

Abstract
Glucocorticoid receptor (GR) gene mutations may cause familial or sporadic generalized glucocorticoid resistance syndrome. Most of the missense forms distribute in the ligand-binding domain and impair its ligand-binding activity and formation of the activation function (AF)-2 that binds LXXLL motif-containing coactivators. We performed molecular dynamics simulations to ligand-binding domain of pathologic GR mutants to reveal their structural defects. Several calculated parameters including interaction energy for dexamethasone or the LXXLL peptide indicate that destruction of ligand-binding pocket (LBP) is a primary character. Their LBP defects are driven primarily by loss/reduction of the electrostatic interaction formed by R611 and T739 of the receptor to dexamethasone and a subsequent conformational mismatch, which deacylcortivazol resolves with its large phenylpyrazole moiety and efficiently stimulates transcriptional activity of the mutant receptors with LBP defect. Reduced affinity of the LXXLL peptide to AF-2 is caused mainly by disruption of the electrostatic bonds to the noncore leucine residues of this peptide that determine the peptide's specificity to GR, as well as by reduced noncovalent interaction against core leucines and subsequent exposure of the AF-2 surface to solvent. The results reveal molecular defects of pathologic mutant receptors and provide important insights to the actions of wild-type GR.
Glucocorticoids, steroid hormones secreted from the adrenal glands as the end products of the hypothalamic-pituitary-adrenal axis, are essential for proper functioning of virtually all organs and tissues of organisms, and play central roles in the regulation of basal and stress-related homeostasis (1, 2). These diverse actions of glucocorticoids are mediated by a single intracellular receptor molecule, the glucocorticoid receptor (GR), which functions as a hormone-activated transcription factor of glucocorticoid target genes (3, 4). The human GR gene consists of 9 exons and expresses 2 splicing variants, GRα and GRβ, from alternative use of terminal exon 9α or 9β (4, 5). GRα is the classic receptor, binding to glucocorticoids and mediating most of their known actions (5). In the absence of ligand, GRα is primarily localized in the cytoplasm, and binding of glucocorticoids induces its rapid nuclear translocation (3). GRα in the nucleus then binds glucocorticoid-response elements in the regulatory region of glucocorticoid-responsive genes and initiates transcription of downstream coding sequences (3, 4). GRα consists of 3 functionally distinct domains, the N-terminal or immunogenic domain, central DNA-binding domain (DBD), and C-terminal ligand-binding domain (LBD) (4). GRα LBD composes of 12 α-helices and 4 β-sheets and forms a ligand-binding pocket (LBP) with its helices including helix-3, helix-4, helix-11, and helix-12 (6). Upon ligand binding, LBD creates the activation function (AF)-2 transactivation domain with several helical structures, including helix-3, helix-4, and helix-12 (6). The newly formed AF-2 then attracts cofactor molecules to stimulate transcription of glucocorticoid-responsive genes in cooperating with another transactivation domain AF-1 located in N-terminal or immunogenic domain (7). Among the cofactors attracted to ligand-bound and DNA-associated GRα, p160-type histone acetyltransferase coactivators (or nuclear receptor coactivators [NCoAs]) play an essential role by binding directly to the AF-2 surface of GRα with their nuclear receptor box (NRB) and by attracting other cofactor molecules. They consist of 3 subfamilies, NCoA-1, NCoA-2, and NCoA-3, each of which also comprises several different isoforms (7). All p160-type coactivators contain 3–4 amphipathic LXXLL motifs in their NRB, which bind directly to the AF-2 surface created on GRα LBD upon ligand binding (8). It is known that each nuclear receptor including GRα preferentially uses specific LXXLL motif(s) for interaction with these coactivators to stimulate transcription (9).
Familial or sporadic glucocorticoid resistance syndrome (FGRS) or Chrousos syndrome is a rare condition characterized by generalized, partial end-organ insensitivity to glucocorticoids (10). Affected subjects have compensatory elevation of serum concentrations of cortisol and plasma ACTH concentrations (10). These subjects do not usually demonstrate overt clinical evidence of hypo- or hyper-cortisolism, whereas their excess ACTH secretion often results in increased production of adrenal steroids with androgenic and/or mineralocorticoid activity; therefore, they may present with hirsutism, infertility, hypertension, and hypokalemia (10). Molecular basis of FGRS has been ascribed to mutations in the GR gene, which impair the molecular action(s) of produced GRα protein (10). Eighteen inactivating mutations have been described previously within all subdomains of GRα, including a 4-base pair deletion at the 3′-boundary of exon 6 of the human GR gene; however, majority of reported nonsynonymous single point mutations (12 cases) are found in LBD of the receptor (10–12).
In this study, we performed computer simulations on the human GRα LBD to understand the structural ramifications of 10 reported pathologic GRα mutant receptors harboring single amino acid replacement. We found that defective LBP is a primary character of these mutant receptors. The simulations suggested novel rules for explaining the mechanisms of decreased functions shared by these pathologic GRα mutants. The results also help elucidating detailed molecular actions of wild-type GRα.
Materials and Methods
Computer-based structural analysis on the GRα LBD
Initial models of the human GRα LBD were drawn from the reported crystal structure of this domain (PDB ID, 1M2Z), which also employed as the LXXLL peptide a peptide sequence PVSPKKKENALLRYLLDKDDT that contains the third LXXLL motif of the human GR-interacting protein 1 (GRIP1), one of the p160-type coactivators or NCoAs. The structure of wild-type GRα LBD was prepared for explicit solvent simulations with periodic boundary conditions, and substitutions were made to create structures for all mutant GRα LBDs. Simulations were performed using the CHARMm force field in the NAMD software (13). 1) The interaction energy of LBP to dexamethasone and of the LXXLL peptide to AF-2; 2) the root mean square deviation (RMSD) of dexamethasone and the LXXLL peptide; 3) the degree of buried area of dexamethasone in LBP; 4) the amount of contacting surface area between the LXXLL peptide and its AF2-binding region; 5) the volume of LBP; and 6) the surface properties of LBP and AF-2 were calculated for wild-type GRα and all mutant receptors with appropriate methods as explained in Supplemental Materials and Methods. All metrics for pathologic GRα mutants were compared against those for wild-type GRα using ANOVA followed by Dunnett-corrected t tests.
Plasmids
All plasmids used in this manuscript are listed and explained in Supplemental Materials and Methods. All GRα- or GRIP1-related molecules expressed from their carrying plasmids were equally expressed in our hands (Supplemental Figure 1) or were validated previously (14).
Reporter assay
Human colon carcinoma HCT116 cells defective in endogenous GRα were transfected with indicated plasmids as previously described (15). Dexamethasone, cortisol (hydrocortisone), deacylcortivazol, or vehicle ethanol was then added to media, and cell lysates were harvested for examining the activity of firefly or Renilla luciferase.
Whole-cell dexamethasone binding assay
Whole-cell dexamethasone binding assay was performed in the HCT116 cells transfected with wild-type or mutant GRα-expressing plasmids by incubating them with [1,2,4,6,7-3H] dexamethasone (GE Healthcare Bioscience Corp), as previously described (16, 17). Scatchard analysis was performed with the GraphPad Prism version 6 (GraphPad Software) for evaluating the affinity (Kd) of GRαs to dexamethasone.
Mammalian 2-hybrid assay
HCT116 cells were transfected with the plasmid expressing GAL4 DBD-fused wild-type or mutant GRα LBD and the plasmid expressing VP16AD-GRIP1 NRB, together with pGAL4-E1B-TK-Luc and pGL4.73[hRluc/SV40], as previously described (17–19). The cells were incubated with 10−5M dexamethasone, and firefly and Renilla luciferase assays were performed. Fold binding activity was calculated by dividing the value obtained in the presence of VP16AD-GRIP1 NRB and dexamethasone with the value obtained in the absence of this protein and in the presence of dexamethasone.
Yeast 2-hybrid assay
Yeast strain EGY48 (Clontech Laboratories, Inc) was transformed with plasmids, treated with 10−6M dexamethasone, and were used for β-galactosidase activity, as previously described (19, 20). β-Galactosidase activity was normalized for cell density determined with absorbance at 600 nm.
Statistical analysis
Statistical analysis for experimental results was performed by using the Student's t test with 2-tailed value. The linear regression analysis was performed for examining correlation of selected 2 groups. All statistical analyses were performed using the GraphPad Prism version 6.
For more details, please see Supplemental Materials and Methods.
