

Abstract
Physiological development requires precise spatiotemporal regulation of cellular and molecular processes. Disruption of these key events can generate developmental toxicity in the form of teratogenesis or mortality. The mechanism behind many developmental toxicants remains unknown. While recent work has focused on the unfolded protein response (UPR), oxidative stress, and apoptosis in the pathogenesis of disease, few studies have addressed their relationship in developmental toxicity. Redox regulation, UPR, and apoptosis are essential for physiological development and can be disturbed by a variety of endogenous and exogenous toxicants to generate lethality and diverse malformations. This review examines the current knowledge of the role of oxidative stress, UPR, and apoptosis in physiological development as well as in developmental toxicity, focusing on studies and advances in vertebrates model systems.
1. INTRODUCTION
Normal development is a tightly regulated, complex, and spatiotemporal process which, when disrupted, can lead to developmental toxicity causing birth defects or embryonic mortality. The cellular and molecular mechanisms behind many teratogens remain unknown and increasing effort is being applied toward their elucidation. Recently, significant research has focused around the roles of oxidative stress, the unfolded protein response (UPR), and apoptosis in the pathogenesis of human disease; however, few studies have investigated their intersection in developmental toxicity. Oxidative stress, the UPR, and apoptosis all play key physiological roles in vertebrate development—in diverse processes from early cell proliferation to late organogenesis and morphogenesis. While many classical teratogens or developmental toxicants have very specific molecular targets, stresses caused by alterations in redox state, protein folding, or apoptosis tend to have more general effects, resulting in a wide array of malformations. Both exogenous and endogenous toxicants can disrupt these key processes and while these pathways have all been implicated in teratogenesis individually, recent work and future research will examine the intersection between the key roles these events play in cell signaling and cellular fate. This review will examine the current knowledge of the role of oxidative stress, the UPR, and apoptosis in physiological development as well as in developmental toxicity, focusing on studies and advances in vertebrates model systems.
2. OXIDATIVE STRESS
Oxidative stress was defined originally in 1985 by Sies as “disturbances in the pro-oxidant/antioxidant systems in favor of the former,” however, in2006, Jones suggested a new definition as “a disruption of redox signaling and control.” Considering that the current literature recognizes the integral role of redox in all forms of aerobic life, this new definition represents a more nuanced view. Exogenous toxicants can alter the redox environment to disrupt development and cause teratogenesis. Although researchers often consider exogenous sources of reactive oxygen species (ROS) in developmental toxicity, ROS are produced endogenously and redox regulation plays a major role in normal vertebrate development. In order to understand the mechanisms behind oxidative teratogens, it is necessary to first grasp the physiological role of ROS in vertebrate development.
2.1 Generation of Endogenous ROS
Redox plays a critical role in cell signaling and homeostasis. This is particularly true during development, when processes are precisely timed and executed with little room for error. ROS and reactive nitrogen species (RNS) are the primary oxidants produced endogenously. Endogenous ROS include super oxide anion ( ), hydroxyl radical (OH•), and hydrogen peroxide (H2O2), while RNS are mainly nitric oxide (NO−) and peroxynitrite (ONOO−) (Figure 1).
Figure 1. Endogenous formation of reactive oxygen species/reactive nitrogenspecies.
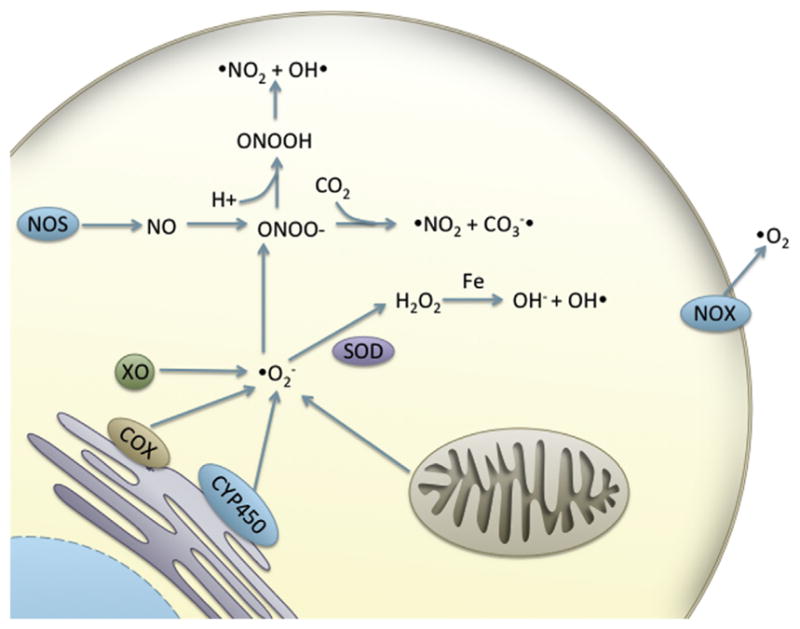
Abbreviations: COX, cyclooxygenase; CYP450, cytochrome P 450; NOX, NADPH oxidase; NOS, nitric oxide synthase; SOD, superoxide dismutase; XO, xanthine oxidase.
It has been estimated that 90% of ROS are generated by the mitochondria (Balaban et al., 2005). Oxidative phosphorylation occurs in the mitochondria and is the primary source of ATP in aerobic organisms, providing the majority of cellular energy. In the electron transport chain, electrons are passed between complexes, resulting in the ejection of hydrogen atoms from the inner mitochondrial space. This generates a membrane potential used to drive the formation of ATP. Leakage of electrons from complexes I and III generates superoxide (Balaban et al., 2005). Superoxide can then dismutate to form hydrogen peroxide, a reaction that occurs spontaneously or is catalyzed by superoxide dismutase (SOD) (Maier and Chan, 2002).
Another nonenzymatic reaction generating ROS is the Fenton reaction. In the Fenton reaction, hydrogen peroxide reacts with mutivalence transition metals (iron, copper, manganese) to form the hydroxyl radical and the hydroxyl anion. The hydroxyl radical is highly reactive with a shorter half-life than other free radicals at 10−9 s, and often reacts with molecules in the vicinity of its creation (Pryor, 1986). Furthermore, in the Haber–Weiss reaction, superoxide can increase hydroxyl radical production by oxidizing [4Fe-4S] cluster containing enzymes, making more Fe(II) available for Fenton chemistry (Leonard et al., 2004).
ROS/RNS can be generated enzymatically via enzymes involved in cell signaling, defense, or biosynthesis such as nitric oxide synthase (NOS), NADPH oxidase (NOX), xanthine oxidase (XO), lipoxygenases (LOX), and cyclooxygenases (COX). XO is a cytosolic enzyme catalyzing the conversion of hypoxanthine to xanthine and xanthine to uric acid, which uses oxygen as a cofactor and can generate superoxide and the hydroxyl radical (McCord and Fridovich, 1968; Kuppusamy and Zweier, 1989). LOX enzymes are involved in eicosanoid metabolism and catalyze the formation of peroxides on fatty acid substrates (Brash, 1999). 5-Lipoxygenase catalyzes the conversion of arachidonic acid to leukotrienes can create ROS in lymphocytes, contributing to activation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) (Soberman and Christmas, 2003). While it was initially believed that NOX was expressed exclusively in macrophages, it has since been discovered in tissues throughout the body and is another well-known generator of superoxide (DeCoursey and Ligeti, 2005). In response to foreign stimuli, these enzymes catalyze the conversion of largeamounts of O2 to superoxide, a process that has been called respiratory oroxidative burst (DeCoursey and Ligeti, 2005). This superoxide contributes toinflammation and is dismutated to H2O2 (DeCoursey and Ligeti, 2005). Cytochrome P 450 (CYP450) monooxygenase enzymes also contribute to ROS generation (Gillette et al., 1957; Nordblom and Coon, 1977). CYP450s are a large class of microsomal, drug metabolizing enzymes that have several endogenous substrates. Uncoupling of the oxygen bound to the heme active site can generate superoxide, which rapidly forms H2O2 (Narasimhulu, 1971; Grinkova et al., 2013). NO is an important signaling molecule generated by one of three tissue specific isoforms of nitric oxide synthase (iNOS (immune cells), eNOS (endothelial cells), and nNOS (neurons)). NO will react readily with superoxide to form the peroxynitrite (ONOO−) anion (Bergendi et al., 1999). In acidic media, (ONOO−)can react with hydrogen and fragment forming the hydroxyl radical, which is extremely reactive (Bergendi et al., 1999).
2.2 Oxidative Damage from ROS/RNS
When ROS/RNS exceed detoxification capacities, oxidative damage to lipids, nucleic acids, and proteins may occur. If this damage is irreparable, the cell will undergo cell death, in the form of either apoptosis (programmed cell death) or necrosis, depending on the extent of the damage. OH• is the most reactive ROS to DNA, and can damage DNA via H abstraction or alkene addition. Hydrogen abstraction occurs from the methyl group of thymine and from the C–H bonds on 2-deoxyribose, leading to multiple oxidized products (Cooke et al., 2003). One major product of oxidative DNA damage is 8-hydroxyguanine, which has been shown to cause G to T and A to C substitutions (Cheng et al., 1992). Oxidatively modified bases can also cause double strand breaks when reacting with C–H bonds on 2-deoxyribose. If DNA repair mechanisms are impaired or overwhelmed, these products can lead to adverse cellular effects, including mutations and cytotoxicity.
Lipids are also susceptible to oxidation by free radicals, particularly polyunsaturated fatty acids with two cis-double bonds separated by a methylene group (Niki, 2014). Lipid peroxidation may occur as a chain reaction separated into three basic steps: initiation, propagation, and termination (Porter et al., 1995). Initiation begins with hydrogen abstraction from a bis-allylic carbon, which rearranges to form relatively a stable cis, trans-pentadienyl radical. The addition of oxygen then forms a lipid peroxylradical, which begins propagation by abstracting a second H from adjacent lipids. The process continues until termination, when two peroxyl radicals react together to form nonradical products or the products are scavenged by cellular antioxidants. Lipid peroxides can fragment to form reactive aldehydes and can alter membrane stability and permeability of key organelles, such as the mitochondria (Sultana et al., 2013). These cellular level effects translate to organ-level pathologies and are implicated in a variety of diseases, including heart disease (Anderson et al., 2012), ocular degeneration (Njie-Mbye et al., 2013), and Alzheimer’s disease (Sultana et al., 2013).
Oxidative damage to proteins can take several forms. Oxidation of the protein backbone following H abstraction by OH• can result in protein fragmentation, as can oxidation of glutamyl, aspartyl, and prolyl side chains (Berlett and Stadtman, 1997). Sulfur-containing amino acids, cysteine, and methionine are particularly sensitive to oxidation (Berlett and Stadtman, 1997). These modifications can result in the loss of enzyme or protein function and the accumulation of unfolded proteins, which can impair cell function, leading to the UPR (Radak et al., 2001).
2.3 Protection against ROS/RNS
Considering the many endogenous sources of ROS, aerobic organisms have developed enzymatic and nonenzymatic responses to mediate ROS levels (Figure 2). Important enzymatic mechanisms include SOD and catalase (CAT). There have been three isozymes of SOD identified in mammals, which are localized into different cellular compartments. Cu-ZnSOD is located in the cytosol, MnSOD is in the mitochondria, and ECSOD is in the extracellular space (thus far, ECSOD has only been identified in mice) (Maier and Chan, 2002). In general, SODs stoichiometrically catalyze the dismutation of two superoxide anions with two hydrogen atoms to for mH2O2 and O2 (Maier and Chan, 2002). CAT and other peroxidases then reduce two molecules of H2O2 to two water molecules and molecularoxygen (Kirkman and Gaetani, 2006).
Figure 2. Damage and protection from reactive oxygen species.

Abbreviations: CAT, catalase; GSH, glutathione; GR, glutathione reductase; Grx, glutaredoxin; GSSG, glutathione disulfide; Prx, peroxiredoxin; R–SH, protein thiol group; R–SS–R, protein with disulfide bond; SOD, superoxide dismutase; Trx, thioredoxin; TrxR, thioredoxin reductase.
Nonenzymatic mechanisms of detoxification include antioxidants, which scavenge free radicals in the cell and/or reduce oxidized thiols. Glutathione (GSH) is a tripeptide of glycine, cysteine, and glutamate, in which the glutamate is connected to the cysteine via a gamma-carboxyl linkage (Lu, 1999). It exists in two forms, a reduced form (GSH) and an oxidized disulfide form (GSSG). GSH is primarily located in the cytosol, with about 10% in the mitochondria. GSH is present in the cell at millimolar concentrations and is the most abundant nonprotein intracellular thiol (DeLeve and Kaplowitz, 1991). It can reduce oxidants (ROS or strong electrophiles) either enzymatically or spontaneously through its thiol group. Glutathione S-transferase can enzymatically add GSH to electrophiles and disulfides. Glutathione reductase then regenerates GSH from oxidized glutathione (GSSG) with NADPH as a cofactor. To combat ROS, selenoprotein glutathione peroxidases (GPx) can reduce H2O2 with two GSH, generating water and GSSG. There are four members of the GPx family, GPx1, GPx2, GPx3, and GPx4 (Imai and Nakagawa, 2003). GPx1 and GPx2 are localized to the cytoplasm, whereas GPx3 is primarily in the plasma of the kidney (Imai and Nakagawa, 2003). GPx4 is distinct from the other GPx isoforms and is the only GPx that can reduce phospholipid hydrogen peroxides (Imai and Nakagawa, 2003).
Other important antioxidants include thioredoxins (Trxs), glutaredoxins (Grx), peroxiredoxins (Prx), and nucleoredoxins. These small proteins are key regulators of redox signaling through the reduction of disulfides and H2O2. Trxs contain the active site motif Cys-Gly-Pro-Cys (Holmgren, 1968). Electrons from NADPH are transferred to thioredoxin reductase (TrxR), which reduces Trx to reduce disulfide bonds (Hanschmann et al., 2013). TrxR is another selenoprotein with a wide substrate specificity and is regulated by nuclear factor (erythroid-derived 2)-like 2 (Nrf2) (Kim et al., 2001). There are four Grxs with different cellular localization in mammals (Hanschmann et al., 2013). Each antioxidant is ultimately part of the GSH pathway and the resulting oxidants are eventually reduced by GSH (Hanschmann et al., 2013). The six mammalian Prxs are localized to a wide variety of cellular compartments (cytosol, nucleus, mitochondria, peroxisomes, lysosomes, and vesicles) (Hanschmann et al., 2013). In contrast to Grxs and Trxs, Prxs reduce H2O2 to water and are regenerated by Trx or GSH (Hanschmann et al., 2013). Finally, nucleoredoxins were first discovered in yeast in 1997 (Kurooka et al., 1997); however, homologs have since been identified across vertebrates (Funato and Miki, 2007). They are localized to the cytoplasm and nucleus, and are involved in the regulation of several redox-regulated transcription factors (Funato and Miki, 2007).
2.4 Physiological Role of ROS in Signaling and Development
Redox plays a key physiological role in vertebrate development; however, little progress has been made concerning the detailed function and nature of redox signaling in developmental processes (Table 1). Alterations in the redox environment of developing embryos can lead to teratogenesis through dysregulation of cell signaling at lower levels and cell damage at higher levels. Here, we will examine the current knowledge base concerning the physiological role of ROS/RNS in development and teratogens that have been associated with oxidative stress.
Table 1.
Embryonic lethality resulting from knock out of redox-related genes
| Category | Essential | Nonessential |
|---|---|---|
| Transcription factors | ||
|
| ||
| p65 | Nrf2 | |
| HIF | RelB | |
| Ref-1 | p50 | |
| p52 | ||
| c-rel | ||
|
| ||
| Antioxidants | ||
|
| ||
| Nrx2 | CAT | |
| GSH | Prx | |
| Trx1 | GPx1 | |
| Trx2 | GPx2 | |
| GPx4 | GPx3 | |
|
| ||
| ROS inducers | ||
|
| ||
| LOX | ||
| COX | ||
| NOX | ||
| NOS | ||
| XO | ||
2.4.1 Oxygen availability and metabolism in mammalian development
The redox environment in the embryo is directly influenced by oxygen availability, which fluctuates throughout development. In the mammalian oviduct, oxygen concentrations are relatively high, and decrease gradually until implantation in the uterus (Fischer and Bavister, 1993). Thus, metabolic pathways vary according to the oxygen gradient. Oxidative phosphorylation predominates preimplantation when glucose levels are low and oxygen high, while glycolysis predominates postimplantation, when glucose levels are high and oxygen low (Ufer et al., 2010). By shifting from oxidative phosphorylation to glycolysis, the embryo is able to minimize the production of ROS from the electron transport chain during this vulnerable period (Leese, 1995). Oxygen concentrations remain low until maternal circulation to the placenta begins at the end of the first trimester, inducing a threefold increase in oxygen concentrations (Burton, 2009). These fluctuations in oxygen levels are critical to normal embryonic development and studies have shown that low levels of H2O2 will induce cell proliferation, while higher levels arrest cell growth(Davies, 1999).
2.4.2 Redox-regulated transcription factors
In addition to oxygen availability, redox regulation of key transcription factors is critical to development (Figure 3). The NF-κB family of transcription factors control expression of inflammatory cytokines, growth factors, redox-regulated enzymes, and apoptotic regulators. Knockout mice of various NF-κB family members are born with several defects, including postnatal immune defects (p50−/− c-Rel−/−), multiorgan inflammation (RelB−/−), disrupted splenic architecture (p52−/−), and prenatal embryonic lethality (p65) (Ufer et al., 2010).
Figure 3. Redox-regulated transcription factors.

Abbreviations: AP-1, activator protein 1; ATF, activating transcription factor; HIF-1α, hypoxia-inducible factor 1α, Keap1, Kelch-like ECH-associated protein 1; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; Nrf2, nuclear factor (erythroid-derived 2)-like 2; PHD, prolyl hydroxylase; Ref-1, redox effector factor 1.
Nrf2 is a redox activated transcription factor controlling gene expression through the antioxidant response element. Under normal unstressed conditions, Nrf2 is targeted for degradation by Kelch-like ECH-associated protein 1 (Keap1) ubiquitylation. However, Keap1 has several reactive cysteine residues, that impair its capacity for ubiquitylation when oxidized, allowing Nrf2 to perform its function as a transcription factor (Nguyen et al., 2009). Nrf2 regulates genes related to detoxification and antioxidants, as well as lipid carbohydrate and heme metabolism (Hayes and Dinkova-Kostova, 2014). While not embryonically lethal, Nrf2 knockout mice display autoimmune-mediated lesions in multiple tissues and premature postnatal death (Ma et al., 2006).
Hypoxia-inducible factors (HIF) are important regulators of gene expression under hypoxia, a condition known to generate oxidative stress and discussed further below. HIFs consist of two subunits. The β subunit (HIFβ, also called aryl hydrocarbon receptor (AhR) nuclear translocator, a critical component of the AhR pathway) is stable, while the α subunit is mediated by O2 levels. Under homeostatic conditions, the α subunit ishydroxylated by an HIF-specific prolyl hydroxylase (PHD), targeting it for ubiquitylation and degradation. PHD has low O2 affinity and is thus its activity is highly dependent on O2 levels. Under hypoxic conditions, HIFα is not hydroxylated and forms a heterodimer with HIFβ to modulate expression of target genes (Majmundar et al., 2010). Although the precise mechanism remains unclear, ROS also negatively regulate PHD, implicating HIF activity under oxidative stress (Majmundar et al., 2010; Eom et al., 2013). HIF regulates genes involved in glucose and iron metabolism, erythropoiesis, angiogenesis, and cell proliferation and differentiation (Maxwell et al., 2001; Yoon et al., 2006). In embryonic development, HIF is essential for vascular development during hypoxic conditions in neurogenesis and organogenesis (Trollmann and Gassmann, 2009). Furthermore, all HIF subunits have been shown to be essential for development as knockouts die between E10.5 and E12.5 (Ufer et al., 2010). HIF-1α null embryos exhibited abnormal neural tube formation, increased apoptosis, and a reduction in the number of somites, as well as impaired erythropoiesis (Ryan et al., 1998; Yoon et al., 2006).
Redox effector factor 1 (Ref-1), also called apurinic/apyrimidinic endonuclease-1 contributes to the base excision repair pathway of DNA lesions; however, it is also involved in modulation of redox sensitive transcription factors (Tell et al., 2009). Oxidation of key cysteine residues impairs DNA-binding capacity of Ref-1 but induces its ability to oxidize several transcription factors, including activator protein 1, NF-κB, and the activating transcription factor (ATF)/CREB (cAMP response element binding protein) family (Tell et al., 2009). Trx can then restore reduced Ref-1 (Tell et al., 2009). Homozygous Ref-1 knockout embryos die by E6, while heterozygous embryos display increased markers of oxidative stress, which can be rescued by the addition of antioxidants (Ufer et al., 2010).
2.4.3 Antioxidants in development
Considering the variable oxygen availability throughout development as well as the potential onslaught from exogenous sources, it is necessary for vertebrates to have redundant antioxidant responses with high capacity to combat ROS. The redox potential of the developing embryo is closely linked to the GSH/GSSG and Trx potentials. GSH is essential for vertebrate development; mice lacking gamma-glutamylcysteine synthetase, an essential enzyme for GSH synthesis, die before E8.5 from apoptotic cell death (Shi et al., 2000). GSH levels are high in the maturing oocyte but steadily decrease following fertilization (Gardiner and Reed, 1994). In developing zebrafish (Danio rerio) embryos, the redox potential as measured through the GSH/GSSG ratio fluctuated greatly (Timme-Laragy et al., 2013). The cellular environment was reducing at fertilization (−225 mV), became increasingly oxidized until 12-hour postfertilization (hpf) (−170 mV), after which point it fluctuated between 24 and 48 hpf, increasing to a stable reduced state at hatch at 72 hpf (−225 mV) (Timme-laragy et al., 2013).
The Trx family has been shown to play a key role in development. During mouse gastrulation (E7), Trx1 mRNA expression was 11-fold higher than adults, but steadily decreased to adult levels by late gestation (Jurado et al., 2003). Trx1 and Trx2 knockout mice undergo embryonic lethality. Trx1 mice die around implantation (E3.5) following reduced proliferation of the inner cell mass (Matsui et al., 1996). In contrast, Trx2 mice develop normally until E8.5, but display an open neural tube at E10.5 and are reabsorbed at E12.5 (Nonn et al., 2003). This corresponds with the onset of mitochondrial maturation and oxidative phosphorylation, suggesting a key role for Trx in regulation by endogenous ROS (Nonn et al., 2003). Trx reductase null mutations are also embryonically lethal, with TrxR2 being essential for proper cardiac development, and TrxR1 not having a role(Jakupoglu et al., 2005). Prxs are also essential for H2O2 metabolism and while null mice are viable, Prx1 plays a key role in motor neuron differentiation through reduction of a disulfide bond in GDE2 (glycerophosphodiester phosphodiesterase 2) (Yan et al., 2009). Another member of the Trx family, Grx also plays a complex role in vertebrate development. A study in zebrafish found Grx2 regulation of CRMP2 (Collapsin response mediator protein 2) thiols-controlled axonal outgrowth, neuron survival, and brain development (Brautigam et al., 2011).
Many enzymes that diminish oxidative stress, such as CAT, SOD, GSH reductase, and GPx, are present at lower levels in developing embryos than in adult organisms, further complicating the embryo’s ability to cope with oxidative insult (Ufer et al., 2010). CAT null mice are viable with no visible defects, yet reduced CAT levels may indicate reduced ability of the embryo to cope with ROS-generating toxicants (Ho et al., 2004). GPx enzymes are important components of the GSH system. While GPx1, GPx2, and GPx3 knockout embryos are viable with no visible defects, indicating that they are dispensable for embryonic development, GPx4 plays a more complex role (Ufer and Wang, 2011). GPx4 null embryos are embryonic lethal, undergoing resorption by day E8.5 (Imai et al., 2003). GPx4 mRNA expression has been detected beginning at E6.5, and at E8.0 in the neural tube, extending in to the developing brain (Borchert et al., 2006). It is later detected in the developing organs and limbs (Schneider et al., 2006). GPx4’s role in these tissues appears to be the prevention of mitochondrial membrane oxidation and subsequent apoptosis-inducing factor-dependent apoptosis (Ufer and Wang, 2011). However, it has also been shown to modulate Nrf2 and NF-κB activity, indicating possible contributions to cell signaling (Ufer and Wang, 2011).
