

Abstract
Neuroblastoma (NB) is one of the most common and deadly childhood solid tumors. These tumors are characterized by clinical heterogeneity, from spontaneous regression to relentless progression, and the Trk family of neurotrophin receptors plays an important role in this heterogeneous behavior. We wanted to determine if entrectinib (RXDX-101, Ignyta, Inc.), an oral Pan-Trk, Alk and Ros1 inhibitor was effective in our NB model. In vitro effects of entrectinib, either as a single agent or in combination with the chemotherapeutic agents Irinotecan (Irino) and Temozolomide (TMZ), were studied on an SH-SY5Y cell line stably transfected with TrkB. In vivo growth inhibition activity was studied in NB xenografts, again as single agent or in combination with Irino-TMZ. Entrectinib significantly inhibited the growth of TrkB-expressing NB cells in vitro, and it significantly enhanced the growth inhibition of Irino-TMZ when used in combination. Single agent therapy resulted in significant tumor growth inhibition in animals treated with entrectinib compared to control animals [p<0.0001 for event-free survival (EFS)]. Addition of entrectinib to Irino-TMZ also significantly improved the EFS of animals compared to vehicle or Irino-TMZ treated animals [p<0.0001 for combination vs. control, p=0.0012 for combination vs. Irino-TMZ]. We show that entrectinib inhibits growth of TrkB expressing NB cells in vitro and in vivo, and that it enhances the efficacy of conventional chemotherapy in in vivo models. Our data suggest that entrectinib is a potent Trk inhibitor and should be tested in clinical trials for NBs and other Trk-expressing tumors.
Keywords: Neuroblastoma, TrkB, NTRK2, entrectinib, RXDX-101, kinase inhibitor, xenograft
1. Introduction
Receptor tyrosine kinases (RTKs) play important roles in normal development [1], and they are frequent targets of activating mutations, rearrangements or overexpression in human cancers [2]. Thus, there is considerable interest in identifying the RTKs that contribute to tumorigenesis in specific tumors, as these present attractive targets for biologically-based therapy. Neuroblastoma (NB) is a tumor of the peripheral nervous system in children, and several RTK genes have been implicated in malignant transformation or progression of these tumors, including ALK, NTRKs, RET, EGFR and IGFR [reviewed in [3]]. We have focused primarily on the Trk family of neurotrophin receptors (TrkA encoded by NTRK1, TrkB encoded by NTRK2, TrkC encoded by NTRK3) because of the critical role they play in regulating both favorable and unfavorable clinical behavior in NBs [4, 5].
TrkA is the receptor for nerve growth factor (NGF), and high TrkA expression is associated with clinically and biologically favorable tumors that have a propensity to undergo spontaneous regression or differentiation [6–11]. Conversely, TrkB is the cognate receptor for brain-derived neurotrophic factor (BDNF), and high expression of TrkB plus BDNF is found in the majority of high-risk NBs, especially those with MYCN amplification [8]. Tumors that co-express TrkB and BDNF are more likely to be invasive, metastatic, angiogenic and drug resistant [12–18]. TrkC is also expressed in primary NBs, but these tumors appear to be a subset of TrkA-expressing tumors [19–21]. Thus, targeting Trk receptors, especially TrkB, should be an effective therapeutic strategy in NBs [3–5]. Furthermore, Trks are activated by translocation or autocrine overexpression in a number of common pediatric and adult cancers [22, 23], so a potent and selective Trk inhibitor would be of interest for the treatment of a variety of cancers.
We have shown previously that inhibition of the Trk signaling pathway with lestaurtinib (CEP-101, Cephalon, Inc.), a pan-Trk inhibitor, resulted in inhibition of growth of TrkB-expressing NB cells in vitro and in vivo, and cotreatment with lestaurtinib and chemotherapy resulted in enhanced anti-tumor efficacy [13, 24–26]. Indeed, this agent was used in a phase 1 clinical trial, and half the patients treated at a biologically effective dose had durable clinical benefit (partial responses, stable disease) for a mean of over 10 months [27]. However, support for clinical development of this agent was discontinued after a corporate takeover. We tested additional pan-Trk inhibitors, including AZ64 (AstraZeneca, Inc.) and GNF-4256 (Genomics Institute of the Novartis Research Foundation) in vitro and in our NB xenograft model [28]. Both were effective inhibitors of TrkA/B/C activation in the low nanomolar range, and both inhibited growth of NB xenografts as single agents. Also, both enhanced the efficacy of chemotherapy with Irinotecan (Irino) and Temozolomide (TMZ), without additional toxicity [28, 29]. However, support for clinical development of these agents was also discontinued.
In this study, we tested the efficacy of entrectinib (RXDX-101; Ignyta, Inc.), a selective pan-Trk, Alk and Ros tyrosine kinase inhibitor, to inhibit the growth of TrkB-expressing NB cells in vitro and in vivo. The compound was tested both as a single agent, and in combination therapy with the relapsed NB chemotherapy regimen, Irino-TMZ. We saw significant inhibition of NB growth both in vitro and in vivo with entrectinib as a single agent. In fact, this agent was more potent than lestaurtinib, which served as a positive control for these studies. Furthermore, the combination of entrectinib with Irino-TMZ resulted in significantly increased EFS compared to the group receiving chemotherapy alone. Therefore, entrectinib is a promising agent that inhibits activated TRK receptors, and we are moving this agent forward to phase 1 clinical trials.
2. Materials and methods
2.1. Compounds
Entrectinib (RXDX-101, Ignyta, Inc.) is an orally available small molecule inhibitor of pan-Trk, Alk and Ros1 tyrosine kinases. It was dissolved in DMSO to obtain stocks for in vitro studies. For in vivo experiments, it was reconstituted in 0.5% methylcellulose (Sigma-Aldrich, viscosity 400cP, 2% in H2O) containing 1% Tween 80 at a final dosing volume of 10 ml/kg (e.g., 0.2 ml for a 20 gm mouse). Entrectinib solution was stirred at RT for 30 min, and then sonicated in a water bath sonicator for 20 min. This formulation was made fresh every week. Animals were dosed BID, 7 days/week at 60 mg/kg.
Temozolomide (Temodar—TMZ, Teva, 20 mg/capsule) was obtained from the pharmacy at The Children's Hospital of Philadelphia (CHOP). The compound was reconstituted in saline at a concentration of 1 mg/ml. Animals were dosed once a day PO at 7.5 mg/kg Monday through Friday of each week (except for the groups that received the compound every other week). Irinotecan (Camptosar—Irino, Novaplus, 20 mg/ml) was diluted in saline and dosed once a day PO at 0.63 mg/kg Monday through Friday of each week.
2.2. Cell Lines and Authentication
Parental NLF and SH-SY5Y cells were obtained from ATCC and cultured as per ATCC guidelines and instructions. Trk-null SH-SY5Y cells (ALK-mutated, F1174L) were stably transfected with TrkB (SY5Y-TrkB, clone BR6) and NLF cells (ALK-WT) stably transfected with TrkB (NLF-TrkB, clone #6). We tested the integrity and authenticity of these cell lines for endotoxins, mycoplasmas, bacterial and other viral contaminations as well as genetic variations by multiplex PCR techniques. These tests were performed on annual basis at the cell center services facility of University of Pennsylvania. These cell lines were used for in vitro and in vivo experiments to determine the effect of entrectinib on TrkB phosphorylation. SH-SY5Y cells were used as Trk-null controls. Cells were grown in RPMI-1640 medium (Gibco) containing 10% fetal bovine serum (Cellgro), 0.4 mg/ml Penicillin/Streptomycin (Gibco), and maintained in 150 cm3 Costar culture flasks in a humidified atmosphere of 95% air and 5% CO2. Transfected cells were maintained in media containing 0.3 mg/ml G418 sulfate (stock solution: 50 mg/ml; Corning). Cells were harvested using 0.2% tetrasodium EDTA in phosphate buffered saline (PBS). Xenograft studies were conducted with SH-SY5Y cells stably transfected with TrkB.
2.3. Animals
Six-week-old athymic nu/nu mice were obtained from Jackson Laboratories. Mice were maintained at five per cage under humidity- and temperature-controlled conditions in a light/dark cycle that was set at 12-hr intervals. The Institutional Animal Care Committee of the Children's Hospital of Philadelphia (CHOP) Research Institute approved the animal studies described herein.
2.4. In Vitro Experiments and Western Blot Analysis
To determine the inhibitory effect of entrectinib on TrkB phosphorylation, cells were grown in 10 cm3 dishes to 70–80% confluence under standard culture conditions. Cells were serum starved in 2% FBS medium for 2 hr before being exposing to different concentrations of entrectinib (10 - 200 nM) for 1 hr. Cells were stimulated with 100 ng/mL of the TrkB ligand, BDNF (PeproTech, Rocky Hill, NJ) for 15 minutes before total protein was harvested for analysis by Western blots. Trk expression was confirmed using anti-Phospho Trk antibody (p-Trk, Tyr-490, Cell Signaling Technology, Danvers, MA) or anti-Pan-Trk antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA). Downstream signaling inhibition was analyzed using anti-phospho-Akt, anti-phospho-Erk1/2 antibodies, total Akt and anti-Erk1/2 (Cell Signaling Technology, Danvers, MA) and actin (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) was used as loading control.