Results
Pathologic GRα mutants demonstrated various degrees of defects in binding to dexamethasone and the LXXLL peptide
In this study, we investigated 10 known pathologic GRα mutant receptors harboring single amino acid replacement in their LBDs: GRα I559N, V571A, V575G, D641V, G679S, R714Q, V729I, F737L, I747M, and L773P (Table 1) (17, 21–31). Because the ligand-binding activity and the AF-2-mediated transcriptional activity are 2 major functions of GRα LBD (4), we first calculated the energy levels required for these mutant receptors to interact with dexamethasone or the LXXLL peptide (Figure 1, A and B). Affinity of the mutant receptors to dexamethasone determined in whole-cell dexamethasone binding assays was correlated with corresponding calculated energy levels (Figure 1C and Supplemental Figure 2). To examine binding strength of their LBD to the GRIP1 NRB domain, we employed mammalian 2-hybrid assays in which we calculated fold binding activity of the wild-type GRα or mutant receptor LBD fused with GAL4 DBD to the VP16 AD-fused GRIP1 NRB in the presence of a high amount (10−5M) of dexamethasone to minimize the influence of their various affinities to this steroid (Supplemental Figure 3). We transfected grading amounts of the VP16AD-GRIP1 NRB-expressing plasmid that provided overall linear expression of this protein (Supplemental Figure 3B), and calculated slops of the fold binding activity of these receptors with the linear regression analysis. As expected, the slops of wild-type and mutant GRαs correlated with the calculated mean energy required for binding to the LXXLL peptide (Figure 1D). The fold binding activity obtained with the highest expression of VP16 AD-fused GRIP1 NRB in mammalian 2-hybrid assays, as well as the fold binding activity of radiolabeled receptors to bacterially produced glutathione S-transferase-fused-GRIP1 NRB in glutathione S-transferase-pull-down assays were also correlated with the calculated mean energy for the LXXLL peptide (data not shown). Taken together, these results confirmed that the results obtained in our computer-based simulation reflected molecular defects of pathologic GRα mutants.
Table 1.
Reported Characteristics and Calculated Energy Required for Interacting With DEX or the LXXLL Peptide in Pathologic GRα Mutants
| Pathologic GRα Mutant | Position in cDNAa | Location in Helix | Interaction Energy for DEX | Interaction Energy for LXXLL | Type | References |
|---|---|---|---|---|---|---|
| I559N | 1676T>A | 5 | c | n.s. | LBP | 1, 2 |
| V571A | 1712T>C | 5 | n.s. | n.s. | — | 3 |
| V575G | 1724T>G | 5 | n.s. | c | AF-2 | 4 |
| D641V | 1922A>T | 6–7 | c | n.s. | LBP | 5 |
| G679S | 2035G>A | 8–9 | c | b | Both | 1, 7 |
| R714Q | 2141G>A | 10 | c | n.s. | LBP | 8 |
| V729I | 2185G>A | 10 | c | n.s. | LBP | 9 |
| F737L | 2209T>G | 11 | c | c | Both | 10 |
| I747M | 2241T>G | 11–12 | c | b | Both | 11 |
| L773P | 2318T>C | 12< | c | n.s. | LBP | 12 |
DEX, dexamethasone; LXXLL, LXXLL peptide; n.s., not significant.
Adenine of the translation start site is numbered as number 1.
P < .001, compared with wild-type GRα.
P < .0001, compared with wild-type GRα.
Figure 1.
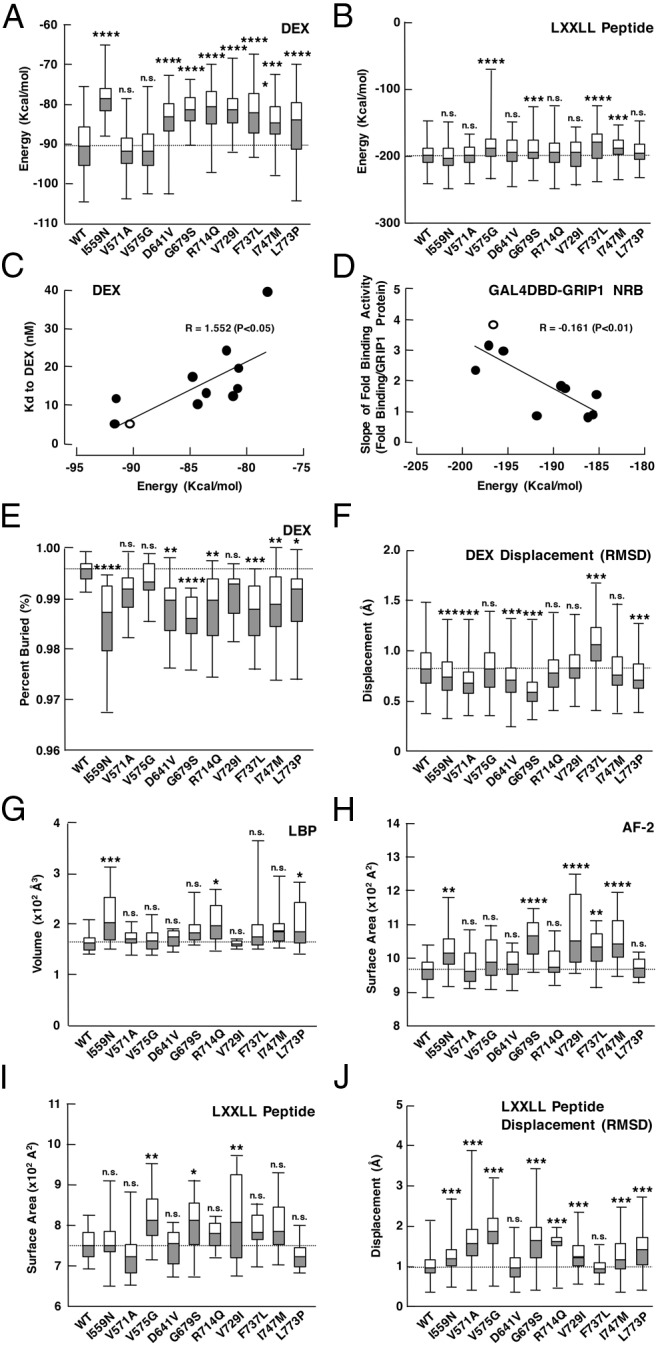
Pathologic GRα mutants demonstrate changes in the energy required for interacting with dexamethasone (DEX) or the LXXLL peptide, their average displacement over time, the percent buried area (partitioning from the surrounded solvent) of DEX, the volume of LBP, and the surface contacting area of AF-2 and the LXXLL peptide. A, Box plots of the interaction energy for DEX. Most of the mutant receptors increase the energy required for interacting with DEX compared with wild-type (WT) GRα. Mutants V571A and V575G show no statistical difference (n = 200). B, Box plots of the interaction energy for the LXXLL peptide. Mutant receptors V575G, G679S, F737L, and I747M increase the energy required for interacting with the LXXLL peptide (n = 200). C, Correlation between affinity (Kd) of the pathologic GRα mutants to DEX and their calculated energy required for interacting with DEX. Kd values of WT GRα and its pathologic mutants obtained in whole-cell DEX binding assays were compared with their calculated mean energy required for interacting with DEX observed in simulation. Open and closed circles indicate results of the WT GRα and pathologic GRα mutants, respectively. DEX titration curves for bound radioactive DEX and subsequent Scatchard plots for WT GRα and its mutant receptors are shown in Supplemental Figure 2. D, Correlation between slopes of the fold binding activity of WT GRα and its mutant receptors to GRIP1 NRB obtained in the mammalian 2-hybrid assay and their calculated energy required for interacting with the LXXLL peptide. Mammalian 2-hybrid assays employing WT GRα or mutant receptor LBD fused with GAL4 DBD and grading amounts of the GRIP1 NRB fused with VP16 AD in the presence or absence of 10−5M DEX were performed, and slopes of their fold binding activity obtained with linear regression analysis (Supplemental Figure 3A) were compared with their calculated mean energy required for interacting with the LXXLL peptide observed in simulation. Open and closed circles indicate results of the WT GRα and pathologic GRα mutants, respectively. Note that the levels of expressed VP16 AD-fused GRIP1 NRB are almost linear in the range of its transfected plasmid amounts employed in this assay (Supplemental Figure 3B). Similar expression of GAL4DBD-GRα LBDs is shown in Supplemental Figure 1B. Representative results of the mammalian 2-hybrid assay using GAL4DBD-GRα LBD (WT) are shown in Supplemental Figure 3C. E, Box plots of the percent buried area of DEX. All pathologic GRα mutants except V571V, V575G, and V729I have significantly less buried area of DEX than WT GRα (n = 20). F, Box plots of the DEX displacement shown with RMSD. Mutant receptors I559N, V571A, D641V, G679S, and V773P have significantly less distance to DEX than WT GRα, whereas F737L has increased distance (n = 20). G, Box plots of the LBP volume. Mutant receptors I559N, R714Q, and L773P have significantly larger LBP volume than WT GRα, suggesting a less snug fit to DEX (n = 20). H and I, Box plots of the surface contacting area of AF-2 and the LXXLL peptide. Mutant receptors I559N, G679S, V729I, F737L, and I747 have significantly more surface contacting area of their AF-2 to the LXXLL peptide than WT GRα (H), whereas the mutant receptors, V575G, G689S, and V729I, have more contacting surface area of the LXXLL peptide to their AF-2 (I) (n = 20). J, Box plots of the LXXLL peptide displacement shown with RMSD. Mutant receptors I559N, V571A, V575G, G679S, R714Q, V729I, I747M, and L773P have significantly more distance to the LXXLL peptide than wild-type GRα (n = 20). Box plots of each panel illustrate the medians, and upper and lower quartiles, whereas the whiskers indicate range of distribution. The median value of WT GRα in each analysis is indicated with a dotted line. *, P < .05; **, P < .01; ***, P < .001; ****, P < .001; n.s., not significant, compared with WT GRα. WT, wild-type.