2.4.4 ROS/RNS-generating enzymes in development
Knock out of many oxidative enzymes, such as NOX, NOS, XO, and some isoforms of LOX and COX produce viable offspring, indicating that they are not essential for embryonic development (Ufer et al., 2010). However, 12-LOX knockout mice displayed skin defects that led to neonatal death, implicating 12-LOX in development of skin structure (Epp et al., 2007). Furthermore, COX-2 knockout embryos displayed defective kidney development and postnatal myocardial fibrosis, accompanied by an increase of neonatal mortality (Dinchuk et al., 1995). NOS null embryos were viable even though NO is an important signaling molecule, and has been shown to be involved in physiological vascular development (Nath et al., 2004). eNOS expression increased steadily throughout development from approximately 3000 to 14,000 copies of mRNA/106 copies of GAPDH (glyceraldehyde-3-phosphate dehydrogenase) mRNA, while iNOS and nNOS were expressed between 50 and 400 copies of mRNA/106 copies of GAPDH mRNA (Ufer et al., 2010).
2.5 Oxidative Stress and Teratogenesis
The complex nature of redox signaling and control throughout development indicates that even relatively minor alterations to redox potential may generate significant developmental effects. While mild imbalances in the redox potential may impact redox-regulated signaling, higher levels of oxidative stress can induce teratogenesis through cytotoxicity. Oxidative stress is implicated as the mechanism of action of many developmental toxicants (Hansen, 2006). One well-studied example is the anticonvulsant drug, phenytoin. Phenytoin generates facial abnormalities and neurological disorders in infants exposed during pregnancy. Its toxicity is believed to be caused following bioactivation by prostaglandin H synthase to form a free radical intermediate (Parman et al., 1998) or by CYP450 to form protein adducts (Munns et al., 1997). Phenytoin has been shown to oxidize DNA to form 8-hydroxyguanine (Liu and Wells, 1995) and increase homologous recombination (Winn et al., 2003). It can also generate lipid peroxidation and protein oxidation in embryonic cells (Liu and Wells, 1994). DNA oxidation and embryopathies in mice can be reduced with the addition of CAT (Abramov and Wells, 2011) and the GSH precursor, N-acetylcysteine (Wong and Wells, 1988). DNA, protein, and lipid damage are believed to generate the developmental toxicity observed with phenytoin.
In addition to oxidative stress generated by pharmaceuticals, environmental exposures to chemicals can also cause oxidative damage leading to developmental toxicity. Many pesticides, such as organophosphates, cause acute toxicity via inhibition of acetylcholinesterase, and are also developmental toxicants. For instance, neonatal rats are more sensitive to the organophosphate, chlorpyrifos, than adults (Chakraborti et al., 1993). Chlorpyrifos has been shown to cause neurobehavioral defects following early exposure in mammals (Chakraborti et al., 1993; Chanda and Pope, 1996). It has also been linked to aberrant behavior, spinal curvature, pericardial edema, and mortality in zebrafish embryos (Kienle et al., 2009), whereas exposed frogs displayed tail flexure and decreased neuromuscular activity (Bonfanti et al., 2004).
The mechanisms of chlorpyrifos developmental toxicity are far more complex than its acute toxicity. In addition to acetylcholinesterase inhibition, organophosphates have also been shown to interfere with neuronal cell differentiation and replication, synapse formation, neurite cell growth, and other neurodevelopmental processes (Flaskos and Sachana, 2010). Additionally, oxidative stress has been implicated in chlorpyrifos toxicity. In procaspase (PC) 12 cells, a model for neurodifferentiation, the addition of 2–50 μM chlorpyrifos significantly induced ROS (Crumpton et al., 2000). This effect was ameliorated with vitamin E (Slotkin et al., 2007). Chlorpyrifos treatment during gestation and on postnatal days one to four did not increase lipid peroxidation in rats; however, oxidative stress was generated following treatment during the peak of neuronal cell differentiation and synaptogenesis (second postnatal week) (Slotkin et al., 2005). It was further discovered that 2 mg/mL chlorpyrifos induced expression of oxidative stress-related genes following treatments to 7-day-old rats (Ray et al., 2010). Understanding the mechanism of action of chlorpyrifos and phenytoin teratogenicity informs future drug and pesticide design to mitigate potential negative effects. However, many toxicants act via a range of mechanisms and can disrupt other normal cellular processes both directly and via oxidative stress, including protein folding.
3. UNFOLDED PROTEIN RESPONSE
The endoplasmic reticulum (ER) is the major site of posttranslational processing of proteins, which amounts to about 30% of the proteome, and performs several other critical functions including calcium storage and metabolic processes (Hetz, 2012). The fidelity of the ER must be able to handle large secretory loads. Secretory cells produce high volumes of proteins for processing and the same can be said of cells at certain embryonic stages. Disruption of proper ER function can lead to the accumulation of unfolded proteins, a state called ER stress. ER stress results in the activation of a complex signaling pathway called the UPR. Through alterations of gene expression and protein translation, the UPR optimizes the ER to correctly manage protein folding and initiate apoptosis or autophagy in cells that are irreversibly damaged. UPR mediators and signaling proteins respond not only to exogenous toxicants that disturb ER function, but also play key roles in embryonic development and organ physiology (Cornejo et al., 2013). In this section, we address the physiological role of the UPR in vertebrate development and how induction and perturbation of the UPR can result in teratogenesis.
3.1 Signal Transduction of Three UPR Branches
There are three branches of the UPR, each with a distinct signal transduction pathway culminating in transcription of UPR target genes (Figure 4). The branches are controlled by inositol-required enzyme 1 (IRE1, α and β), protein kinase RNA-like ER kinase (PERK), and ATF6 (α and β).
Figure 4. The three branches of the unfolded protein response.

Abbreviations: ATF4, activating transcription factor 4; ATF6, activating transcription factor 6; BiP, immunoglobulin heavy-chain-binding protein; eIF2α, eukaryotic initiation factor 2α, IRE1, inositol-required enzyme 1; PERK, protein kinase RNA-like ER kinase; RIDD, regulated IRE1-dependent decay; SP1/2, serine proteases 1 and 2; XBP-1, X-box protein 1.
The most extensively studied branch of the UPR, IRE1α is also the most evolutionarily conserved branch and is the only branch characterized in yeast (Mori, 2009). IRE1α is both a kinase and an endonuclease (Tirasophon et al., 1998). When activated by the UPR, IRE1α dimerizes and undergoes autotransphosphorylation, leading to activation of the cytosolic RNase domain (Hetz, 2012). It then splices a 26-base-pair region from the center of X-box binding protein 1 (XBP-1) mRNA, allowing the translated protein to fulfill its role as a transcription factor (Calfon et al., 2002; Yoshida et al., 2001). XBP-1 targets a wide variety of genes, 29% of which are involved in protein trafficking, processing, and secretion and 14.8% of which are involved in protein biosynthesis and posttranslational modifications (Acosta-Alvear et al., 2007). Thus, through XBP-1, the IRE1α branch of the pathway is primarily responsible for the secretion and degradation of unfolded proteins through ER-associated degradation (ERAD). However, in addition to genes impacting ER function, XBP-1 also controls expression of genes for cell growth and differentiation, RNA processing, signal transduction, ion channels, DNA replication, DNA repair, and redox potential (Acosta-Alvear et al., 2007).
IRE1 further contributes to the UPR through regulated IRE1-dependent decay (RIDD), a process by which IRE1 mediates decay of certain mRNA within the ER (Hollien and Weissman, 2006). It is through the RIDD pathway that IRE1 regulates cell fate during ER stress (Chen and Brandizzi, 2013).
ATF6α and β are part of the CREB/ATF family of proteins and are constitutively expressed transmembrane ER proteins (Yoshida et al., 1998a). When unfolded proteins accumulate in the ER, ATF6α translocates to the golgi apparatus, where it is cleaved by site-1 and site-2 proteases into its active form (Nadanaka et al., 2004; Ye et al., 2000). It then can enter the nucleus to act as a transcription factor for ERSE-regulated genes (ER stress element) (Okada et al., 2003; Yoshida et al., 1998a). These include ERAD proteins, various ER chaperones, and XBP-1.
Finally, the PERK branch of the UPR is primarily responsible for translational attenuation in order to reduce the load of unfolded proteins. Upon activation, PERK oligomerizes and phosphorylates the translation initiation factor eIF2α (eukaryotic initiation factor 2), inhibiting its activity and attenuating protein translation (Harding et al., 2000). Additionally, PERK regulates other transcription factors and UPR mediators. Without available eIF2α, some mRNAs with specific sequences are preferentially translated, including ATF4, a member of the same family of transcription factors as ATF6 (Walter and Ron, 2011).
ATF4 has a wide variety of gene targets and can function as both a transcriptional activator and repressor. Its repressor activities are less well studied and include negative regulation of the cAMP response element (Karpinski et al., 1992) and memory storage (Chen et al., 2003). ATF4 can function as a transcriptional activator both alone and as a heterodimer with other transcription factors. ATF4’s transcriptional targets are wide ranging and include genes involved in amino acid transport and metabolism, transcription, mitochondrial function, and redox/detoxification (Ameri and Harris. 2008).
3.2 Induction and Regulation of UPR
ER stress can be generated from disruption of calcium homeostasis, ER-localized oxidative stress, or impaired vesicular trafficking, or protein degradation (Hetz, 2012). In studies of ER stress, pharmacological agents are generally used to induce the UPR. These include thapsigargin (Tg), which inhibits Ca2+-dependent ATPase, tunicamycin (Tm), a protein glycosylation inhibitor, and dithiothreitol (DTT), a disruptor of disulfide bonds (DuRose et al., 2006).
The dynamic nature of the response to ER stress is in part due to varied forms of regulation at several levels. It was originally proposed that the UPR was governed by the inducible ER resident chaperone BiP (Immunoglobulin heavy-chain-binding protein, also known as GRP78 or HSPA5). An early and pioneering study by Bertolotti et al. (2000) demonstrated BiP was able to complex with the luminal domains of IRE1 and PERK under ER homeostasis and dissociated from the complex under ER stress. Furthermore, the activities of IRE1 and PERK were attenuated when BiP was overexpressed (Bertolotti et al., 2000). In the case of ATF6, BiP binding masks golgi localization signals, preventing constitutive transactivation (Shen et al., 2002). Once the UPR is activated, BiP then dissociates from each branch to perform its duties as a chaperone for unfolded proteins.
However, further research suggests that BiP is not the only regulator of UPR signaling. For example, it has been suggested that decreased glycosylation of ATF6 may control its own activation. This mechanism of ATF6 regulation was proposed following observations that depletion of Ca2+ in the ER induced newly synthesized and partially glycosylated ATF6 (Hong et al., 2004). This under-glycosylated ATF6 was no longer able to associate with calreticulin, resulting in faster translocation of ATF6 to the golgi and stronger UPR activation (Hong et al., 2004). In addition, reduction of disulfide bonds within ATF6 also facilitated its transport to the golgi, but was not sufficient for its activation (Nadanaka et al., 2007).
Several studies have been performed characterizing the kinetics of UPR activation. In one study, DTT caused 100% activation of PERK, ATF6, and IRE1 within 1 h following treatment. PERK and IRE1 were equally as sensitive to Tg, while ATF6 was not (DuRose et al., 2006). Furthermore, all three branches took longer (2–5 h) to reach full activation following treatment with Tm (DuRose et al., 2006). PERK signaling was also maintained under prolonged ER stress caused by Tm and Tg, while IRE1α and ATF6 signaling peaked at 4 h posttreatment then declined (Lin et al., 2007).
3.3 UPR in Physiological Development
Although once thought to merely function as a stress response, evidence supporting a role for the UPR in developmental physiology is increasing. UPR mediators have been implicated in diverse developmental pathways and many are essential for embryonic development, demonstrated by nonviable embryonic knockouts. While the primary role of the UPR is mediation of protein folding and load in secretory cells, various UPR transcription factors and signal transduction pathways are involved in several other processes (Table 2). UPR mediators have been implicated in hematopoiesis, osteogenesis, chondrogenesis, angiogenesis, neurogenesis, hepatogenesis, as well as pancreatic and lens cell development. In this section, we will examine each branch of the UPR and its role in physiological development.
Table 2.
Role of unfolded protein response genes in physiological development
| Gene name | Embryonic lethal knockout? | Developmental process |
|---|---|---|
| IREα | Yes | Immune cell differentiation, hepatogenesis, chondrogenesis, adipogenesis |
| IRE1β | No | |
| XBP-1 | Yes | Immune cell differentiation, hepatogenesis, zymogen cell differentiation, adipogenesis |
| ATF6α/β | Individual KO of α or β—No Double α/βKO—Yes | Neurogenesis, hepatogenesis |
| PERK | No | Pancreatic β-cell differentiation, osteogenesis |
| ATF4 | Yes | Osteogenesis, lens formation, hematopoiesis |
| BiP | Yes | Early development, neurogenesis |
3.3.1 Inositol-required enzyme 1/X-box binding protein 1
The function of the IRE1 and XBP-1 axis in development is the best studied of the three pathways. IRE1α knockout mice are embryonic lethal and full IRE1α knockout embryos die between E12.5 and E13 (Zhang et al., 2005). Embryos at E13 demonstrated fetal liver hypoplasia and decreased proliferation of hematopoietic stem cells (HSCs) (Zhang et al., 2005). Subsequent studies on IRE1α null mice have shown severe defects to the labyrinth in the placenta mediated by XBP-1-independent decreased expression of vascular endothelial growth factor (VEGF)-A (Iwawaki et al., 2009). Interestingly, following selective IRE1α expression in the placenta, embryos displayed no liver hypoplasia and were viable (Iwawaki et al., 2009). In contrast to IRE1α, IRE1β knockout mice are viable (Bertolotti et al., 2001). However, a study in Xenopus laevis frog embryos found IRE1β to be required for mesoderm development, suggesting evolutionary alterations in function (Yuan et al., 2008).
XBP-1 mRNA can be detected in the nucleus and cytoplasm as early as the one-cell stage (Zhang et al., 2012). Similar to IRE1α null mice, XBP-1−/− mice are not viable, with liver hypoplasia and lethality beginning at E12.5 (Reimold et al., 2000). Livers from knockout mice had decreased proliferation and increased apoptosis of hepatocytes (Reimold et al., 2000). This corresponded to decreased levels in several acute-phase proteins (Reimold et al., 2000).
The UPR has been recognized as a key regulator in secretory cells, and IRE1 and XBP-1 have been implicated in immune cell development and function. While rag−/− mice transplanted with IRE1α−/− HSCs were able to produce pro-B cells, as well as erythroid, myeloid, and thrombocyte lineages, B-cell receptors were not detected and a reduction in B-cell Ig VDJ recombination was observed (Zhang et al., 2005). Furthermore, though spliced XBP-1 was not necessary for early B lymphocyte differentiation, it was necessary and sufficient for terminal plasma cell differentiation (Reimold et al., 2001; Zhang et al., 2005). Ectopic XBP-1 expression induced B-cell differentiation into plasma cells (Reimold et al., 2001) and induction of XBP-1 spliced mRNA correlated with Ig heavy chain secretion during plasma cell differentiation (Zhang et al., 2005) and induced interleukin-6 (Iwakoshi et al., 2003). The PERK branch of the response was not necessary for this response (Zhang et al., 2005).
The XBP-1 branch of the UPR has also been implicated in several other developmental pathways. For example, XBP-1 is responsible for the development of zymogen cells in the gastric epithelium (Huh et al., 2010). IRE1 regulated chondrocyte differentiation, via inhibition of UPR-induced apoptosis following induction by BMP2 (bone morphogenic protein 2) (Han et al., 2013). Finally, XBP-1 transcription is induced by C/EBPβ (CCAAT-enhancer-binding protein beta), an early adipogenic factor, and plays a key role in adipogenesis, as cells lacking XBP-1 and IRE1α display defects in adipogenesis (Sha et al., 2009). It is clear that the IRE1/XBP-1 signal transduction pathway plays a key role in physiological development through both modulation of secretory cell differentiation and function, and through transcriptional regulation.
Considering the diverse network of genes regulated by XBP-1 and the large number of mRNA’s subject to RIDD, the possible roles for IRE1 and XBP-1 in vertebrate development are limitless. XBP-1 targets include genes involved in cell growth and differentiation, including several cyclin-dependent kinases (Acosta-Alvear et al., 2007). The role of RIDD in physiological development is still unclear. However, several genes essential for embryonic development are RIDD targets. For example, homeo box B4 is an RIDD target (Hollien et al., 2009) and is essential for skeletal development in mice (Ramirez-Solis et al., 1993), and HSC differentiation from embryonic stem cells (Fan et al., 2012).
3.3.2 Activating transcription factor 6
ATF6α and ATF6β single knockouts develop normally, while embryos with ATF6α/β double knockouts are not viable, suggesting some overlap in function (Yamamoto et al., 2007). Mice lacking ATF6α, but not ATF6β, develop liver steatosis when challenged with ER stress and have a depressed UPR (Yamamoto et al., 2010). ATF6α/β double knockouts have been studied in the model fish, the Japanese medaka (Ishikawa et al., 2013). Double knockouts were found to undergo more severe physiological ER stress in the brain, otic vesicles, and notochord, accompanied by significant decreases in expression of BiP and other chaperones (Ishikawa et al., 2013). Double knockout embryos had severely degenerated notochords in comparison to single knockouts and displayed a similar phenotype to BiP knockout embryos (Ishikawa et al., 2013). Thus, the role for ATF6 in physiological development appears to be tied to the reduced ability of secretory cells to induce chaperone proteins, thus leading to degeneration.
Studies concerning the role of ATF6 in physiological development are limited. In myoblasts, ATF6 was exclusively responsible for the induction of apoptosis during muscle development (Nakanishi et al., 2005). Other ER localized members of the ATF6 family sharing structural similarities have also been shown to play a role in development. OASIS (old astrocyte specifically induced substance) knockout mice display defects in bone formation and bone weakness caused by a decrease in a type I collagen, Col1a1 (Murakami et al., 2009). OASIS was found to bind to a UPR response element sequence in the osteoblast Col1a1 promoter (Murakami et al., 2009). Furthermore, OASIS plays a key role in astrocyte differentiation (Kondo et al., 2005). Thus, the ATF6 family may play a greater role in development than previously realized and further research is required to fully understand its role in physiological development.
3.3.3 Protein kinase RNA-like ER kinase/eukaryotic initiation factor 2α/activating transcription factor 4
The PERK pathway is essential for many facets of embryonic development. PERK knockout mice develop severe hyperglycemia and lose pancreatic insulin-secreting β cells postnatally (Zhang et al., 2006). PERK is required for β-cell proliferation and differentiation during development, but not for maintenance in the adult stage (Zhang et al., 2006). Knockouts also display skeletal dysplasia, reduced growth, and impaired locomotor activity, accompanied by decreased bone mineralization and abnormal expression of collagen 1 (Zhang et al., 2002). These skeletal defects may be related to the important role of ATF4 in skeletal development.
ATF4−/− mice have reduced and delayed bone mineralization, and reduced adult bone mass (Yang et al., 2004). ATF4 deficiency also reduced Type 1 collagen, as well as osteocalcin, which is not only a late-stage marker for osteoblast differentiation, but also possesses an ATF4 binding site in its promotor (Yang et al., 2004). Similarly, BMP2 also induced mild ER stress through the PERK/ATF4 pathway in osteogenesis (Saito et al., 2011). Alternatively, ATF4 may modulate osteogenesis through RSK2 (Ribosomal S6 protein kinase 2) (Yang et al., 2004) or regulate growth plate chondrocyte proliferation and differentiation as a transcription factor of Indian hedgehog, a factor required for skeletal development (Wang et al., 2012a).
In addition to skeletal deformities, mice lacking ATF4 exhibited severe microphthalmia caused by a complete absence of the lens (Tanaka et al., 1998; Hettmann et al., 2000). They displayed normal lens development until E14.5, at which point the lens degenerated due to severe apoptosis, suggesting a role for ATF4 in later stages of lens fiber cell differentiation (Tanaka et al., 1998). Recent studies have confirmed that UPR is activated during physiological lens development (Firtina and Duncan, 2011). Other abnormalities associated with ATF4-deficient mice include defects in post-natal hair growth, reduced body size, and impaired hematopoiesis resulting in severe anemia (Masuoka and Townes, 2002).
3.3.4 Immunoglobulin heavy-chain-binding protein
As an ER chaperone, BiP plays diverse physiological roles apart from its important role in the UPR. BiP expression in mouse embryonic development begins at the two-cell stage and its expression is barely detectable until the blastocyst stage when BiP protein levels increase significantly (Kim et al., 1990). BiP null mutations were lethal to embryos at the peri-implantation stage (E7.5) and exhibited decreased proliferation and increased apoptosis of the inner cell mass in comparison to wild-type embryos (Luo et al., 2006). This suggests an important role for BiP in early development that is potentially linked to the increased secretory protein load during this crucial period of proliferation.
Little research has been performed on the role of BiP in physiological development past the peri-implantation stage. Mimura et al. (2007) generated knock-in BiP mice with a mutant retrieval sequence that disrupted BiP return to the ER from the secretory pathway. Embryos containing this mutant BiP were unable to secrete pulmonary surfactants from alveolar cells and died shortly after birth from respiratory failure. Furthermore, these mice displayed significantly smaller brains, and defective neocortical stratification possibly resulting from decreased reelin secretion, a glycoprotein that regulates neuronal migration (Mimura et al., 2008). A genetic screen for defects in thalamocortical development found that mutated BiP-delayed axon extension, caused over fasciculation and impaired corticostriatal boundary crossing (Favero et al., 2013). These studies indicate an important role for BiP in neural development, yet more research is needed to fully understand its precise functions.
3.4 UPR in Developmental Toxicity
Considering the diverse role of the UPR in all stages of development, toxicants that interfere with the physiological UPR or agents that induce the UPR in developing embryos have the potential to generate developmental toxicity and birth defects. Induction of ER stress is not limited to exogenous toxicants; the UPR also plays a key role in chondrodysplasia caused by mutations in key extracellular matrix (ECM) components (Patterson and Dealy, 2014). Mutations in ECM components such as type II collagen, collagen X, and cartilage-matrix oligomeric protein, often result in impaired protein folding, processing, or export. As healthy chondrocytes typically secrete large volumes of these ECM proteins, the mutated unfolded proteins are retained in the ER and generate ER stress (Arnold and Fertala, 2013). Activation of the UPR typically results in reduced chondrocyte proliferation, increased apoptosis, or altered differentiation, culminating in the observed short stature and deformities of the face and joints in many types of chondrodysplasia (Patterson and Dealy, 2014). For instance, a key glycine to serine mutation in collagen 2A1 results in shortening of the femurs and humeri, suggesting disruptions in endochondral bone ossification (Liang et al., 2014). Researchers observed significant increases in the gene expression of UPR mediators, decreased proliferation and increased apoptosis resulting in elimination of the hypertrophic zone of developing endochondral bones (Liang et al., 2014).
Although there are few studies currently investigating the effects of exogenous toxicants on the UPR in chondrodysplasia, they may also have the potential to induce similar effects. ATF4 has been shown to play a role in physiological chondrocyte differentiation (Wang et al., 2012a); alteration of that pathway could result in toxicity to chondrocytes. For example, the UPR was induced in zebrafish displaying spinal curvature following treatment with silver nanoparticles (Christen et al., 2013). Furthermore, induction of ER stress following targeted mutation of collagen 10a1 in terminally differentiated hypertrophic chondrocytes causes reversion into predifferentiated cells, which have delayed ossification, generating a chondrodysplasia phenotype (Tsang et al., 2007).
4. APOPTOSIS
Programmed cell death, also known as apoptosis, is a tightly regulated process by which multicellular organisms dispose of unwanted cells. Apoptosis can occur naturally during embryogenesis or as a result of cell damage or stress. Apoptotic cells are characterized by overall cell shrinkage and membrane blebbing into small apoptotic bodies. This is accompanied by degradation of intracellular components, including condensation and fragmentation of DNA, fragmentation of organelles, and proteolysis of many proteins (Taylor et al., 2008).
