gynecol Oncol. 2012;125:158–62. doi: 10.1016/j.ygyno.2011.12.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Long HJ, 3rd, Bundy BN, Grendys EC, Jr, Benda JA, McMeekin DS, Sorosky J, et al. Randomized phase III trial of cisplatin with or without topotecan in carcinoma of the uterine cervix: A Gynecologic Oncology Group Study. J Clin Oncol. 2005;23:4626–33. doi: 10.1200/JCO.2005.10.021. [DOI] [PubMed] [Google Scholar]
- 11.Alberts DS, Garcia D, Mason-Liddil N. Cisplatin in advanced cancer of the cervix: An update. Semin Oncol. 1991;18:11–24. [PubMed] [Google Scholar]
- 12.Ahmad FB, Holdsworth DK. Traditional medicinal plants of Sabah, Malaysia. Part III. The Rungus people of Kudat. Int J Pharmacogn. 1995;33:262–4. [Google Scholar]
- 13.Ibrahim FH, Hamzah N. The use of medicinal plant species by the Temuan tribe of Ayer Hitam Forest, Selangor, Peninsular Malaysia. Pertanika J Trop Agric Sci. 1999;22:85–94. [Google Scholar]
- 14.Mat-Salleh K, Latiff A. Bangi: Universiti Kebangsaan Malaysia; 2002. Medicinal Plants of Malaysia (Tumbuhan Ubatan Malaysia). Pusat Pengurusan Penyelidikan. [Google Scholar]
- 15.Johnny L, Yusuf UK, Rulit N. The effects of herbal plant extracts on the growth and sporulation of Colletotrhichum gloesporioides. J Appl Biosci. 2010;34:2218–24. [Google Scholar]
- 16.Wiart C, Mogana S, Khalifah S, Mahan M, Ismail S, Buckle M, et al. Antimicrobial screening of plants used for traditional medicine in the state of Perak, Peninsular Malaysia. Fitoterapia. 2004;75:68–73. doi: 10.1016/j.fitote.2003.07.013. [DOI] [PubMed] [Google Scholar]
- 17.Muliawan SY. Effect of Dillenia suffruticosa extract on dengue virus type 2 replication. Univ Med. 2008;27:1–5. [Google Scholar]
- 18.Foo JB, Yazan LS, Tor YS, Armania N, Ismail N, Imam MU, et al. Induction of cell cycle arrest and apoptosis in caspase-3 deficient MCF-7 cells by Dillenia suffruticosa root extract via multiple signalling pathways. BMC Complement Altern Med. 2014;14:197. doi: 10.1186/1472-6882-14-197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tor YS, Yazan LS, Foo JB, Armania N, Cheah YK, Abdullah R, et al. Induction of apoptosis through oxidative stress-related pathways in MCF-7, human breast cancer cells, by ethyl acetate extract of Dillenia suffruticosa. BMC Complement Altern Med. 2014;14:55. doi: 10.1186/1472-6882-14-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Armania N, Yazan LS, Musa SN, Ismail IS, Foo JB, Wei CK, et al. Dillenia suffruticosa exhibited antioxidant and cytotoxic activity through induction of apoptosis and G (2)/M cell cycle arrest. J Ethnopharmacol. 2013;23:13320–39. doi: 10.1016/j.jep.2013.01.017. [DOI] [PubMed] [Google Scholar]
- 21.Armania N, Latifah SY, Musa SN, Ismail IS, Foo JB, Wei CK, et al. Dillenia suffruticosa exhibited antioxidant and cytotoxic activity through induction of apoptosis and G (2)/M cell cycle arrest. J Ethnopharmacol. 2013;14:525–35. doi: 10.1016/j.jep.2013.01.017. [DOI] [PubMed] [Google Scholar]
- 22.Zeiss CJ. The apoptosis-necrosis continuum: Insights from genetically altered mice. Vet Pathol. 2003;40:481–95. doi: 10.1354/vp.40-5-481. [DOI] [PubMed] [Google Scholar]
- 23.Herr I, Debatin KM. Cellular stress response and apoptosis in cancer therapy. Blood. 2001;98:2603–14. doi: 10.1182/blood.v98.9.2603. [DOI] [PubMed] [Google Scholar]
- 24.Woyengo TA, Ramprasath VR, Jones PJ. Anticancer effects of phytosterols. Eur J Clin Nutr. 2009;63:813–20. doi: 10.1038/ejcn.2009.29. [DOI] [PubMed] [Google Scholar]
- 25.Mizoroki T, Meshitsuka S, Maeda S, Murayama M, Sahara N, Takashima A. Aluminum induces tau aggregation in vitro but not in vivo. J Alzheimers Dis. 2007;11:419–27. doi: 10.3233/jad-2007-11401. [DOI] [PubMed] [Google Scholar]