Most of the pathologic GRα mutants tested demonstrated significant increase in required energies for this steroid and peptide, whereas some showed characteristic changes; All pathologic mutant receptors except GRα V571A and V575G, demonstrated elevated energy requirement for dexamethasone. GRα I559N, D641V, R714Q, V729I, and L773P had highly elevated energy levels for dexamethasone but did not show the elevation for the LXXLL peptide; thus, they are “pure” mutant receptors defective in ligand binding (LBP type: I559N, D641V, R714Q, V729I, and L773P). GRα V575G showed an opposite phenotype: elevated energy for the LXXLL peptide but not for dexamethasone, behaving as a pure AF-2-defective mutant (AF-2 type, V757G). This is consistent with a previous report, which characterized the human GRα V575M as a mutant receptor with marked decrease in transcriptional activity but with only a minimal reduction in ligand binding (32). The other mutant receptors, GRα G679S, F737L, and I747M, demonstrated elevated energy levels for both dexamethasone and the LXXLL peptide (both type: G679S, F737L, and I747M). Thus, the pathologic GRα mutants harboring 1 amino acid replacement in LBD can be categorized into 3 groups (Table 1). Because most of these mutant receptors demonstrated energy increase for dexamethasone, destruction of LBP is a primary and common character, whereas defective AF-2 is secondary.
We compared the above-indicated molecular phenotypes of pathologic GRα mutants with clinical manifestations of their carrying patients (Supplemental Table 1). Although case numbers are small, the patients harboring the mutant receptors defective in ligand-binding activity or LBP may be associated with presence of mineralocorticoid excess. Because GRα V571A was reported to be pathologic, we were surprised to see this mutant receptor demonstrated no significant changes in the interaction energy for dexamethasone or the LXXLL peptide. These results suggest that impact of V571A mutation appears to be mild and patient's glucocorticoid resistance emerged only in a special condition of carrying another mutation in the 21-hydorxylase (CPY21A2) gene, which causes significant reduction of cortisol and aldosterone but tremendous increase in androgen production (23).
We analyzed additional metrics to explore more details of defects in LBP and AF-2 of the pathologic GRα mutants. These metrics included the average displacement of dexamethasone over time (a measure of the “tightness” of binding), as well as the percent buried area of this steroid and the LBP volume (Figure 1, E–G, and Supplemental Table 2A). As expected, the mutant receptors, which demonstrated energy elevation for dexamethasone binding (I559N, D641V, G679S, R714Q, V729I, F737L, L747M, and L773P), showed significantly less percent buried area of dexamethasone, suggesting their greater interaction with solvent and lower ligand affinity. Compared with wild-type GRα, some of these mutant receptors (I559N, D641V, G679S, and L773P) were also associated with reduced dexamethasone displacement in RMSD (thus reduced tightness). R714Q had a larger LBP volume, indicating a less snug fit to dexamethasone and lower ligand affinity. In contrast, V575G, a pure AF-2-defective mutant, did not show significant alteration in these analyses. V571A showed statistically significant reduction in dexamethasone displacement (RMSD) only. These results suggest that the alteration we found in energy requirement for dexamethasone binding is caused by changes in size, and possibly, shape of LBP.
We also calculated the surface contacting area of AF-2 and the LXXLL peptide, and the average displacement of the LXXLL peptide over time, for further elucidating defects observed in the molecular interaction between AF-2 of pathologic GRα mutants and the LXXLL peptide (Figure 1, H–J, and Supplemental Table 2B). The mutant receptors, which demonstrated energy elevation for binding to the LXXLL peptide (V575G, G679S, F737L, and I747M), had significantly more contacting surface area of AF-2 and/or the LXXLL peptide than wild-type GRα, whereas their changes in displacement of the LXXLL peptide were variable. As expected, the mutant receptors that did not demonstrate energy elevation for binding to the LXXLL peptide (I559N, V571A, D641V, R714Q, V729I, and L773P) showed minimal changes in these analyses. Thus, defects of pathologic GRα mutants in their binding to the LXXLL peptide are associated with (or based on) the increase in binding surface of their AF-2 and/or the LXXLL peptide. It is interesting to note that distribution of the significantly altered surface contacting area (AF-2 and the LXXLL peptide) of the indicated mutant receptors was much larger than that of wild-type GRα (Figure 1, I and J), suggesting a variety of configurations and interactions, and therefore, less binding affinity. V571A demonstrated reduced displacement of both dexamethasone and the LXXLL peptide; thus, this mutant receptor has little but obvious molecular changes.
Gross visual analysis on LBP and AF-2 of pathologic GRα mutants
To gain further structural insights into the LBP and the AF-2 surface of pathologic GRα mutants, we created Cα-trace 3-dimensional (3D) models of wild-type and all mutant LBDs bound with dexamethasone and the LXXLL peptide (Figure 2 and Supplemental Movie 1). These images are mostly overlap with each other, indicating that topology of their peptide backbones is highly preserved. This result also suggests that alteration in property and/or positioning of the side chain of replaced amino acids is rather crucial for developing molecular defects. As expected, the termini of LBD and the bound LXXLL peptide moved most.
Figure 2.
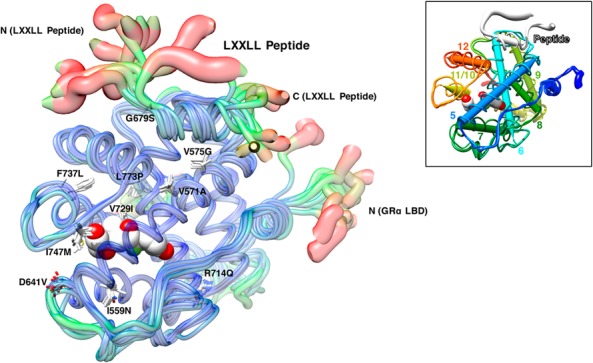
Averaged trajectories reveal slight changes in overall conformation of pathologic GRα mutant LBDs and their interaction with the LXXLL peptide. Thickness and color of the overlaid Cα-traces of mutant receptor LBDs indicate the areas of least (thin and blue) to most (thick and red) motion over the course of simulation. Locations and side chains of the mutated amino acids are indicated, whereas dexamethasone (shown with the white and red spheres of space-filling model) is located inside LBP. In the right upper inset, helix-5 to helix-12 of the wild-type GRα LBD are shown with colored dipoles. The movie rotating this image is available as Supplemental Movie 1.
Many of the mutant receptors harboring the amino acid replacement located relatively close to dexamethasone (or LBP) (eg, I559N, D641V, V729I, I747M, and L737P) and to the LXXLL peptide (or AF-2) (eg, V575G and G679S) had defective LBP and AF-2, respectively (Supplemental Movie 1), supporting the idea that proximity of the mutations to LBP or AF-2 may in some degree help predicting their impact on these structures. On the other hand, R714Q and L773P, the mutant receptors with defective LBP, were located far distant to this module, suggesting strong allosteric effects of their amino acid replacements consistent with our previous report (17). Although V571A was positioned relatively close to both LBP and AF-2, it did not affect their functions much, possibly due to little structural impact of its amino acid replacement (valine to alanine).
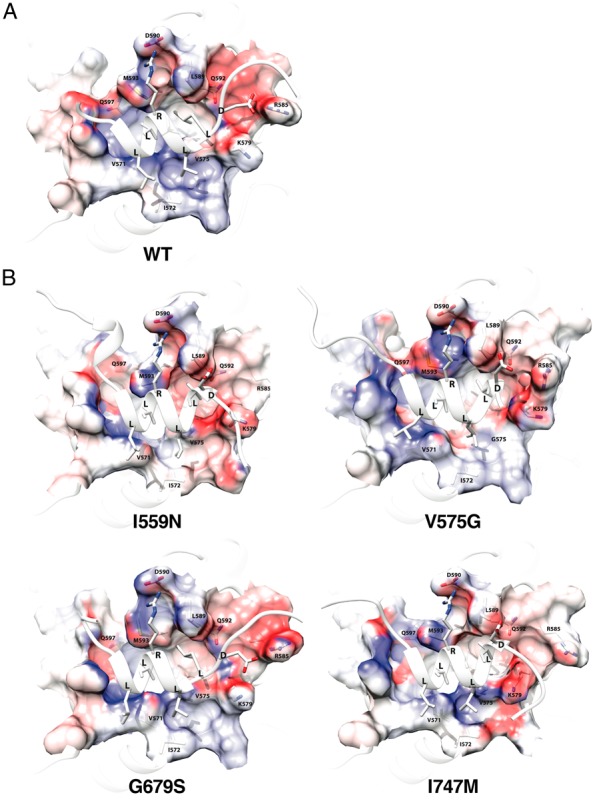
To evaluate gross impact of the mutations on LBP further, we created 3D LBP models with influence of surface electrostatic potential for 4 representative pathologic GRα mutants (I559N, LBP type; V575G, AF-2 type; G679S and I747M, Both type) along with wild-type GRα (Figure 3). LBP of wild-type GRα (Figure 3A) was a closed volume with little positive or negative electrostatic potential (shown with blue and red, respectively). LBPs of pathologic GRα mutants (Figure 3B) had in many instances openings due to side chain rearrangements, notably at R611, and the electrostatic potential of the mutant receptors with defective LBP (I559N, G679S, and I747M) became more negative (red), suggesting a change in the polar character of their LBPs, and thus, a likely decrease in ligand affinity, as dexamethasone is also negatively charged in its large surface. As expected, electrostatic potential of V575G LBP (AF-2 type) did not show obvious alteration compared with wild-type GRα.