The primary effectors of the apoptotic response are caspases, cysteine proteases that cleave their substrates following an aspartic acid residue (Kumar, 2007). All caspases are present in cells as inactive zymogens and exist as initiator or effector caspases. Initiator caspases 9, 2, 8, and 10 are the first to become activated in the apoptotic pathway (Fuentes-Prior and Salvesen, 2004). The N-terminal sequences of initiator caspases contain caspase recruitment domains or death effector domains, which allow them to form oligomeric complexes with adaptor proteins Fas-associated death domain (FADD) or apoptotic protease activating factor 1 (Apaf-1) (Fuentes-Prior and Salvesen, 2004). It has been proposed that dimerization of initiator PC monomers allows for activation and cleavage into the caspase form (Boatright et al., 2003). The active form will then go on to process the effector caspases.
Effector caspases 3, 7, and 6 lack the recruitment domains of the initiators and are responsible for the proteolytic processing of proteins during the apoptotic response. There have been nearly 1000 natural substrates of effector caspases identifieded (Crawford and Wells, 2011). Proteolysis by effector caspases can lead to protein loss of function, such as with structural proteins and proteins involved in transcription and translation, gain of function, such as upstream apoptotic modulators, change of function, and change of localization (Crawford and Wells, 2011). Furthermore, many targets are thought to be merely bystander proteins, whose cleavage has no immediate effect on the apoptotic response (Crawford and Wells, 2011). Caspase substrates have been found to be involved in cell adhesion, cell structure, nuclear structure, cell cycle, DNA synthesis, cleavage and repair, DNA transcription, RNA synthesis and splicing, protein translation, modification and degradation, lipid metabolism, and neurodegeneration (Fischer et al., 2003).
4.1 Extrinsic and Intrinsic Apoptosis
Apoptosis can be initiated by both intracellular and extracellular signals. The extrinsic pathway is initiated by natural killer cells, which release proapoptotic ligands that bind to death receptors on the surface of infected or damaged target cells (Ashkenazi, 2008). This event initiates a signal cascade that recruits PC 8, PC 10 (humans only), and FADD to form the death-inducing signaling complex (DISC) (Ashkenazi, 2008). The aggregation of several molecules of caspase 8 and 10 within DISC induces their autoprocessing. These initiator caspases are then able to cleave the effector caspases 3 and 7 and propagate the apoptotic response (Figure 5).
Figure 5. Extrinsic and intrinsic apoptosis.

Abbreviations: Apaf-1, apoptotic protease activating factor 1; BAK, Bcl-2 homologous antagonist/killer; BAX, BCL2-associated X; Bcl-2, B-cell lymphoma 2; c-FLIP, cellular FLICE (FADD-like IL-1β-converting enzyme)-inhibitory protein; Casp, caspase; CytC, cytochrome C; FADD, Fas-associated protein with death domain; FAS-L, FAS ligand; PC, procaspase.
In contrast, the intrinsic pathway of apoptosis, also called the mitochondrial pathway, is initiated by cell stress or damage, such as oxidative stress or DNA damage (Figure 5). The antiapoptotic Bcl-2 (B-cell lymphoma 2) family of proteins tightly regulates this pathway. This family inhibits the proapoptotic family of proteins, BH3-only (Bcl-2 homology 3 domain only). These proapoptotic molecules regulate another family of BH (Bcl-2 homology) domain proteins, which include BCL2-associated X (BAX) and Bcl-2 homologous antagonist/killer (BAK). As BH3-only protein signaling overcomes Bcl-2 inhibition, BAX and BAK assemble into oligomers in the mitochondrial membrane, promoting the release of cytochrome C into the cytoplasm (Taylor et al., 2008). Cytochrome C release initiates assembly of the apoptosome, containing Apaf-1 and oligomers of PC 9, which then goes on to activate effector caspases and propagate the response.
4.2 Apoptosis in Physiological Development
Apoptosis regulates many developmental processes including morphogenesis, deletion of vestigial structures, cell number regulation, and elimination of dangerous cells as the broad functions of cell death in development (Fuchs and Stellar, 2011) (Figure 6). These categories characterize the variety of important roles that apoptosis can play in development. Apoptosis in limb development is mediated by BMP and signaling forms the joints and the separation of the digits (Mori et al., 1995; Zou and Niswander, 1996; Macias et al., 1997). Apoptosis also shapes structures by removing cells to form cavities and the four-chambered heart from the endocardial cushion (Abdelwahid et al., 2002). Furthermore, inendochondral bone formation, chondrocytes undergo apoptosis to stimulate calcification and blood vessel perfusion (Gibson, 1998).
Figure 6.

Apoptosis in physiological development.
Overproduction of cells during development is common and apoptosis is necessary to regulate appropriate cell number. During immune system development lymphocytes are subjected to multiple checkpoints. Random genetic recombination generates unique antigen receptors, which must be properly aligned and evaluated for autoreactivity (Rathmell and Thompson, 2002). If the junctions are not joined in frame or the receptor is autoreactive, the developing T or B cell will undergo apoptosis (Rathmell and Thompson, 2002). Apoptosis also plays an important role in the development of the vertebrate nervous system. From 30% to 80% of neural and glial cells undergo apoptosis throughout the course of development (Buss et al., 2006). It has been suggested that cell death in the developing nervous system functions to ensure sufficient innervation, regulate cell number, and correct errors in cell location or axonal pathfinding (Buss et al., 2006).
On the cellular level, knockouts of apoptotic mediators and effectors can result in embryonic mortality or deformities, demonstrating the key role for apoptosis in development. For instance, knockout of caspase 3, caspase 9, and Apaf-1 results in mortality and forebrain malformations, suggesting an important caspase-dependent role for intrinsic apoptosis on brain development (Yoshida et al., 1998b; Kuida et al., 1996, 1998). In the extrinsic pathway, knockout of caspase 8 or the FAS adaptor protein, FADD, generates cardiac defects that result in embryonic lethality (Yeh et al., 1998; Varfolomeev et al., 1998).
4.3 Apoptosis in Developmental Toxicity
Although apoptosis is a key event in development, cell death can severely impact formation and growth during development by acting on either actively proliferating cells or by expanding the area of cells already programmed for apoptosis. In early development, different cell lineages may confer differing susceptibility to apoptosis, depending on their metabolic requirements and microenvironments (Pampfer, 2000). It is generally accepted that if cell damage is too severe and ATP stores entirely depleted, the cell will undergo necrosis. However, apoptosis under less severe exogenous stress in development is still common. Thus, although apoptosis resulting from a developmental toxicant may be the final outcome for a cell or tissue, apoptosis is not the initiating event of toxicity. One interesting example of this is the potent immunosuppressant and chemotherapeutic agent, cyclophosphamide.
Cyclophosphamide is a well-studied teratogen, causing growth deficiencies, craniofacial abnormalities, and limb malformations (Vaux et al., 2003). As a treatment for cancer, cytotoxicity is the goal of cyclophosphamide’s drug use; thus, it is no surprise that embryonic development is particularly susceptible to toxicity. CYP450s activate cyclophosphamide to a variety of toxic metabolites (Mirkes, 1985). These metabolites are well known to cause DNA strand breaks, cross-linking, and adducts (Ozolins, 2010). The DNA damage has been shown to induce extensive cell death to embryonic limbs (Moallem and Hales, 1998), tail (Torchinsky et al., 1995), neural tube (Xiao et al., 2007), facial structures, and somites (Chernoff et al., 1989). Illustrating the varying vulnerability of embryonic tissues to apoptosis, the embryonic heart appears resistant to cyclophosphamide apoptosis, while neural tissues are particularly sensitive (Mirkes and Little, 1998; Soleman et al., 2003). However, the mechanism behind this resistance remains unknown.
The volume of research focusing on the pathway of apoptosis from cyclophosphamide has demonstrated its complexity. Caspase 3A and poly (ADP-ribose) polymerase, a polymerase associated with DNA repair, were found to be activated in mouse embryos treated with cyclophosphamide (Mirkes and Little, 1998). A role for p53 has been associated with cyclophosphamide apoptosis, as embryonic mice expressing functional p53 have apoptosis in their limbs, while those with a p53 knockout undergo extensive necrosis (Moallem and Hales, 1998). p53 protein also accumulates in the head and trunk of murine embryos exposed on E9, but not in the heart (Hosako et al., 2007). Cytochrome C release also occurred in these cells (Mirkes and Little, 2000), as did activation of caspase 9 (Soleman et al., 2003), indicating a role for intrinsic apoptosis. Others found an increase in BAX expression; however, this was not accompanied by a decrease in Bcl-2 (Mammon et al., 2006). NF-κB and TNFα (tumor necrosis factor α) have also been implicated in cyclophosphamide teratogenicity. A decrease in NF-κB activity has been observed (Torchinsky et al., 2002; Molotski et al., 2008); however, others have reported an increase (Pekar et al., 2007), following embryonic cyclophosphamide treatment. TNFα may also play a protective role, as knockouts exhibited greater malformations than wild type (Torchinsky et al., 2003). Studies on cyclophosphamide as a model developmental toxicant have provided a wealth of knowledge concerning the mechanisms behind other teratogens and into the nature of development itself.
Environmental toxicants also pose a risk for developmental toxicity caused by apoptosis. TCDD (2,3,7,8-tetrachlorodibenzo-para-dioxin) is a member of the dioxin family and enters the environment following use in herbicide manufacturing and the combustion of organic materials. In mammals, TCDD causes fetal mortality, growth retardation, edema, lymphoid organ hypoplasia, and cleft palate at concentrations below the threshold for maternal toxicity (Birnbaum, 1995). In fish, it causes yolk sac edema, craniofacial abnormalities, hemorrhage, and mortality (King-Heiden et al., 2012). TCDD is an ideal ligand for the AhR, a transcription factor regulating xenobiotic metabolism, cell–cell interactions, cell cycle control, and the endocrine system (King-Heiden et al., 2012). It is believed that TCDD exerts its primary effects through the AhR, but oxidative stress has also been suggested as an additional mechanism of toxicity.
Several studies have implicated apoptosis in TCDD developmental toxicity. In Japanese medaka (Oryzias latipes), apoptosis was detected in the brain and heart of late-stage embryos treated with 4 pg of TCDD at fertilization (Cantrell et al., 1996). These effects were eliminated following treatment with a CYP450 inhibitor and decreased with antioxidants (Cantrell et al., 1996). Treatment of Fundulus heteroclitus embryos with TCDD generated apoptosis in the brain, eye, gill, kidney, tail, heat, intestine, and blood vessels (Toomey et al., 2001). Apoptosis was detected in the dorsal midbrain of zebrafish embryos following treatment with0.3 ppb TCDD, and was inhibited by coexposure with antioxidants and CYP450 inhibitors (Dong et al., 2002).
Exact mechanisms of TCDD-induced apoptosis are unclear. Several studies suggest a role for the extrinsic pathway in TCDD-induced immunotoxicity. Apoptosis in thymocytes of TCDD-treated mice was inhibited in Fas and Fas-L knockouts (Rhile et al., 1996; Kamath et al., 1999). Furthermore, increased levels of Fas, TRAIL (TNF-related apoptosis-inducing ligand), and DR5 (death receptor 5) mRNA were observed in postnatal mice treated with TCDD during gestation, while no changes in BAX or Bcl-2 expression was observed (Camacho et al., 2004). Prenatal exposure to TCDD also caused a significant decrease in microRNAs with sequences specific for Fas, Fas-L, and CYP1A1 and AhR in thymocytes (Singh et al., 2012).
5. INTEGRATION OF OXIDATIVE STRESS, UPR, AND APOPTOSIS
5.1 Positive Feedback in Oxidative Stress/UPR Signaling
As ER fidelity and redox homeostasis are essential for nearly all cellular processes, it stands to reason that a complex interplay between the UPR and oxidative stress would exist (Figure 7). Oxidative stress can interfere with protein folding via disruption of disulfide bonds or through inhibition of Ca2+ATPase, resulting in inactivation of enzymes or important signaling molecules. If oxidative stress in the vicinity of the ER leads to the accumulation of these misfolded proteins, the UPR can be activated to combat the response. Furthermore, the UPR itself has been implicated in the generation of ROS (Harding et al., 2003), resulting in a positive feedback loop between the UPR and oxidative stress. In contrast to the reducing environment of the cytosol, the ER lumen is oxidizing, which promotes formation of disulfide bonds (Van der Vlies et al., 2003). Protein disulfide isomerase (PDI) is a member of the Prx superfamily and catalyzes disulfide bond formation, isomerization, and reduction of proteins within the ER (Ferrari and Soling, 1999). Following oxidation of protein thiols, the reduced form of PDI is oxidized by ER oxidoreductins (ERO), ERO1α, and ERO1β (Pagani et al., 2000). ERO1 uses a flavin-dependent mechanism to transfer electrons to molecular oxygen, which may generate ROS (as superoxide and H2O2) as a result of electron uncoupling (Tu and Weissman, 2004).
Figure 7. Interplay between the unfolded protein response and redox potential.

Abbreviations: ATF4, activating transcription factor 4; BiP, immunoglobulin heavy-chain-binding protein; CHOP, C/EBP homologous protein; eIF2α, eukaryotic initiation factor 2α; ERO1, ER oxidoreductin 1; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; Nrf2, nuclear factor (erythroid-derived 2)-like two; PDI, protein disulfide isomerase; PERK, protein kinase RNA-like ER kinase.
In addition to this causative association between oxidative damage and the UPR, other more complex interactions between redox and the UPR function in networks of gene expression and signal transduction throughout embryonic development. Many UPR transcription factors have gene targets that are involved in the response to oxidative stress or are redox regulated. The association between oxidative stress and the UPR has recently been established in several human diseases (Van der Vlies et al., 2003; Cao and Kaufman, 2014). However, their relationship with physiological embryonic development and developmental toxicity has yet to be thoroughly explored.
One connection between the UPR and oxidative stress lies within the PERK pathway. Although eIF2α is the primary target of PERK, PERK has the capacity to phosphorylate other substrates, one of which is Nrf2 (Cullinan et al., 2003). PERK phosphorylation promotes release of Nrf2 from Keap1 and its translocation into the nucleus, independently of eIF2α (Cullinan et al., 2003). Furthermore, ER-stressed cell survival was reduced in Nrf2 knockouts (Cullinan et al., 2003). Nrf2−/− mice challenged with ER stress by Tm had changes in expression of genes involved in apoptosis, cell cycle, glucose biosynthesis, calcium homeostasis, ER/golgi transport and biosynthesis, and drug metabolism and transport (Nair et al., 2007). Another component of the PERK pathway, ATF4 is an Nrf2 interacting protein, specifically in the regulation of heme oxygenase-1. This is consistent with data indicating that fibroblasts increase ATF4 DNA-binding in response to anoxia (Estes et al., 1995) and the fact that ATF4−/− cells are more susceptible to oxidative stress (Harding et al., 2003).
Another important redox-regulated transcription factor, NF-κB is also regulated by mediators of the UPR (Pahl and Baeuerle, 1995). However, this signal transduction pathway is thought to be separate from the UPR and has been designated the ER overload response (Jiang et al., 2003). In PERK signaling, phosphorylation of eIF2α is required for activation of NF-κB under ER stress and stress caused by amino acid starvation (Jiang et al., 2003) possibly via translational inhibition of NF-κB inhibitor, IκBα (Deng et al., 2004). The IRE1 and ATF6 branches of the response are also thought to be involved in NF-κB signaling (Hu et al., 2006; Yamazaki et al., 2009).
5.2 Oxidative Stress-Induced Apoptosis
The delicate balance between endogenous ROS and ROS defense systems can be tipped by exogenous toxicants to generate the cellular damage discussed above. Oxidative damage can initiate apoptosis through a complex web of pathways and mechanisms. Here we will give a brief overview of apoptosis resulting from ROS-induced mitochondrial damage, disruptions in calcium homeostasis, and DNA damage (Figure 8).
Figure 8. Oxidative stress-induced apoptosis.
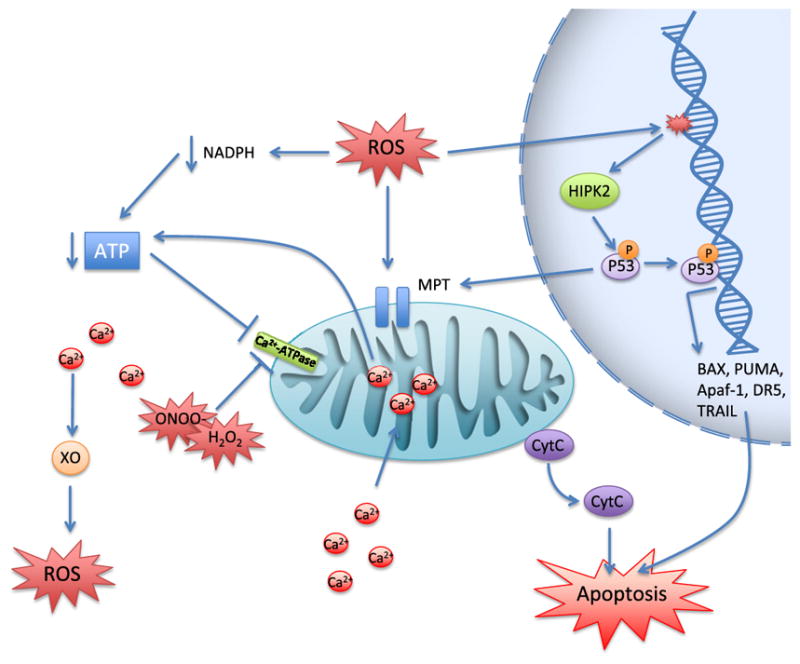
Abbreviations: Apaf-1, apoptotic protease activating factor 1; BAX, BCL2-associated X; CytC, cytochrome C; DR5, death receptor 5; HIPK2, homeodomain interacting protein kinase 2; MPT, mitochondrial permeability transition; PUMA, p53-upregulated modulator of apoptosis; TRAIL, TNF-related apoptosis-inducing ligand; XO, xanthine oxidase.
Mitochondrial damage can result in mitochondrial permeability transition (MPT; pores through the inner and outer mitochondrial membrane), which causes release of cytochrome C and activation of the intrinsic pathway. MPT is induced by ROS (Kim et al., 2003) and oxidation of mitochondrial proteins exposes hydrophilic moieties, which leads to protein aggregation and pore formation in the membrane (Kowaltowski et al., 2001). MPT can also be induced by NADPH oxidation, which can occur as ROS deplete NADPH through the GSH pathway (Kowaltowski et al., 2001). Furthermore, lipid peroxidation can destroy the mitochondrial membrane, causing a release of intracellular Ca2+ loss of membrane potential and attenuation of ATP production (Kowaltowski and Vercesi, 1999), leading to MPT.
ROS can also induce apoptosis via disruption of calcium homeostasis. Calcium is sequestered in the extracellular space, ER, and mitochondria in order to keep cytoplasmic Ca2+ levels low and maintain membrane potential. Peroxynitrite and H2O2 can inactivate Ca2+ ATPase causing accumulation of cytoplasmic Ca2+ (Viner et al., 1996; Zaidi et al., 2003). Furthermore, ROS can induce ATP depletion by consumption of NADPH and inactivation of the electron transport chain depriving Ca2+ ATPase of substrate, leading to inactivation. Elevation of intracellular Ca2+ can harm the cell in several ways. Ca2+ uptake by the mitochondria can cause ATP depletion via disruption of membrane potential eventually leading to MPT (Kim et al., 2003). Elevated Ca2+ can also induce proteolytic cleavage of hydrolases that activate XO, leading to increased ROS, providing a positive feedback loop between hypercalcemia and ROS production (Harrison, 2002).
Alternatively, ROS can cause oxidative damage to DNA. When this damage is irreparable, the apoptotic response is mediated by the transcription factor p53. p53 phosphorylation by homeodomain interacting protein kinase 2 results in its activation and transcription of its proapoptotic target genes, BAX, Apaf-1, PUMA (p53-upregulated modulator of apoptosis), and p53AIP1 (p53-regulated apoptosis-inducing protein 1) (Surova and Zhivotovsky, 2013). In addition to these intrinsic proteins, p53 can also transcribe various factors of the extrinsic pathway, including TRAIL and DR5. p53 can further function outside its role as a nuclear transcription factor and has been shown to cause caspase activation, Bcl-2 inhibition, and MPT in the cytoplasm (Surova and Zhivotovsky, 2013).
5.3 ER Stress-Induced Apoptosis
Apoptosis induced by the UPR has been shown to proceed via both the intrinsic pathway and extrinsic pathway (Timmins et al., 2009; Hu et al., 2006). Exact mechanisms resulting in apoptosis from the UPR are unknown; however, several UPR mediators have shown to regulate cell death (Figure 9). IRE1α has both endoribonuclease and kinase functions, and several mechanisms have been proposed for IRE1α modulation of apoptosis. One study found that RIDD was a significant contributor to apoptosis (Han et al., 2009). Furthermore, IRE1α was found to cleave select microRNAs that repress translation of proapoptotic caspase 2, an initiator caspase (Upton et al., 2012). IRE1α cleavage of five microRNAs resulted in significant increases in caspase 2 protein levels to regulate apoptosis (Upton et al., 2012).
Figure 9. Oxidative stress induced by the unfolded protein response.

Abbreviations: ASK1, apoptosis signal-regulating kinase 1; ATF4, activating transcription factor 4; BAX, BCL2-associated X; Bcl-2, B-cell lymphoma 2; Casp, caspase; CHOP, C/EBP homologous protein; eIF2α, eukaryotic initiation factor 2α, ERO1, ER oxidoreductin 1; IRE1, inositol-required enzyme 1; JNK, c-Jun N-terminal kinase; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; PERK, protein kinase RNA-like ER kinase; RIDD, regulated IRE1-dependent decay; TRAF2, TNF receptor-associated factor 2; XBP-1, X-box protein 1.
Conversely, several important apoptosis-related proteins have been shown to be IRE1α interactors (Chen and Brandizzi, 2013), indicating an integral and complex role for IRE1 in UPR-mediated cell death. Specifically, IREα was shown to be able to bind proapoptotic factors, BAX, and BAK (Hetz et al., 2006). Mice lacking BAX-BAK that are challenged with Tm have fewer apoptotic cells and display normal PERK and BiP signaling, but have defects in the IRE1 pathway, indicating an important role for IRE1 and BAX-BAK in UPR-induced cell death (Hetz et al., 2006). IRE1α was also shown to interact with BAX-BAK and binding increased under ER stress and may modulate IRE1α activation through their proapoptotic BH3 and BH1 domains (Hetz et al., 2006). Furthermore, expression of several BH3-only proteins has been shown to increase under ER stress (Sovolyova et al., 2014).
Another potential mechanism of IRE1α-mediated apoptosis occurs through the c-Jun N-terminal kinase (JNK) pathway, a well-known initiator of apoptosis. ER stress has been shown to induce JNK in an IRE1-dependent manner through IRE1 interaction with TRAF2 (tumor necrosis factor receptor (TNFR)-associated factor 2), which is mediated through its kinase domain (Urano et al., 2000). Another study demonstrated that apoptosis signal-regulating kinase 1 (ASK1) is also another essential component of this complex for the induction of apoptosis; primary neurons lacking ASK1 were resistant to ER stress and apoptosis (Nishitoh et al., 2002).
The PERK pathway has been shown to be involved in apoptosis, through the ATF4 target gene, CHOP (C/EBP homologous protein), a leucine zipper transcription factor activated during cellular stress. As with IRE1, multiple associations between CHOP and ER stress-induced apoptosis have been drawn. In vivo, CHOP-deficient mice had decreased cardiomyocyte apoptosis following transverse aortic constriction in comparison to controls (Fu et al., 2010). Much of the CHOP regulation of apoptosis has been associated with oxidative stress. Elevated CHOP expression has been linked to decreases in Bcl-2 downregulation, which is accompanied by a depletion of GSH and increased ROS production in mouse embryonic fibroblasts (McCullough et al., 2001).