- 26.Li AP, Bode C, Sakai Y. A novel in vitro system, the integrated discrete multiple organ cell culture (IdMOC) system, for the evaluation of human drug toxicity: Comparative cytotoxicity of tamoxifen towards normal human cells from five major organs and MCF-7 adenocarcinoma breast cancer cells. Chem Biol Interact. 2004;150:129–36. doi: 10.1016/j.cbi.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 27.Waterfield CJ, Delaney J, Kerai MD, Timbrell JA. Correlations between in vivo and in vitro effects of toxic compounds: Studies with hydrazine. ToxicolIn Vitro. 1997;11:217–27. doi: 10.1016/s0887-2333(97)00012-x. [DOI] [PubMed] [Google Scholar]
- 28.Spiridon K, Maria B. Boca Raton, Florida, USA: CRC Press; 2004. Plants and Cancer.Plants that Fight Cancer; pp. 15–34. [Google Scholar]
- 29.Hartwell LH, Kastan MB. Cell cycle control and cancer. Science. 1994;266:1821–8. doi: 10.1126/science.7997877. [DOI] [PubMed] [Google Scholar]
- 30.Bratton DL, Fadok VA, Richter DA, Kailey JM, Guthrie LA, Henson PM. Appearance of phosphatidylserine on apoptotic cells requires calcium-mediated nonspecific flip-flop and is enhanced by loss of the aminophospholipid translocase. J Biol Chem. 1997;272:26159–65. doi: 10.1074/jbc.272.42.26159. [DOI] [PubMed] [Google Scholar]
- 31.Fadok VA, Henson PM. Apoptosis: Giving phosphatidylserine recognition an assist – With a twist. Curr Biol. 2003;13:R655–7. doi: 10.1016/s0960-9822(03)00575-x. [DOI] [PubMed] [Google Scholar]
- 32.van Engeland M, Nieland LJ, Ramaekers FC, Schutte B, Reutelingsperger CP. Annexin V-affinity assay: A review on an apoptosis detection system based on phosphatidylserine exposure. Cytometry. 1998;31:1–9. doi: 10.1002/(sici)1097-0320(19980101)31:1<1::aid-cyto1>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 33.McNutt NS, Saenz-Santamaría C, Volkenandt M, Shea CR, Albino AP. Abnormalities of p53 protein expression in cutaneous disorders. Arch Dermatol. 1994;130:225–32. [PubMed] [Google Scholar]
- 34.Miyashita T, Reed JC. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell. 1995;80:293–9. doi: 10.1016/0092-8674(95)90412-3. [DOI] [PubMed] [Google Scholar]
- 35.Hockenbery D, Nuñez G, Milliman C, Schreiber RD, Korsmeyer SJ. Bcl-2 is an inner mitochondrial membrane protein that blocks programmed cell death. Nature. 1990;348:334–6. doi: 10.1038/348334a0. [DOI] [PubMed] [Google Scholar]
- 36.Chao DT, Korsmeyer SJ. BCL-2 family: Regulators of cell death. Annu Rev Immunol. 1998;16:395–419. doi: 10.1146/annurev.immunol.16.1.395. [DOI] [PubMed] [Google Scholar]
- 37.Strasser A, Harris AW, Jacks T, Cory S. DNA damage can induce apoptosis in proliferating lymphoid cells via p53-independent mechanisms inhibitable by Bcl-2. Cell. 1994;79:329–39. doi: 10.1016/0092-8674(94)90201-1. [DOI] [PubMed] [Google Scholar]
- 38.Miyashita T, Krajewski S, Krajewska M, Wang HG, Lin HK, Liebermann DA, et al. Tumor suppressor p53 is a regulator of bcl-2 and bax gene expression in vitro and in vivo. Oncogene. 1994;9:1799–805. [PubMed] [Google Scholar]
- 39.Wilson CM, High S. Studying endoplasmic reticulum function in vitro using siRNA. Methods Mol Biol. 2010;619:389–402. doi: 10.1007/978-1-60327-412-8_23. [DOI] [PubMed] [Google Scholar]
- 40.Ni M, Lee AS. ER chaperones in mammalian development and human diseases. FEBS Lett. 2007;581:3641–51. doi: 10.1016/j.febslet.2007.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006;7:880–5. doi: 10.1038/sj.embor.7400779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004;11:381–9. doi: 10.1038/sj.cdd.4401373. [DOI] [PubMed] [Google Scholar]
- 43.Rasheva VI, Domingos PM. Cellular responses to endoplasmic reticulum stress and apoptosis. Apoptosis. 2009;14:996–1007. doi: 10.1007/s10495-009-0341-y. [DOI] [PubMed] [Google Scholar]
- 44.Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: Disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2008;7:1013–30. doi: 10.1038/nrd2755. [DOI] [PubMed] [Google Scholar]