Figure 3.

Four representative pathologic GRα mutants change conformational and chemical properties of their LBP. LBP of the wild-type GRα (A) and 4 representative pathologic GRα mutants (I559N, ligand-binding defective; V575G, AF-2 defective; G679S and I747M, both defective) (B) are shown. Positive and negative electrostatic potential are indicated with blue and red, respectively. The key residues of the receptors making important molecular interactions to dexamethasone are incorporated. The structures shown and their calculated biochemical properties are those of the averaged trajectories. DEX, dexamethasone; WT, wild type.
Similar analysis was applied to AF-2 of wild-type GRα and a same set of the pathologic GRα mutants (Figure 4). In wild-type GRα, the AF-2 groove holding 3 leucines of the core LXXLL motif was neutral in electrostatic potential, whereas its periphery had high electronic charge (Figure 4A). AF-2 surface of the mutant receptors including V575G (AF-2 type) displayed minimal changes in electrostatic potential compared with LBP (Figure 4B), indicating that alteration in surface electrostatic potential does not seem to contribute significantly to the development of their AF-2 defects.
Figure 4.
Four representative pathologic GRα mutants change conformational and chemical properties of their AF-2 surface. The AF-2 surface of wild-type GRα (A) and 4 representative pathologic GRα mutants (I559N, ligand-binding defective; V575G, AF-2 defective; G679S and I747M, both defective) (B) are shown. Positive and negative electrostatic potential are indicated with blue and red, respectively. Key residues of the receptors and the LXXLL peptide that make important molecular interactions are incorporated. The structures shown and their calculated biochemical properties are those of the averaged trajectories. WT, wild type.
Loss of electrostatic interaction between LBP and dexamethasone explains reduced affinity of pathologic GRα mutants to this steroid
We examined modes and strength of the molecular interaction between LBP and dexamethasone. Figure 5A demonstrates simplified models of such interactions in wild-type and pathologic GRα mutants where only the interactions dramatically altered are shown (full interaction models are shown in Supplemental Figure 4). Wild-type GRα formed numerous interactions supported by electrostatic (hydrogen bond and salt bridge) and noncovalent (van der Waals) bonds formed between dexamethasone and side chains of the amino acids located mainly in helix-2, helix-4, and helix-9. Key interactions between wild-type GRα and dexamethasone are also shown in 3D models and in a structural diagram (Figure 5B). Mutant receptors lost some or many of these interactions and/or developed new interactions not recognized in wild-type GRα. The observed changes were rather complicated and quite different between one mutant receptor to the other, but some tendency emerged; noncovalent bonds were largely preserved throughout the pathologic mutant receptors although some demonstrated changes in their strength, whereas their electrostatic interactions showed relatively large change. Indeed, the electrostatic bond is known to be a much stronger molecular interaction than the noncovalent bond (33). In wild-type GRα, N564, Q570, R611, and T739 formed electrostatic bonds with dexamethasone (Figure 5B, right bottom panel): N564 and T739 interacted with one end of dexamethasone, the former created bonds to the hydroxyl group of carbon-11 and carbon-21 of dexamethasone, whereas the latter to the carbonyl oxygen of carbon-20. Q570 and R611 on the other hand interacted with the other end of dexamethasone, both forming a bond to the carbonyl oxygen at carbon-3 of dexamethasone. Thus, LBP of wild-type GRα holds dexamethasone from its both ends with multiple electrostatic bonds. Among these electrostatic interactions observed in wild-type GRα, huge reduction/disappearance existed at the one formed by R611 and, to less extent, by T739 of the mutant receptors, whereas those of N564 and Q570 showed minimal changes (Figure 5A). Based on the changes in the electrostatic interaction formed by R611 and T739, pathologic mutant receptors can be categorized into 3 groups; group A (V571A and V575G) have increased R611 bond and reduced T739 bond, group B (G679S) has no R611 bond but increased T739 bond, and group C (I559N, D641V, R714Q, V729I, F737L, I747M, and L773P) shows reduced or no R611 and T739 bond. Thus, alteration in the electrostatic bond formed by R611, and in some part by T739, provides a potential mechanism of differential binding and reduction in the affinity of mutant receptors to this steroid. In superimposed LBP images of wild-type and all mutant receptors (Figure 5C), the side chain of R611 most significantly shifted among the other residues, whereas that of T739 showed some deviation similar to those of N564 and Q570. In 3D LBP images of 3 representative mutant receptors with defective LBP (I559N, G679S, and I747M) (Figure 3), they demonstrated significant shift of R611 and subsequent opening of LBP at this portion. Alteration in the electrostatic bond by T739 is likely to contribute to the changes in surface electrostatic potential of their LBPs. Thus, loss of the electrostatic bond by R611 and, to some extent, by T739 through allosteric effects of the amino acid replacements of mutant receptors appears to provide structural background for their reduced affinity to dexamethasone.
Figure 5.

Alteration of the electrostatic bond formed by R611 and T739 of pathologic GRα mutants may largely explain the reduced affinity of many pathologic GRα mutants to this steroid. A, Diagram of individual molecular interactions between dexamethasone and wild-type (WT) GRα or pathologic GRα mutants. Purple and orange lines indicate electrostatic and noncovalent bonds, respectively. Line thickness indicates frequency of the observed interaction (thus strength of interaction), whereas dotted lines indicate no statistical change in the frequency of interaction compared with WT GRα. Only the interactions dramatically altered in the mutant receptors are shown. Arrows are for attracting readers' attention. Based on the strength of interaction created by R611 and T739 of receptors, pathologic GRα mutants can be categorized into 3 groups (groups A, B, and C). Full interaction diagrams are shown in Supplemental Figure 4. B, 3D or schematic models (residues only) of the molecular interaction between WT GRα LBP and dexamethasone. Top left and top right panels demonstrate 3D interaction images of dexamethasone and the key residues of WT GRα that create important contacts to this steroid. Bottom right panel shows schematic molecular interaction between WT GRα and dexamethasone. Purple and orange arrows indicate electrostatic and noncovalent bonds, respectively. C, Superimposed 3D images of dexamethasone and the key residues of all pathologic GRα mutants. Panels demonstrate superimposed 3D interaction images of dexamethasone and the key residues of all pathologic GRα mutants. Among the key amino acids of pathologic mutants participating in interaction with dexamethasone, R611 is largely deviated in these mutant receptors, which underlies reduced/disappeared electrostatic interaction between this residue and carbonyl oxygen at carbon-3 of dexamethasone. Q570 and N564 are omitted from these panels. DEX, dexamethasone; WT, wild type.
Deacylcortivazol is a more potent agonist for pathologic GRα mutants with LBP defect than dexamethasone by filling the space created by the deviated side chain of R611
Deacylcortivazol, a pyrazilosteroid, stimulates GRα transcriptional activity acting as a ligand by binding to its LBP (34, 35). This steroid has a large phenylpyrazole group at its steroid A-ring where dexamethasone and other glucocorticoids have carbonyl oxygen (at carbon-3) (Figure 6A) (34). Deacylcortivazol has much higher affinity to GRα than dexamethasone through creation of additional contacts to GRα with its phenylpyrazole group (35). This reported evidence suggests that the portion of GRα LBP around R611 has high molecular flexibility, whereas our simulation results indicate that it is highly susceptible to the allosteric changes caused by pathologic GRα mutations. Thus, we hypothesized that deacylcortivazol could act as a potent (and thus potentially beneficial) ligand for some pathologic GRα mutants by filling this space with its bulky phenylpyrazole group. Indeed in the reported crystal structure of GRα LBD associated with deacylcortivazol (PBD ID, 3BQD) (35), this compound filled LBP by shifting the side chain of R611 to the direction similar to its deviation observed in pathologic mutant receptors (Figure 6B).
Figure 6.