2.5. Sulforhodamine B (SRB) assay
Sulforhodamine B (SRB) assays were performed to determine the effect of entrectinib as a single agent and in combination with Irino-TMZ on the survival and growth of TrkB-expressing NB cells. NLF, NLF-TrkB, SY5Y or SY5Y-TrkB cells (5×103/per well) were plated in 96 well plates, and they were exposed to drug at different concentrations (1, 5, 10, 20, 30, 50 and 100 nM of entrectinib, 1.5 μM Irino and 50 μM TMZ, respectively) for one hr followed by addition of 100 ng/mL of BDNF. Plates were harvested at 24, 48, and 72 hr following addition of drug. The plates were processed and cell viability was analyzed using a standard SRB assay protocol [30]. All in vitro experiments were performed in triplicate and repeated at least 3 times.
2.6. In Vivo Experiments
For the xenograft studies, animals were injected subcutaneously in the flank with 1 × 107 SY5Y-TrkB cells in 0.1 ml of Matrigel (BD Bioscience, Palo Alto, CA). Tumors were measured 2 times per week in 3 dimensions, and the volume calculated as follows: [(0.523xLxWxW)/1000]. Body weights were measured at least twice a week, and the dose of compound was adjusted accordingly. Treatment with entrectinib, Irino and TMZ started about 15–17 days after tumor inoculation when the average tumor size was 0.2 cm3. Mice were sacrificed when tumor volume reached 3 cm3. Tumors were harvested and flash frozen on dry ice for analysis of protein expression using Western blot. Tumor lysates were obtained using Fast Prep 24 System (MP Biomedicals) in the presence of a protease inhibitor cocktail (EMD Millipore) and phosphatase inhibitor cocktail (EMD Millipore). The following antibodies were used for the Western blot (all were from Cell Signaling Technology, unless otherwise specified): anti-TrkB (Abcam), anti-phospho- TrkB (Tyr816); anti-Trk (pan-Trk); anti-phospho-Akt (Ser473); anti-Akt; anti-phosphop44/42 Erk (Thr202/Tyr204); anti-p44/42 Erk; anti-Phospho-PLCγ1 (Tyr783) and anti- PLCγ1. Plasma was obtained at different times points after dosing for PK/PD studies.
2.7. Pharmacokinetic studies
Entrectinib was dosed at 60 mg/kg BID, for the entire duration of the study. After the final dose was given, the blood samples were drawn from 4 mice per time point via retro-orbital bleeding and collected in heparinized tubes on wet ice. The plasma was then separated by centrifugation at 1200 g for 10 minutes at 4°C. The concentration of entrectinib (free base) was measured by LC-MS-MS. The pharmacokinetic analysis was performed using the Watson system (v. 7.4, Thermo Fisher Scientific, Waltham, MA, USA), and plotted using GraphPad Prism (mean ±SD).
2.8. Statistical Analysis
Linear mixed effects model was used to test the difference in the rate of tumor volume changing over time between different groups. The model included group, day, and group-by-day interaction as fixed effects, and included a random intercept and a random slope for each mouse. A significant group-by-day interaction would suggest that the tumor volume changes at different rates for the two comparison groups. The model used the control group as the reference group and created separate group indicators and interaction terms for other groups. Appropriate contrast statements were created to compare the two groups other than control group. Event free survival (EFS) curves were estimated using Kaplan-Meier method and compared using log-rank test. Event includes death and sacrifice of mice due to tumor burden. Statistical analyses on the western blot images were performed using the Prism two-way ANOVA method followed by a Sidak post-test. Each experiment was performed at least three times either in triplicate or quadruplicate sets. Results for phosphoprotein expression was normalized against each total protein detection antibody.
3. Results
3.1. Effect of entrectinib on TrkB activation in vitro
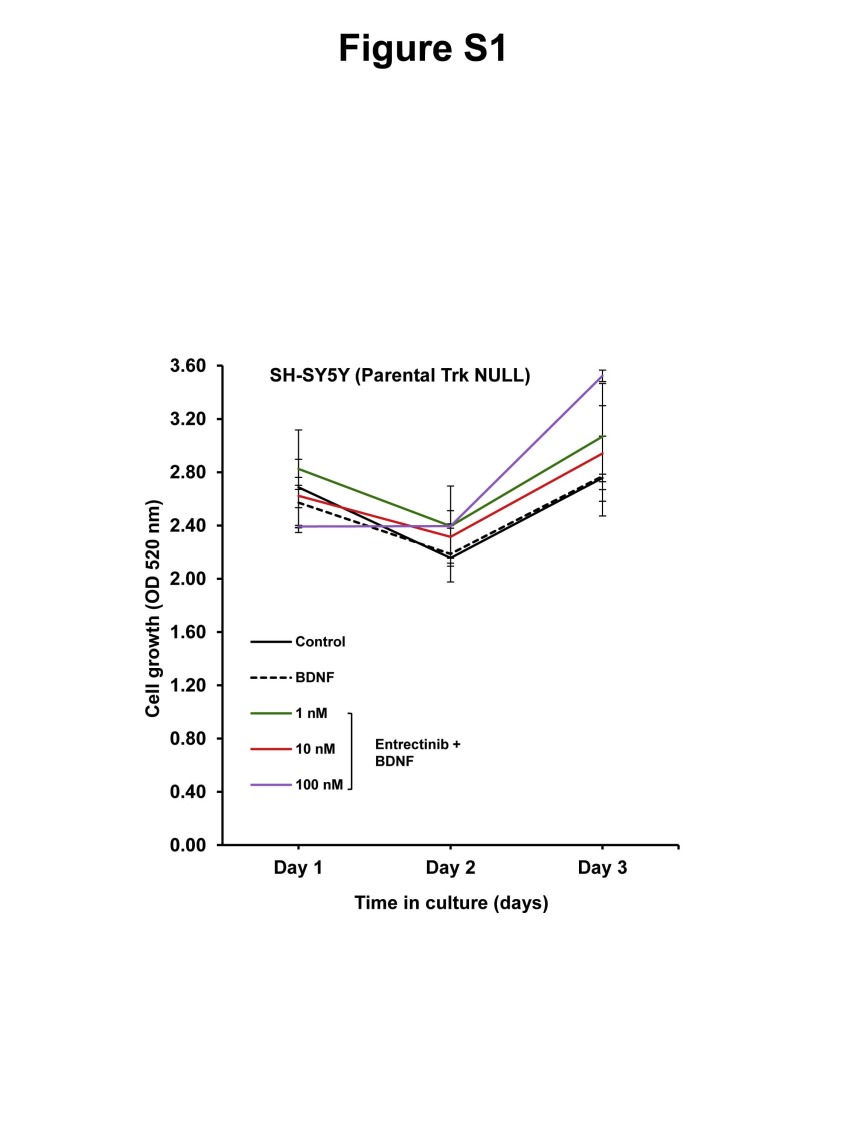
In order to understand the inhibitory effect of entrectinib on Trk phosphorylation we performed western blot analysis using SY5Y-TrkB and NLF-TrkB clones. The addition of exogenous BDNF produced rapid phosphorylation of TrkB, with maximum phosphorylation observed at about 15 minutes after BDNF addition both in SY5Y-TrKB and NLF-TrkB cells. Increasing concentrations of entrectinib produced a dose-dependent inhibition of phosphorylation in SY5Y-TrkB cells upon entrectinib treatment (Fig. 1A). Substantial inhibition of Trk phosphorylation was observed at 1 nM concentration of entrectinib, and almost complete inhibition at 10 nM or higher concentrations (Fig. 1A). We also observed similar results in NLF-TrkB clones (NLF-TrkB clone 6), with almost complete inhibition of TrkB phosphorylation at 50 nM entrectinib upon entrectinib treatment (Fig. 2A). This differential sensitivity may be due genomic differences, as NLF cells are MYCN amplified, p53 mutated whereas SY5Y cells are non-MYCN amplified and p53 WT. SRB assays showed a decrease in cell number with increasing concentrations of entrectinib in SY5Y-TrkB (Fig. 1B) and NLF-TrkB (Fig. 2B). At concentrations of ≥50 nM, the cell growth was equal to or lower than the baseline control (without ligand). No effect of entrectinib was observed in the Trk-null parental SH-SY5Y (Fig. S1) or parental NLF cells (data not shown) with SRB assay. Similar results were obtained when we extended these studies in other NB cell lines that had endogenous expression of TrkB (NB69: MYCN amplified, ALK-WT; (Fig. S3) and NB1643: MYCN amplified, ALK-R1275Q (data not shown). Although endogenous TrkB levels in these cells were relatively low compared to transfected SY5Y and NLF cells, both showed growth inhibition in response to entrectinib treatment. These results demonstrate a dose-dependent growth inhibition of BDNF-activated TrkB by entrectinib in TrkB-expressing NB cell lines.
Fig. 1.