Furthermore, ERO1α is a transcriptional target of CHOP (Marciniak et al., 2004). ERO1α has been implicated in Ca2+ regulation (Anelli et al., 2012) and has been shown to be involved in the release of ER Ca2+ stores during ER stress. Increases in cytoplasmic calcium have been linked to Fas death receptor induction and the extrinsic pathway of apoptosis through calcium/calmodulin-dependent protein kinase IIγ (CaMKIIγ) (Timmins et al., 2009). CaMKIIγ was also linked to intrinsic apoptosis through accumulation of mitochondrial Ca2+ leading to cytochrome c release during ER stress (Timmins et al., 2009). This was further connected to an increase in ROS via activation of NOX, which was able to induce CHOP through a positive feedback loop (Li et al., 2010). Thus, CHOP-induced oxidative stress and Ca2+ signaling may play a role in ER stress-induced apoptosis. Recent work has suggested an integral role for caspase 8 and DR5 in ER stress-induced apoptosis (Lu et al., 2014). ER stress was shown to upregulate DR5 transcripts via CHOP and depletion of caspase 8 and DR5 inhibited ER stress-induced apoptosis (Lu et al., 2014). The role of DR5 was shown to be independent of ligand binding (Lu et al., 2014).
6. INTERSECTION OF UPR, APOPTOSIS, AND OXIDATIVE STRESS IN DEVELOPMENTAL TOXICITY
After considering the importance of each pathway throughout development, the overlapping role of redox, the UPR, and apoptosis in cell signaling and developmental processes indicates the potential for interplay in developmental toxicity. While the majority of studies concerning the role of the UPR, redox, and apoptosis in physiological development focus on the use of knockouts, toxicants that disturb development may function via either inhibition or activation of these processes. For instance, toxicants that induce oxidative stress via redox cycling in the vicinity of the ER may lead to the induction of the UPR, interfering with normal signaling, and causing apoptosis. As secretory protein load increases throughout embryonic development, the induction of oxidative stress may impair folding of these proteins, which the UPR is unable to correct, potentially leading to apoptosis. The ability of oxidative stress and the UPR to induce each other and apoptosis in concert or individually, suggests a wide range of possible developmental toxicities from exogenous toxicants. There are few studies concerning the combined role of oxidative stress, the UPR, and apoptosis in developmental toxicity. However, more recent work can draw parallels between these processes in birth defects that are not well understood. Here, we will discuss the potential interplay of the UPR, oxidative stress, and apoptosis in pathologies induced by a physical stressor, hypoxia; a nonchemical stressor, diabetic embryopathy; and a chemical stressor, fetal alcohol syndrome.
6.1 Hypoxia-Induced Developmental Toxicity
While mammalian embryos experience hypoxia during the early postimplantation stages of development, allowing for a reduction in ROS generated from oxidative phosphorylation, hypoxia at later stages and excess hypoxia at earlier stages can lead to developmental toxicity and birth defects. Fetal hypoxia can be induced by a number of factors, including severe maternal anemia, gestation at high altitudes, vasodilator inhibitors, drug-induced uterine vessel constriction, and drugs causing embryonic heart brachycardia (Webster and Abela, 2007). Drugs that induce uterine vessel constriction include cocaine and nicotine, while those causing embryonic brachycardia are drugs used as therapy for cardiac arrhythmia, such as almokalant and dofetilide (Webster and Abela, 2007). Hypoxia can generate a range of congenital malformations, the most common of which are growth retardation, limb defects, neurological disorders, hypoplasia of genitalia or face, cardiac defects, and eye defects (Webster and Abela, 2007).
The cellular response to hypoxia is regulated by HIF transcription factors, and HIF-1 in particular is crucial for oxygen homeostasis. HIF-1 protein levels are maintained low during normoxic conditions following degradation of the HIF-1α subunit (Wang et al., 1995) by PHD. Under hypoxic conditions, degradation of HIF-1α is attenuated and HIF-1 binds to the hypoxia response element (HRE) in its target genes (Wang et al., 1995). HIF-1 targets include genes involved in apoptosis, angiogenesis (e.g., VEGF), cellular metabolism, and differentiation (Liu et al., 2012).
6.1.1 Oxidative stress generated by hypoxia
Oxidative stress is a known contributor of developmental toxicity from hypoxia during pregnancy and malondialdehyde, a product of lipid peroxidation, is often used as a biomarker of hypoxia in newborns (Plank et al., 2008). Furthermore, it was found that GPx1 overexpression was protective against hypoxia in neonatal mouse brains, although SOD overexpression was not (Sheldon et al., 2008). Three sources of hypoxia-induced oxidative stress have been identified (Figure 10). First, ROS are generated by the mitochondria during the first few minutes of hypoxia in astrocytes and neural cells, whereas later in hypoxia and during reoxygenation, XO generates large numbers of ROS, and finally, NOX produces further oxidative stress during reoxygenation (Abramov et al., 2007).
Figure 10. The unfolded protein response and oxidative stress induction by hypoxia.
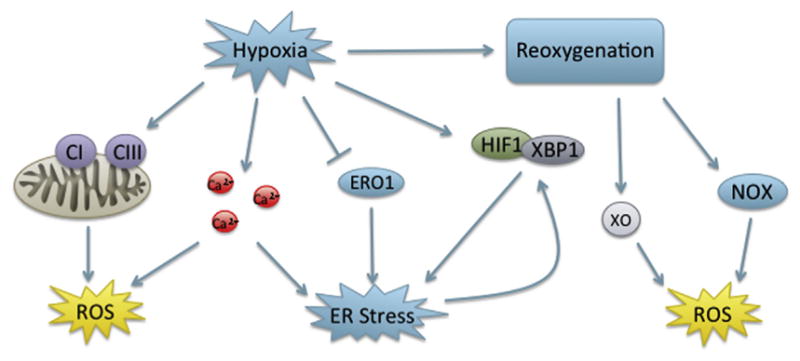
Abbreviations: CI and CIII, complex I and III; ERO1, ER oxidoreductin 1; HIF, hypoxia-inducible factor; NOX, NADPH oxidase; XO, xanthine oxidase; XBP-1, X-box protein 1.
As mentioned above, the mitochondria are the primary producers of physiological ROS and research has shown that it contributes significantly to oxidative stress following hypoxia. In cardiomyocytes, the mitochondria are responsible for ROS generation at complexes I and III during ischemia, while ROS were not generated by XO, NOS, and NOX (Becker et al., 1999). Work on neonatal mice found that the C1q component of the complement pathway was responsible for ROS produced by the mitochondria following hypoxic-ischemia in the brain (Ten et al., 2010).
The contribution of the mitochondria to oxidative stress generated by hypoxia is under debate, as studies have found inhibition of mitochondrial complexes has no effect on overall tissue injury (Abramov et al., 2007). It has been traditionally recognized that the primary mechanism of ROS induction in hypoxia occurs during reoxygenation rather than under hypoxic conditions. As oxygen levels decrease, the cell switches from oxidative phosphorylation to glycolysis. During hypoxia, ATP is degraded to hypoxanthine, which is then oxidized by XO during reoxygenation (Collard and Gelman, 2001; Engerson et al., 1987). Studies in neonatal rats found that treatment with allopurinol, an XO inhibitor, significantly decreased pulmonary hypertension in a similar manner as ROS scavengers (Jankov et al., 2008). Similar results were demonstrated in newborn piglets subjected to hypoxia-ischemia (Peeters-Scholte et al., 2003). Piglets treated with allopurinol had significantly less cerebral edema; however, no significant difference in apoptosis was observed (Peeters-Scholte et al., 2003).
A third contributor to oxidative damage from hypoxia-ischemia is NOX. Although NOX was once thought to localize exclusively in phagocytes, it has since been discovered in tissues throughout the body. NOX4 is an HIF-1 target gene and NOX4 levels increase significantly along with ROS in pulmonary artery smooth muscle cells (Diebold et al., 2010). Additionally, studies in neonatal rats found that inhibition of NOX significantly decreased ROS and brain injury and that NOX inhibition in hippocampal slices decreased ROS and apoptosis (Lu et al., 2012).
6.1.2 Hypoxia and UPR
UPR induction by hypoxia has been long recognized and is recently under further investigation for its role in fetal hypoxia. Hypoxia in human fibro-blasts resulted in phosphorylation of eIF2α and protein translational attenuation (Koumenis et al., 2002) and BiP expression increased in the neurons of ischemic rat brains (Ito et al., 2001). In astrocytes, sustained increases in CHOP expression were found to be necessary for astrocyte cell death following oxygen glucose deprivation. In vivo, CHOP knockout rats had decreased renal apoptosis and ROS following hypoxia-reperfusion (Chen et al., 2014b). Specific to neonatal hypoxia, Carloni et al. (2014) observed an increase in BiP, CHOP, spliced XBP-1, and phosphorylated eIF2α in neonatal rat brains following hypoxia. One study found that nonnative women living at high altitudes (3100 m) had increased placental phosphorylated eIF2α (Yung et al., 2012).
It is believed that ER stress in hypoxia results from the inability of PDI and ERO1 to generate disulfide bonds (Figure 10). Because ERO1 uses oxygen as its cofactor, it is unable to form disulfide bonds during hypoxia (Tu and Weissman, 2002). Another hypothesis is that hypoxia causes a release of Ca2+ stores from the ER and mitochondria, generating ER stress. Astrocytes demonstrated a release of ER Ca2+ accompanied by mitochondrial release of cytochrome C and increased phosphorylation of eIF2α during reoxygenation following oxygen/glucose depravation (Liu et al., 2010). Release of ER Ca2+ was abolished when treated with a Na+-K+-Cl− inhibitor or a Na+/Ca2+ inhibitor, suggesting a role for these transporters in these effects (Liu et al., 2010). Another study found that this release in ER Ca2+ was linked to the G protein-coupled receptor, the calcium sensing receptor, which induced the UPR and apoptosis in cardiomyocytes (Lu et al., 2010).
Furthermore, connections between the UPR and HIF-1 have recently been established. The UPR has been found to potentiate HIF-1 under hypoxia, which led to an increase in VEGF-A, an important factor in angiogenesis (Pereira et al., 2014). Another study found XBP-1 to interact physically with HIF-1α in the nucleus of hypoxic 293T cells and co-occupy common targets (Chen et al., 2014a).
6.1.3 Apoptosis from hypoxia
Apoptosis and necrosis have both been cited as mechanisms of cell death causing tissue damage following hypoxia in neonates (Northington et al., 2001). However, the mechanism behind hypoxia-induced apoptosis is complex, with involvement from both the intrinsic and extrinsic pathways depending on cell type and duration of stress. Weinmann et al. (2004) demonstrated that the extrinsic pathway was expendable to hypoxia-induced apoptosis in Jurkat cells, while the intrinsic pathway was essential. In a chick embryo model, hypoxia resulted in apoptosis of optic tectum cells, which was characterized by cytochrome C release and caspase 9 cleavage, but not caspase 8 activation (Pozo Devoto et al., 2013), also indicating involvement of the intrinsic pathway. Furthermore, inhibition of caspase 9 in neonatal rat cortical neurons decreased apoptosis and BiP levels, providing a connection between intrinsic apoptosis and the UPR (Zhang et al., 2013). Although cleavage of both caspase 8 and caspase 3 were observed in renal tissues of rats following hypoxia reoxygenation, no increase in BAX or Bcl-2 was observed (Chen et al., 2014b). Hypoxia-ischemia-treated neonatal mice had increased TRAIL and DR5 levels in the brain after 8 and 24 h (Kichev et al., 2014). Additionally, the Fas death receptor was upregulated in the brains of neonatal rats following hypoxia, and apoptosis was blocked by caspase 8 inhibition (Felderhoff-Mueser et al., 2000). Still, upregulation of Fas in the brains of neonatal mice following hypoxia was associated with both intrinsic and extrinsic apoptosis, suggesting that the interplay between the two in neonatal hypoxia is complex (Graham et al., 2004).
The function of HIF in hypoxia-induced apoptosis is unclear, as studies reporting both a role in inhibition and induction of apoptosis have been published. The proapoptotic protein BNIP3 (Bcl-2/adenovirus EIB 19kD-interacting protein 3) and its homolog Nip-3-like protein 3 have both been found to be targets of HIF-1 (Sowter et al., 2001). Furthermore, HREs have been found in the promoters of caspase 3 and caspase 9 (Liu et al., 2002; Nishiyama et al., 2001). Studies on neonatal rat cardiomyocytes found that inhibition of HIF-1α resulted in decreased apoptosis, accompanied by decreased caspase 3 and BNIP3 (Wang et al., 2012b). However, others have found that overexpression of HIF-1α in cardiac fibroblasts resulted in decreased apoptosis (Yang et al., 2014). Thus, the mechanism of apoptosis following hypoxia is varied and may incorporate both the intrinsic and extrinsic forms of apoptosis depending on the duration, location, and intensity of the hypoxia.
6.2 Metabolic Disorders and Diabetic Embryopathy
Considering the important role for the UPR in pancreatic β-cell development as well as in processing of key metabolic proteins, it is clear that alterations in these processes during development could result in metabolic disorders and developmental toxicity. Diabetic embryopathy is a condition whereby offspring of diabetic mothers are born with congenital abnormalities of the brain, spinal cord, heart, kidneys, gut, and skeleton (Loeken, 2008). The diverse array of malformations associated with maternal diabetes makes isolating a single teratogenic mechanism difficult. However, oxidative stress, ER stress, and apoptosis have all been implicated separately and in conjunction in the condition.
6.2.1 Oxidative stress-induced diabetic embryopathy
In the 1990s, studies began investigating the role of ROS in diabetic embryopathy. It was found that malformations in embryos cultured in a high-glucose environment could be prevented with the addition of free radical scavenging enzymes, SOD, CAT, and GPx (Eriksson and Borg, 1991). Further studies found increased ROS and significantly decreased GSH in embryos cultured from diabetic rats (Menegola et al., 1996; Sakamaki et al., 1999), and induced lipid peroxidation (Wenztel et al., 1999). The observed increase in oxidative stress was generated by several factors (Figure 11). Early research demonstrated that developmental defects were not due directly to maternal insulin, but were due instead to maternal hyperglycemia (Widness et al., 1983; Miller et al., 1981). In one study, hyperglycemia generated increased glucose flux was linked to an increase in glucosamine-6-phosphate, an inhibitor of glucose-6-phosphate dehydrogenase, which is responsible for the regeneration of NADPH (Horal et al., 2004). As a reducing equivalent for GSH, decreases in NADPH lead to depleted levels of reduced GSH.
Figure 11. Hyperglycemic induction of oxidative stress.

Abbreviations: GSH, glutathione; iNOS, inducible nitric oxide synthase; JNK, c-Jun N-terminal kinases; NOX, NADPH oxidase; PKC, protein kinase C.
Hypoxia may also play a role in hyperglycemic ROS production. Increased glucose levels during early development, during which the environment is naturally hypoxic, could result in increased glycolysis, depleting the already low oxygen levels (Li et al., 2005). This hypoxia, followed by reoxygenation during vascularization could result in increased ROS. Others have suggested that hyperglycemia leads to increased activity of ROS generating enzymes such as NOX and NOS. Hyperglycemia has been shown to induce protein kinase C (PKC) signaling through increased production of diacylglycerol in mice models of diabetic embryopathy (Hiramatsu et al., 2002). Studies in mesanglial cells have demonstrated the ability of PKC signaling to induce NOX expression when cultured in high glucose, which significantly contributed to ROS production (Hua et al., 2003). Expression of iNOS has also been shown to increase in the murine yolk sac vasculature following exposure to high glucose (Nath et al., 2004) with associated increases in RNS (Yang et al., 2010) and was ameliorated by exogenous SOD1 (Weng et al., 2012). Increased iNOS activity in mouse embryos has been shown to be modulated by the proapoptotic JNK pathway (Yang et al., 2010).
6.2.2 Apoptosis and diabetic embyropathy
Oxidative stress generated by maternal diabetes has been linked to apoptosis in several different embryopathies, including neural tube, cardiac, and kidney defects (Phelan et al., 1997; Reinking et al., 2009). Characteristic neural tube defects have been linked to ROS-mediated inhibition of Pax3 (paired box 3), a transcription factor necessary for neural tube closure, leading to apoptosis (Phelan et al., 1997; Chang et al., 2003). Apoptosis resulting from Pax3 inhibition is rescued in p53 loss of function mouse embryos (Pani et al., 2002).
Recent research has further elucidated the apoptotic pathway in diabetes-induced neural tube defects. Preliminary studies have shown the importance of caspase 8 in hyperglycemia-induced apoptosis, as caspase 8 knockouts have significantly decreased apoptosis and deformities (Zhao et al., 2009). Others have demonstrated that the MAP3K (Mitogen-activated protein kinase kinase kinase) ASK1 induction of FoxO3a (Forkhead box O3a), and further induction of caspase 8 was responsible for observed neural tube defects (Yang et al., 2013). Furthermore, this entire pathway could be blocked by the addition of Trx, emphasizing the importance of oxidative stress in hyperglycemic apoptosis (Yang et al., 2013).
Associations between oxidative stress and apoptosis have also been formed through the JNK pathway. JNK signaling has been shown to increase in diabetic embryopathy and knock out of JNK activity prevented malformations caused by maternal diabetes (Yang et al., 2007) and yolk sac vasculopathy (Yang et al., 2008). Furthermore, overexpression of SOD1 abrogates hyperglycemic induction of JNK, suggesting that this proapoptotic JNK activity is modulated by increases in ROS (Li et al., 2012).
6.2.3 UPR and diabetic embryopathy
ER stress generated by hyperglycemia has been implicated in diabetes (Thomas et al., 2010), as illustrated by the importance of PERK to β-cell development. Recent research has begun to connect ER stress and the UPR to malformations from maternal diabetes. ER stress was first examined during cardiogenesis in embryos from hyperglycemic female mice (Zhao, 2012). Researchers found increased phosphorylated eIF2α and calnexin in the endocardial cushions and myocardium of embryos from diabetic mothers (Zhao, 2012). Further work on neural tube defects found increased levels of these factors along with phosphorylated PERK in the neural tubes of mice with neural tube defects (Zhao et al., 2012). Treatment of mice with the NOS-2 inhibitor, L-N6-(1-iminoethyl)lysine, eliminated these effects and significantly decreased cleaved caspase 8 (Zhao et al., 2012).
The details of the UPR in diabetic embryopathy have only recently begun to be investigated. A key paper by Li et al. (2013) found increases in ER chaperone gene expression, including BiP; increased CHOP; phosphorylated PERK and spliced XBP-1, indicating a role for each of the branches of the UPR. The researchers connected these events to the activation of JNK1 and 2, as JNK knockouts significantly decreased UPR activation. Furthermore, treatment of embryos cultured in high glucose with 4-phenylbutyric acid, an ER stress inhibitor, significantly decreased the observed JNK activation and neural tube defects. Finally, treatment with 4-PBA also significantly decreased caspase 3 and 8 activation. In complement to this study, SOD1 overexpression was also found to abolish UPR activation (Wang et al., 2013). This work illustrates the complex, intertwined nature of oxidative stress, the UPR, and apoptosis in diabetic embryopathy.
6.3 Fetal Alcohol Spectrum Disorders and Developmental Neurotoxicity
Alcohol consumption during pregnancy can cause a variety of pathological conditions ranging from birth defects to neurological disorders. FASD is a complex set of disorders with many potential contributors to observed toxicity, including disruptions in cellular energetics, cell–cell interactions, cell cycle, and cell signaling pathways (Goodlett et al., 2005).
6.3.1 Oxidative stress in FASD
Oxidative stress generated in ethanol metabolism is one hypothesized mechanism of FASD (Figure 12). Fetal alcohol exposure has been shown to lead to an increase in lipid peroxidation in the brain and liver (Chen et al., 1997). There have been several studies documenting the use of antioxidants or ROS-clearing enzymes to decrease the rates of alcohol-induced malformations (Chen et al., 2004; Shirpoor et al., 2009; Patten et al., 2013; Parnell et al., 2010). However, others have noted that the corresponding decrease in oxidative stress and apoptosis may not be sufficient to correct some neurological disorders caused by prenatal alcohol exposure (Marino et al., 2004).
Figure 12. Potential mechanisms of alcohol-induced ER stress and oxidativestress.
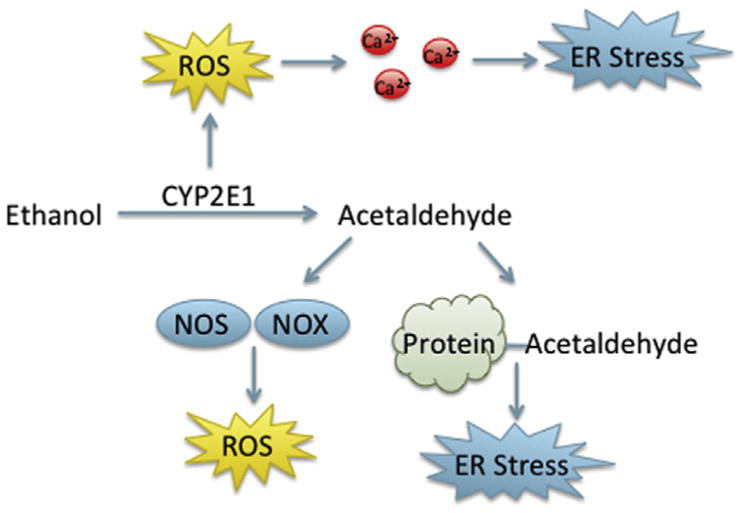
Abbreviations: NOS, nitric oxide synthase; NOX, NADPH oxidase.
Several potential mechanisms of alcohol-induced oxidative stress have been postulated. Ethanol is oxidatively metabolized first into acetaldehyde then to acetate by alcohol dehydrogenase and acetaldehyde dehydrogenase, respectively. However, fetal brain and liver CYP2E1 are also able to catalyze the conversion of ethanol to acetaldehyde, leading to an increase in ROS through enzyme uncoupling (Koop, 1992). Furthermore, CYP2E1 levels have been found to increase in the livers of alcoholics and ethanol-fed rats (Buhler et al., 1991) and CYP2E1 activity increased following chronic ethanol exposure in fetal guinea pigs (Hewitt et al., 2010). Acetaldehyde has been implicated in additional generation of ROS through transcriptional activation of NOX and NOS (Haorah et al., 2008). Alcohol-induced NOX transcriptional regulation has also been shown mouse embryos and treatment with a NOX inhibitor resulted in decreased oxidative stress and apoptosis (Dong et al., 2010). In these ethanol-treated mice, increased NO production was also observed (Dong et al., 2010).
6.3.2 Alcohol-induced ER stress
In addition to oxidative stress, a role for ER stress in FASD is also under investigation. To date, only one study directly examining alcohol-induced ER stress during development has been published (Ke et al., 2011). In this study, 7-day-old mice (equivalent to the human third trimester) were injected with 5 g/kg ethanol, which resulted in an increase in active caspase 3, BiP, CHOP and phosphorylated eIF2α, phosphorylated IRE1α, phosphorylated PERK, ATF6, and caspase 12 protein levels in mouse cerebral cortexes (Ke et al., 2011). However, CHOP knockout did not decrease neuronal apoptosis in these mice as in hepatocytes, suggesting that the UPR is not the only mechanism of neuronal apoptosis in FASD (Ke et al., 2011; Ji et al., 2005).
Additional research on the UPR and ER stress in alcohol-induced organ injury is available. In yeast, the original model for the UPR, ethanol exposure results in accumulation of unfolded proteins and subsequent activation of UPR (Miyagawa et al., 2014). In alcoholic liver disease, increased BiP, CHOP, and caspase 12 mRNA expression has been observed in the livers of alcohol-fed mice, and CHOP knockout mice had significantly less alcohol-induced apoptosis than controls (Ji and Kaplowitz, 2003; Ji et al., 2005). In zebrafish models, ethanol treatment-induced steatosis, which was preceded by increased BiP, XBP-1 splicing, and phosphorylated eIF2α (Tsendensodnom et al., 2013). Furthermore, ethanol reduced liver secretory capacity. These changes were accompanied by an increase in ROS, and were abrogated after cotreatment with antioxidants. A zebrafish model of fatty liver disease found ATF6 to be necessary and sufficient for ethanol-induced steatosis and ATF6 overexpression altered lipogenesis and lipid export (Howarth et al., 2014). The UPR has also been implicated in other organ systems following alcohol exposure, including the brain (Dlugos, 2014) and pancreas (Pandol et al., 2010).