Deacylcortivazol (DAC) is a more potent agonist for the pathologic GRα mutants with defective LBP than dexamethasone (DEX) by filling the space created by deviated R611 with its bulky phenylpyrazole structure. A, Molecular structure model of DAC. DAC has a large phenylpyrazole group at its steroid A-ring. B, Superimposed 3D LBP models of wild-type (WT) GRα bound with DEX or DAC. 3D image of WT GRα LBP bound with DEX or DAC are shown (white, the reported crystallographic image with DEX [PDB ID, 1M2Z]; green, our simulation image associated with DEX; and orange, the reported crystallographic image with DAC [PDB ID, 3BQD]). Note that the former 2 images highly overlap with each other. Cα anchor position of R611 does not change between the green and the orange image, while its side chain significantly shifts to the similar direction as those observed in pathologic GRα mutants (shown with thin gray lines, obtained with the simulation for the mutant LBDs associated with DEX), forming a space filled with a large phenylpyrazole group of this compound. Electrostatic interactions of DEX or DAC to GRα residues are shown with purple broken lines. C, DEX, cortisol, and DAC differentially stimulate the transcriptional activity of GRα V729I, D641V, and V575G. HCT116 cells were transfected with WT or indicated mutant GRα-expressing plasmid together with pOLDO-MMTV-Luc and pGL4.73[hRluc/SV40] in the presence of 0M, 10−10M, 10−8M, or 10−6M DEX (left panel), cortisol (middle panel), or DAC (right panel). Bars represent mean ± SE values of firefly luciferase activity normalized for Renilla luciferase activity. WT, wild type. *, P < .05; **, P < .01; n.s., not significant, compared with the values of WT GRα treated with the same concentrations of compound (n = 3). D, DEX and DAC differentially stimulates the transcriptional activity of pathologic GRα mutants at its 2 concentrations. HCT116 cells were transfected with WT or indicated mutant GRα-expressing plasmid together with pOLDO-MMTV-Luc and pGL4.73[hRluc/SV40] in the presence of 0M, 10−10M, or 10−6M DEX (top panel) or DAC (bottom panel). Bars represent mean ± SE values of firefly luciferase activity normalized for Renilla luciferase activity. WT, wild type. *, P < .05; **, P < .01; n.s., not significant, compared with the values of WT GRα treated with the same concentrations of compound (n = 3). E, DAC improves the transcriptional activity of pathologic GRα mutants in contrast to DEX. Pathologic GRα mutants and WT GRα are categorized into 2 groups, based on their property of the electrostatic bond at R611: no or weak electrostatic bond (inactive) (<30% of the WT GRα): I559N, D641V, G679S, V729I, F737L, and I747M; and active bond (active) (>90% of the WT GRα): V571A, V575G, and WT GRα. Their relative transcriptional activity against WT GRα at 10−10M or 10−6M DEX (left panel) or DAC (right panel) was then compared. Bars represent mean ± SE values of fold transcriptional activity of GRαs (firefly luciferase activity normalized for Renilla luciferase activity) against that of the WT GRα. WT, wild type. *, P < .05; n.s., not significant, compared with the conditions indicated (inactive group, n = 6 and active group, n = 3).
To address this hypothesis, we examined reporter assay-based titration profiles of dexamethasone, cortisol, and deoxycortivazol for 3 representative pathologic GRα mutants characterized, respectively, as pure LBP-type (V729I and D641V) and pure AF-2-type (V575G) using GRα-defective HCT116 cells and a mouse mammary tumor virus (MMTV) glucocorticoid-response element-containing glucocorticoid-responsive promoter construct (Figure 6C). We chose V729I and D641V as pure LBP-type mutants, because V729I has no electrostatic bond of R611, whereas D641V maintains it weakly (<30% of wild-type GRα). V575G preserves this interaction. All these mutant receptors demonstrated significantly reduced transcriptional activity compared with wild-type GRα in the presence of dexamethasone or cortisol. Wild-type GRα and these mutant receptors showed right and downward shift of their titration curves for cortisol compared with dexamethasone (Figure 6C, left and middle panel). The titration curve of deacylcortivazol for wild-type GRα shifted left and upward compared with dexamethasone and cortisol, consistent with the previous report (35). Interestingly, V729I and D641V, which are defective in their LBP, demonstrated the similar transcriptional activity to wild-type GRα at 10−8M and 10−6M deacylcortivazol, whereas these receptors showed reduced transcriptional activity at 10−10M of this compound compared with wild-type GRα (Figure 6C, right panel). In contrast to these mutant receptors, V575G, a mutant receptor defective only in AF-2, demonstrated less transcriptional activity throughout the titration of deacylcortivazol, whereas its activity at 10−10M deacylcortivazol was higher than those of V729I and D641V. We therefore hypothesized that deacylcortivazol can stimulate the transcriptional activity of the mutant receptors defective in LBP at its higher concentrations to the degree equivalent to its effect on wild-type GRα, whereas this compound cannot activate strongly the mutant receptors defective in AF-2. Indeed, the same reporter assay employing all pathologic GRα mutants in the presence of these concentrations of deacylcortivazol revealed that pure LBP-type mutant receptors, such as D641V, R714Q, V729I, and L773P, demonstrated the transcriptional activity equivalent to wild-type receptor at 10−6M deacylcortivazol. However, rest of the mutant receptors defective in AF-2 or both LBP and AF-2 demonstrated blunted transcriptional activity (Figure 6D, bottom panel). At 10−10M deacylcortivazol, all mutant receptors demonstrated weaker transcriptional activity compared with wild-type GRα. Only V571A and V575G, which harbor intact LBP activated the transcription relatively strongly compared with the other mutant receptors. As a control, we performed the same experiment using dexamethasone, and found that all pathologic GRα mutants demonstrated reduced transcriptional activity both at 10−6M and at 10−10M dexamethasone (Figure 6D, top panel). These results suggest that deacylcortivazol at its relatively high concentrations can efficiently stimulate the transcriptional activity of LBP-type mutant receptors possibly by keeping high affinity to these mutant receptors in contrast to dexamethasone.
To further examine the contribution of R611 to dexamethasone- or deacylcortivazol-induced transcriptional activity of pathologic GRα mutants, we compared strength of the electrostatic bond created between R611 and carbonyl oxygen at carbon-3 of dexamethasone (thus, it acts as an indicator for molecular shift of the R611 side chain) with the transcriptional activity of pathologic GRα mutants in the presence of 10−10M or 10−6M dexamethasone or deacylcortivazol (Figure 6E). We divided wild-type GRα and pathologic GRα mutants into 2 groups based on the intensity of the electrostatic bond created by R611. One group with “no or weak” bond (inactive group) (<30% of the wild-type GRα): I559N, D641V, G679S, V729I, F737L, and I747M; and the other group with “active” bond (active group) (>90% of the wild-type GRα): V571A, V575G, and wild-type GRα. In this examination, the effect of dexamethasone was blunted in “inactive” group compared with active group at both concentrations. Deacylcortivazol efficiently stimulated the transcriptional activity of inactive group to the similar degree as active group at 10−6M of its concentration, although it still demonstrated a difficulty to stimulate the transcriptional activity of the former group at 10−10M. These results suggest that deacylcortivazol, in contrast to dexamethasone, can stimulate the transcriptional activity of the pathologic GRα mutants harboring significant shift of the R611 side chain at its higher concentrations by filling the space with its large phenylpyrazole group. Deacylcortivazol, however, has a difficulty to stimulate their transcriptional activity at its lower concentrations, possibly due to its improved, but yet reduced affinity to these receptors compared with wild-type GRα. Taken together, these results further indicate that deacylcortivazol at its higher concentrations may be beneficial for the patients expressing the mutant receptors with LBP defect.