Effect of entrectinib on TrkB phosphorylation in SY5Y-TrkB cells (clone BR6). A. Cells were exposed to BDNF in the presence or absence of increasing concentrations of entrectinib. Substantial inhibition of phosphorylation was observed at as low as 1nM concentration of entrectinib. Inhibition of Akt phosphorylation was also observed with increasing concentrations of entrectinib. Pan-Trk, Akt, pMAPK and actin showed no effect of inhibition. B. Growth profile of SY5Y-TrkB cells. Representative SRB assay indicating dose dependent inhibition of SY5Y-TrkB cells upon entrectinib treatment as indicated. Each assay was performed in triplicate, and at least three independent experiments were conducted.
Fig. 2.

Effect of entrectinib on TrkB phosphorylation in NLF-TrkB cells. A. Western blot analysis of NLF-TrkB upon entrectinib treatment. Cells were exposed to BDNF in the presence or absence of increasing concentrations of entrectinib. Substantial inhibition of phosphorylation was observed at as low as 50 nM concentration of entrectinib. Inhibition of Akt phosphorylation was also seen with increasing concentrations of entrectinib. B. Growth profile of NLF-TrkB cells. Representative SRB assay indicating dose-dependent inhibition of NLF-TrkB cells (clone 6) upon entrectinib treatment. Each assay was performed in triplicate, and at least three independent experiments were conducted.
3.2. Effect of entrectinib on SY5Y-TrkB NB xenografts
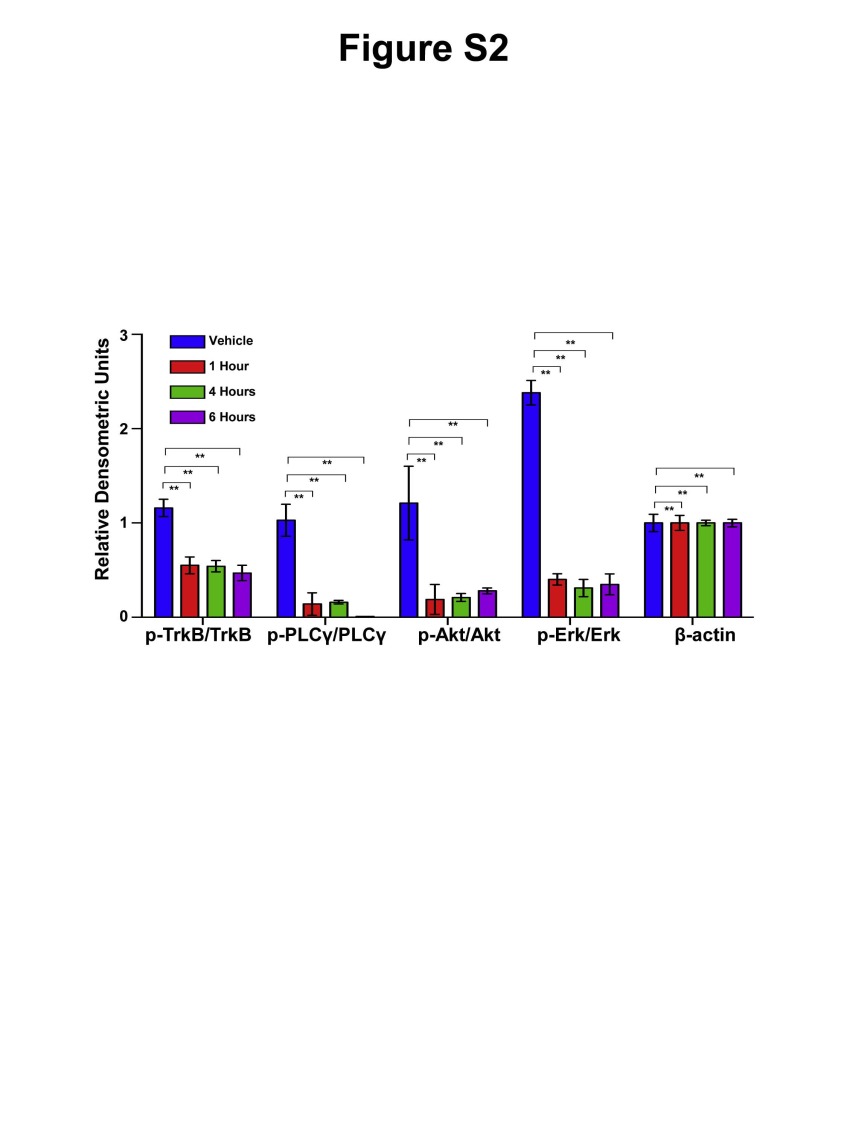
As SY5Y-TrkB clones showed differential sensitivity and more effective response at very low entrectinib concentrations, we focused our further study using this clone. In order to test the ability of entrectinib to inhibit SY5Y-TrkB cells grown as xenografts in athymic nu/nu mice, we injected mice on the flank with 1 × 107 cells suspended in matrigel. Animals were randomized into control and treatment groups when the average tumor size measured 0.2 cm3. Animals in the control group were treated with saline, PO, BID × 7 days/week. Animals in the treatment group received entrectinib at 60 mg/kg PO, BID × 7 days/week. Tumors and animal weights were measured at least twice a week, and the drug dose was adjusted accordingly. Significant tumor growth inhibition was observed with entrectinib treatment compared to vehicle (p<0.0001, Fig. 3A). Entrectinib treated mice also demonstrated a significantly prolonged EFS (p<0.0001, Fig. 3B). Analysis of the tumors harvested at different time points after treatment (1, 4 and 6 hr, respectively) showed inhibition of TrkB phosphorylation in tumors treated with entrectinib compared to vehicle controls. Inhibition of phosphorylation was also seen for p-PLCγ, p-Akt and p-Erk in entrectinib treated tumors (Fig. 3C). Densitometric analysis of Western blot images was also performed to indicate the statistical significance of phosphoprotein expression in xenografts upon entrectinib treatment (Fig. S2). Actin levels were unchanged indicating that there is no global inhibition of protein. These results show that entrectinib significantly inhibited the growth of SY5Y-TrkB xenograft as well as Trk phosphorylation in vivo.
Fig. 3.
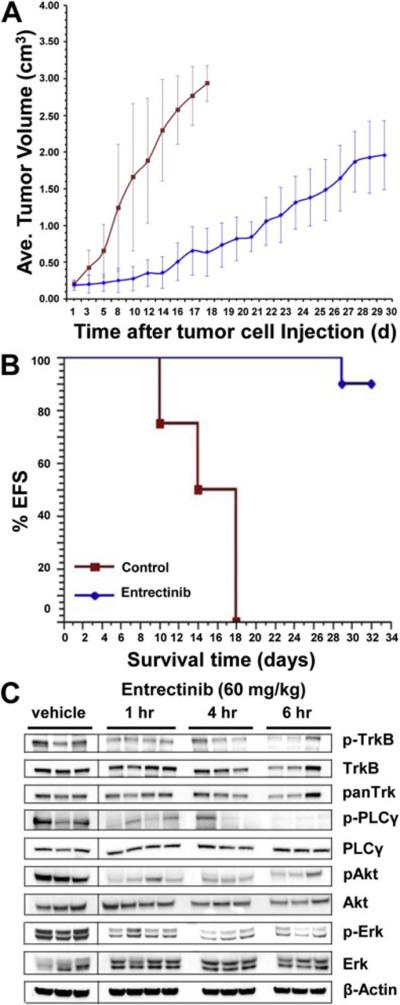
Effect of entrectinib on the growth of SY5Y-TrkB (BR6) xenografts. A. Growth pattern of SY5Y-TrkB xenografts. Entrectinib significantly decreases the growth of SY5Y-TrkB cells grown as xenografts in athymic nu/nu mice (p<0.0001). B. Kaplan-Meier survival curves of SY5Y-TrkB xenografts. Significantly prolonged EFS was also observed in the entrectinib treated group compared to control (p<0.0001). C. Western blot analysis of tumor samples at various times. Analysis of tumor samples shows inhibition of p-TrkB, p-PLCγ, p-Akt and p-Erk in tumors of animals treated with entrectinib. The control group showed no significant inhibition.
3.3. Pharmacokinetic studies on entrectinib treated xenograft tumors
In order to determine plasma concentrations at different time points after entrectinib treatment, plasma concentrations of blood from tumors were measured at various intervals. Analysis of entrectinib (free base) in plasma at different time points after treatment showed that peak plasma concentrations were achieved at around 6 hr after dosing, with subsequent drop in concentration at later time points (Fig. 4). Thus, highest entrectinib levels were observed around 6 hr of treatment, supporting our BID dosing schedule.
Fig. 4.