- 45.Wajant H. The Fas signaling pathway: More than a paradigm. Science. 2002;296:1635–6. doi: 10.1126/science.1071553. [DOI] [PubMed] [Google Scholar]
- 46.Martinez JA, Zhang Z, Svetlov SI, Hayes RL, Wang KK, Larner SF. Calpain and caspase processing of caspase-12 contribute to the ER stress-induced cell death pathway in differentiated PC12 cells. Apoptosis. 2010;15:1480–93. doi: 10.1007/s10495-010-0526-4. [DOI] [PubMed] [Google Scholar]
- 47.Tan Y, Dourdin N, Wu C, De Veyra T, Elce JS, Greer PA. Ubiquitous calpains promote caspase-12 and JNK activation during endoplasmic reticulum stress-induced apoptosis. J Biol Chem. 2006;281:16016–24. doi: 10.1074/jbc.M601299200. [DOI] [PubMed] [Google Scholar]
- 48.Xu D, Perez RE, Rezaiekhaligh MH, Bourdi M, Truog WE. Knockdown of ERp57 increases BiP/GRP78 induction and protects against hyperoxia and tunicamycin-induced apoptosis. Am J Physiol Lung Cell Mol Physiol. 2009;297:L44–51. doi: 10.1152/ajplung.90626.2008. [DOI] [PubMed] [Google Scholar]
- 49.Li Y, Camacho P. Ca2+-dependent redox modulation of SERCA 2b by ERp57. J Cell Biol. 2004;164:35–46. doi: 10.1083/jcb.200307010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chay D, Cho H, Lim BJ, Kang ES, Oh YJ, Choi SM, et al. ER-60 (PDIA3) is highly expressed in a newly established serous ovarian cancer cell line, YDOV-139. Int J Oncol. 2010;37:399–412. doi: 10.3892/ijo_00000688. [DOI] [PubMed] [Google Scholar]
- 51.Pressinotti NC, Klocker H, Schäfer G, Luu VD, Ruschhaupt M, Kuner R, et al. Differential expression of apoptotic genes PDIA3 and MAP3K5 distinguishes between low- and high-risk prostate cancer. Mol Cancer. 2009;8:130. doi: 10.1186/1476-4598-8-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Celli CM, Jaiswal AK. Role of GRP58 in mitomycin C-induced DNA cross-linking. Cancer Res. 2003;63:6016–25. [PubMed] [Google Scholar]
- 53.Leys CM, Nomura S, LaFleur BJ, Ferrone S, Kaminishi M, Montgomery E, et al. Expression and prognostic significance of prothymosin-alpha and ERp57 in human gastric cancer. Surgery. 2007;141:41–50. doi: 10.1016/j.surg.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 54.Ayshamgul H, Ma H, Ilyar S, Zhang LW, Abulizi A. Association of defective HLA-I expression with antigen processing machinery and their association with clinicopathological characteristics in Kazak patients with esophageal cancer. Chin Med J (Engl) 2011;124:341–6. [PubMed] [Google Scholar]
- 55.Mehta AM, Jordanova ES, Kenter GG, Ferrone S, Fleuren GJ. Association of antigen processing machinery and HLA class I defects with clinicopathological outcome in cervical carcinoma. Cancer Immunol Immunother. 2008;57:197–206. doi: 10.1007/s00262-007-0362-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tanaka T, Kuramitsu Y, Fujimoto M, Naito S, Oka M, Nakamura K. Downregulation of two isoforms of ubiquitin carboxyl-terminal hydrolase isozyme L1 correlates with high metastatic potentials of human SN12C renal cell carcinoma cell clones. Electrophoresis. 2008;29:2651–9. doi: 10.1002/elps.200700847. [DOI] [PubMed] [Google Scholar]
- 57.Lwin ZM, Yip GW, Chew FT, Bay BH. Downregulation of ER60 protease inhibits cellular proliferation by inducing G1/S arrest in breast cancer cells in vitro. Anat Rec (Hoboken) 2012;295:410–6. doi: 10.1002/ar.22413. [DOI] [PubMed] [Google Scholar]
- 58.Corazzari M, Lovat PE, Armstrong JL, Fimia GM, Hill DS, Birch-Machin M, et al. Targeting homeostatic mechanisms of endoplasmic reticulum stress to increase susceptibility of cancer cells to fenretinide-induced apoptosis: The role of stress proteins ERdj5 and ERp57. Br J Cancer. 2007;96:1062–71. doi: 10.1038/sj.bjc.6603672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chung H, Cho H, Perry C, Song J, Ylaya K, Lee H, et al. Downregulation of ERp57 expression is associated with poor prognosis in early-stage cervical cancer. Biomarkers. 2013;18:573–9. doi: 10.3109/1354750X.2013.827742. [DOI] [PubMed] [Google Scholar]