Defects in electrostatic interaction against nonleucine amino acids of the LXXLL peptide as well as reduced noncovalent interaction to the core leucine motif explain defective AF-2 activity of pathologic GRα mutants
We next explored details of the molecular defects observed in AF-2 of pathologic GRα mutants. We again examined modes and strength of the interaction between their AF-2 and the LXXLL peptide. Figure 7A demonstrates simplified models of such interactions in wild-type GRα and mutant receptors where only the interactions dramatically changed in the mutant receptors are shown. The models demonstrating all detected interactions are shown in Supplemental Figure 5. For this analysis, we employed the third (C-terminal) LXXLL motif sequence (NALLRYLLDKD) of the human GRIP1, as GRα is known to use this motif preferentially for its binding to and transcriptional activation with GRIP1 (9). Wild-type GRα interacted with the LXXLL peptide through its multiple residues: the residues, such as L589, V575, M592, L596, and M752, formed noncovalent bonds to core leucines of the LXXLL motif, whereas the residues, such as R585, D590, Q597, and N759, created electrostatic bonds against noncore leucine amino acids located in and around the motif, such as R746, D750, and D752 (Supplemental Figure 5). All mutant receptors showed significant alteration in these interactions, which are more complicated than those observed in the interaction between their LBP and dexamethasone, but some patterns again emerged: electrostatic bonds to noncore residues of the LXXLL motif showed relatively large changes throughout the mutant receptors, whereas noncovalent bonds against the core LXXLL motif became weaker but were more or less preserved. Among the electrostatic bonds, the greatest variability existed at the interaction formed between R585 of the receptor and D750 of the LXXLL peptide, and all mutant receptors can be categorized into 3 groups by the changes in these interactions. Some mutant receptors (group II: I559N, D641V, V729I, F737L, and L737P; and group III: V571A and I747M) lost this bond, with marked increase (group II) or loss (group III) of the electrostatic bond between their K579 and D752 of the LXXLL peptide. Group I receptors (V575G, G679S, and R714Q) preserved a bond between R585 (receptor) and D750 (the LXXLL peptide). Although relatively minor, some changes were observed in the noncovalent bonds formed between L589, M593, and V575 of the mutant receptors and core leucines (especially the first and third leucines) of the LXXLL peptide. These results suggest that reduction or disappearance of the electrostatic bond formed between R585 (mutant receptors) and D750 (noncore residue of LXXLL peptide) and some reciprocal changes in the electrostatic interaction between K579 (mutant receptors) and D752 (noncore residue of LXXLL peptide) may largely contribute to the defective molecular interaction between the AF-2 surface of pathologic GRα mutants and the LXXLL peptide. Reduced but yet preserved noncovalent bonds against the core LXXLL motif help maintaining their interaction, and thus, underlies the partially defective transactivation activity of pathologic mutant receptors (10).
Figure 7.

Loss/reduction of the electrostatic bond between R585 of pathologic GRα mutants and noncore LXXLL residues (especially D750) of the LXXLL peptide contributes to the reduced AF-2 activity of these mutant receptors. A, Diagram of individual molecular interactions between the LXXLL peptide and wild-type (WT) GRα or pathologic GRα mutants. Panels are created by following the same rules as that employed in Figure 5A. For model demonstration, tyrosine (Y) located 1 amino acid N-terminally to the third core leucine is omitted, as its side chain protrudes into free space opposite to the AF-2 surface and it does not make any interactions with AF-2. All mutant receptors can be categorized into 3 groups based on the 2 interactions indicated with brackets (groups I, II, and III). Full interaction diagrams are shown in Supplemental Figure 5. B, 3D interaction images of WT GRα AF-2 and the LXXLL peptide. The AF-2 surface of WT GRα has 3 large pockets into which core leucines (L745, L748, and L749) of the LXXLL peptide deeply bury themselves. There are additional intermolecular contacts that are important for peptide binding, including the electrostatic bonds created between 1) R746 (LXXLL peptide) and D590 (receptor), 2) D750 (LXXLL peptide) and R585 (receptor), and 3) D752 (LXXLL peptide) and K579 (receptor). C, Deviation of the receptor residues forming electrostatic bonds against noncore LXXLL residues. 3D image indicates molecular interaction between the LXXLL peptide (peptide) and key residues of WT GRα. The LXXLL peptide forms important electrostatic bonds with its noncore leucine residues (R746, D750, and D752) against the receptor residues (D590, R585, and K579, respectively). Pathologic GRα mutants demonstrated significant shift of the side chains of some of these receptor residues, among which the side chain of R585 demonstrated the most significant deviation. Molecular interaction and side chain shift of groups I, II, and III receptors are shown in right insets. Top left inset contains the minimally deviated side chains of D590 (receptor) and their interacting partner R746 (LXXLL peptide). D, The third (C-terminal) LXXLL motif of GRIP1 NRB contributes most significantly to the enhancement of GRα transcriptional activity by this coactivator. HCT116 cells were transfected with indicated GRIP1-expressing plasmids together with WT GRα-expressing plasmid, pMMTV-Luc and pGL4.73[hRluc/SV40], and were incubated in the presence or absence of 10−6M dexamethasone (DEX). Bars represent mean ± SE values of firefly luciferase activity normalized for Renilla luciferase activity. L Mut, GRIPI mutant defective in indicated LXXLL motif(s). **, P < .01, compared with the condition with WT GRIP1 and DEX (n = 3). E, R746 and D750, but not D752, of GRIP1 contributes to the enhancement of GRα transcriptional activity by this coactivator. HCT116 cells were transfected with indicated GRIP1-expressing plasmids together with WT GRα-expressing plasmid, pMMTV-Luc and pGL4.73[hRluc/SV40], and were incubated in the presence or absence of 10−6M DEX. Bars represent mean ± SE values of firefly luciferase activity normalized for Renilla luciferase activity. **, P < .01; n.s., not significant, compared with the condition with WT GRIP1 and DEX (n = 3). F, R746, D750, and D752 of GRIP1 contribute to the binding of GRIP1 NRB to WT GRα LBD in a yeast 2-hybrid assay. EGY48 yeast cells were transformed with p8Op-LacZ, pLexA-GRα (480–777) and indicated GRIP1 NRB-expressing pB42AD-derived plasmids. Bars represent mean ± SE values of β-galactosidase activity corrected for cell density. *, P < .05; **, P < .01, compared with the condition with WT GRIP1 NRB and DEX (n = 3). G, Damaged electrostatic bond of some pathologic GRα mutants to GRIP1 D750 underlies the defective enhancement of their transcriptional activity by GRIP1. HCT116 cells were transfected with WT GRIP1- or GRIP1 D750A-expressing plasmids together with WT GRα- or its mutant receptor-expressing plasmid, pMMTV-Luc and pGL4.73[hRluc/SV40], and were incubated in the presence or absence of 10−6M DEX. Bars represent mean ± SE values of firefly luciferase activity normalized for Renilla luciferase activity. Presence or absence of the electrostatic bond between D750 (LXXLL) and R585 (receptor) are demonstrated under x-axis. *, P < .05; **, P < .01; n.s., not significant, compared with the condition in the presence of WT GRIP1 and DEX, or the 2 conditions indicated (n = 3).
To visualize these changes observed in the above-indicated simplified interaction models, we again created the 3D simulation images for the wild-type GRα and mutant receptors. In wild-type GRα, the LXXLL peptide was buried in the groove of AF-2 to which core leucines of the LXXLL peptide fitted tightly (Figure 7B). In addition to this major contact, AF-2 of the wild-type GRα had small indentations for the side chain of R746, D750, and D752, which positioned very close to the side chain of their interaction partners D590, R585, and K579 of the receptor. In superimposed images of the local interaction between AF-2 and the LXXLL peptide, the side chain of R585 of mutant receptors shifted most, whereas that of D590 (receptors) showed relatively minor changes (Figure 7C, insets). The side chain of K579 (receptor) also shifted dramatically in coordinating with the changes in R585 mainly due to drift of the LXXLL peptide, consistent with the results of the changes in their electrostatic interactions. In individual 3D images of AF-2, 3 pathologic GRα mutants (V575G, G679S, and I747M) defective in AF-2 activity had a wider AF-2 groove, losing tight contact to core leucine residues of the LXXLL peptide, and thus, allowing water molecules to enter into the gap created between them (Figure 4B): this change was coincident with the alteration observed in noncovalent interactions formed against core leucine residues by these mutant receptors (Figure 7A and Supplemental Figure 5). R585 of the mutant receptors that formed an electrostatic bond against D750 of the LXXLL peptide in the wild-type receptor deviated dramatically in the mutant receptors G679S and I747M, losing close contact to D750 of the LXXLL peptide. In contrast, position of D590 was preserved in the mutant receptors, maintaining its close proximity to R746 of the LXXLL peptide (Figure 4B), again consistent with changes in the electrostatic interaction formed between these amino acids (Figure 7A and Supplemental Figure 5) and those observed in the superimposed images of their interaction (Figure 7C). We therefore concluded that reduction of the AF-2 activity observed in pathologic GRα mutants is largely caused by molecular deviation of their R585 and K579 against D750 and D752 of the LXXLL peptide, which is coincident with the disappeared/reduced electrostatic interaction between them, as well as loose contact of the LXXLL peptide to the AF-2 groove caused by reduced noncovalent interaction against core leucines of the LXXLL motif.
Although V575G is a pure AF-2 type mutant receptor, it relatively maintained electrostatic interaction formed by its Q597, D590, and R585 (Figure 7A and Supplemental Figure 5) but still demonstrated defective AF-2 and reduction of binding energy to the LXXLL peptide (Table 1). In this exceptional mutant receptor, a major defect resided in replacement of valine at position 575 by glycine: V575 is a key amino acid forming noncovalent bonds against the second and the third core leucine residues (Supplemental Figure 5); thus, replacement of this amino acid with glycine directly damaged the AF-2 activity by reducing these interactions, and hence, by changing structure of the AF-2 groove consistent with our previous report (31), in contrast to the other mutant receptors whose amino acid replacements allosterically damage their AF-2.