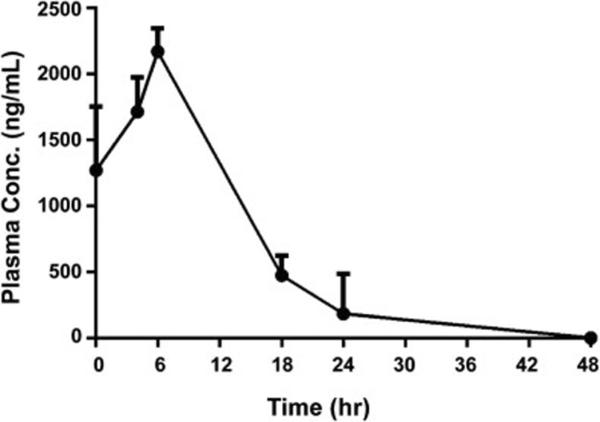
Pharmacokinetic analysis of entrectinib treated tumors. Representative graph indicating peak plasma concentrations (ng/mL) at about 6 hr post administration of the drug. After the final dose (60 mg/kg BID) was given, the blood samples were drawn from 4 mice per time point via retro-orbital bleeding, collected in heparinized tubes on wet ice, and plasma concentrations were measured as described in methods.
3.4. Effect of entrectinib in combination with Irino-TMZ on SY5Y-TrkB cell growth in vitro
Entrectinib was used in combination with Irino-TMZ to determine whether entrectinib would enhance the efficacy of the chemotherapeutic agents. SY5Y-TrkB cells were treated with entrectinib alone (10, 20 and 50 nM), Irino alone (1.5 μM), TMZ alone (50 μM), Irino-TMZ, or a combination of entrectinib (10, 20 and 50 nM) + Irino-TMZ. SRB assays showed a significantly enhanced inhibition of cell growth when entrectinib was used in combination with Irino-TMZ compared to either entrectinib or Irino-TMZ alone (Fig. 5).
Fig. 5.

Effect of entrectinib in combination with Irino-TMZ on SY5Y-TrkB cell growth. Increased growth inhibition was observed for cells treated with the combination of entrectinib + Irino-TMZ compared to single agent, Irino alone, TMZ alone or Irino-TMZ group.
3.5. Effect of entrectinib in combination with Irino-TMZ on SY5Y-TrkB NB xenografts
Entrectinib was tested in our xenograft model in combination with Irino-TMZ to determine if the combination therapy showed enhanced tumor control in vivo compared to either entrectinib or Irino-TMZ alone. Animals were randomized into control (saline, PO, 7×/week), entrectinib alone (PO, 60 mg/kg, BID, 7×/week), Irino (0.63 mg/kg) + TMZ (7.5 mg/kg) (PO, QD, 5×/week), or entrectinib + Irino-TMZ treatment groups. Three different regimens were followed for the entrectinib + Irino-TMZ combinations: Regimen 1—entrectinib + Irino-TMZ (throughout the study); Regimen 2—entrectinib (weeks 1, 3 and 5 only) + Irino-TMZ (weeks 2, 4 and 6 only); and Regimen 3—entrectinib (throughout the study) + Irino-TMZ weeks 1, 3 and 5 only). Statistical analysis of differences in the change rate of log tumor volumes showed that entrectinib treatment groups (single agent or combination) were significantly better than either control or Irino-TMZ alone groups (Fig. 6A). Analysis of EFS showed significant differences between control vs. entrectinib alone (p = 0.0001); control vs. Irino-TMZ alone (p = 0.0206); control vs. Regimen 1 (p<0.0001); control vs. Regimen 2 (p = 0.0030); and control vs. Regimen 3 (p<0.0001). Statistically significant differences were also observed for the following comparisons: entrectinib alone vs. Irino-TMZ alone (p = 0.0209); entrectinib alone vs. Regimen 2 (p = 0.0097); Regimen 1 vs. Irino-TMZ (5×/week) (p = 0.0012); Regimen 1 vs. Regimen 2 (p = 0.0002); Regimen 2 vs. Regimen 3 (p<0.0001). Thus, co-treatment of entrectinib with Irino-TMZ significantly enhanced the effect of Irino-TMZ treatment, but the dosing schedule was important in determining the overall survival advantage (Fig. 6B).
Fig. 6.

Effect of entrectinib in combination with Irino-TMZ on SY5Y-TrkB xenograft growth. A. Growth pattern of SY5Y-TrkB xenografts. Significant inhibition of tumor growth was observed in the entrectinib (single agent or combination with Irino-TMZ) compared to control or Irino-TMZ group. B. Kaplan-Meier survival curves of SY5Y-TrkB xenografts. Statistically significant differences in EFS were observed in the combination treated groups compared to Irino-TMZ alone.
4. Discussion
Neuroblastomas are known to have heterogeneous clinical behaviors, including spontaneous regression, differentiation, or relentless progression despite intensive, multimodality therapy. Patients under the age of 12–18 months tend to have better outcomes than older patients. Unfortunately, over half of the NBs detected clinically are metastatic at the time of diagnosis, and 5-year overall survival is less than 50% [31–33]. Furthermore, use of multimodality therapy has reached the limits of tolerability, due to serious acute- and long-term toxicity associated with these treatments [31–33].
Data from our laboratory and others suggest that Trk receptors play important roles in determining the clinical behavior of NBs [4–11, 13–15, 20, 21, 34–36]. TrkA is expressed in prognostically favorable tumors that are likely to regress or differentiate, whereas TrkB plus BDNF are co-expressed in clinically aggressive tumors, especially those with MYCN amplification. Co-expression of TrkB and BDNF constitutes an autocrine survival pathway promoting invasion, metastasis, angiogenesis and drug resistance [4, 5, 9, 13–15, 34–36].
We have shown previously that targeted inhibition of TrkB using lestaurtinib (CEP-701), AZ64, or GNF-4256 was effective in inhibiting TrkB auto-phosphorylation and subsequently the growth of these cells as xenografts in mice [24–26, 28, 29]. Phase 1 data confirmed that biologically relevant doses of lestaurtinib resulted in protracted responses and significantly inhibited NB, with limited toxicity [27]. Despite these promising results, none of these agents are being supported for future clinical trials. Therefore, a potent and selective Trk inhibitor with a favorable safety profile is needed to treat NBs and other tumors with activation of Trk receptors.
Entrectinib (RXDX-101, Ignyta, Inc.) is a small molecule inhibitor of all TRKs, ALK and ROS1 tyrosine kinases at low nanomolar concentrations. It is a competitive inhibitor of the ATP binding site in the kinase domain of these receptors [37]. Although it has been previously evaluated for its efficacy against Alk and Ros1 [38–42], we have evaluated entrectinib for its ability to inhibit TrkB-expressing NBs using both in vitro and in vivo models. We have demonstrated that entrectinib is a potent inhibitor of TrkB autophosphorylation and cell growth in vitro, and that it has no effect on growth of the Trk-null parental cells. Entrectinib also caused significant inhibition of tumor growth in a xenograft mouse model compared to vehicle control, with no apparent toxicities. Peak plasma concentrations were achieved at about 6 hr after dosing, with subsequent drop in concentration of the drug in plasma at later time points.
The combination of entrectinib with Irino-TMZ resulted in enhanced inhibition of cell growth in our in vitro experiments when compared to either entrectinib or Irino-TMZ alone. Xenograft studies using entrectinib in combination with Irino-TMZ showed decreased tumor growth and a significant increase in EFS for the combination arms compared to Irino-TMZ alone. However, the schedule of drug administration seemed to play a role in tumor growth control and enhanced survival. Regimen 1 dosing appeared to be most efficacious, with Regimen 3 closely following, and both Regimens 1 and 3 were significantly superior to Regimen 2 dosing. Nevertheless, entrectinib significantly enhanced the efficacy of chemotherapy in causing inhibition of NB tumor growth in NB xenograft models, regardless of the schedule employed.
We used entrectinib to target TrkB-BDNF autocrine activation, but this agent also inhibits Alk and Ros1. There is no evidence to date for ROS1 gene activation in neuroblastomas, but germline activation of ALK accounts for the majority of cases with hereditary NB predisposition, and ALK activation by mutation or amplification occurs in 8–10% of primary NBs [43–48]. The parental Trk-null NB line SY5Y carries an activating ALK mutation at F1174L, but we saw no effect of entrectinib on this line, suggesting that entrectinib is not effective against this mutation, similar to crizotinib [49–51]. Thus, the growth inhibition of entrectinib seen in our study can be attributed to its effects on TrkB-BDNF autocrine pathway, and not on activated Alk receptor or off-target effects.
Entrectinib is being currently studied in phase 1 clinical trials in adults as a single agent in patients with solid tumors harboring molecular alterations in NTRK, ALK and ROS1 genes (ALKA-372-001, STARTRK-1) to determine the maximum tolerated dose (MTD) and/or recommended Phase 2 dose. So far, 67 patients have been dosed across both clinical trials, and entrectinib has been well tolerated [52, 53]. No treatment-related serious adverse events were reported. The most relevant frequent adverse events were fatigue, dysgeusia, constipation, nausea and paresthesia, all of which were reversible after stopping the drug. Pharmacokinetic measurements indicate a half-life compatible with once daily dosing in humans. Indeed, of 11 patients enrolled across both clinical trials who demonstrated presence of NTRK1/2/3, ROS1 or ALK fusions and were treated at or above the recommended phase 2 dose, 10 achieved objective clinical response, yielding an overall response rate of 91% in this population, and 9 of these patients remained in the study with durable responses up to 16 cycles of treatment [52, 53]. There is evidence for TRK expression, alteration, and/or autocrine activation in many other pediatric and adult cancers, including Wilms tumor [54], medulloblastoma [55–58], Ewing sarcoma [59], infantile fibrosarcoma [60–62], breast cancer [63–69], prostate cancer [70–74], colorectal cancer [38, 75–80], pancreatic cancer [81–85], lung cancer [86–89] and others. Suppression of TRK expression in these tumors has shown similar effects on cell growth and metastatic potential, and entrecitnib has specifically been tested in both colorectal cancer and non-small-cell lung cancer [37, 38, 42, 52, 53, 75–77, 86]. Given these adult phase 1 results, and our data described here, entrectinib is clearly a promising agent to be carried forward into future clinical trials for NBs, as well as other pediatric and adult tumors with evidence of TRK activation.