- 60.Liao CJ, Wu TI, Huang YH, Chang TC, Wang CS, Tsai MM, et al. Glucose-regulated protein 58 modulates cell invasiveness and serves as a prognostic marker for cervical cancer. Cancer Sci. 2011;102:2255–63. doi: 10.1111/j.1349-7006.2011.02102.x. [DOI] [PubMed] [Google Scholar]
- 61.Zong WX, Li C, Hatzivassiliou G, Lindsten T, Yu QC, Yuan J, et al. Bax and Bak can localize to the endoplasmic reticulum to initiate apoptosis. J Cell Biol. 2003;162:59–69. doi: 10.1083/jcb.200302084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, et al. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403:98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- 63.Shiraishi H, Okamoto H, Yoshimura A, Yoshida H. ER stress-induced apoptosis and caspase-12 activation occurs downstream of mitochondrial apoptosis involving Apaf-1. J Cell Sci. 2006;119:3958–66. doi: 10.1242/jcs.03160. [DOI] [PubMed] [Google Scholar]
- 64.Jimbo A, Fujita E, Kouroku Y, Ohnishi J, Inohara N, Kuida K, et al. ER stress induces caspase-8 activation, stimulating cytochrome c release and caspase-9 activation. Exp Cell Res. 2003;283:156–66. doi: 10.1016/s0014-4827(02)00033-2. [DOI] [PubMed] [Google Scholar]
- 65.Morishima N, Nakanishi K, Takenouchi H, Shibata T, Yasuhiko Y. An endoplasmic reticulum stress-specific caspase cascade in apoptosis.Cytochrome c-independent activation of caspase-9 by caspase-12. J Biol Chem. 2002;277:34287–94. doi: 10.1074/jbc.M204973200. [DOI] [PubMed] [Google Scholar]
- 66.Bedard K, Szabo E, Michalak M, Opas M. Cellular functions of endoplasmic reticulum chaperones calreticulin, calnexin, and ERp57. Int Rev Cytol. 2005;245:91–121. doi: 10.1016/S0074-7696(05)45004-4. [DOI] [PubMed] [Google Scholar]
- 67.Mosmann T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 68.Hafiza WA, Latifah SY. Potential implications of GRP58 expression and susceptibility of cervical cancer to cisplatin and thymoquinone-based therapy. Onco Targets Ther. 2014;7:1375–87. doi: 10.2147/OTT.S62928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cazanave SC, Elmi NA, Akazawa Y, Bronk SF, Mott JL, Gores GJ. CHOP and AP-1 cooperatively mediate PUMA expression during lipoapoptosis. Am J Physiol Gastrointest Liver Physiol. 2010;299:G236–43. doi: 10.1152/ajpgi.00091.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kaser A, Lee AH, Franke A, Glickman JN, Zeissig S, Tilg H, et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell. 2008;134:743–56. doi: 10.1016/j.cell.2008.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chérasse Y, Maurin AC, Chaveroux C, Jousse C, Carraro V, Parry L, et al. The p300/CBP-associated factor (PCAF) is a cofactor of ATF4 for amino acid-regulated transcription of CHOP. Nucleic Acids Res. 2007;35:5954–65. doi: 10.1093/nar/gkm642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Willis D, Li KW, Zheng JQ, Chang JH, Smit AB, Kelly T, et al. Differential transport and local translation of cytoskeletal, injury-response, and neurodegeneration protein mRNAs in axons. J Neurosci. 2005;25:778–91. doi: 10.1523/JNEUROSCI.4235-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Le Pennec S, Mirebeau-Prunier D, Boutet-Bouzamondo N, Jacques C, Guillotin D, Lauret E, et al. Nitric oxide and calcium participate in the fine regulation of mitochondrial biogenesis in follicular thyroid carcinoma cells. J Biol Chem. 2011;286:18229–39. doi: 10.1074/jbc.M110.217521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rahman HS, Rasedee A, Abdul AB, Zeenathul NA, Othman HH, Yeap SK, et al. Zerumbone-loaded nanostructured lipid carrier induces G2/M cell cycle arrest and apoptosis via mitochondrial pathway in a human lymphoblastic leukemia cell line. Int J Nanomedicine. 2014;9:527–38. doi: 10.2147/IJN.S54346. [DOI] [PMC free article] [PubMed] [Google Scholar]