D750 in the third LXXLL motif of GRIP1 significantly contributes to binding and transcriptional enhancement to wild-type GRα, and defective interaction of receptor R585 to this peptide residue participates in the reduced AF-2 activity of pathologic GRα mutants
In our structural simulation, we found that AF-2 of the wild-type GRα LBD created electrostatic bonds against R746, D750, and D752 of the LXXLL motif. We therefore evaluated importance of R746, D750, and D752 of the LXXLL motif in GRIP1-mediated enhancement of wild-type GRα transcriptional activity and physical binding between these 2 molecules (Figure 7, D–F). We first examined contribution of the 3 LXXLL motifs of GRIP1 to GRα-induced transcriptional activity by using a transient transfection-based reporter assay employing the glucocorticoid-responsive MMTV promoter and HCT116 cells. Wild-type GRIP1 strongly enhanced GRα-induced transcriptional activity in a dexamethasone-dependent fashion. Although all GRIP1 mutants defective in the LXXLL motif(s) showed less enhancing effects, loss of the third (C-terminal) LXXLL motif caused the most significant reduction (Figure 7D), indicating that GRα and GRIP1 preferentially uses the latter's third LXXLL motif. We then examined importance of R746, D750, and D752 of the third GRIP1 LXXLL motif in wild-type GRα-induced transcriptional activity. Replacement of arginine at 746 (R746A) or asparatic acid at 750 (D750A) with alanine significantly reduced GRIP1-induced enhancement of GRα transcriptional activity. Impact of the D750A replacement appears to be stronger than the R746A replacement (Figure 7E). In contrast, D752A replacement did not influence enhancement of GRα transcriptional activity by GRIP1, consistent with the finding that D752 creates a weak electrostatic bond to histidine 585 of the wild-type GRα (Supplemental Figure 5). R746- or D750-defective GRIP1 NRB mutants demonstrated significantly less binding activity to GRα LBD than the wild-type NRB in a yeast 2-hybrid assay, whereas impact of D752A replacement was minor, as expected (Figure 7F). Taken together, these results indicate that R746 and D750 of GRIP1 located outside of the core LXXLL motif contribute to GRIP1-mediated transcriptional enhancement of GRα transcriptional activity as well as to the binding of this peptide to GRα AF-2. R746 and D750 are specific to the third LXXLL motif among the 3 motifs of the human GRIP1, and are preserved in 3 human NCoA subtypes and in mouse NCoA2 (GRIP1) (Supplemental Table 3). Although D752 is also preserved among these NCoA molecules, we found its weak contribution to the binding to the wild-type GRα LBD, but not to its transcriptional activity in our assay system. It appears that R746 and D750, the nonleucine amino acids specific to the third LXXLL motif of NCoAs, explain preferential use of this LXXLL motif by GRα, contributing strongly to their physical interaction.
Because the electrostatic bond created between R585 (receptor) and D750 (LXXLL) is frequently damaged in pathologic GRα mutants, we examined the effect of the GRIP1 D750A mutant on the transcriptional activity of all pathologic GRα mutants and wild-type GRα in a reporter assay employing GRα-deficient HCT116 cells to address the contribution of this bond defect to the transcriptional activity of mutant receptors (Figure 7G). As expected, all pathologic GRα mutants demonstrated reduced transcriptional activity in the presence of wild-type GRIP1 compared with wild-type GRα, and GRIP1 D750A was less active on wild-type GRα than its intact form. Interestingly, the mutant receptors, which lost or reduced their electrostatic bond against D750, such as V571A, D641V, V729I, and I747M, responded to GRIP1 D750A to the similar degree as wild-type GRIP1, whereas those preserving the bond, such as V575G, G679S, and R714Q, demonstrated reduced response to this mutant GRIP1. Although F737L and L773P do not create an electrostatic bond to D750A, they still demonstrated blunted response to GRIP1 D750A, possibly due to the indirect influence of the D750A mutation to their reciprocally emerged strong electrostatic bond against D752. Taken together, these results suggest that loss of reduced electrostatic bonds created by nonleucine resides in the third LXXLL motif of GRIP1 significantly contributes to the transcriptional defects observed in some pathologic GRα mutants.
Discussion
In this study, we examined by using computer-based simulation the structural bases for the functional defects observed in the pathologic GRα mutants harboring 1 amino acid replacement at LBD, focusing on molecular defects in their LBP and AF-2. We found that structural alteration in these modules was rather mild with minimal changes in their peptide backbones. Pathologic GRα mutants can be categorized into 3 groups, those defective in 1) LBP (I559N, D641V, R714Q, V729I, and L773P), 2) AF-2 (V575G), and 3) both (G679S, F737L, and I747M), and loss of ligand-binding activity (or defective LBP) of mutant receptors appears to be a primary cause of glucocorticoid resistance observed in FGRS. Through the analysis on molecular interaction of these receptors with dexamethasone and the LXXLL peptide, defective electrostatic interaction formed by R611, and to lesser extent, by T739 well explained the reduced LBP activity of pathologic GRα mutants. We also found that loss/reduction of the electrostatic interaction formed between R585 (and K579) of the mutant receptors and D750 (and D752) of the LXXLL peptide as well as reduced noncovalent interaction to the core LXXLL motif were responsible for their defective AF-2.
Our analysis on the link between molecular phenotypes to clinical manifestations provided an interesting tendency: defective LBP appears to cause more obvious glucocorticoid resistance and manifestations of mineralocorticoid excess. We think the above tendency may be caused by specific impact of damaged LBP to the GRα signaling system.
Our current analysis indicated that the electrostatic bond formed by R611 to dexamethasone was highly damaged in the mutant receptors defective in LBP, through massive deviation of its side chain. Deacylcortivazol, which has a large phenylpyrazole group at its steroid A-ring, stimulated more efficiently the transcriptional activity of some pathologic mutant receptors than dexamethasone and cortisol. This compound recovered the transcriptional activity of the pathologic GRα mutants defective in LBP at its higher concentrations, possibly by filling the space created by the shifted R611 side chain with its phenylpyrazole group. However, this steroid still demonstrated a difficulty to stimulate the transcriptional activity of these mutant receptors, probably due to yet reduced affinity against these mutant receptors compared with wild-type GRα. Taken together, these results suggest a possibility of developing tailored treatment compounds to patients with FSGR using structural simulations.
Structural defects observed in AF-2 of pathologic GRα mutants are more complicated than those seen in their LBP. Noncovalent interactions against the LXXLL peptide play a primary role in the association of this peptide to AF-2 of wild-type GRα. Pathologic GRα mutants defective in AF-2 activity lost or reduced some of such interactions especially those against the first and third core leucines of the LXXLL motif, which was also evident in the structural changes of the AF-2 groove that holds side chain of these core leucines. In addition to these leucines previously known to support the interaction between nuclear receptors and NRB of p160-type coactivators (8), R746 and D750, respectively, located next to the second and third core leucines of this LXXLL motif, contribute to the physical association of the LXXLL peptide to GRα AF-2 through electrostatic interactions, and explains preferential use of the third LXXLL motif of p160-type coactivators for the enhancement of GRα transcriptional activity. Importantly, the interaction between R585 of receptors and D750 of the LXXLL peptide was frequently damaged in the pathologic GRα mutants harboring defective AF-2, whereas the changes observed in this interaction contribute to defective transcriptional activity of some mutant receptors by compromising their association to NCoAs. These results explain not only details of the molecular defects observed in AF-2 of pathologic GRα mutants, but also provide novel human evidence for the contribution of nonleucine amino acids resided around the core LXXLL motif to the recognition and the binding of nuclear receptors to specific LXXLL motifs, and ultimately, to the transcriptional activity of GRα.
Additional material
Supplementary data supplied by authors.
Acknowledgments
We thank Dr R. M. Evans (Salk Institute, La Jolla, CA), Dr G. L. Hager (National Cancer Institute, Bethesda, MD), Dr J. N. Miner (Ligand Pharmaceuticals, San Diego, CA), Dr J. H. Segars (Eunice Kennedy Shriver National Institute of Child Health and Human Development, Bethesda, MD), Dr S. S. Simons, Jr. (National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD), and Dr M. G. Stallcup (University of Southern California, Los Angeles, CA) for providing plasmids or reagents, and Mr E. K. Zachman for superb technical assistance.
This work was supported by the Intramural Research Program of Eunice Kennedy Shriver National Institute of Child Health and Human Development (Z01 HD008732-05 HNT), National Institutes of Health, and Sirda Medical and Research Center. S.S. was supported by the Asahikawa Medical University.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- AF
- activation function
- 3D
- 3-dimensional
- DBD
- DNA-binding domain
- FGRS
- familial or sporadic glucocorticoid resistance syndrome
- GR
- glucocorticoid receptor
- GRIP1
- GR-interacting protein 1
- LBD
- ligand-binding domain
- LBP
- ligand-binding pocket
- MMTV
- mouse mammary tumor virus
- NCoA
- nuclear receptor coactivator
- NRB
- nuclear receptor box
- RMSD
- root mean square deviation
- WT
- wild-type.