Supplementary Material
Supplementary Fig. S1. Effect of entrectinib on Growth profile of parental SY5Y (Trk-NULL) cells. Representative SRB assay indicating no significant response to entrectinib treatment as there is no expressed Trk in these cells. Each assay was performed in triplicate, and at least three independent experiments were conducted.
{kind=link}
Supplementary Fig. S2. Densitometric and statistical analysis of Western blot results presented in Figure 3C. All individual western blot images were scanned, normalized and statistical analysis was performed. Significant inhibition of phosphoprotein expression was observed for p-Trk, p-PLCγ, p-Akt and p-Erk after 1 hr, 4 hr and 6 hr of entrectinib treatment when compared with untreated vehicle controls. Actin levels remain unchanged, indicating no global inhibition of protein expression. P-values were calculated (**<0.01) by two-way ANOVA using Prism followed by Sidak post-test.
{kind=link}
Supplementary Fig. S3. Effect of entrectinib growth profile of NB69 cells. Representative SRB assay indicating dose dependent inhibition of NB69 cells upon entrectinib treatment as indicated. Each assay was performed in triplicate.
{kind=link}
Highlights.
Autocrine overexpression of TrkB and its ligand are found in over half of high-risk neuroblastomas.
Entrectinib (RXDX-101) is a potent, nontoxic inhibitor of TrkB-expressing neuroblastoma growth in vivo in mouse xenografts.
Entrectinib significantly enhances the efficacy of chemotherapy when used in combination, without additional toxicity.
Entrectinib is a very promising agent to treat TrkB-expressing neuroblastomas and other tumor with Trk receptor activation
A phase 1 clinical trial for recurrent/refractory pediatric solid tumors is planned.
Acknowledgments
This work was supported in part by NIH grant CA094194; Ignyta, Inc.; the Alex's Lemonade Stand Foundation; and the Audrey E. Evans Endowed Chair.
Disclosure of Potential Conflicts of Interest This work was supported in part by a sponsored research agreement between Ignyta, Inc., and Dr. Garrett Brodeur's Laboratory.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141:1117–1134. doi: 10.1016/j.cell.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].McDonell LM, Kernohan KD, Boycott KM, Sawyer SL. Receptor tyrosine kinase mutations in developmental syndromes and cancer: two sides of the same coin. Human molecular genetics. 2015;24:R60–66. doi: 10.1093/hmg/ddv254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Brodeur GM, Iyer R, Croucher JL, Zhuang T, Higashi M, Kolla V. Therapeutic targets for neuroblastomas. Expert opinion on therapeutic targets. 2014;18:277–292. doi: 10.1517/14728222.2014.867946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Brodeur GM, Minturn JE, Ho R, Simpson AM, Iyer R, Varela CR, Light JE, Kolla V, Evans AE. Trk receptor expression and inhibition in neuroblastomas. Clin Cancer Res. 2009;15:3244–3250. doi: 10.1158/1078-0432.CCR-08-1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Thiele CJ, Li Z, McKee AE. On Trk--the TrkB signal transduction pathway is an increasingly important target in cancer biology. Clin Cancer Res. 2009;15:5962–5967. doi: 10.1158/1078-0432.CCR-08-0651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Kogner P, Barbany G, Dominici C, Castello MA, Raschella G, Persson H. Coexpression of messenger RNA for TRK protooncogene and low affinity nerve growth factor receptor in neuroblastoma with favorable prognosis. Cancer Res. 1993;53:2044–2050. [PubMed] [Google Scholar]
- [7].Nakagawara A, Arima M, Azar CG, Scavarda NJ, Brodeur GM. Inverse relationship between trk expression and N-myc amplification in human neuroblastomas. Cancer Res. 1992;52:1364–1368. [PubMed] [Google Scholar]
- [8].Nakagawara A, Azar CG, Scavarda NJ, Brodeur GM. Expression and function of TRK-B and BDNF in human neuroblastomas. Mol Cell Biol. 1994;14:759–767. doi: 10.1128/mcb.14.1.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Nakagawara A, Arima-Nakagawara M, Scavarda NJ, Azar CG, Cantor AB, Brodeur GM. Association between high levels of expression of the TRK gene and favorable outcome in human neuroblastoma. N Engl J Med. 1993;328:847–854. doi: 10.1056/NEJM199303253281205. [DOI] [PubMed] [Google Scholar]
- [10].Nakagawara A, Brodeur GM. Role of neurotrophins and their receptors in human neuroblastomas: a primary culture study. Eur J Cancer. 1997;33:2050–2053. doi: 10.1016/s0959-8049(97)00280-3. [DOI] [PubMed] [Google Scholar]
- [11].Suzuki T, Bogenmann E, Shimada H, Stram D, Seeger RC. Lack of high-affinity nerve growth factor receptors in aggressive neuroblastomas. Journal of the National Cancer Institute. 1993;85:377–384. doi: 10.1093/jnci/85.5.377. [DOI] [PubMed] [Google Scholar]
- [12].Acheson A, Conover JC, Fandl JP, DeChiara TM, Russell M, Thadani A, Squinto SP, Yancopoulos GD, Lindsay RM. A BDNF autocrine loop in adult sensory neurons prevents cell death. Nature. 1995;374:450–453. doi: 10.1038/374450a0. see comments. [DOI] [PubMed] [Google Scholar]
- [13].Ho R, Eggert A, Hishiki T, Minturn JE, Ikegaki N, Foster P, Camoratto AM, Evans AE, Brodeur GM. Resistance to chemotherapy mediated by TrkB in neuroblastomas. Cancer Res. 2002;62:6462–6466. [PubMed] [Google Scholar]
- [14].Matsumoto K, Wada RK, Yamashiro JM, Kaplan DR, Thiele CJ. Expression of brain-derived neurotrophic factor and p145TrkB affects survival, differentiation, and invasiveness of human neuroblastoma cells. Cancer Res. 1995;55:1798–1806. [PubMed] [Google Scholar]
- [15].Nakamura K, Martin KC, Jackson JK, Beppu K, Woo CW, Thiele CJ. Brain-derived neurotrophic factor activation of TrkB induces vascular endothelial growth factor expression via hypoxia-inducible factor-1alpha in neuroblastoma cells. Cancer Res. 2006;66:4249–4255. doi: 10.1158/0008-5472.CAN-05-2789. [DOI] [PubMed] [Google Scholar]
- [16].Ivanov SV, Panaccione A, Brown B, Guo Y, Moskaluk CA, Wick MJ, Brown JL, Ivanova AV, Issaeva N, El-Naggar AK, Yarbrough WG. TrkC signaling is activated in adenoid cystic carcinoma and requires NT-3 to stimulate invasive behavior. Oncogene. 2013;32:3698–3710. doi: 10.1038/onc.2012.377. [DOI] [PubMed] [Google Scholar]
- [17].Lee J, Jiffar T, Kupferman ME. A novel role for BDNF-TrkB in the regulation of chemotherapy resistance in head and neck squamous cell carcinoma. PLoS ONE. 2012;7:e30246. doi: 10.1371/journal.pone.0030246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ng YK, Wong EY, Lau CP, Chan JP, Wong SC, Chan AS, Kwan MP, Tsao SW, Tsang CM, Lai PB, Chan AT, Lui VW. K252a induces anoikis-sensitization with suppression of cellular migration in Epstein-Barr virus (EBV)--associated nasopharyngeal carcinoma cells. Invest New Drugs. 2012;30:48–58. doi: 10.1007/s10637-010-9513-4. [DOI] [PubMed] [Google Scholar]
- [19].Ryden M, Sehgal R, Dominici C, Schilling FH, Ibanez CF, Kogner P. Expression of mRNA for the neurotrophin receptor trkC in neuroblastomas with favourable tumour stage and good prognosis. Br J Cancer. 1996;74:773–779. doi: 10.1038/bjc.1996.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Yamashiro DJ, Liu XG, Lee CP, Nakagawara A, Ikegaki N, McGregor LM, Baylin SB, Brodeur GM. Expression and function of Trk-C in favourable human neuroblastomas. Eur J Cancer. 1997;33:2054–2057. doi: 10.1016/s0959-8049(97)00309-2. [DOI] [PubMed] [Google Scholar]