Several mechanisms for UPR induction in alcoholic organ damage have been postulated (Figure 12) (Ji, 2012). These include oxidative stress-induced disruptions in calcium homeostasis, formation of acetaldehyde adducts that inhibit protein folding, homocysteine toxicity, and epigenetic regulation of UPR components (Ji, 2012).
The contribution of the UPR to alcohol-induced pathologies indicates the potential for a greater role for the UPR in FASD than previously recognized. This is supported by growing consideration for the UPR in various neurodegenerative diseases, including ALS, Parkinson’s, and Alzheimer’s disease (Hetz and Mollereau, 2014). Furthermore, the potential for the UPR and oxidative stress to cause malformations in cartilage or skeletal structures may be responsible for the malformations in addition to the neurological disorders associated with FASD. Additional research into the combined role of oxidative stress and the UPR in FASD is merited.
7. CONCLUSIONS
The complex spatiotemporal orchestration of development requires great precision, leaving the developing fetus vulnerable to slight perturbations that can result in teratogenesis or lethality. To date, the mechanism of action of many teratogens and developmental toxicants remain unknown. Understanding these mechanisms impacts both development of safer pharmaceuticals and of therapeutics to combat teratogens, such as those designed to prevent developmental toxicity resulting from hypoxia. While the mode of action of developmental toxicants was once thought to require specificity, it is becoming increasingly clear that many toxicants may impact several key cellular processes, leading to varied outcomes depending on dose, duration, and developmental stage.
Processes such as the UPR and redox-regulated signaling play complex and diverse roles in physiological development. Xenobiotic-induced perturbations in these events can generate oxidative stress or ER stress, which can lead to damage and apoptosis through a variety of mechanisms. As demonstrated, these events can exert their effects both independently and in concert, resulting in the potentiation of cellular damage (Figure 13). Although oxidative stress and apoptosis have long been regarded as mechanisms of developmental toxicity, the role of the UPR is only just coming under scrutiny. Much of our current understanding of the UPR in developmental toxicity comes from its newly recognized role in disease. As research elucidating its role in developmental toxicity becomes available, the UPR, like oxidative stress, will become a target for new therapeutics.
Figure 13.
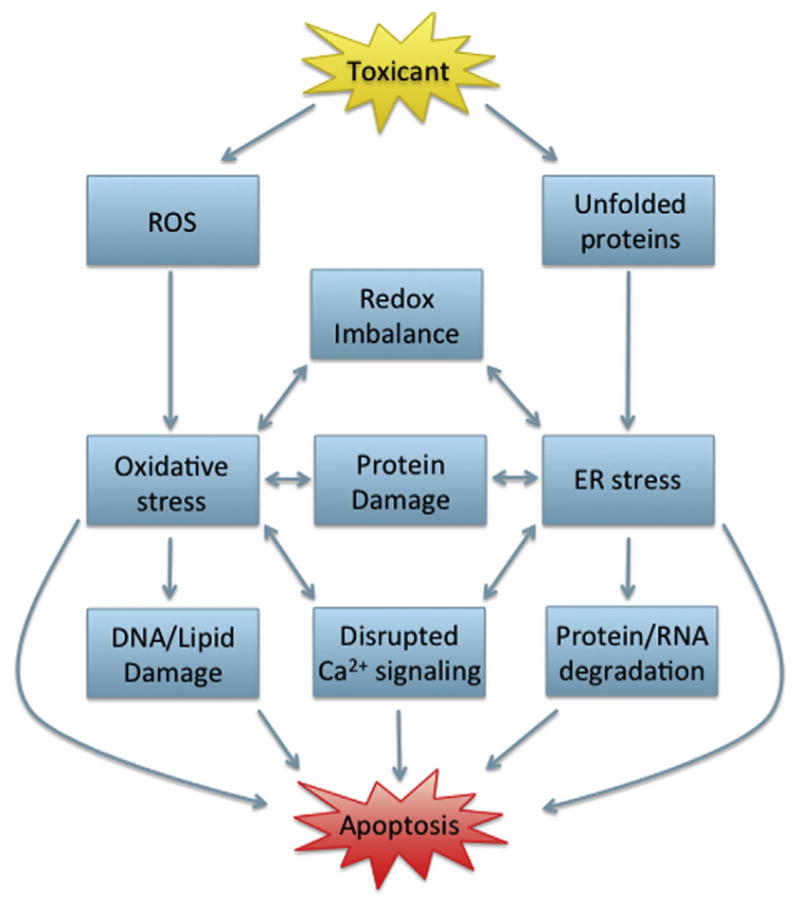
Interplay between the unfolded protein response, oxidative stress, and apoptosis in developmental toxicity.
Acknowledgments
A. Kupsco was supported by a National Research Service Award Institutional Training Grant (T32 ES018827).
Abbreviations
- AIF
Apoptosis-inducing factor
- Apaf-1
Apoptotic protease activating factor 1
- ARE
Antioxidant response element
- ARNT
Arylhydrocarbon nuclear translocator
- ASK1
Apoptosis signal-regulating kinase 1
- ATF4
Activating transcription factor 4
- ATF6
Activating transcription factor 6
- Bcl-2
B-cell lymphoma 2
- BH3-only
Bcl-2 homology 3 domain only
- BiP
Immunoglobulin heavy-chain-binding protein
- BMP2
Bone morphogenic protein 2
- BNIP-3
Bcl-2/adenovirus EIB 19kD-interacting protein 3
- CaMKIIγ
Calcium/calmodulin-dependent protein kinase IIγ
- CARD
Caspase recruitment domains
- CAT
Catalase
- CHOP
C/EBP homologous protein
- COMP
Cartilage-matrix oligomeric protein
- COX
Cyclooxygenase
- CYP450
Cytochrome P450
- DED
Death effector domain
- DISC
Death-inducing signaling complex
- DR5
Death receptor 5
- DTT
Dithiothreitol
- ECM
Extracellular matrix
- eIF2α
Eukaryotic initiation factor 2α
- ER
Endoplasmic reticulum
- ERAD
ER-associated degradation
- ERO
ER oxidoreductin
- FADD
Fas-associated death domain
- FASD
Fetal alcohol spectrum disorders
- GPx
Glutathione peroxidase
- Grx
Glutaredoxin
- GSH
Glutathione
- GST
Glutathione S-transferase
- H2O2
Hydrogen peroxide
- HIF
Hypoxia-inducible factor
- HO
Heme oxygenase
- HOXB4
Homeo box B4
- HRE
Hypoxia response element
- HSC
Hematopoietic stem cells
- IRE1
Inositol-required enzyme 1
- JNK
c-Jun N-terminal kinase
- LOX
Lipoxygenase
- MPT
Mitochondrial permeability transition
- NF-κB
Nuclear factor kappa-light-chain-enhancer of activated B cells
- NO−
Nitric oxide
- NOS
Nitric oxide synthase
- NOX
NADPH oxidase
- Nrf2
Nuclear factor (erythroid-derived 2)-like 2
- Nrx
Nucleoredoxin
- O2−
Superoxide anion
- OASIS
Old astrocyte specifically induced substance
- OH
Hydroxyl radical
- ONOO−
Peroxynitrite
- PARP
Poly (ADP-ribose) polymerase
- PDI
Protein disulfide isomerase
- PERK
Protein kinase RNA-like ER kinase
- PHD
Prolyl hydroxylase
- PHS
Prostaglandin H synthase
- PKC
Protein kinase C
- Prx
Peroxiredoxins
- PUMA
p53-upregulated modulator of apoptosis
- Ref-1
Redox effector factor 1
- RIDD
Regulated IRE1-dependent decay
- RNS
Reactive nitrogen species
- ROS
Reactive oxygen species
- SOD
Superoxide dismutase
- TCDD
2,3,7,8-Tetrachlorodibenzo-para-dioxin
- Tg
Thapsigargin
- Tm
Tunicamycin
- TNFα
Tumor necrosis factor α
- TRAF2
Tumor necrosis factor receptor (TNFR)-associated factor 2
- TRAIL
TNF-related apoptosis-inducing ligand
- Trx
Thioredoxin
- TrxR
Thioredoxin reductase
- UPR
Unfolded protein response
- XBP-1
X-box binding protein 1
- XO
Xanthine oxidase
References
- Abdelwahid E, Pelliniemi LJ, Jokinen E. Cell death and differentiation in the development of the endocardial cushion of the embryonic heart. Microsc Res Tech. 2002;58:395–403. doi: 10.1002/jemt.10159. [DOI] [PubMed] [Google Scholar]
- Abramov JP, Wells PG. Embryonic catalase protects against endogenous and phenytoin-enhanced DNA oxidation and embryopathies in acatalasemic and human catalase-expressing mice. FASEB J. 2011;25:2188–2200. doi: 10.1096/fj.11-182444. [DOI] [PubMed] [Google Scholar]
- Abramov AY, Scorziello A, Duchen MR. Three distinct mechanisms generate oxygen free radicals in neurons and contribute to cell death during anoxia and reoxygenation. J Neurosci. 2007;27:1129–1138. doi: 10.1523/JNEUROSCI.4468-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acosta-Alvear D, Zhou Y, Blais A, Tsikitis M, Lents NH, Arias C, Lennon CJ, Kluger Y, Dynlacht BD. XBP1 controls diverse cell type- and condition-specific transcriptional regulatory networks. Mol Cell. 2007;27:53–66. doi: 10.1016/j.molcel.2007.06.011. [DOI] [PubMed] [Google Scholar]
- Ameri K, Harris AL. Activating transcription factor 4. Int J Biochem Cell Biol. 2008;40:14–21. doi: 10.1016/j.biocel.2007.01.020. [DOI] [PubMed] [Google Scholar]
- Anderson EJ, Katunga LA, Willis MS. Mitochondria as a source and target of lipid peroxidation products in healthy and diseased heart. Clin Exp Pharmacol Physiol. 2012;39:179–193. doi: 10.1111/j.1440-1681.2011.05641.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anelli T, Bergamelli L, Margittai E, Rimessi A, Fagioli C, Malgaroli A, Pinton P, Ripamonti M, Rizzuto R, Sitia R. Ero1α regulates Ca(2+) fluxes at the endoplasmic reticulum-mitochondria interface (MAM) Antioxid Redox Signal. 2012;16:1077–1087. doi: 10.1089/ars.2011.4004. [DOI] [PubMed] [Google Scholar]
- Arnold WV, Fertala A. Skeletal diseases caused by mutations that affect collagen structure and function. Int J Biochem Cell Biol. 2013;45:1556–1567. doi: 10.1016/j.biocel.2013.05.017. [DOI] [PubMed] [Google Scholar]
- Ashkenazi A. Directing cancer cells to self-destruct with pro-apoptotic receptor agonists. Nat Rev Drug Discov. 2008;7:1001–1012. doi: 10.1038/nrd2637. [DOI] [PubMed] [Google Scholar]
- Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Becker LB, Vanden Hoek TL, Shao ZH, Li CQ, Schumacker PT. Generation of superoxide in cardiomyocytes during ischemia before reperfusion. Am J Physiol. 1999;277:H2240–H2246. doi: 10.1152/ajpheart.1999.277.6.H2240. [DOI] [PubMed] [Google Scholar]
- Bergendi L, Benes L, Durackova Z, Ferencik M. Chemistry, physiology, and pathology of free radicals. Life Sci. 1999;65:1865–1874. doi: 10.1016/s0024-3205(99)00439-7. [DOI] [PubMed] [Google Scholar]
- Berlett BS, Stadtman ER. Protein oxidation in aging, disease, and oxidative stress. J Biol Chem. 1997;272:20313–20316. doi: 10.1074/jbc.272.33.20313. [DOI] [PubMed] [Google Scholar]
- Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol. 2000;2:326–332. doi: 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- Bertolotti A, Wang X, Novoa I, Jungreis R, Schlessinger K, Cho JH, West AB, Ron D. Increased sensitivity to dextran sodium sulfate colitis in IRE1b-deficient mice. J Clin Invest. 2001;107:585–593. doi: 10.1172/JCI11476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birnbaum LS. Developmental effects of dioxins. Environ Health Perspect. 1995;103:89–94. doi: 10.1289/ehp.95103s789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boatright KM, Renatus M, Scott FL, Sperandio S, Shin H, Pedersen IM, Ricci JE, Edris WA, Sutherlin DP, Green DR, Salvesen GS. A unified model for apical caspase activation. Mol Cell. 2003;11:529–541. doi: 10.1016/s1097-2765(03)00051-0. [DOI] [PubMed] [Google Scholar]
- Bonfanti P, Colombo A, Orsi F, Nizzetto I, Andrioletti M, Bacchetta R, Mantecca P, Fascio U, Vailati G, Vismara C. Comparative teratogenicity of Chlorpyrifos and Malathion on Xenopus laevis development. Aquat Toxicol. 2004;70:189–200. doi: 10.1016/j.aquatox.2004.09.007. [DOI] [PubMed] [Google Scholar]
- Borchert A, Wang CC, Ufer C, Schiebel H, Savaskan NE, Kuhn H. The role of phospholipid hydroperoxide glutathione peroxidase isoforms in murine embryogenesis. J Biol Chem. 2006;281:19655–19664. doi: 10.1074/jbc.M601195200. [DOI] [PubMed] [Google Scholar]
- Brash AR. Lipoxygenases: occurrence, functions, catalysis, and acquisition of substrate. J Biol Chem. 1999;274:23679–23682. doi: 10.1074/jbc.274.34.23679. [DOI] [PubMed] [Google Scholar]
- Brautigam L, Schutte LD, Godoy JR, Prozorovski T, Gellert M, Hauptmann G, Holmgren A, Lillig CH, Berndt C. Vertebrate-specific glutaredoxin is essential for brain development. Proc Natl Acad Sci USA. 2011;108:20532–20537. doi: 10.1073/pnas.1110085108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bühler R, Lindros KO, von Boguslawsky K, Kärkkäinen P, Mäkinen J, Ingelman-Sundberg M. Perivenous expression of ethanol-inducible cytochrome P450 IIE1 in livers from alcoholics and chronically ethanol-fed rats. Alcohol Alcohol Suppl. 1991;1:311–315. [PubMed] [Google Scholar]
- Burton GJ. Oxygen, the Janus gas, its effects on human placental development and function. J Anat. 2009;215:27–35. doi: 10.1111/j.1469-7580.2008.00978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buss RR, Sun W, Oppenheim RW. Adaptive roles of programmed cell death during nervous system development. Annu Rev Neurosci. 2006;29:1–35. doi: 10.1146/annurev.neuro.29.051605.112800. [DOI] [PubMed] [Google Scholar]
- Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415:92–96. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- Camacho IA, Nagarkatti M, Nagarkatti PS. Evidence for induction of apoptosis in T Cells from murine fetal thymus following perinatal exposure to 2,3,7,8-tetrachlorodi-benzo-p-dioxin (TCDD) Toxicol Sci. 2004;78:96–106. doi: 10.1093/toxsci/kfh048. [DOI] [PubMed] [Google Scholar]
- Cantrell SM, Lutz LH, Tillitt DE, Hannink M. Embryotoxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD): the embryonic vasculature is a physiological target for TCDD-induced DNA damage and apoptotic cell death in Medaka (Orizias latipes) Toxicol Appl Pharmacol. 1996;141:23–34. doi: 10.1006/taap.1996.0256. [DOI] [PubMed] [Google Scholar]
- Cao SS, Kaufman RJ. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid Redox Signal. 2014;21:396–413. doi: 10.1089/ars.2014.5851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carloni S, Albertini MC, Galluzzi L, Buonocore G, Proiette F, Balduini W. Increased autophagy reduces endoplasmic reticulum stress after neonatal hypoxia–ischemia: role of protein synthesis and autophagic pathways. Exp Neurol. 2014;255:103–112. doi: 10.1016/j.expneurol.2014.03.002. [DOI] [PubMed] [Google Scholar]
- Chakraborti TK, Farrar JD, Pope CN. Comparative neurochemical and neuro-behavioral effects of repeated chlorpyrifos exposures in young and adult rats. Pharmacol Biochem Behav. 1993;46:219–224. doi: 10.1016/0091-3057(93)90344-s. [DOI] [PubMed] [Google Scholar]
- Chanda SM, Pope CN. Neurochemical and neurobehavioral effects of repeated gestational exposure to chlorpyrifos in maternal and developing rats. Pharmacol Biochem Behav. 1996;53:771–776. doi: 10.1016/0091-3057(95)02105-1. [DOI] [PubMed] [Google Scholar]
- Chang TI, Horal M, Jain SK, Wang F, Patel R, Loeken MR. Oxidant regulation of gene expression and neural tube development: Insights gained from diabetic pregnancy on molecular causes of neural tube defects. Diabetologia. 2003;46:538–545. doi: 10.1007/s00125-003-1063-2. [DOI] [PubMed] [Google Scholar]
- Chen Y, Brandizzi F. IRE1: ER stress sensor and cell fate executor. Trends Cell Biol. 2013;23:547–555. doi: 10.1016/j.tcb.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JJ, Schenker S, Henderson GI. 4-Hydroxynonenal levels are enhanced in fetal liver mitochondria by in utero ethanol exposure. Hepatology. 1997;25:142–147. doi: 10.1002/hep.510250126. [DOI] [PubMed] [Google Scholar]
- Chen A, Muzzio IA, Malleret G, Bartsch D, Verbitsky M, Pavlidis P, Yonan AL, Vronskaya S, Grody MB, Cepeda I, Gilliam TC, Kandel ER. Inducible enhancement of memory storage and synaptic plasticity in transgenic mice expressing an inhibitor of ATF4 (CREB-2) and C/EBP proteins. Neuron. 2003;39:655–669. doi: 10.1016/s0896-6273(03)00501-4. [DOI] [PubMed] [Google Scholar]
- Chen SY, Dehart DB, Sulik KK. Protection from ethanol-induced limb malformations by the superoxide dismutase/catalase mimetic, EUK-134. FASEB J. 2004;18:1234–1236. doi: 10.1096/fj.03-0850fje. [DOI] [PubMed] [Google Scholar]
- Chen X, Iliopoulos D, Zhang Q, Tang Q, Greenblatt MB, Hatziapostolou M, Lim E, Tam WL, Ni M, Chen Y, Mai J, Shen H, Hu DZ, Adoro S, Hu B, Song M, Tan C, Landis MD, Ferrari M, Shin SJ, Brown M, Chang JC, Liu XS, Glimcher LH. XBP1 promotes triple negative breast cancer by controlling the HIF1 α pathway. Nature. 2014a;508:103–107. doi: 10.1038/nature13119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen BL, Sheu ML, Tsai KS, Lan KC, Guan SS, Wu CT, Chen LP, Hung KY, Huang JW, Chiang CK, Liu SH. CCAAT-enhancer-binding protein homologous protein deficiency attenuates oxidative stress and renal ischemia-reperfusion injury. Antioxid Redox Signal. 2014b doi: 10.1089/ars.2013.5768. in press. [DOI] [PubMed] [Google Scholar]
- Cheng KC, Cahill DS, Kasais H, Nishimura S, Loeb LA. 8-Hydroxyguanine, an abundant form of oxidative DNA damage, causes G–T and A–C substitutions. J Biol Chem. 1992;267:166–172. [PubMed] [Google Scholar]
- Chernoff N, Rogers JM, Alles AJ, Zucker RM, Elstein KH, Massaro EJ, Sullik KK. Cell cycle alterations and cell death in cyclophosphamide teratogenesis. Teratog Carcinog Mutagen. 1989;9:199–209. doi: 10.1002/tcm.1770090403. [DOI] [PubMed] [Google Scholar]
- Christen V, Capelle M, Fent K. Silver nanoparticles induce endoplasmatic reticulum stress response in zebrafish. Toxicol Appl Pharmacol. 2013;272:519–528. doi: 10.1016/j.taap.2013.06.011. [DOI] [PubMed] [Google Scholar]
- Collard CD, Gelman S. Pathophysiology, clinical manifestations, and prevention of ischemia–reperfusion injury. Anesthesiology. 2001;94:1133–1138. doi: 10.1097/00000542-200106000-00030. [DOI] [PubMed] [Google Scholar]
- Cooke MS, Evans MD, Dizdaroglu M, Lunec J. Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J. 2003;17:1195–1214. doi: 10.1096/fj.02-0752rev. [DOI] [PubMed] [Google Scholar]
- Cornejo VH, Pihan P, Vidal RL, Hetz C. Role of the unfolded protein response in organ physiology: lessons from mouse models. IUBMB Life. 2013;65:962–975. doi: 10.1002/iub.1224. [DOI] [PubMed] [Google Scholar]
- Crawford ED, Wells JA. Caspase substrates and cellular remodeling. Annu Rev Biochem. 2011;80:1055–1087. doi: 10.1146/annurev-biochem-061809-121639. [DOI] [PubMed] [Google Scholar]
- Crumpton TL, Seidler FJ, Slotkin TA. Is oxidative stress involved in the developmental neurotoxicity of chlorpyrifos? Dev Brain Res. 2000;121:189–195. doi: 10.1016/s0165-3806(00)00045-6. [DOI] [PubMed] [Google Scholar]
- Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ, Diehl JA. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol Cell Biol. 2003;23:7198–7209. doi: 10.1128/MCB.23.20.7198-7209.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies KJA. The broad spectrum of responses to oxidants in proliferating cells: a new paradigm for oxidative stress. Life. 1999;48:41–47. doi: 10.1080/713803463. [DOI] [PubMed] [Google Scholar]
- DeCoursey TE, Ligeti E. Regulation and termination of NADPH oxidase activity. Cell Mol Life Sci. 2005;62:2173–2193. doi: 10.1007/s00018-005-5177-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLeve LD, Kaplowitz N. Glutathione metabolism and its role in hepatotoxicity. Pharmac Ther. 1991;52:287–307. doi: 10.1016/0163-7258(91)90029-l. [DOI] [PubMed] [Google Scholar]
- Deng J, Lu PD, Zhang Y, Scheuner D, Kaufman RJ, Sonenberg N, Harding HP, Ron D. Translational repression mediates activation of nuclear factor kappa B by phosphorylated translation initiation factor 2. Mol Cell Biol. 2004;24:10161–10168. doi: 10.1128/MCB.24.23.10161-10168.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diebold I, Petry A, Hess J, Gorlach A. The NADPH oxidase subunit NOX4 is a new target gene of the hypoxia-inducible factor-1. Mol Biol Cell. 2010;21:2087–2096. doi: 10.1091/mbc.E09-12-1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinchuk JE, Car BD, Focht RJ, Johnston JJ, Jaffee BD, Covington MB, Contel NR, Eng VM, Collins RJ, Czerniak PM, et al. Renal abnormalities and an altered inflammatory response in mice lacking cyclooxygenase II. Nature. 1995;378:406–409. doi: 10.1038/378406a0. [DOI] [PubMed] [Google Scholar]
- Dlugos CA. ATF6 and caspase 12 expression in purkinje neurons in acute slices from adult, ethanol-fed rats. Brain Res. 2014;1577:11–20. doi: 10.1016/j.brainres.2014.06.025. [DOI] [PubMed] [Google Scholar]
- Dong W, Teraoka H, Yamazaki K, Tsukiyama S, Imani S, Imagawa T, Stegeman JJ, Peterson RE, Hiraga T. 2,3,7,8-Tetrachlorodibenzo-p-dioxin toxicity in the zebrafish embryo: local circulation failure in the dorsal midbrain is associated with increased apoptosis. Toxicol Sci. 2002;69:191–201. doi: 10.1093/toxsci/69.1.191. [DOI] [PubMed] [Google Scholar]
- Dong J, Sulik KK, Chen SY. The role of NOX enzymes in ethanol-induced oxidative stress and apoptosis in mouse embryos. Toxicol Lett. 2010;193:94–100. doi: 10.1016/j.toxlet.2009.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuRose JB, Tam AB, Niwa M. Intrinsic capacities of molecular sensors of the unfolded protein response to sense alternate forms of endoplasmic reticulum stress. Mol Biol Cell. 2006;17:3095–3107. doi: 10.1091/mbc.E06-01-0055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engerson TD, McKelvey TG, Rhyne DB, Boggio EB, Snyder SJ, Jones HP. Conversion of xanthine dehydrogenase to oxidase in ischemic rat tissues. J Clin Invest. 1987;79:1564–1570. doi: 10.1172/JCI112990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eom H, Ahn J, Kim Y, Choi J. Hypoxia inducible factor-1 (HIF-1)–flavin containing monooxygenase-2 (FMO-2) signaling acts in silver nanoparticles and silver ion toxicity in the nematode, Caenorhabditis elegans. Toxicol Appl Pharmacol. 2013;270:106–113. doi: 10.1016/j.taap.2013.03.028. [DOI] [PubMed] [Google Scholar]
- Epp N, Furstenberger G, Muller K, de Juanes S, Leitges M, Hausser I, Thieme F, Liebisch G, Schmitz G, Krieg P. 12R-lipoxygenase deficiency disrupts epidermal barrier function. J Cell Biol. 2007;177:173–182. doi: 10.1083/jcb.200612116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson UJ, Borg LA. Protection by free oxygen radical scavenging enzymes against glucose-induced embryonic malformations in vitro. Diabetologia. 1991;34:325–331. doi: 10.1007/BF00405004. [DOI] [PubMed] [Google Scholar]
- Estes SD, Stoler DL, Anderson GR. Normal fibroblasts induce the C/EBP beta and ATF-4 bZIP transcription factors in response to anoxia. Exp Cell Res. 1995;220:47–54. doi: 10.1006/excr.1995.1290. [DOI] [PubMed] [Google Scholar]