References
- 1. Chrousos GP. Stress and disorders of the stress system. Nat Rev Endocrinol. 2009;5:374–381. [DOI] [PubMed] [Google Scholar]
- 2. Kino T, Chrousos GP. Glucocorticoid effect on gene expression. In: Steckler T, Kalin NH, Reul JMHM, eds. Handbook on Stress and the Brain. Amsterdam, The Netherlands: Elsevier BV; 2005:295–312. [Google Scholar]
- 3. Chrousos GP, Kino T. Intracellular glucocorticoid signaling: a formerly simple system turns stochastic. Sci STKE. 2005;2005:pe48. [DOI] [PubMed] [Google Scholar]
- 4. Kino T. Glucocorticoid receptor. In: Chrousos GP, ed. Adrenal Disease and Function. South Dartmouth, MA: Endotext; 2000. [Google Scholar]
- 5. Kino T, Su YA, Chrousos GP. Human glucocorticoid receptor isoform β: recent understanding of its potential implications in physiology and pathophysiology. Cell Mol Life Sci. 2009;66:3435–3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bledsoe RK, Montana VG, Stanley TB, et al. Crystal structure of the glucocorticoid receptor ligand binding domain reveals a novel mode of receptor dimerization and coactivator recognition. Cell. 2002;110:93–105. [DOI] [PubMed] [Google Scholar]
- 7. McKenna NJ, Lanz RB, O'Malley BW. Nuclear receptor coregulators: cellular and molecular biology. Endocr Rev. 1999;20:321–344. [DOI] [PubMed] [Google Scholar]
- 8. Heery DM, Kalkhoven E, Hoare S, Parker MG. A signature motif in transcriptional co-activators mediates binding to nuclear receptors. Nature. 1997;387:733–736. [DOI] [PubMed] [Google Scholar]
- 9. Darimont BD, Wagner RL, Apriletti JW, et al. Structure and specificity of nuclear receptor-coactivator interactions. Genes Dev. 1998;12:3343–3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Charmandari E, Kino T, Ichijo T, Chrousos GP. Generalized glucocorticoid resistance: clinical aspects, molecular mechanisms, and implications of a rare genetic disorder. J Clin Endocrinol Metab. 2008;93:1563–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Charmandari E, Kino T, Chrousos GP. Primary generalized familial and sporadic glucocorticoid resistance (Chrousos syndrome) and hypersensitivity. Endocr Dev. 2013;24:67–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nicolaides NC, Geer EB, Vlachakis D, et al. A novel mutation of the hGR gene causing Chrousos syndrome. Eur J Clin Invest. 2015;45:782–791. [DOI] [PubMed] [Google Scholar]
- 13. Phillips JC, Braun R, Wang W, et al. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ding XF, Anderson CM, Ma H, et al. Nuclear receptor-binding sites of coactivators glucocorticoid receptor interacting protein 1 (GRIP1) and steroid receptor coactivator 1 (SRC-1): multiple motifs with different binding specificities. Mol Endocrinol. 1998;12:302–313. [DOI] [PubMed] [Google Scholar]
- 15. Kino T, Jaffe H, Amin ND, et al. Cyclin-dependent kinase 5 modulates the transcriptional activity of the mineralocorticoid receptor and regulates expression of brain-derived neurotrophic factor. Mol Endocrinol. 2010;24:941–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kino T, Hurt DE, Ichijo T, Nader N, Chrousos GP. Noncoding RNA gas5 is a growth arrest- and starvation-associated repressor of the glucocorticoid receptor. Sci Signal. 2010;3:ra8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nader N, Bachrach BE, Hurt DE, et al. A novel point mutation in helix 10 of the human glucocorticoid receptor causes generalized glucocorticoid resistance by disrupting the structure of the ligand-binding domain. J Clin Endocrinol Metab. 2010;95:2281–2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nader N, Chrousos GP, Kino T. Circadian rhythm transcription factor CLOCK regulates the transcriptional activity of the glucocorticoid receptor by acetylating its hinge region lysine cluster: potential physiological implications. FASEB J. 2009;23:1572–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kino T, Gragerov A, Slobodskaya O, Tsopanomichalou M, Chrousos GP, Pavlakis GN. Human immunodeficiency virus type 1 (HIV-1) accessory protein Vpr induces transcription of the HIV-1 and glucocorticoid-responsive promoters by binding directly to p300/CBP coactivators. J Virol. 2002;76:9724–9734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kino T, Ichijo T, Amin ND, et al. Cyclin-dependent kinase 5 differentially regulates the transcriptional activity of the glucocorticoid receptor through phosphorylation: clinical implications for the nervous system response to glucocorticoids and stress. Mol Endocrinol. 2007;21:1552–1568. [DOI] [PubMed] [Google Scholar]
- 21. Karl M, Lamberts SW, Koper JW, et al. Cushing's disease preceded by generalized glucocorticoid resistance: clinical consequences of a novel, dominant-negative glucocorticoid receptor mutation. Proc Assoc Am Physicians. 1996;108:296–307. [PubMed] [Google Scholar]
- 22. Kino T, Stauber RH, Resau JH, Pavlakis GN, Chrousos GP. Pathologic human GR mutant has a transdominant negative effect on the wild-type GR by inhibiting its translocation into the nucleus: importance of the ligand-binding domain for intracellular GR trafficking. J Clin Endocrinol Metab. 2001;86:5600–5608. [DOI] [PubMed] [Google Scholar]
- 23. Mendonca BB, Leite MV, de Castro M, et al. Female pseudohermaphroditism caused by a novel homozygous missense mutation of the GR gene. J Clin Endocrinol Metab. 2002;87:1805–1809. [DOI] [PubMed] [Google Scholar]
- 24. Hurley DM, Accili D, Stratakis CA, et al. Point mutation causing a single amino acid substitution in the hormone binding domain of the glucocorticoid receptor in familial glucocorticoid resistance. J Clin Invest. 1991;87:680–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ruiz M, Lind U, Gafvels M, et al. Characterization of two novel mutations in the glucocorticoid receptor gene in patients with primary cortisol resistance. Clin Endocrinol (Oxf). 2001;55:363–371. [DOI] [PubMed] [Google Scholar]
- 26. Charmandari E, Kino T, Ichijo T, Zachman K, Alatsatianos A, Chrousos GP. Functional characterization of the natural human glucocorticoid receptor (hGR) mutants hGRαR477H and hGRαG679S associated with generalized glucocorticoid resistance. J Clin Endocrinol Metab. 2006;91:1535–1543. [DOI] [PubMed] [Google Scholar]
- 27. Malchoff DM, Brufsky A, Reardon G, et al. A mutation of the glucocorticoid receptor in primary cortisol resistance. J Clin Invest. 1993;91:1918–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Charmandari E, Kino T, Ichijo T, et al. A novel point mutation in helix 11 of the ligand-binding domain of the human glucocorticoid receptor gene causing generalized glucocorticoid resistance. J Clin Endocrinol Metab. 2007;92:3986–3990. [DOI] [PubMed] [Google Scholar]
- 29. Vottero A, Kino T, Combe H, Lecomte P, Chrousos GP. A novel, C-terminal dominant negative mutation of the GR causes familial glucocorticoid resistance through abnormal interactions with p160 steroid receptor coactivators. J Clin Endocrinol Metab. 2002;87:2658–2667. [DOI] [PubMed] [Google Scholar]
- 30. Charmandari E, Raji A, Kino T, et al. A novel point mutation in the ligand-binding domain (LBD) of the human glucocorticoid receptor (hGR) causing generalized glucocorticoid resistance: the importance of the C terminus of hGR LBD in conferring transactivational activity. J Clin Endocrinol Metab. 2005;90:3696–3705. [DOI] [PubMed] [Google Scholar]
- 31. Nicolaides NC, Roberts ML, Kino T, et al. A novel point mutation of the human glucocorticoid receptor gene causes primary generalized glucocorticoid resistance through impaired interaction with the LXXLL motif of the p160 coactivators: dissociation of the transactivating and transreppressive activities. J Clin Endocrinol Metab. 2014;99:E902–E907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kunz S, Sandoval R, Carlsson P, Carlstedt-Duke J, Bloom JW, Miesfeld RL. Identification of a novel glucocorticoid receptor mutation in budesonide-resistant human bronchial epithelial cells. Mol Endocrinol. 2003;17:2566–2582. [DOI] [PubMed] [Google Scholar]
- 33. Lodish H, Berk A, A KC, et al. Chemical foundations. In: Ahr K, ed. Molecular Cell Biology. 4th ed New York, NY: W. H. Freeman and Company; 2006:31–62. [Google Scholar]
- 34. Schlechte JA, Simons SS, Jr, Lewis DA, Thompson EB. [3H]cortivazol: a unique high affinity ligand for the glucocorticoid receptor. Endocrinology. 1985;117:1355–1362. [DOI] [PubMed] [Google Scholar]
- 35. Suino-Powell K, Xu Y, Zhang C, et al. Doubling the size of the glucocorticoid receptor ligand binding pocket by deacylcortivazol. Mol Cell Biol. 2008;28:1915–1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.