- [21].Yamashiro DJ, Nakagawara A, Ikegaki N, Liu XG, Brodeur GM. Expression of TrkC in favorable human neuroblastomas. Oncogene. 1996;12:37–41. [PubMed] [Google Scholar]
- [22].Chen Z, Akbay E, Mikse O, Tupper T, Cheng K, Wang Y, Tan X, Altabef A, Woo SA, Chen L, Reibel JB, Janne PA, Sharpless NE, Engelman JA, Shapiro GI, Kung AL, Wong KK. Co-clinical trials demonstrate superiority of crizotinib to chemotherapy in ALK-rearranged non-small cell lung cancer and predict strategies to overcome resistance. Clin Cancer Res. 2014;20:1204–1211. doi: 10.1158/1078-0432.CCR-13-1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Vaishnavi A, Le AT, Doebele RC. TRKing down an old oncogene in a new era of targeted therapy. Cancer discovery. 2015;5:25–34. doi: 10.1158/2159-8290.CD-14-0765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Evans AE, Kisselbach KD, Liu X, Eggert A, Ikegaki N, Camoratto AM, Dionne C, Brodeur GM. Effect of CEP-751 (KT-6587) on neuroblastoma xenografts expressing TrkB. Medical & Pediatric Oncology. 2001;36:181–184. doi: 10.1002/1096-911X(20010101)36:1<181::AID-MPO1043>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- [25].Evans AE, Kisselbach KD, Yamashiro DJ, Ikegaki N, Camoratto AM, Dionne CA, Brodeur GM. Antitumor activity of CEP-751 (KT-6587) on human neuroblastoma and medulloblastoma xenografts. Clin Cancer Res. 1999;5:3594–3602. [PubMed] [Google Scholar]
- [26].Iyer R, Evans AE, Qi X, Ho R, Minturn JE, Zhao H, Balamuth N, Maris JM, Brodeur GM. Lestaurtinib enhances the antitumor efficacy of chemotherapy in murine xenograft models of neuroblastoma. Clin Cancer Res. 2010;16:1478–1485. doi: 10.1158/1078-0432.CCR-09-1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Minturn JE, Evans AE, Villablanca JG, Yanik GA, Park JR, Shusterman S, Groshen S, Hellriegel ET, Bensen-Kennedy D, Matthay KK, Brodeur GM, Maris JM. Phase I trial of lestaurtinib for children with refractory neuroblastoma: a new approaches to neuroblastoma therapy consortium study. Cancer chemotherapy and pharmacology. 2011;68:1057–1065. doi: 10.1007/s00280-011-1581-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Croucher JL, Iyer R, Li N, Molteni V, Loren J, Gordon WP, Tuntland T, Liu B, Brodeur GM. TrkB inhibition by GNF-4256 slows growth and enhances chemotherapeutic efficacy in neuroblastoma xenografts. Cancer Chemother Pharmacol. 2015;75:131–141. doi: 10.1007/s00280-014-2627-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Iyer R, Varela CR, Minturn JE, Ho R, Simpson AM, Light JE, Evans AE, Zhao H, Thress K, Brown JL, Brodeur GM. AZ64 inhibits TrkB and enhances the efficacy of chemotherapy and local radiation in neuroblastoma xenografts. Cancer Chemother Pharmacol. 2012;70:477–486. doi: 10.1007/s00280-012-1879-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Vichai V, Kirtikara K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nature protocols. 2006;1:1112–1116. doi: 10.1038/nprot.2006.179. [DOI] [PubMed] [Google Scholar]
- [31].Brodeur GM, Maris JM. Neuroblastoma. In: Pizzo PA, Poplack DG, editors. Principles and Practice of Pediatric Oncology. Lippincott, Williams and Wilkins; Philadelphia: 2010. pp. 886–922. [Google Scholar]
- [32].Maris JM, Hogarty MD, Bagatell R, Cohn SL. Neuroblastoma. Lancet. 2007;369:2106–2120. doi: 10.1016/S0140-6736(07)60983-0. [DOI] [PubMed] [Google Scholar]
- [33].Modak S, Cheung NK. Neuroblastoma: Therapeutic strategies for a clinical enigma. Cancer treatment reviews. 2010;36:307–317. doi: 10.1016/j.ctrv.2010.02.006. [DOI] [PubMed] [Google Scholar]
- [34].Eggert A, Grotzer MA, Ikegaki N, Liu XG, Evans AE, Brodeur GM. Expression of the neurotrophin receptor TrkA down-regulates expression and function of angiogenic stimulators in SH-SY5Y neuroblastoma cells. Cancer Res. 2002;62:1802–1808. [PubMed] [Google Scholar]
- [35].Lucarelli E, Kaplan D, Thiele CJ. Activation of trk-A but not trk-B signal transduction pathway inhibits growth of neuroblastoma cells. Eur J Cancer. 1997;33:2068–2070. doi: 10.1016/s0959-8049(97)00266-9. [DOI] [PubMed] [Google Scholar]
- [36].Jaboin J, Kim CJ, Kaplan DR, Thiele CJ. Brain-derived neurotrophic factor activation of TrkB protects neuroblastoma cells from chemotherapy-induced apoptosis via phosphatidylinositol 3'-kinase pathway. Cancer Res. 2002;62:6756–6763. [PubMed] [Google Scholar]
- [37].Rolfo C, Ruiz R, Giovannetti E, Gil-Bazo I, Russo A, Passiglia F, Giallombardo M, Peeters M, Raez L. Entrectinib: a potent new TRK, ROS1, and ALK inhibitor. Expert opinion on investigational drugs. 2015;24:1493–1500. doi: 10.1517/13543784.2015.1096344. [DOI] [PubMed] [Google Scholar]
- [38].Amatu A, Somaschini A, Cerea G, Bosotti R, Valtorta E, Buonandi P, Marrapese G, Veronese S, Luo D, Hornby Z, Multani P, Murphy D, Shoemaker R, Lauricella C, Giannetta L, Maiolani M, Vanzulli A, Ardini E, Galvani A, Isacchi A, Sartore-Bianchi A, Siena S. Novel CAD-ALK gene rearrangement is drugable by entrectinib in colorectal cancer. Br J Cancer. 2015;113:1730–1734. doi: 10.1038/bjc.2015.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Aveic S, Pantile M, Seydel A, Esposito MR, Zanon C, Li G, Tonini GP. Combating autophagy is a strategy to increase cytotoxic effects of novel ALK inhibitor entrectinib in neuroblastoma cells. Oncotarget. 2015 doi: 10.18632/oncotarget.6778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Awad MM, Shaw AT. ALK inhibitors in non-small cell lung cancer: crizotinib and beyond. Clin Adv Hematol Oncol. 2014;12:429–439. [PMC free article] [PubMed] [Google Scholar]
- [41].Iragavarapu C, Mustafa M, Akinleye A, Furqan M, Mittal V, Cang S, Liu D. Novel ALK inhibitors in clinical use and development. J Hematol Oncol. 2015;8:17. doi: 10.1186/s13045-015-0122-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Lee J, Kim HC, Hong JY, Wang K, Kim SY, Jang J, Kim ST, Park JO, Lim HY, Kang WK, Park YS, Lee J, Lee WY, Park YA, Huh JW, Yun SH, Do IG, Kim SH, Balasubramanian S, Stephens PJ, Ross JS, Li GG, Hornby Z, Ali SM, Miller VA, Kim KM, Ou SH. Detection of novel and potentially actionable anaplastic lymphoma kinase (ALK) rearrangement in colorectal adenocarcinoma by immunohistochemistry screening. Oncotarget. 2015;6:24320–24332. doi: 10.18632/oncotarget.4462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Chen Y, Takita J, Choi YL, Kato M, Ohira M, Sanada M, Wang L, Soda M, Kikuchi A, Igarashi T, Nakagawara A, Hayashi Y, Mano H, Ogawa S. Oncogenic mutations of ALK kinase in neuroblastoma. Nature. 2008;455:971–974. doi: 10.1038/nature07399. [DOI] [PubMed] [Google Scholar]
- [44].George RE, Sanda T, Hanna M, Frohling S, Luther W, 2nd, Zhang J, Ahn Y, Zhou W, London WB, McGrady P, Xue L, Zozulya S, Gregor VE, Webb TR, Gray NS, Gilliland DG, Diller L, Greulich H, Morris SW, Meyerson M, Look AT. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature. 2008;455:975–978. doi: 10.1038/nature07397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Janoueix-Lerosey I, Lequin D, Brugieres L, Ribeiro A, de Pontual L, Combaret V, Raynal V, Puisieux A, Schleiermacher G, Pierron G, Valteau-Couanet D, Frebourg T, Michon J, Lyonnet S, Amiel J, Delattre O. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature. 2008;455:967–970. doi: 10.1038/nature07398. [DOI] [PubMed] [Google Scholar]