- Fan R, Bonde S, Gao P, Sotomayor B, Chen C, Mouw T, Zavazava N, Tan K. Dynamic HoxB4-regulatory network during embryonic stem cell differentiation to hematopoietic cells. Blood. 2012;119:e139–e147. doi: 10.1182/blood-2011-12-396754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favero CB, Henshaw RN, Grimsley-Myers CM, Shrestha A, Beier DR, Dwyer ND. Mutation of the BiP/GRP78 gene causes axon outgrowth and fasciculation defects in the thalamocortical connections of the mammalian forebrain. J Comp Meurol. 2013;521:677–696. doi: 10.1002/cne.23199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felderhoff-Mueser U, Taylor DL, Greenwood K, Kozma M, Stibenz D, Joashi UC, Edwards DA, Mehmet H. Fas/CD95/APO-1 can function as a death receptor for neuronal cells in vitro and in vivo and is upregulated following cerebral hypoxic-ischemic injury to the developing rat brain. Brain Pathol. 2000;10:17–29. doi: 10.1111/j.1750-3639.2000.tb00239.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari DM, Soling HD. The protein disulphide-isomerase family: unravelling a string of folds. Biochem J. 1999;339:1–10. [PMC free article] [PubMed] [Google Scholar]
- Firtina Z, Duncan MK. Unfolded Protein Response (UPR) is activated during normal lens development. Gene Expr Patterns. 2011;11:135–143. doi: 10.1016/j.gep.2010.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer B, Bavister BD. Oxygen tension in the oviduct and uterus of rhesus monkeys, hamsters and rabbits. J Reprod Fertil. 1993;99:673–679. doi: 10.1530/jrf.0.0990673. [DOI] [PubMed] [Google Scholar]
- Fischer U, Janicke RU, Schulze-Osthoff K. Many cuts to ruin: a comprehensive update of caspase substrates. Cell Death Differ. 2003;10:76–100. doi: 10.1038/sj.cdd.4401160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaskos J, Sachana M. Developmental neurotoxicity of anticholinesterase pesticides. In: Satoh T, Guptah RC, editors. Anticholinesterase Pesticides: Metabolism, Neurotoxicity, and Epidemiology. John Wiley & Sons; Hoboken: 2010. pp. 203–223. [Google Scholar]
- Fu HY, Okada K, Liao Y, Tsukamoto O, Isomura T, Asai M, Sawada T, Okuda K, Asano Y, Sanada S, Asanuma H, Asakura M, Takashima S, Komuro I, Kitakaze M, Minamino T. Ablation of C/EBP homologous protein attenuates endoplasmic reticulum-mediated apoptosis and cardiac dysfunction induced by pressure overload. Circulation. 2010;122:361–369. doi: 10.1161/CIRCULATIONAHA.109.917914. [DOI] [PubMed] [Google Scholar]
- Fuchs Y, Steller H. Programmed cell death in animal development and disease. Cell. 2011;147:742–758. doi: 10.1016/j.cell.2011.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuentes-Prior P, Salvesen GS. The protein structures that shape caspase activity, specificity, activation and inhibition. Biochem J. 2004;384:201–232. doi: 10.1042/BJ20041142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funato Y, Miki H. Nucleoredoxin, a novel thioredoxin family member involved in cell growth and differentiation. Antioxid Redox Signal. 2007;9:1035–1057. doi: 10.1089/ars.2007.1550. [DOI] [PubMed] [Google Scholar]
- Gardiner CS, Reed DJ. Status of glutathione during oxidant-induced oxidative stress in the preimplantation mouse embryo. Biol Reprod. 1994;51:1307–1314. doi: 10.1095/biolreprod51.6.1307. [DOI] [PubMed] [Google Scholar]
- Gibson G. Active role of chondrocyte apoptosis in endochondral ossification. Microsc Res Tech. 1998;43:191–204. doi: 10.1002/(SICI)1097-0029(19981015)43:2<191::AID-JEMT10>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Gillette JR, Brodie BB, La Du BN. The oxidation of drugs by liver microsomes: on the role of TPNH and oxygen. J Pharmacol Exp Ther. 1957;119:532–540. [PubMed] [Google Scholar]
- Goodlett CR, Horn KH, Zhou FC. Alcohol teratogenesis: mechanisms of damage and strategies for intervention. Exp Biol Med. 2005;230:394–406. doi: 10.1177/15353702-0323006-07. [DOI] [PubMed] [Google Scholar]
- Graham EM, Sheldon RA, Flock DL, Ferriero DM, Martin LJ, O’Riordan DP, Northington FJ. Neonatal mice lacking functional Fas death receptors are resistant to hypoxic-ischemic brain injury. Neurobiol Dis. 2004;17:89–98. doi: 10.1016/j.nbd.2004.05.007. [DOI] [PubMed] [Google Scholar]
- Grinkova YV, Denisov IG, McLean MA, Sligar SG. Oxidase uncoupling in heme monooxygenases: human cytochrome P450 CYP3A4 in nanodiscs. Biochem Biophys Res Commun. 2013;430:1223–1227. doi: 10.1016/j.bbrc.2012.12.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han D, Lerner AG, Walle LV, Upton JP, Xu W, Hagen A, Backes BJ, Oakes SA, Papa FR. IRE1α kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell. 2009;138:562–575. doi: 10.1016/j.cell.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, Zhou J, Zhang P, Song F, Jiang R, Li M, Xia F, Guo FJ. IRE1α dissociates with BiP and inhibits ER stress-mediated apoptosis in cartilage development. Cell Signal. 2013;25:2136–2146. doi: 10.1016/j.cellsig.2013.06.011. [DOI] [PubMed] [Google Scholar]
- Hanschmann E, Godoy JR, Berndt C, Hudemann C, Lillig CH. Thioredoxins, glutaredoxins, and peroxiredoxins—molecular mechanisms and health significance: from cofactors to antioxidants to redox signaling. Antioxid Redox Signal. 2013;19:1539–1605. doi: 10.1089/ars.2012.4599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen JM. Oxidative stress as a mechanism of teratogenesis. Birth Defects Res C Embryo Today. 2006;78:293–307. doi: 10.1002/bdrc.20085. [DOI] [PubMed] [Google Scholar]
- Haorah J, Ramirez SH, Floreani N, Gorantla S, Morsey B, Persidsky Y. Mechanism of alcohol-induced oxidative stress and neuronal injury. Free Radic Biol Med. 2008;45:1542–1550. doi: 10.1016/j.freeradbiomed.2008.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapiro M, Ron D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell. 2000;6:1099–1108. doi: 10.1016/s1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, Stojdl DF, Bell JC, Hettmann T, Leiden JM, Ron D. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell. 2003;11:619–633. doi: 10.1016/s1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- Harrison R. Structure and function of xanthine oxidoreductase: where are we now? Free Radic Biol Med. 2002;33:774–797. doi: 10.1016/s0891-5849(02)00956-5. [DOI] [PubMed] [Google Scholar]
- Hayes JD, Dinkova-Kostova AT. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem Sci. 2014;39:199–218. doi: 10.1016/j.tibs.2014.02.002. [DOI] [PubMed] [Google Scholar]
- Hettmann T, Barton K, Leiden JM. Microphthalmia due to p53-mediated apoptosis of anterior lens epithelial cells in mice lacking the CREB-2 transcription factor. Dev Biol. 2000;222:110–123. doi: 10.1006/dbio.2000.9699. [DOI] [PubMed] [Google Scholar]
- Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Mol Cell Biol. 2012;13:89–102. doi: 10.1038/nrm3270. [DOI] [PubMed] [Google Scholar]
- Hetz C, Mollereau B. Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat Rev Neurosci. 2014;15:233–249. doi: 10.1038/nrn3689. [DOI] [PubMed] [Google Scholar]
- Hetz C, Bernasconi P, Fisher J, Lee AH, Bassik MC, Antonsson B, Brandt GS, Iwakoshi NN, Schinzel A, Glimcher LH, Korsmeyer SJ. Proapoptotic BAX and BAK modulate the unfolded protein response by a direct interaction with IRE1alpha. Science. 2006;312:572–576. doi: 10.1126/science.1123480. [DOI] [PubMed] [Google Scholar]
- Hewitt AJ, Walker KR, Kobus SM, Poklewska-Koziell M, Reynolds JN, Brien JF. Differential effects of chronic ethanol exposure on cytochrome P450 2E1 and the hypothalamic—pituitary–adrenal axis in the maternal–fetal unit of the guinea pig. Neurotoxicol Teratol. 2010;32:164–170. doi: 10.1016/j.ntt.2009.12.002. [DOI] [PubMed] [Google Scholar]
- Hiramatsu Y, Sekiguchi N, Hayashi M, Isshiki K, Yokota T, King GL, Loeken MR. Diacylglycerol production and protein kinase C activity are increased in a mouse model of diabetic embryopathy. Diabetes. 2002;51:2804–2810. doi: 10.2337/diabetes.51.9.2804. [DOI] [PubMed] [Google Scholar]
- Ho Y, Xiong Y, Ma W, Spector A, Ho DS. Mice lacking catalase develop normally but show differential sensitivity to oxidant tissue injury. J Biol Chem. 2004;279:32804–32812. doi: 10.1074/jbc.M404800200. [DOI] [PubMed] [Google Scholar]
- Hollien J, Lin JH, Li H, Stevens N, Walter P, Weissman JS. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J Cell Biol. 2009;186:323–331. doi: 10.1083/jcb.200903014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollien J, Weissman JS. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science. 2006;313:104–107. doi: 10.1126/science.1129631. [DOI] [PubMed] [Google Scholar]
- Holmgren A. Thioredoxin. 6 The amino acid sequence of the protein from Escherichia coli. B Eur J Biochem. 1968;6:475–484. doi: 10.1111/j.1432-1033.1968.tb00470.x. [DOI] [PubMed] [Google Scholar]
- Hong M, Luo S, Baumeister P, Huang J, Gogia RK, Li M, Lee AS. Under-glycosylation of ATF6 as a novel sensing mechanism for activation of the unfolded protein response. J Biol Chem. 2004;279:11354–11363. doi: 10.1074/jbc.M309804200. [DOI] [PubMed] [Google Scholar]
- Horal M, Zhang Z, Stanton R, Virkamaki A, Loeken MR. Activation of the hexosamine pathway causes oxidative stress and abnormal embryo gene expression: involvement in diabetic teratogenesis. Birth Defects Res A. 2004;70:519–527. doi: 10.1002/bdra.20056. [DOI] [PubMed] [Google Scholar]
- Hosako H, Little SA, Barrier M, Mirkes PE. Teratogen-induced activation of p53 in early postimplantation mouse embryos. Toxicol Sci. 2007;95:257–269. doi: 10.1093/toxsci/kfl143. [DOI] [PubMed] [Google Scholar]
- Howarth DL, Lindtner C, Vacaru AM, Sachidanandam R, Tsedensodnom O, Vasilkova T, Buettner C, Sadler KC. Activating transcription factor 6 is necessary and sufficient for alcoholic fatty liver disease in zebrafish. PLoS Genet. 2014;10:e1004335. doi: 10.1371/journal.pgen.1004335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu P, Han Z, Couvillon AD, Kaufman RJ, Exton JH. Autocrine tumor necrosis factor alpha links endoplasmic reticulum stress to the membrane death receptor pathway through IRE1alpha-mediated NF-kappaB activation and down-regulation of TRAF2 expression. Mol Cell Biol. 2006;26:3071–3084. doi: 10.1128/MCB.26.8.3071-3084.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua H, Munk S, Goldberg H, Fantus IG, Whiteside CI. High glucose-suppressed Endothelin-1 Ca2+ signaling via NADPH oxidase and diacylglycerol-sensitive protein kinase C isozymes in mesangial cells. J Biol Chem. 2003;278:33951–33962. doi: 10.1074/jbc.M302823200. [DOI] [PubMed] [Google Scholar]
- Huh WJ, Esen E, Geahlen JH, Bredemeyer AJ, Lee A, Shi G, Konieczny SF, Glimcher LH, Mills JC. XBP1 controls maturation of gastric zymogenic cells by induction of MIST1 and expansion of the rough endoplasmic reticulum. Gastroenterology. 2010;139:2038–2049. doi: 10.1053/j.gastro.2010.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai H, Nakagawa Y. Biological significance of phospholipid hydroperoxide glutathione peroxidase (phgpx, gpx4) in mammalian cells. Free Radic Biol Med. 2003;34:145–169. doi: 10.1016/s0891-5849(02)01197-8. [DOI] [PubMed] [Google Scholar]
- Imai H, Hirao F, Sakamoto T, Sekine K, Mizukura Y, Saito M, Kitamoto T, Hayasaka M, Hanaoka K, Nakagawa Y. Early embryonic lethality caused by targeted disruption of the mouse PHGPx gene. Biochem Biophys Res Commun. 2003;305:278–286. doi: 10.1016/s0006-291x(03)00734-4. [DOI] [PubMed] [Google Scholar]
- Ishikawa T, Okada T, Ishikawa-Fujiwara T, Todo T, Kamei Y, Shigenobu S, Tanaka M, Saito TL, Yoshimura J, Morishita S, Toyoda A, Sakaki Y, Taniguchi Y, Takeda S, Mori K. ATF6alpha/beta-mediated adjustment of ER chaperone levels is essential for development of the notochord in medaka fish. Mol Biol Cell. 2013;24:1387–1395. doi: 10.1091/mbc.E12-11-0830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito D, Tanaka K, Suzuki S, Dembo T, Kosakai A, Fukuuchi Y. Up-regulation of the Ire1-mediated signaling molecule, Bip, in ischemic rat brain. Mol Neurosci. 2001;12:4023–4028. doi: 10.1097/00001756-200112210-00034. [DOI] [PubMed] [Google Scholar]
- Iwakoshi NN, Lee AH, Vallabhajosyula P, Otipoby KL, Rajewsky K, Glimcher LH. Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP-1. Nat Immunol. 2003;4:321–329. doi: 10.1038/ni907. [DOI] [PubMed] [Google Scholar]
- Iwawaki T, Akai R, Yamanaka S, Kohno K. Function of IRE1a in the placenta is essential for placental development and embryonic viability. Proc Natl Acad Sci USA. 2009;106:16657–16662. doi: 10.1073/pnas.0903775106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakupoglu C, Przemeck GK, Schneider M, Moreno SG, Mayr N, Hatzopoulos AK, de Angelis MH, Wurst W, Bornkamm GW, Brielmeier M, Conrad M. Cytoplasmic thioredoxin reductase is essential for embryogenesis but dispensable for cardiac development. Mol Cell Biol. 2005;25:1980–1988. doi: 10.1128/MCB.25.5.1980-1988.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankov RP, Kantores C, Pan J, Belik J. Contribution of xanthine oxidase-derived superoxide to chronic hypoxic pulmonary hypertension in neonatal rats. Am J Physiol Lung Cell Mol Physiol. 2008;294:L233–L245. doi: 10.1152/ajplung.00166.2007. [DOI] [PubMed] [Google Scholar]
- Ji C. Mechanisms of alcohol-induced endoplasmic reticulum stress and organ injuries. Biochem Res Int. 2012;2012:216450. doi: 10.1155/2012/216450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji C, Kaplowitz N. Betaine decreases hyperhomocysteinemia, endoplasmic reticulum stress, and liver injury in alcohol-fed mice. Gastroenterology. 2003;124:1488–1499. doi: 10.1016/s0016-5085(03)00276-2. [DOI] [PubMed] [Google Scholar]
- Ji C, Mehrian-Shai R, Chan C, Hsu YH, Kaplowitz N. Role of CHOP in hepatic apoptosis in the murine model of intragastric ethanol feeding. Alcohol Clon Exp Res. 2005;29:1496–1503. doi: 10.1097/01.alc.0000174691.03751.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang HY, Wek SA, McGrath BC, Scheuner D, Kaufman RJ, Cavener DR, Wek RC. Phosphorylation of the α subunit of eukaryotic initiation factor 2 is required for activation of NF-κB in response to diverse cellular stresses. Mol Cell Biol. 2003;23:5651–5663. doi: 10.1128/MCB.23.16.5651-5663.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DP. Redefining oxidative stress. Antioxid Redox Signal. 2006;8:1865–1879. doi: 10.1089/ars.2006.8.1865. [DOI] [PubMed] [Google Scholar]
- Jurado J, Prieto-Alamo M, Madrid-Risquez J, Pueyo C. Absolute gene expression patterns of thioredoxin and glutaredoxin redox systems in mouse. J Biol Chem. 2003;278:45546–45554. doi: 10.1074/jbc.M307866200. [DOI] [PubMed] [Google Scholar]
- Kamath AB, Camacho I, Nagarkatti PS, Nagarkatti M. Role of Fas–Fas ligand interactions in 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-induced immunotoxicity: increased resistance of thymocytes from Fas-deficient (lpr) and fas ligand-defective (gld) mice to TCDD-induced toxicity. Toxicol Appl Pharmacol. 1999;160:141–155. doi: 10.1006/taap.1999.8753. [DOI] [PubMed] [Google Scholar]
- Karpinshi BA, Morle GD, Huggenvik J, Uhler MD, Leiden JM. Molecular cloning of human CREB-2: an ATF/CREB transcription factor that can negatively regulate transcription from the cAMP response element. Proc Natl Acad Sci USA. 1992;89:4820–4824. doi: 10.1073/pnas.89.11.4820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke Z, Wang X, Liu Y, Fan Z, Chen G, Xu M, Bower KA, Frank JA, Li M, Fang S, Shi X, Luo J. Ethanol induces endoplasmic reticulum stress in the developing brain. Alcohol Clin Exp Res. 2011;35:1574–1583. doi: 10.1111/j.1530-0277.2011.01503.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kichev A, Rousset CI, Baburamani AA, Levison SW, Wood TL, Gressens P, Thornton C, Hagberg H. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) signaling and cell death in the immature central nervous system after hypoxia-ischemia and inflammation. J Biol Chem. 2014;289:9430–9439. doi: 10.1074/jbc.M113.512350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kienle C, Kohler H, Gerhardt A. Behavioural and developmental toxicity of chlorpyrifos and nickel chloride to zebrafish (Danio rerio) embryos and larvae. Ecotox Environ Safe. 2009;72:1740–1747. doi: 10.1016/j.ecoenv.2009.04.014. [DOI] [PubMed] [Google Scholar]
- Kim KS, Kim YK, Lee AS. Expression of the glucose-regulated proteins (GRP94 and GRP78) in differentiated and undifferentiated mouse embryonic cells and the use of the GRP78 promoter as an expression system in embryonic cells. Differentiation. 1990;42:153–159. doi: 10.1111/j.1432-0436.1990.tb00756.x. [DOI] [PubMed] [Google Scholar]
- Kim YC, Masutani H, Yamaguchi Y, Itoh K, Yamamoto M, Yodoi J. Hemin-induced activation of the thioredoxin gene by Nrf2. A differential regulation of the antioxidant responsive element by a switch of its binding factors. J Biol Chem. 2001;276:18399–18406. doi: 10.1074/jbc.M100103200. [DOI] [PubMed] [Google Scholar]
- Kim JS, He L, Lemasters JJ. Mitochondrial permeability transition: a common pathway to necrosis and apoptosis. Biochem Biophys Res Commun. 2003;304:463–470. doi: 10.1016/s0006-291x(03)00618-1. [DOI] [PubMed] [Google Scholar]
- King-Heiden TC, Mehta V, Xiong KM, Lanham KA, Antkiewicz DS, Ganser A, Heideman W, Peterson RE. Reproductive and developmental toxicity of dioxin in fish. Mol Cell Endocrinol. 2012;354:121–138. doi: 10.1016/j.mce.2011.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkman HN, Gaetani GF. Mammalian catalase: a venerable enzyme with new mysteries. Trends Biochem Sci. 2006;32:44–50. doi: 10.1016/j.tibs.2006.11.003. [DOI] [PubMed] [Google Scholar]
- Kondo S, Murakami T, Tatsumi K, Ogata M, Kanemoto S, Otori K, Iseki K, Wanaka A, Imaizumi K. OASIS, a CREB/ATF-family member, modulates UPR signalling in astrocytes. Nat Cell Biol. 2005;7:186–194. doi: 10.1038/ncb1213. [DOI] [PubMed] [Google Scholar]
- Koop DR. Oxidative and reductive metabolism by cytochrome P450 2E1. FASEB J. 1992;6:724–730. doi: 10.1096/fasebj.6.2.1537462. [DOI] [PubMed] [Google Scholar]
- Koumenis C, Naczki C, Koritzinsky M, Rastani S, Diehl A, Sonenberg N, Koromilas A, Wouters BG. Regulation of protein synthesis by hypoxia via activation of the endoplasmic reticulum kinase PERK and phosphorylation of the translation initiation factor eIF2α Mol. Cell Biol. 2002;22:7405–7416. doi: 10.1128/MCB.22.21.7405-7416.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowaltowski AJ, Vercesi AE. Mitochondrial damage induced by conditions of oxidative stress. Free Radic Biol Med. 1999;26:463–471. doi: 10.1016/s0891-5849(98)00216-0. [DOI] [PubMed] [Google Scholar]
- Kowaltowski AJ, Castilho RF, Vercesi AE. Mitochondrial permeability transition and oxidative stress. FEBS Lett. 2001;495:12–15. doi: 10.1016/s0014-5793(01)02316-x. [DOI] [PubMed] [Google Scholar]
- Kuida K, Haydar TF, Kuan CY, Gu Y, Taya C, Karasuyama H, Su MS, Rakic P, Flavell RA. Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell. 1998;94:325–337. doi: 10.1016/s0092-8674(00)81476-2. [DOI] [PubMed] [Google Scholar]
- Kuida K, Zheng TS, Na S, Kuan C, Yang D, Karasuyama H, Su MS, Rakic P, Flavell RA. Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature. 1996;384:368–372. doi: 10.1038/384368a0. [DOI] [PubMed] [Google Scholar]
- Kumar S. Caspase function in programmed cell death. Cell Death Differ. 2007;14:32–43. doi: 10.1038/sj.cdd.4402060. [DOI] [PubMed] [Google Scholar]
- Kuppusamy P, Zweier JL. Characterization of free radical generation by xanthine oxidase. J Biol Chem. 1989;264:9880–9884. [PubMed] [Google Scholar]
- Kurooka H, Kato K, Minoguchi S, Takahashi Y, Ikeda J, Habu S, Osawa N, Buchberg AM, Moriwaki K, Shisa H, Honjo T. Cloning and characterization of the nucleoredoxin gene that encodes a novel nuclear protein related to thioredoxin. Genomics. 1997;39:331–339. doi: 10.1006/geno.1996.4493. [DOI] [PubMed] [Google Scholar]
- Leese HJ. Metabolic control during preimplantation mammalian development. Hum Reprod Update. 1995;1:63–72. doi: 10.1093/humupd/1.1.63. [DOI] [PubMed] [Google Scholar]
- Leonard SS, Harris GK, Shi X. Metal-induced oxidative stress and signal transduction. Free Radic Biol Med. 2004;37:1921–1942. doi: 10.1016/j.freeradbiomed.2004.09.010. [DOI] [PubMed] [Google Scholar]
- Li R, Chase M, Jung SK, Smith PJ, Loeken MR. Hypoxic stress in diabetic pregnancy contributes to impaired embryo gene expression and defective development by inducing oxidative stress. Am J Physiol Endocrinol Metab. 2005;289:E591–E599. doi: 10.1152/ajpendo.00441.2004. [DOI] [PubMed] [Google Scholar]
- Li G, Scull C, Ozcan L, Tabas I. NADPH oxidase links endoplasmic reticulum stress, oxidative stress, and PKR activation to induce apoptosis. J Cell Biol. 2010;191:1113–1125. doi: 10.1083/jcb.201006121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Xu C, Yang P. c-Jun NH2-terminal kinase 1/2 and endoplasmic reticulumstress as interdependent and reciprocal causation in diabetic embryopathy. Diabetes. 2013;62:599–608. doi: 10.2337/db12-0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang G, Lian C, Huang D, Gao W, Liang A, Peng Y, Ye W, Wu Z, Su S, Huang D. Endoplasmic reticulum stress-unfolding protein response-apoptosis cascade causes chondrodysplasia in a col2a1 p. Gly1170Ser mutated mouse model. PLoS One. 2014;9:e86894. doi: 10.1371/journal.pone.0086894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JH, Li H, Yasamura D, Cohen HR, Zhang C, Panning B, Shokat KM, LaVail MM, Walter P. IRE1 signaling affects cell fate during the unfolded protein response. Science. 2007;318:944–949. doi: 10.1126/science.1146361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Wells PG. In vivo phenytoin-initiated oxidative damage to proteins and lipids in murine maternal hepatic and embryonic tissue organelles: potential molecular targets mediating chemical teratogenesis. Toxicol Appl Pharmacol. 1994;125:247–255. doi: 10.1006/taap.1994.1070. [DOI] [PubMed] [Google Scholar]
- Liu L, Wells PG. DNA oxidation as a potential molecular mechanism mediating drug-induced birth defects: phenytoin and structurally related teratogens initiate the formation of 8-hydroxy-2′-deoxyguanosine in vitro and in vivo in murine maternal hepatic and embryonic tissues. Free Radic Biol Med. 1995;19:639–648. doi: 10.1016/0891-5849(95)00082-9. [DOI] [PubMed] [Google Scholar]
- Liu W, Wang G, Yaklovlev AG. Identification and functional analysis of the rat caspase-3 gene promoter. J Biol Chem. 2002;277:8273–8278. doi: 10.1074/jbc.M110768200. [DOI] [PubMed] [Google Scholar]
- Liu Y, Kinter DB, Begum G, Algharabli J, Cengiz P, Shull GE, Liu XJ, Sun D. Endoplasmic reticulum Ca2+ signaling and mitochondrial Cyt C release in astrocytes following oxygen and glucose deprivation. J Neurochem. 2010;114:1436–1446. doi: 10.1111/j.1471-4159.2010.06862.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Shen SM, Zhao XY, Chen GQ. Targeted genes and interacting proteins of hypoxia inducible factor-1. Int J Biochem Mol Biol. 2012;3:165–178. [PMC free article] [PubMed] [Google Scholar]
- Loeken MR. Challenges in understanding diabetic embryopathy. Diabetes. 2008;57:3187–3188. doi: 10.2337/db08-1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S. Regulation of hepatic glutathione synthesis: current concepts and controversies. FASEB J. 1999;13:1169–1183. [PubMed] [Google Scholar]
- Lu F, Tian Z, Zhang W, Zhao Y, Li H, Ren H, Zheng H, Liu C, Hu G, Tian Y, Yang BF, Wang R, Xu C. Calcium-sensing receptors regulate cardiomyocyte Ca2+ signaling via the sarcoplasmic reticulum-mitochondrion interface during hypoxia/reoxygenation. J Biomed Sci. 2010;17:50. doi: 10.1186/1423-0127-17-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Q, Wainwright MS, Harris VA, Aggarwal S, Hou Y, Rau T, Poulsen DJ, Black SM. Increased NADPH oxidase derived superoxide is involved in the neuronal cell death induced by hypoxia ischemia in neonatal hippocampal slice cultures. Free Radic Biol Med. 2012;53:1139–1151. doi: 10.1016/j.freeradbiomed.2012.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M, Lawrence DA, Marsters S, Acosta-Alvear D, Kimmig P, Mendez AS, Paton AW, Paton JC, Walter P, Ashkenazi A. Opposing unfolded-protein-response signals converge on death receptor 5 to control apoptosis. Science. 2014;345:98–101. doi: 10.1126/science.1254312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo S, Mao C, Lee B, Lee AS. GRP78/BiP is required for cell proliferation and protecting the inner cell mass from apoptosis during early mouse embryonic development. Mol Cell Biol. 2006;26:5688–5697. doi: 10.1128/MCB.00779-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Q, Battelli L, Hubbs AF. Multiorgan autoimmune inflammation, enhanced lymphoproliferation, and impaired homeostasis of reactive oxygen species in mice lacking the antioxidant-activated transcription factor Nrf2. Am J Pathol. 2006;168:1960–1974. doi: 10.2353/ajpath.2006.051113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macias D, Ganan Y, Sampath TK, Piedra ME, Ros MA, Hurle JM. Role of BMP-2 and OP-1 (BMP-7) in programmed cell death and skeletogenesis during chick limb development. Development. 1997;124:1109–1117. doi: 10.1242/dev.124.6.1109. [DOI] [PubMed] [Google Scholar]
- Maier CM, Chan PK. Role of superoxide dismutases in oxidative damage and neurodegenerative disorders. Neuroscientist. 2002;8:323–334. doi: 10.1177/107385840200800408. [DOI] [PubMed] [Google Scholar]
- Majmundar AJ, Wong WJ, Simon MC. Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell. 2010;40:294–309. doi: 10.1016/j.molcel.2010.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammon K, Savion S, Keshert R, Aroch I, Orenstein H, Fein A, Torchinsky A, Toder V. Expression of apoptosis-associated molecules in the fetoplacental unit of cyclophosphamide-treated mice. Reprod Toxicol. 2006;22:774–782. doi: 10.1016/j.reprotox.2006.07.009. [DOI] [PubMed] [Google Scholar]
- Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, Jungreis R, Nagata K, Harding HP, Ron D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004;18:3066–3077. doi: 10.1101/gad.1250704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino MD, Aksenov MY, Kelly SJ. Vitamin E protects against alcohol-induced cell loss and oxidative stress in the neonatal rat hippocampus. Int J Dev Neurosci. 2004;22:363–377. doi: 10.1016/j.ijdevneu.2004.04.005. [DOI] [PubMed] [Google Scholar]
- Masuoda HC, Townes TM. Targeted disruption of the activating transcription factor 4 gene results in severe fetal anemia in mice. Blood. 2002;99:736–745. doi: 10.1182/blood.v99.3.736. [DOI] [PubMed] [Google Scholar]
- Matsui M, Oshima M, Oshima H, Takaku K, Maruyama T, Yodoi J, Taketo MM. Early embryonic lethality caused by targeted disruption of the mouse thioredoxin gene. Dev Biol. 1996;178:179–185. doi: 10.1006/dbio.1996.0208. [DOI] [PubMed] [Google Scholar]
- Maxwell PH, Pugh CW, Ratcliff PJ. Activation of the HIF pathway in cancer. Curr Opin Genet Dev. 2001;11:293–299. doi: 10.1016/s0959-437x(00)00193-3. [DOI] [PubMed] [Google Scholar]
- McCord JM, Fridovich I. The reduction of cytochrome c by milk xanthine oxidase. J Biol Chem. 1968;243:5753–5760. [PubMed] [Google Scholar]
- McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol. 2001;21:1249–1259. doi: 10.1128/MCB.21.4.1249-1259.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menegola E, Broccia ML, Prati M, Ricolfi R, Giavini E. Glutathione status in diabetes-induced embryos. Biol Neonate. 1996;69:293–297. doi: 10.1159/000244323. [DOI] [PubMed] [Google Scholar]
- Miller E, Hare JW, Cloherty JP, Dunn PJ, Gleason RE, Soeldner JS, Kitzmiller JL. Elevated maternal hemoglobin A1c in early pregnancy and major congenital anomalies in infants of diabetic mothers. N Engl J Med. 1981;304:1331–1334. doi: 10.1056/NEJM198105283042204. [DOI] [PubMed] [Google Scholar]
- Mimura N, Hamada H, Kashio M, Jin H, Toyama Y, Kimura K, Iida M, Goto S, Saisho H, Toshimori K, Koseki H, Aoe T. Quality control in the endoplasmic reticulum impairs the biosynthesis of pulmonary surfactant in mice expressing mutant BiP. Cell Death Differ. 2007;14:1475–1485. doi: 10.1038/sj.cdd.4402151. [DOI] [PubMed] [Google Scholar]
- Mimura N, Yuasa S, Soma M, Jin H, Kimura K, Goto S, Koseki H, Aoe T. Altered quality control in the endoplasmic reticulum causes cortical dysplasia in knock-in mice expressing a mutant BiP. Mol Cell Biol. 2008;28:293–301. doi: 10.1128/MCB.00473-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirkes PE. Cyclophosphamide teratogenesis: a review. Teratog Carcinog Mutagen. 1985;5:75–88. doi: 10.1002/tcm.1770050202. [DOI] [PubMed] [Google Scholar]
- Mirkes PE, Little SA. Teratogen-induced cell death in postimplantation mouse embryos: differential tissue sensitivity and hallmarks of apoptosis. Cell Death Differ. 1998;5:592–600. doi: 10.1038/sj.cdd.4400390. [DOI] [PubMed] [Google Scholar]
- Mirkes PE, Little SA. Cytochrome c release from mitochondria of early postimplantaiton murine embryos exposed to 4-hydroperoxycyclophosphamide, heat shock and staurosporine. Toxicol Appl Pharmacol. 2000;162:197–206. doi: 10.1006/taap.1999.8849. [DOI] [PubMed] [Google Scholar]
- Miyagawa KI, Ishiwata-Kimata Y, Kohna H, Kimata Y. Ethanol stress impairs protein folding in the endoplasmic reticulum and activates Ire1 in Saccharomyces cerevisiae. Biosci Biotech Biochem. 2014;78:1389–1391. doi: 10.1080/09168451.2014.921561. [DOI] [PubMed] [Google Scholar]
- Moallem SA, Hales BF. The role of P53 and cell death by apoptosis and necrosis in 4-hyroxycyclophosphamide-induced limb malformations. Development. 1998;125:3225–3234. doi: 10.1242/dev.125.16.3225. [DOI] [PubMed] [Google Scholar]
- Molotski N, Savion S, Gerchikov N, Fein A, Toder V, Torchinsky A. Teratogen-induced distortions in the classical NF-κB activation pathway: correlation with the ability of embryos to survive teratogenic stress. Toxicol Appl Pharmacol. 2008;229:197–205. doi: 10.1016/j.taap.2008.01.011. [DOI] [PubMed] [Google Scholar]
- Mori K. Signaling pathways in the unfolded protein response: development from yeast to mammals. J Biochem. 2009;146:743–750. doi: 10.1093/jb/mvp166. [DOI] [PubMed] [Google Scholar]
- Mori C, Nakamura N, Kimura S, Irie H, Takigawa T, Shiota K. Programmed cell death in the interdigital tissue of the fetal mouse limb is apoptosis with DNA fragmentation. Anat Rec. 1995;242:103–110. doi: 10.1002/ar.1092420114. [DOI] [PubMed] [Google Scholar]
- Munns AJ, De Voss JJ, Hooper WD, Dickinson RG, Gillam EMJ. Bioactivation of phenytoin by human cytochrome P450: characterization of the mechanism and targets of covalent adduct formation. Chem Res Toxicol. 1997;10:1049–1058. doi: 10.1021/tx9700836. [DOI] [PubMed] [Google Scholar]
- Murakami T, Saito A, Hino S, Kondo S, Kanemoto S, Chihara K, Sekiya H, Tsumagari K, Ochiai K, Yoshinaga K, Saitoh M, Nishimura R, Yoneda T, Kou I, Furuichi T, Ikegawa S, Ikawa M, Okabe M, Wanaka A, Imaizumi K. Signalling mediated by the endoplasmic reticulum stress transducer OASIS is involved in bone formation. Nat Cell Biol. 2009;11:1205–1211. doi: 10.1038/ncb1963. [DOI] [PubMed] [Google Scholar]
- Nadanaka S, Yoshida H, Kano F, Murata M, Mori K. Activation of mammalian unfolded protein response is compatible with the quality control system operating in the endoplasmic reticulum. Mol Biol Cell. 2004;15:2537–2548. doi: 10.1091/mbc.E03-09-0693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadanaka S, Okada T, Yoshida H, Mori K. Role of disulfide bridges formed in the luminal domain of ATF6 in sensing endoplasmic reticulum stress. Mol Cell Biol. 2007;27:1027–1043. doi: 10.1128/MCB.00408-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair S, Xu C, Shen G, Hebbar V, Gopalakrishnan A, Hu R, Jain MR, Liew C, Chan JY, Kong AN. Toxicogenomics of endoplasmic reticulum stress inducer tunicamycin in the small intestine and liver of Nrf2 knockout and C57BL/6J mice. Toxicol Lett. 2007;168:21–39. doi: 10.1016/j.toxlet.2006.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi K, Sudo T, Morishima N. Endoplasmic reticulum stress signaling transmitted by ATF6 mediates apoptosis during muscle development. J Cell Biol. 2005;169:555–560. doi: 10.1083/jcb.200412024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narasimhulu S. Uncoupling of oxygen activation from hydroxylation in the steroid C-21 hydroxylare of bovine adrenocortical microsomes. Arch Biochem Biophys. 1971;147:384–390. doi: 10.1016/0003-9861(71)90394-8. [DOI] [PubMed] [Google Scholar]
- Nath AK, Enciso J, Kuniyasu M, Hao XY, Madri JA, Pinter E. Nitric oxide modulates murine yolk sac vasculogenesis and rescues glucose induced vasculopathy. Development. 2004;131:2485–2496. doi: 10.1242/dev.01131. [DOI] [PubMed] [Google Scholar]
- Nguyen T, Nioi P, Pickett CB. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J Biol Chem. 2009;284:13291–13295. doi: 10.1074/jbc.R900010200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niki E. Biomarkers of lipid peroxidation in clinical material. Biochim Biophys Acta. 2014;1840:809–817. doi: 10.1016/j.bbagen.2013.03.020. [DOI] [PubMed] [Google Scholar]
- Nishitoh H, Matsuzawa A, Tobiume K, Saegusa K, Takeda K, Inoue K, Hori S, Kakizuka A, Ichijo H. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 2002;16:1345–1355. doi: 10.1101/gad.992302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama J, Yi X, Venkatachalam MA, Dong Z. cDNA cloning and promoter analysis of rat caspase-9. Biochem J. 2001;360:49–56. doi: 10.1042/0264-6021:3600049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Njie-Mbye YF, Kulkarni-Chitnis M, Opere CA, Barrett A, Ohia SE. Lipid peroxidation: pathophysiological and pharmacological implications in the eye. Front Physiol. 2013;4:1–10. doi: 10.3389/fphys.2013.00366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonn L, Williams RR, Erickson RP, Powis G. The absence of mitochondrial thioredoxin 2 causes massive apoptosis, exencephaly, and early embryonic lethality in homozygous mice. Mol Cell Biol. 2003;23:916–922. doi: 10.1128/MCB.23.3.916-922.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordblom GD, Coon MJ. Hydrogen peroxide formation and stoichiometry of hydroxylation reactions catalyzed by highly purified liver microsomal cytochrome P-450. Arch Biochem Biophys. 1977;180:343–347. doi: 10.1016/0003-9861(77)90047-9. [DOI] [PubMed] [Google Scholar]
- Northington FJ, Ferriero DM, Graham EM, Traystman RJ, Martin LJ. Early neurodegeneration after hypoxia-ischemia in neonatal rat is necrosis while delayed neuronal death is apoptosis. Neurobiol Dis. 2001;8:207–219. doi: 10.1006/nbdi.2000.0371. [DOI] [PubMed] [Google Scholar]
- Okada T, Haze K, Nadanaka S, Yoshida H, Seidah NG, Hirano Y, Sato R, Negishi M, Mori K. J Biol Chem. 2003;278:31024–31032. doi: 10.1074/jbc.M300923200. [DOI] [PubMed] [Google Scholar]
- Ozolins TR. Cyclophosphamide and the teratology society: an awkward marriage. Birth Defects Res B. 2010;89:289–299. doi: 10.1002/bdrb.20255. [DOI] [PubMed] [Google Scholar]
- Pagani M, Fabbri M, Benedetti C, Fassio A, Pilati S, Bulleid NJ, Cabibbo A, Sitia R. Endoplasmic reticulum oxidoreductin 1-lbeta (ERO1-Lbeta), a human gene induced in the course of the unfolded protein response. J Biol Chem. 2000;275:23685–23692. doi: 10.1074/jbc.M003061200. [DOI] [PubMed] [Google Scholar]
- Pahl HL, Baeuerle PA. A novel signal transduction pathway from the endoplasmic reticulum to the nucleus is mediated by transcription factor NF-κB. EMBO J. 1995;14:2580–2588. doi: 10.1002/j.1460-2075.1995.tb07256.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pampfer S. Apoptosis in rodent peri-implantation embryos: differential susceptibility of inner cell mass and trophectoderm cell lineages—a review. Placenta. 2000;21:S3–S10. doi: 10.1053/plac.1999.0519. [DOI] [PubMed] [Google Scholar]
- Pandol SJ, Gorelick FS, Gerloff A, Lugea A. Alcohol abuse, endoplasmic reticulum stress and pancreatitis. Dig Dis. 2010;28:776–782. doi: 10.1159/000327212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pani L, Horal M, Loekan MR. Rescue of neural tube defects in Pax-3-deficient embryos by p53 loss of function: implications for Pax-3-dependent development and tumorigenesis. Genes Dev. 2002;16:676–680. doi: 10.1101/gad.969302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parman T, Chen G, Wells PG. Free radical intermediates of phenytoin and related teratogens: prostaglandin H synthase-catalyzed bioactivation, electron paramagnetic resonance spectrometry, and photochemical product analysis. J Biol Chem. 1998;273:25079–25088. doi: 10.1074/jbc.273.39.25079. [DOI] [PubMed] [Google Scholar]
- Parnell SE, Sulik KK, Dehart DB, Chen SY. Reduction of ethanol-induced ocular abnormalities in mice through dietary administration of N-acetylcysteine. Alcohol. 2010;44:699–705. doi: 10.1016/j.alcohol.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patten AR, Brocardo PS, Christie BR. Omega-3 supplementation can restore glutathione levels and prevent oxidative damage caused by prenatal ethanol exposure. J Nutr Biochem. 2013;24:760–769. doi: 10.1016/j.jnutbio.2012.04.003. [DOI] [PubMed] [Google Scholar]
- Patterson SE, Dealy CN. Mechanisms and models of endoplasmic reticulum stress in chondrodysplasia. Dev Dyn. 2014;243:875–893. doi: 10.1002/dvdy.24131. [DOI] [PubMed] [Google Scholar]
- Peeters-Scholte C, Braun K, Koster J, Kops N, Blomgren K, Buonocore G, van Buul-Offers S, Hagberg H, Nicolay K, van Bel F, Groenendaal F. Effects of allopurinol and deferoxamine on reperfusion injury of the brain in newborn piglets after neonatal hypoxia-ischemia. Pediatr Res. 2003;54:516–522. doi: 10.1203/01.PDR.0000081297.53793.C6. [DOI] [PubMed] [Google Scholar]
- Pekar O, Molotski N, Savion S, Fein A, Toder V, Torchinsky A. p53 regulates cyclophosphamide teratogenesis by controlling caspases 3, 8, 9 activation and NF-{kappa}B DNA binding. Reproduction. 2007;134:379–388. doi: 10.1530/REP-07-0086. [DOI] [PubMed] [Google Scholar]
- Pereira ER, Frudd K, Awad W, Hendershot LM. Endoplasmic reticulum (ER) stress and hypoxia response pathways interact to potentiate hypoxia-inducible factor 1 (HIF-1) transcriptional activity on targets like vascular endothelial growth factor (VEGF) J Biol Chem. 2014;289:3352–3364. doi: 10.1074/jbc.M113.507194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelan SA, Ito M, Loeken MR. Neural tube defects in embryos of diabetic mice: role of the Pax-3 gene and apoptosis. Diabetes. 1997;46:1189–1197. doi: 10.2337/diab.46.7.1189. [DOI] [PubMed] [Google Scholar]
- Plank MS, Boskovic DS, Sowers LC, Angeles DM. Biochemical markers of neonatal hypoxia. Pediatr Health. 2008;2:485–501. [Google Scholar]
- Porter NA, Caldwell SE, Mills KA. Mechanisms of free radical oxidation of unsaturated lipids. Lipids. 1995;30:277–290. doi: 10.1007/BF02536034. [DOI] [PubMed] [Google Scholar]
- Pozo Devoto VM, Bogetti ME, Fiszer de Plazas S. Developmental and hypoxia-induced cell death share common ultrastructural and biochemical apoptotic features in the central nervous system. Neuroscience. 2013;252:190–200. doi: 10.1016/j.neuroscience.2013.07.065. [DOI] [PubMed] [Google Scholar]
- Pryor W. Oxy-radicals and related species: their formation, lifetimes, and reactions. Ann Rev Physiol. 1986;48:657–667. doi: 10.1146/annurev.ph.48.030186.003301. [DOI] [PubMed] [Google Scholar]
- Radak Z, Kaneko T, Tahara S, Nakamoto H, Pucsok J, Sasvari M, Nyakas C, Goto S. Regular exercise improves cognitive function and decreases oxidative damage in rat brain. Neurochem Int. 2001;38:17–23. doi: 10.1016/s0197-0186(00)00063-2. [DOI] [PubMed] [Google Scholar]
- Ramirez-Solis R, Zheng H, Whiting J, Krumlauf R, Bradley A. Hoxb-4 (Hox-2.6) mutant mice show homeotic transformation of a cervical vertebra and defects in the closure of the sternal rudiments. Cell. 1993;73:279–294. doi: 10.1016/0092-8674(93)90229-j. [DOI] [PubMed] [Google Scholar]
- Rathmell JC, Thompson CB. Pathways of apoptosis in lymphocyte development, homeostasis, and disease. Cell. 2002;109:S97–S107. doi: 10.1016/s0092-8674(02)00704-3. [DOI] [PubMed] [Google Scholar]
- Ray A, Liu J, Ayoubi P, Pope C. Dose-related gene expression changes in fore-brain following acute, low-level chlorpyrifos exposure in neonatal rats. Toxicol Appl Pharmacol. 2010;248:144–155. doi: 10.1016/j.taap.2010.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimold AM, Etkin A, Clauss I, Perkins A, Friend DS, Zhang J, Horton HF, Scott A, Orkin SH, Byrne MC, Grusby MJ, Glimcher LH. An essential role in liver development for transcription factor XBP-1. Genes Dev. 2000;14:152–157. [PMC free article] [PubMed] [Google Scholar]
- Reimold AM, Iwakoshi NN, Manis J, Vallabhajosyula P, Szomolanyi-Tsuda E, Gravallese EM, Friend D, Grusby MJ, Alt F, Glimcher LH. Plasma cell differentiation requires the transcription factor XBP-1. Nature. 2001;412:300–307. doi: 10.1038/35085509. [DOI] [PubMed] [Google Scholar]
- Reinking BE, Wedemeyer EW, Weiss RM, Segar JL, Scholz TD. Cardiomyopathy in offspring of diabetic rats is associated with activation of the MAPK and apoptotic pathways. Cardiovasc Diabetol. 2009;8:43. doi: 10.1186/1475-2840-8-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhile MJ, Nagarkatti M, Nagarkatti PS. Role of Fas apoptosis and MHC genes in 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-induced immunotoxicity of T cells. Toxicology. 1996;110:153–167. doi: 10.1016/0300-483x(96)83962-x. [DOI] [PubMed] [Google Scholar]
- Ryan HE, Lo J, Johnson RS. HIF-1α is required for solid tumor formation and embryonic vascularization. EMBO J. 1998;17:3005–3015. doi: 10.1093/emboj/17.11.3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito A, Ochiai K, Kondo S, Tsumagari K, Murakami T, Cavener DR, Imaizumi K. Endoplasmic reticulum stress response mediated by the PERK-eIF2α-ATF4 pathway is involved in osteoblast differentiation induced by BMP2. J Biol Chem. 2011;286:4809–4818. doi: 10.1074/jbc.M110.152900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamaki H, Akazawa S, Ishibashi M, Izumino K, Takino H, Yamasaki H, Yamaguchi Y, Goto S, Urata Y, Kondo T, Nagataki S. Significance of glutathione-dependent antioxidant system in diabetes-induced embryonic malformations. Diabetes. 1999;48:1138–1144. doi: 10.2337/diabetes.48.5.1138. [DOI] [PubMed] [Google Scholar]