- [46].Mosse YP, Laudenslager M, Longo L, Cole KA, Wood A, Attiyeh EF, Laquaglia MJ, Sennett R, Lynch JE, Perri P, Laureys G, Speleman F, Kim C, Hou C, Hakonarson H, Torkamani A, Schork NJ, Brodeur GM, Tonini GP, Rappaport E, Devoto M, Maris JM. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature. 2008;455:930–935. doi: 10.1038/nature07261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Azarova AM, Gautam G, George RE. Emerging importance of ALK in neuroblastoma. Semin Cancer Biol. 2011;21:267–275. doi: 10.1016/j.semcancer.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Carpenter EL, Mosse YP. Targeting ALK in neuroblastoma-preclinical and clinical advancements. Nat Rev Clin Oncol. 2012;9:391–399. doi: 10.1038/nrclinonc.2012.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Berry T, Luther W, Bhatnagar N, Jamin Y, Poon E, Sanda T, Pei D, Sharma B, Vetharoy WR, Hallsworth A, Ahmad Z, Barker K, Moreau L, Webber H, Wang W, Liu Q, Perez-Atayde A, Rodig S, Cheung NK, Raynaud F, Hallberg B, Robinson SP, Gray NS, Pearson AD, Eccles SA, Chesler L, George RE. The ALK(F1174L) mutation potentiates the oncogenic activity of MYCN in neuroblastoma. Cancer Cell. 2012;22:117–130. doi: 10.1016/j.ccr.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Heuckmann JM, Holzel M, Sos ML, Heynck S, Balke-Want H, Koker M, Peifer M, Weiss J, Lovly CM, Grutter C, Rauh D, Pao W, Thomas RK. ALK mutations conferring differential resistance to structurally diverse ALK inhibitors. Clin Cancer Res. 2011;17:7394–7401. doi: 10.1158/1078-0432.CCR-11-1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Sasaki T, Okuda K, Zheng W, Butrynski J, Capelletti M, Wang L, Gray NS, Wilner K, Christensen JG, Demetri G, Shapiro GI, Rodig SJ, Eck MJ, Janne PA. The neuroblastoma-associated F1174L ALK mutation causes resistance to an ALK kinase inhibitor in ALK-translocated cancers. Cancer Res. 2010;70:10038–10043. doi: 10.1158/0008-5472.CAN-10-2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].De Braud FG, Niger M, Damian S, Bardazza B, Martinetti A, Pelosi G, Marrapese G, Palmeri L, Cerea G, Valtorta E, Veronese S, Santore-Bianchi A, Ardini E, Isachi A, Martignoni M, Galvani A, Luo D, Yeh L, Senderowicz AM, Siena S. Alka-372-001: First-in-human, phase I study of entrectinib - an oral pan-trk, ROS1, and ALK inhibitor – in patients with advanced solid tumors with relevant molecular alterations. ASCO Annual Meeting, Journal of Clinical Oncology, Chicago, IL. 2015;(suppl) abstr 2517. [Google Scholar]
- [53].Patel MR, ABauer TM, Liu SV, Drilon AE, Wheler JJ, Shaw AT, Farago AF, Ou SH, Luo D, Yeh L, Hornby Z, Senderowicz AM, Lim J. STARTRK-1: Phase 1/2a study of entrectinib, an oral Pan-Trk, ROS1, and ALK inhibitor, in patients with advanced solid tumors with relevant molecular alterations. ASCO Annual Meeting, Journal of Clinical Oncology, Chicago, IL. 2015;(suppl) abstr 2596. [Google Scholar]
- [54].Eggert A, Grotzer MA, Ikegaki N, Zhao H, Cnaan A, Brodeur GM, Evans AE. Expression of the neurotrophin receptor TrkB is associated with unfavorable outcome in Wilms' tumor. J Clin Oncol. 2001;19:689–696. doi: 10.1200/JCO.2001.19.3.689. [DOI] [PubMed] [Google Scholar]
- [55].Chou TT, Trojanowski JQ, Lee VM. Neurotrophin signal transduction in medulloblastoma. J Neurosci Res. 1997;49:522–527. doi: 10.1002/(SICI)1097-4547(19970901)49:5<522::AID-JNR2>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- [56].Grotzer MA, Janss AJ, Fung K, Biegel JA, Sutton LN, Rorke LB, Zhao H, Cnaan A, Phillips PC, Lee VM, Trojanowski JQ. TrkC expression predicts good clinical outcome in primitive neuroectodermal brain tumors. J Clin Oncol. 2000;18:1027–1035. doi: 10.1200/JCO.2000.18.5.1027. [DOI] [PubMed] [Google Scholar]
- [57].Kim JY, Sutton ME, Lu DJ, Cho TA, Goumnerova LC, Goritchenko L, Kaufman JR, Lam KK, Billet AL, Tarbell NJ, Wu J, Allen JC, Stiles CD, Segal RA, Pomeroy SL. Activation of neurotrophin-3 receptor TrkC induces apoptosis in medulloblastomas. Cancer Res. 1999;59:711–719. [PubMed] [Google Scholar]
- [58].Segal RA, Goumnerova LC, Kwon YK, Stiles CD, Pomeroy SL. Expression of the neurotrophin receptor TrkC is linked to a favorable outcome in medulloblastoma. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:12867–12871. doi: 10.1073/pnas.91.26.12867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Nogueira E, Navarro S, Pellin A, Llombart-Bosch A. Activation of TRK genes in Ewing's sarcoma. Trk A receptor expression linked to neural differentiation. Diagn Mol Pathol. 1997;6:10–16. doi: 10.1097/00019606-199702000-00003. [DOI] [PubMed] [Google Scholar]
- [60].Knezevich SR, McFadden DE, Tao W, Lim JF, Sorensen PH. A novel ETV6-NTRK3 gene fusion in congenital fibrosarcoma. Nat Genet. 1998;18:184–187. doi: 10.1038/ng0298-184. [DOI] [PubMed] [Google Scholar]
- [61].Liu Q, Schwaller J, Kutok J, Cain D, Aster JC, Williams IR, Gilliland DG. Signal transduction and transforming properties of the TEL-TRKC fusions associated with t(12;15)(p13;q25) in congenital fibrosarcoma and acute myelogenous leukemia. EMBO J. 2000;19:1827–1838. doi: 10.1093/emboj/19.8.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Rubin BP, Chen CJ, Morgan TW, Xiao S, Grier HE, Kozakewich HP, Perez-Atayde AR, Fletcher JA. Congenital mesoblastic nephroma t(12;15) is associated with ETV6-NTRK3 gene fusion: cytogenetic and molecular relationship to congenital (infantile) fibrosarcoma. Am J Pathol. 1998;153:1451–1458. doi: 10.1016/S0002-9440(10)65732-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Jin W, Kim GM, Kim MS, Lim MH, Yun C, Jeong J, Nam JS, Kim SJ. TrkC plays an essential role in breast tumor growth and metastasis. Carcinogenesis. 2010;31:1939–1947. doi: 10.1093/carcin/bgq180. [DOI] [PubMed] [Google Scholar]
- [64].Lagadec C, Meignan S, Adriaenssens E, Foveau B, Vanhecke E, Romon R, Toillon RA, Oxombre B, Hondermarck H, Le Bourhis X. TrkA overexpression enhances growth and metastasis of breast cancer cells. Oncogene. 2009;28:1960–1970. doi: 10.1038/onc.2009.61. [DOI] [PubMed] [Google Scholar]
- [65].Dolle L, Adriaenssens E, El Yazidi-Belkoura I, Le Bourhis X, Nurcombe V, Hondermarck H. Nerve growth factor receptors and signaling in breast cancer. Curr Cancer Drug Targets. 2004;4:463–470. doi: 10.2174/1568009043332853. [DOI] [PubMed] [Google Scholar]
- [66].Dolle L, El Yazidi-Belkoura I, Adriaenssens E, Nurcombe V, Hondermarck H. Nerve growth factor overexpression and autocrine loop in breast cancer cells. Oncogene. 2003;22:5592–5601. doi: 10.1038/sj.onc.1206805. [DOI] [PubMed] [Google Scholar]
- [67].Tognon C, Knezevich SR, Huntsman D, Roskelley CD, Melnyk N, Mathers JA, Becker L, Carneiro F, MacPherson N, Horsman D, Poremba C, Sorensen PH. Expression of the ETV6-NTRK3 gene fusion as a primary event in human secretory breast carcinoma. Cancer Cell. 2002;2:367–376. doi: 10.1016/s1535-6108(02)00180-0. [DOI] [PubMed] [Google Scholar]
- [68].Euhus DM, Timmons CF, Tomlinson GE. ETV6-NTRK3--Trk-ing the primary event in human secretory breast cancer. Cancer Cell. 2002;2:347–348. doi: 10.1016/s1535-6108(02)00184-8. [DOI] [PubMed] [Google Scholar]
- [69].Descamps S, Pawlowski V, Revillion F, Hornez L, Hebbar M, Boilly B, Hondermarck H, Peyrat JP. Expression of nerve growth factor receptors and their prognostic value in human breast cancer. Cancer Res. 2001;61:4337–4340. [PubMed] [Google Scholar]
- [70].Festuccia C, Muzi P, Gravina GL, Millimaggi D, Speca S, Dolo V, Ricevuto E, Vicentini C, Bologna M. Tyrosine kinase inhibitor CEP-701 blocks the NTRK1/NGF receptor and limits the invasive capability of prostate cancer cells in vitro. Int J Oncol. 2007;30:193–200. [PubMed] [Google Scholar]