- Schneider M, Vogt Weisenhorn DM, Seiler A, Bornkamm GW, Brielmeier M, Conrad M. Embryonic expression profile of phospholipid hydroperoxide glutathione peroxidase. Gene Expr Patterns. 2006;6:489–494. doi: 10.1016/j.modgep.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Sha H, He Y, Chen H, Wang C, Zenno A, Shi H, Yang X, Zhang X, Qi L. The IRE1α-XBP1 pathway of the unfolded protein response is required for adipogenesis. Cell Metab. 2009;9:556–564. doi: 10.1016/j.cmet.2009.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldon RA, Christen S, Ferriero DM. Genetic and pharmacologic manipulation of oxidative stress after neonatal hypoxia-ischemia. Int J Dev Neurosci. 2008;26:87–92. doi: 10.1016/j.ijdevneu.2007.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Chen X, Hendershot L, Prywes R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev Cell. 2002;3:99–111. doi: 10.1016/s1534-5807(02)00203-4. [DOI] [PubMed] [Google Scholar]
- Shi ZZ, Osei–Frimpong J, Kala G, Kala SV, Barrios RJ, Habib GM, Lukin D, Danney CM, Matzuk MM, Lieberman MW. Glutathione synthesis is essential for mouse development but not for cell growth in culture. Proc Natl Acad Sci USA. 2000;97:5101–5106. doi: 10.1073/pnas.97.10.5101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirpoor A, Salami S, Khadem-Ansari MH, Minassian S, Yegiazarian M. Protective effect of vitamin E against ethanol-induced hyperhomocysteinemia, DNA damage, and atrophy in the developing male rat brain. Alcohol Clin Exp Res. 2009;33:1181–1186. doi: 10.1111/j.1530-0277.2009.00941.x. [DOI] [PubMed] [Google Scholar]
- Sies H. Oxidative stress: introductory remarks. In: Sies H, editor. Oxidative Stress. Academic Press; London: 1985. pp. 1–8. [Google Scholar]
- Singh NP, Singh UP, Guan H, Nagarkatti PS, Nagarkatti M. Prenatal exposure to TCDD triggers significant modulation of microRNA expression profile in the thymus that affects consequent gene expression. PLoS One. 2012;7:e45054. doi: 10.1371/journal.pone.0045054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slotkin TA, Oliver CA, Seidler FJ. Critical periods for the role of oxidative stress in the developmental neurotoxicity of chlorpyrifos and terbutaline, alone or in combination. Dev Brain Res. 2005;157:172–180. doi: 10.1016/j.devbrainres.2005.04.001. [DOI] [PubMed] [Google Scholar]
- Slotkin TA, MacKillop EA, Ryde IT, Seidler FJ. Ameliorating the developmental neurotoxicity of chlorpyrifos: a mechanisms-based approach in PC12 cells. Environ Health Perspect. 2007;115:1306–1313. doi: 10.1289/ehp.10194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soberman RJ, Christmas P. The organization and consequences of eicosanoid signaling. J Clin Invest. 2003;111:1107–1113. doi: 10.1172/JCI18338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soleman D, Cornel L, Little SA, Mirkes PE. Teratogen-induced activation of the mitochondrial apoptotic pathway in the yolk sac of day 9 mouse embryos. Birth Defects Res A. 2003;67:98–107. doi: 10.1002/bdra.10005. [DOI] [PubMed] [Google Scholar]
- Sovolyova N, Healy S, Samali A, Logue SE. Stressed to death – mechanisms of ER stress-induced cell death. Biol Chem. 2014;395:1–13. doi: 10.1515/hsz-2013-0174. [DOI] [PubMed] [Google Scholar]
- Sowter HM, Ratcliff PJ, Watson P, Greenberg AH, Harris AL. HIF-1-dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Cancer Res. 2001;61:6669–6673. [PubMed] [Google Scholar]
- Sultana R, Perluigi M, Butterfield DA. Lipid peroxidation triggers neurodegeneration: a redox proteomics view into the Alzheimer disease brain. Free Radic Biol Med. 2013;62:157–169. doi: 10.1016/j.freeradbiomed.2012.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surova O, Zhivotovsky B. Various modes of cell death induced by DNA damage. Oncogene. 2013;32:3789–3797. doi: 10.1038/onc.2012.556. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Tsujimura T, Takeda K, Sugihara A, Maekawa A, Terada N, Yoshida N, Akira S. Targeted disruption of ATF4 discloses its essential role in the formation of eye lens fibres. Genes Cells. 1998;3:801–810. doi: 10.1046/j.1365-2443.1998.00230.x. [DOI] [PubMed] [Google Scholar]
- Taylor RC, Cullen SP, Martin SJ. Apoptosis: controlled demolition at the cellular level. Mol Cell Biol. 2008;9:231–241. doi: 10.1038/nrm2312. [DOI] [PubMed] [Google Scholar]
- Tell G, Quadrifoglio F, Tiribelli C, Kelley MR. The Many functions of APE1/Ref-1: not only a DNA repair enzyme. Antioxid Redox Signal. 2009;11:601–691. doi: 10.1089/ars.2008.2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ten VS, Yao J, Ratner V, Sosunov S, Fraser DA, Botto M, Baalasubramanian S, Morgan BP, Silverstein S, Stark R, Polin R, Vannucci SJ, Pinsky D, Starkov AA. Complement component C1q mediates mitochondria-driven oxidative stress in neonatal hypoxic–ischemic brain injury. J Neurosci. 2010;30:2077. doi: 10.1523/JNEUROSCI.5249-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas SE, Dalton LE, Daly ML, Malzer E, Marciniak SJ. Diabetes as a disease of endoplasmic reticulum stress. Diabetes Metab Res Rev. 2010;26:611–621. doi: 10.1002/dmrr.1132. [DOI] [PubMed] [Google Scholar]
- Timme-Laragy AR, Goldstone JV, Imhoff BR, Stegeman JJ, Hahn ME, Hansen JM. Glutathione redox dynamics and expression of glutathione-related genes in the developing embryo. Free Radic Biol Med. 2013;65:89–101. doi: 10.1016/j.freeradbiomed.2013.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmins JM, Ozcan L, Seimon TA, Li G, Malagelada C, Backs J, Backs T, Bassel-Duby R, Olson EN, Anderson ME, Tabas I. Calcium/calmodulin-dependent protein kinase II links ER stress with Fas and mitochondrial apoptosis pathways. J Clin Invest. 2009;119:2925–2941. doi: 10.1172/JCI38857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirasophon W, Welihinda AA, Kaufman RJ. A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev. 1998;12:1812–1824. doi: 10.1101/gad.12.12.1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toomey BH, Bello S, Hahn ME, Cantrell S, Wright P, Tillitt DE, Di Giulio RT. 2,3,7,8-Tetrachlorodibenzo-p-dioxin induces apoptotic cell death and cytochrome P4501A expression in developing Fundulus heteroclitus embryos. Aquat Toxicol. 2001;53:127–138. doi: 10.1016/s0166-445x(00)00161-2. [DOI] [PubMed] [Google Scholar]
- Torchinsky A, Savion S, Gorivodsky M, Shepshelovich J, Zaslavsky Z, Fein A, Toder V. Cyclophosphamide induced teratogenesis in ICR mice: the role of apoptosis. Teratog Carcinog Mutagen. 1995;15:179–190. doi: 10.1002/tcm.1770150404. [DOI] [PubMed] [Google Scholar]
- Torchinsky A, Lishanski L, Wolstein O, Shepshelovich J, Orenstein H, Savion S, Zaslavsky Z, Carp H, Brill A, Dikstein R, Toder V, Fein A. NF-κB DNA-binding activity in embryos responding to a teratogen, cyclophosphamide. BMC Dev Biol. 2002;2:2. doi: 10.1186/1471-213X-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torchinsky A, Shepshelovich J, Orebstein H, Zaslavsky Z, Savion S, Carp H, Fein A, Toder V. TNF-alpha protects embryos exposed to developmental toxicants. Am J Reprod Immunol. 2003;49:159–168. doi: 10.1034/j.1600-0897.2003.01174.x. [DOI] [PubMed] [Google Scholar]
- Trollmann R, Gassmann M. The role of hypoxia-inducible transcription factors in the hypoxic neonatal brain. Brain Dev Jpn. 2009;31:503–509. doi: 10.1016/j.braindev.2009.03.007. [DOI] [PubMed] [Google Scholar]
- Tsang KY, Chan D, Cheslett D, Chan WC, So CL, Melhado IG, Chan TW, Kwan KM, Hunziker EB, Yamada Y, Bateman JF, Cheung KM, Cheah KS. Surviving endoplasmic reticulum stress is coupled to altered chondrocyte differentiation and function. PLoS Biol. 2007;5:e44. doi: 10.1371/journal.pbio.0050044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsendensodnom O, Vacaru AM, Howarth DL, Yin C, Sadler KC. Ethanol metabolism and oxidative stress are required for unfolded protein response activation and steatosis in zebrafish with alcoholic liver disease. Dis Model Mech. 2013;6:1213–1226. doi: 10.1242/dmm.012195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu BP, Weissman JS. The FAD- and O2-dependent reaction cycle of Ero1-mediated oxidative protein folding in the endoplasmic reticulum. Mol Cell. 2002;10:983–994. doi: 10.1016/s1097-2765(02)00696-2. [DOI] [PubMed] [Google Scholar]
- Tu BP, Weissman JS. Oxidative protein folding in eukaryotes: mechanisms and consequences. J Cell Biol. 2004;164:341–346. doi: 10.1083/jcb.200311055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ufer C, Wang CC. The roles of glutathione peroxidases during embryo development. Front Mol Neurosci. 2011;4:1–14. doi: 10.3389/fnmol.2011.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ufer C, Wang CC, Borchert A, Heydeck D, Kuhn H. Redox control in mammalian embryo development. Antioxid Redox Signal. 2010;13:833–874. doi: 10.1089/ars.2009.3044. [DOI] [PubMed] [Google Scholar]
- Upton JP, Wang L, Han D, Wang ES, Huskey NE, Lim L, Tuitt M, McManus, Ruggero D, Goga A, Papa FR, Oakes SA. IRE1α cleaves select micro-RNAs during ER stress to derepress translation of proapoptotic caspase-2. Science. 2012;338:818–822. doi: 10.1126/science.1226191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, Ron D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287:664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- Van Der Vlies D, Makkinje M, Jansens A, Braakman I, Verklejj AJ, Wirtz KW, Andries J. Oxidation of ER resident proteins upon oxidative stress: effects of altering cellular redox/antioxidant status and implications for protein maturation. Antioxid Redox Signal. 2003;5:381–387. doi: 10.1089/152308603768295113. [DOI] [PubMed] [Google Scholar]
- Varfolomeev EE, Schuchmann M, Luria V, Chiannilkulchai N, Beckmannm JS, Mett IL, Rebrikov D, Brodianski VM, Kemper OC, Kollet O, Lapidot T, Soffer D, Sobe T, Avraham KB, Goncharov T, Holtmann H, Lonai P, Wallach D. Targeted disruption of the mouse caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity. 1998;9:267–276. doi: 10.1016/s1074-7613(00)80609-3. [DOI] [PubMed] [Google Scholar]
- Vaux KK, Kahole NC, Jones KL. Cyclophosphamide, methotrexate, and cytarabine embryopathy: is apoptosis the common pathway? Birth Defects Res A Clin Mol Teratol. 2003;67:403–408. doi: 10.1002/bdra.10060. [DOI] [PubMed] [Google Scholar]
- Viner RI, Huhmer AF, Bigelow DJ, Schoneich C. The oxidative inactivation of sarcoplasmic reticulum Ca(2+)-ATPase by peroxynitrite. Free Radic Res. 1996;24:243–259. doi: 10.3109/10715769609088022. [DOI] [PubMed] [Google Scholar]
- Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 isabasic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA. 1995;92:5510–5514. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Lian N, Ma Y, Li L, Gallant RC, Elefteriou F, Yang X. Chondrocytic Atf4 regulates osteoblast differentiation and function via Ihh. Development. 2012a;139:601–611. doi: 10.1242/dev.069575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Ma S, Qi G. Effect of hypoxia-inducible factor 1-alpha on hypoxia/reoxygenation-induced apoptosis in primary neonatal rat cardiomyocytes. Biochem Biophys Res Commun. 2012b;417:1227–1234. doi: 10.1016/j.bbrc.2011.12.115. [DOI] [PubMed] [Google Scholar]
- Wang F, Reece A, Yang P. Superoxide dismutase 1 overexpression in mice abolishes maternal diabetes-induced endoplasmic reticulum stress in diabetic embryopathy. Am J Obstet Gynecol. 2013;209:345.e1–345.e7. doi: 10.1016/j.ajog.2013.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster WS, Abela D. The effect of hypoxia in development. Birth Defects Res C. 2007;81:215–288. doi: 10.1002/bdrc.20102. [DOI] [PubMed] [Google Scholar]
- Weinmann M, Jendrossek V, Handrick R, Guner D, Goecke B, Belka C. Molecular ordering of hypoxia-induced apoptosis: critical involvement of the mitochondrial death pathway in a FADD/caspase-8 independent manner. Oncogene. 2004;23:3757–3769. doi: 10.1038/sj.onc.1207481. [DOI] [PubMed] [Google Scholar]
- Weng H, Li X, Reece EA, Yang P. SOD1 suppresses maternal hyperglycemia-increased iNOS expression and consequent nitrosative stress in diabetic embryopathy. Am J Obstet Gynecol. 2012;206:448.e1–448.e7. doi: 10.1016/j.ajog.2012.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenztel P, Welsh N, Eriksson UJ. Developmental damage, increased lipid peroxidation, diminished cyclooxygenase-2 gene expression, and lowered prostaglandin E2 levels in rat embryos exposed to a diabetic environment. Diabetes. 1999;48:813–820. doi: 10.2337/diabetes.48.4.813. [DOI] [PubMed] [Google Scholar]
- Widness JA, Goldman AS, Susa JB, Oh W, Schwartz R. Impermeability of the rat placenta to insulin during organogenesis. Teratology. 1983;28:327–332. doi: 10.1002/tera.1420280304. [DOI] [PubMed] [Google Scholar]
- Winn LM, Kim PM, Nickoloff JA. Oxidative stress-induced homologous recombination as a novel mechanism for phenytoin-initiated toxicity. J Pharmacol Exp Ther. 2003;306:523–527. doi: 10.1124/jpet.103.052639. [DOI] [PubMed] [Google Scholar]
- Wong M, Wells PG. Effects of N-acetylcysteine on fetal development and on phenytoin teratogenicity in mice. Teratog Carcinog Mutagen. 1988;8:65–79. doi: 10.1002/tcm.1770080202. [DOI] [PubMed] [Google Scholar]
- Xiao R, Yu HL, Zhao HF, Liang J, Feng JF, Wang W. Developmental neurotoxicity role of cyclophosphamide on post-neural tube closure of rodents in vitro and in vivo. Int J Dev Neurosci. 2007;25:531–537. doi: 10.1016/j.ijdevneu.2007.09.012. [DOI] [PubMed] [Google Scholar]
- Yamamoto K, Sato T, Matsui T, Sato M, Okada T, Yoshida H, Harada A, Mori K. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6α and XBP1. Dev Cell. 2007;13:365–376. doi: 10.1016/j.devcel.2007.07.018. [DOI] [PubMed] [Google Scholar]
- Yamamoto K, Takahara K, Oyadomari S, Okada T, Sato T, Harada A, Mori K. Induction of liver steatosis and lipid droplet formation in ATF6α-knockout mice burdened with pharmacological endoplasmic reticulum stress. Mol Biol Cell. 2010;21:2975–2986. doi: 10.1091/mbc.E09-02-0133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki H, Hiramatsu N, Hayakawa K, Tagawa Y, Okamura M, Ogata R, Huang T, Nakajima S, Paton AW, Paton JC, Kitamura M. Activation of the Akt-NF-κB pathway by subtilase cytotoxin through the ATF6 branch of the unfolded protein response. J Immunol. 2009;183:1480–1487. doi: 10.4049/jimmunol.0900017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Y, Sabharwal P, Rao M, Sockanathan S. The antioxidant enzyme Prdx1 controls neuronal differentiation by thiol-redox-dependent activation of GDE2. Cell. 2009;138:1209–1221. doi: 10.1016/j.cell.2009.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Matsuda K, Bialek P, Jacquot S, Masuoka HC, Schinke T, Li L, Brancorsini S, Sassone-Corsi P, Townes TM, Hanauer A, Karsenty G. ATF4 is a substrate of RSK2 and an essential regulator of osteoblast biology; implication for Coffine–Lowry Syndrome. Cell. 2004;117:387–398. doi: 10.1016/s0092-8674(04)00344-7. [DOI] [PubMed] [Google Scholar]
- Yang P, Zhao Z, Reece EA. Involvement of c-Jun N-terminal kinases activation in diabetic embryopathy. Biochem Biophys Res Commun. 2007;357:749–754. doi: 10.1016/j.bbrc.2007.04.023. [DOI] [PubMed] [Google Scholar]
- Yang P, Zhao Z, Reece A. Activation of oxidative stress signaling that is implicated in apoptosis with a mouse model of diabetic embryopathy. Am J Obstet Gynecol. 2008;198:130.e1–130.e7. doi: 10.1016/j.ajog.2007.06.070. [DOI] [PubMed] [Google Scholar]
- Yang P, Cao Y, Li H. Hyperglycemia induces inducible nitric oxide synthase gene expression and consequent nitrosative stress via c-Jun N-terminal kinase activation. Am J Obstet Gynecol. 2010;203:185.e5–185.e11. doi: 10.1016/j.ajog.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang P, Li X, Xu C, Eckert RL, Reece EA, Zielke HR, Wang F. Maternal hyperglycemia activates an ASK1–FoxO3a–caspase 8 pathway that leads to embryonic neural tube defects. Sci Signal. 2013;6:ra74. doi: 10.1126/scisignal.2004020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B, He K, Zheng F, Wan L, Yu X, Wang X, Zhao D, Bai Y, Chu W, Yan S, Lu Y. Over-expression of hypoxia-inducible factor-1 alpha in vitro protects the cardiac fibroblasts from hypoxia-induced apoptosis. J Cardiovasc Med Hagerst. 2014;15:579–586. doi: 10.2459/JCM.0b013e3283629c52. [DOI] [PubMed] [Google Scholar]
- Ye J, Rawson RB, Komuro R, Chen X, Dave UP, Prywes R, Brown MS, Goldstein JL. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol Cell. 2000;6:1355–1364. doi: 10.1016/s1097-2765(00)00133-7. [DOI] [PubMed] [Google Scholar]
- Yeh WC, de la Pompa JL, McCurrach ME, Shu HB, Elia AJ, Shahinian A, Ng M, Wakeham A, Khoo W, Mitchell K, El-Deiry WS, Lowe SW, Goeddel DV, Mak TW. FADD: essential for embryo development and signaling from some, but not all, inducers of apoptosis. Science. 1998;279:1954–1958. doi: 10.1126/science.279.5358.1954. [DOI] [PubMed] [Google Scholar]
- Yoon D, Pastore YD, Divolky V, Liu E, Mlodnicka AE, Rainey K, Ponka P, Semenza GL, Schumacher A, Prchal JT. Hypoxia-inducible Factor-1 deficiency results in dysregulated erythropoiesis signaling and iron homeostasis in mouse development. J Biol Chem. 2006;281:25703–25711. doi: 10.1074/jbc.M602329200. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Haze K, Yanagi H, Yura T, Mori K. Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins, involvement of basic-leucine zipper transcription factors. J Biol Chem. 1998a;273:33741–33749. doi: 10.1074/jbc.273.50.33741. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Kong YY, Yoshida R, Elia AJ, Hakem A, Hakem R, Penninger JM, Mak TW. Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell. 1998b;94:739–750. doi: 10.1016/s0092-8674(00)81733-x. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–891. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- Yuan L, Cao Y, Oswald F, Knochel W. IRE1β is required for mesoderm formation in Xenopus embryos. Mech Dev. 2008;125:207–222. doi: 10.1016/j.mod.2007.11.010. [DOI] [PubMed] [Google Scholar]
- Yung HW, Cox M, Tissot van Patot M, Burton GJ. Evidence of endoplasmic reticulum stress and protein synthesis inhibition in the placenta of non-native women at high altitude. FASEB J. 2012;26:1970–1981. doi: 10.1096/fj.11-190082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidi A, Barron L, Sharov VS, Schoneich C, Michaelis EK, Michaelis ML. Oxidative inactivation of purified plasma membrane Ca2+-ATPase by hydrogen peroxide and protection by calmodulin. Biochemistry. 2003;42:12001–12010. doi: 10.1021/bi034565u. [DOI] [PubMed] [Google Scholar]
- Zhang P, McGrath B, Li S, Frank A, Zambito F, Reinert J, Gannon M, Ma K, McNaughton K, Cavener DR. The PERK eukaryotic initiation factor 2 alpha kinase is required for the development of the skeletal system, postnatal growth, and the function and viability of the pancreas. Mol Cell Biol. 2002;22:3864–3874. doi: 10.1128/MCB.22.11.3864-3874.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Wong HN, Song B, Miller CN, Scheuner D, Kaufman RJ. The unfolded protein response sensor IRE1a is required at 2 distinct steps in B cell lymphopoiesis. J Clin Invest. 2005;115:268–281. doi: 10.1172/JCI21848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Feng D, Li Y, Lida K, McGrath B, Cavener DR. PERK EIF2AK3 control of pancreatic beta cell differentiation and proliferation is required for postnatal glucose homeostasis. Cell Metab. 2006;4:491–497. doi: 10.1016/j.cmet.2006.11.002. [DOI] [PubMed] [Google Scholar]
- Zhang JY, Diao YF, Kim HR, Jin DI. Inhibition of endoplasmic reticulum stress improves mouse embryo development. PLoS One. 2012;7:e40433. doi: 10.1371/journal.pone.0040433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Li H, Liu X, BIJ Effect of caspase-9 inhibition on endoplasmic reticulum stress induced cortical neuronal injury in rats. Int J Clin Exp Med. 2013;6:546–551. [PMC free article] [PubMed] [Google Scholar]
- Zhao Z. Endoplasmic reticulum stress in maternal diabetes-induced cardiac malformations during critical cardiogenesis period. Birth Defects Res B. 2012;95:1–6. doi: 10.1002/bdrb.20330. [DOI] [PubMed] [Google Scholar]
- Zhao Z, Yang P, Eckert RL, Reece EA. Caspase-8: a key role IN the pathogenesis of diabetic embryopathy. Birth Defects Res B Dev Reprod Toxicol. 2009;86:72–77. doi: 10.1002/bdrb.20185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Eckert RL, Reece EA. Reduction in embryonic malformations and alleviation of endoplasmic reticulum stress by nitric oxide synthase inhibition in diabetic embryopathy. Reprod Sci. 2012;19:823–831. doi: 10.1177/1933719111434543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou H, Niswander L. Requirement for BMP signaling in interdigital apoptosis and scale formation. Science. 1996;272:738–741. doi: 10.1126/science.272.5262.738. [DOI] [PubMed] [Google Scholar]