- [71].Weeraratna AT, Dalrymple SL, Lamb JC, Denmeade SR, Miknyoczki S, Dionne CA, Isaacs JT. Pan-trk inhibition decreases metastasis and enhances host survival in experimental models as a result of its selective induction of apoptosis of prostate cancer cells. Clin Cancer Res. 2001;7:2237–2245. [PubMed] [Google Scholar]
- [72].Weeraratna AT, Arnold JT, George DJ, DeMarzo A, Isaacs JT. Rational basis for Trk inhibition therapy for prostate cancer. Prostate. 2000;45:140–148. doi: 10.1002/1097-0045(20001001)45:2<140::aid-pros8>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- [73].Dionne CA, Camoratto AM, Jani JP, Emerson E, Neff N, Vaught JL, Murakata C, Djakiew D, Lamb J, Bova S, George D, Isaacs JT. Cell cycle-independent death of prostate adenocarcinoma is induced by the trk tyrosine kinase inhibitor CEP-751 (KT6587) Clin Cancer Res. 1998;4:1887–1898. [PubMed] [Google Scholar]
- [74].Pflug BR, Dionne C, Kaplan DR, Lynch J, Djakiew D. Expression of a Trk high affinity nerve growth factor receptor in the human prostate. Endocrinology. 1995;136:262–268. doi: 10.1210/endo.136.1.7828539. [DOI] [PubMed] [Google Scholar]
- [75].Sartore-Bianchi A, Ardini E, Bosotti R, Amatu A, Valtorta E, Somaschini A, Raddrizzani L, Palmeri L, Banfi P, Bonazzina E, Misale S, Marrapese G, Leone A, Alzani R, Luo D, Hornby Z, Lim J, Veronese S, Vanzulli A, Bardelli A, Martignoni M, Davite C, Galvani A, Isacchi A, Siena S. Sensitivity to Entrectinib Associated With a Novel LMNA-NTRK1 Gene Fusion in Metastatic Colorectal Cancer. J Natl Cancer Inst. 2016;108 doi: 10.1093/jnci/djv306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Russo M, Misale S, Wei G, Siravegna G, Crisafulli G, Lazzari L, Corti G, Rospo G, Novara L, Mussolin B, Bartolini A, Cam N, Patel R, Yan S, Shoemaker R, Wild R, Di Nicolantonio F, Bianchi AS, Li G, Siena S, Bardelli A. Acquired Resistance to the TRK Inhibitor Entrectinib in Colorectal Cancer. Cancer discovery. 2015 doi: 10.1158/2159-8290.CD-15-0940. [DOI] [PubMed] [Google Scholar]
- [77].Lee SJ, Li GG, Kim ST, Hong ME, Jang J, Yoon N, Ahn SM, Murphy D, Christiansen J, Wei G, Hornby Z, Lee DW, Park JO, Park YS, Lim HY, Hong SN, Kim SH, Kang WK, Park K, Park WY, Kim KM, Lee J. NTRK1 rearrangement in colorectal cancer patients: evidence for actionable target using patient-derived tumor cell line. Oncotarget. 2015;6:39028–39035. doi: 10.18632/oncotarget.5494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Sasahira T, Ueda N, Kurihara M, Matsushima S, Ohmori H, Fujii K, Bhawal UK, Yamamoto K, Kirita T, Kuniyasu H. Tropomyosin receptor kinases B and C are tumor progressive and metastatic marker in colorectal carcinoma. Human pathology. 2013;44:1098–1106. doi: 10.1016/j.humpath.2012.09.016. [DOI] [PubMed] [Google Scholar]
- [79].Fujikawa H, Tanaka K, Toiyama Y, Saigusa S, Inoue Y, Uchida K, Kusunoki M. High TrkB expression levels are associated with poor prognosis and EMT induction in colorectal cancer cells. Journal of gastroenterology. 2012;47:775–784. doi: 10.1007/s00535-012-0532-0. [DOI] [PubMed] [Google Scholar]
- [80].Yu Y, Zhang S, Wang X, Yang Z, Ou G. Overexpression of TrkB promotes the progression of colon cancer. Apmis. 2010;118:188–195. doi: 10.1111/j.1600-0463.2009.02577.x. [DOI] [PubMed] [Google Scholar]
- [81].Sclabas GM, Fujioka S, Schmidt C, Li Z, Frederick WA, Yang W, Yokoi K, Evans DB, Abbruzzese JL, Hess KR, Zhang W, Fidler IJ, Chiao PJ. Overexpression of tropomysin-related kinase B in metastatic human pancreatic cancer cells. Clin Cancer Res. 2005;11:440–449. [PubMed] [Google Scholar]
- [82].Miknyoczki SJ, Wan W, Chang H, Dobrzanski P, Ruggeri BA, Dionne CA, Buchkovich K. The neurotrophin-trk receptor axes are critical for the growth and progression of human prostatic carcinoma and pancreatic ductal adenocarcinoma xenografts in nude mice. Clin Cancer Res. 2002;8:1924–1931. [PubMed] [Google Scholar]
- [83].Schneider MB, Standop J, Ulrich A, Wittel U, Friess H, Andren-Sandberg A, Pour PM. Expression of nerve growth factors in pancreatic neural tissue and pancreatic cancer. J Histochem Cytochem. 2001;49:1205–1210. doi: 10.1177/002215540104901002. [DOI] [PubMed] [Google Scholar]
- [84].Sakamoto Y, Kitajima Y, Edakuni G, Sasatomi E, Mori M, Kitahara K, Miyazaki K. Expression of Trk tyrosine kinase receptor is a biologic marker for cell proliferation and perineural invasion of human pancreatic ductal adenocarcinoma. Oncol Rep. 2001;8:477–484. doi: 10.3892/or.8.3.477. [DOI] [PubMed] [Google Scholar]
- [85].Miknyoczki SJ, Lang D, Huang L, Klein-Szanto AJ, Dionne CA, Ruggeri BA. Neurotrophins and Trk receptors in human pancreatic ductal adenocarcinoma: expression patterns and effects on in vitro invasive behavior. Int J Cancer. 1999;81:417–427. doi: 10.1002/(sici)1097-0215(19990505)81:3<417::aid-ijc16>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- [86].Farago AF, Le LP, Zheng Z, Muzikansky A, Drilon A, Patel M, Bauer TM, Liu SV, Ou SH, Jackman D, Costa DB, Multani PS, Li GG, Hornby Z, Chow-Maneval E, Luo D, Lim JE, Iafrate AJ, Shaw AT. Durable Clinical Response to Entrectinib in NTRK1-Rearranged Non-Small Cell Lung Cancer. J Thorac Oncol. 2015;10:1670–1674. doi: 10.1097/01.JTO.0000473485.38553.f0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Harada T, Yatabe Y, Takeshita M, Koga T, Yano T, Wang Y, Giaccone G. Role and relevance of TrkB mutations and expression in non-small cell lung cancer. Clin Cancer Res. 2011;17:2638–2645. doi: 10.1158/1078-0432.CCR-10-3034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Ricci A, Greco S, Mariotta S, Felici L, Bronzetti E, Cavazzana A, Cardillo G, Amenta F, Bisetti A, Barbolini G. Neurotrophins and neurotrophin receptors in human lung cancer. Am J Respir Cell Mol Biol. 2001;25:439–446. doi: 10.1165/ajrcmb.25.4.4470. [DOI] [PubMed] [Google Scholar]
- [89].Vaishnavi A, Capelletti M, Le AT, Kako S, Butaney M, Ercan D, Mahale S, Davies KD, Aisner DL, Pilling AB, Berge EM, Kim J, Sasaki H, Park SI, Kryukov G, Garraway LA, Hammerman PS, Haas J, Andrews SW, Lipson D, Stephens PJ, Miller VA, Varella-Garcia M, Janne PA, Doebele RC. Oncogenic and drug-sensitive NTRK1 rearrangements in lung cancer. Nature medicine. 2013;19:1469–1472. doi: 10.1038/nm.3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig. S1. Effect of entrectinib on Growth profile of parental SY5Y (Trk-NULL) cells. Representative SRB assay indicating no significant response to entrectinib treatment as there is no expressed Trk in these cells. Each assay was performed in triplicate, and at least three independent experiments were conducted.
Supplementary Fig. S2. Densitometric and statistical analysis of Western blot results presented in Figure 3C. All individual western blot images were scanned, normalized and statistical analysis was performed. Significant inhibition of phosphoprotein expression was observed for p-Trk, p-PLCγ, p-Akt and p-Erk after 1 hr, 4 hr and 6 hr of entrectinib treatment when compared with untreated vehicle controls. Actin levels remain unchanged, indicating no global inhibition of protein expression. P-values were calculated (**<0.01) by two-way ANOVA using Prism followed by Sidak post-test.
Supplementary Fig. S3. Effect of entrectinib growth profile of NB69 cells. Representative SRB assay indicating dose dependent inhibition of NB69 cells upon entrectinib treatment as indicated. Each assay was performed in triplicate.