

Abstract
A novel HAuCl4@UiO-66-NH2 material has been obtained and utilized as a heterogeneous Au(III) catalyst. This Au(III) catalyst was able to promote the formation of a variety of dihydrochalcones starting from 2H-chromenes in moderate to good yields. A tandem hydride shift/hydration reaction sequence has been proposed based on deuterium labeling studies, which revealed a 1,5-hydride shift reaction pathway. A flavone intermediate has been synthesized to further support the proposed mechanism. Furthermore, the HAuCl4@UiO-66-NH2 catalyst can be recycled several times without compromising the catalytic activity.
Introduction
Metal-organic frameworks (MOFs) are being utilized as a novel class of microporous materials in the fields of gas storage/separation, sensors, proton conductivity and drug delivery.1 MOFs can also serve as excellent heterogeneous supports for the synthesis of a variety of solid and recyclable catalysts, taking advantage of their large surface area, porous nature and tunable structure.2 Despite the limited activities promoted by MOF catalysts, the additional catalytic activity of MOFs can be facilely introduced via post-synthetic modification (PSM) strategies within the pores of MOF structures.3 This new functionalization strategy allows the introduction of new catalytically active sites to be attached to a common MOF scaffold, which is highly desirable as it provides an alternative route to achieve heterogeneous catalysts rapidly.4 To date, transition metals with various catalytic activities, such as Cu, Pd, Mn, etc., were incorporated into the porous structures of post-synthetically modified MOFs.5 However, the synthesis of these M@MOF catalysts usually takes two or more steps. A simpler approach to access M@MOF would be desirable. As a result, a one-step synthetic method for direct transition metal PSM of M@MOF catalysts without extra introduction of organic groups is worth developing.
Natural and synthetic dihydrochalcones were found to have various interesting biological activities.6 The dihydrochalcones phloretin and phloridzin, which are major phenolic constituents of apples, have strong antioxidant activity.7 C-benzylated dihydrochalcones also show interesting cytotoxicity toward human promyelocytic leukemia HL-60 cells.8 Furthermore, dihydrochalcones can act as intermediates for the synthesis of bioactive chiral 2-substituted 2,3-dihydrobenzofuran.9 Because of their interesting biological activities, there have been several approaches to generate dihydrochalcone skeletons for library synthesis and biological studies.10 The most common method for synthesizing a dihydrochalcone is the reduction of a chalcone, which can be generated through Claisen–Schmidt condensation reaction.11 However, strong alkaline conditions are generally required for the Claisen–Schmidt condensation.12 Alternatively, dihydrochalcones can be synthesized through a concise Pd-mediated Heck-type reaction, which was previously reported by Wagner and coworkers.13 The Maiti group first investigated the conversion of 2H-chromenes to dihydrochalcones.14 Being the only example in this field, this hydride shift/hydration reaction sequence is a unique approach for accessing dihydrochalcone structures.
Herein, we report the development of a general post-synthetic modification strategy to introduce Au(III) to well-defined amino-functionalized MOFs. Nobel metal salts, such as HAuCl4 and HPdCl4 can be easily incorporated. This method provides an easy handle to access a metal-supported heterogeneous MOF catalyst under mild conditions. The as-synthesized catalyst exhibited good catalytic activities for the transformation of a variety of 2H-chromenes to dihydrochalcones under mild conditions. Deuterium labeling studies have been performed to lend further insight into the reaction mechanism, which reveals a hydride shift/hydration sequence of 2H-chromene catalyzed by a HAuCl4@UiO-66-NH2 catalyst. Furthermore, the as-synthesized catalyst can be recycled several times without loss of catalytic activity.
Results and discussion
Scanning electron microscope (SEM) images suggest the well dispersed nanoparticles of UiO-66-NH2 and HAuCl4@UiO-66-NH2 materials. The morphology of UiO-66-NH2 crystal appears to be octahedral at about 150 nm in terms of size. The nanomorphology was retained after HAuCl4 post-synthetic modification as shown in Fig. S1. The great dispersion of HAuCl4@UiO-66-NH2 nanoparticles is the advantage for utilizing such a material for catalysis.
The powder X-ray diffraction (XRD) of UiO-66-NH2 revealed the material was carried, which is in agreement with the literature (Figure 1).15 The structural integrity of the HAuCl4@UiO-66-NH2 catalyst was also proved by the powder XRD (Fig. 1). UiO-66-NH2 shows excellent acidic stability in the presence of HAuCl4 during our investigation. However, several other MOFs, IRMOF-3 for example, were not able to survive upon the treatment of HAuCl4 and partial structural decomposition was observed according to XRD characterization. The structural instability of MOF supports towards acid and heat is known in the literature.16
Fig. 1.
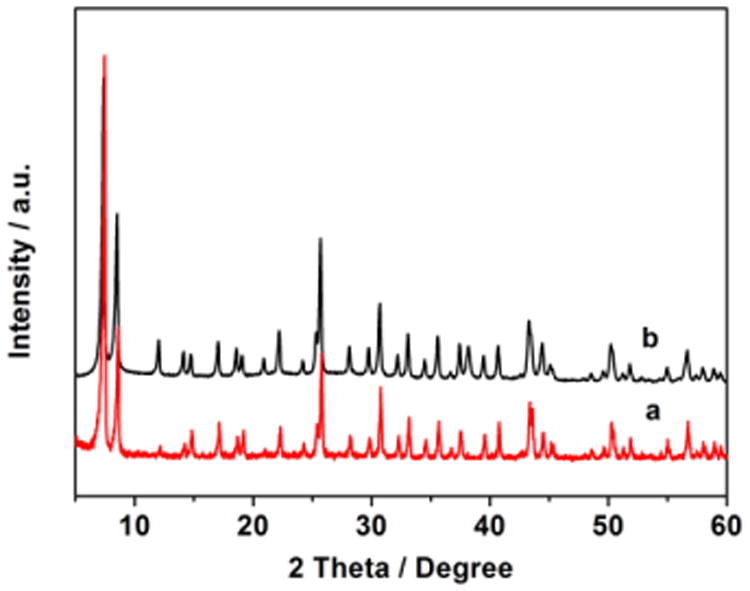
Powder XRD patterns of (a) UiO-66-NH2 and (b) HAuCl4@UiO-66-NH2.
Fourier Transform infrared spectroscopy (FTIR) of HAuCl4@UiO-66-NH2 shows strong peaks at 1569, 1433 and 1384 cm-1, which correspond to the UiO-66-NH2 support. However, the peak of HAuCl4 is not visible in the FTIR spectrum of HAuCl4@UiO-66-NH2 due to the low loading (Fig. 2).
Fig. 2.
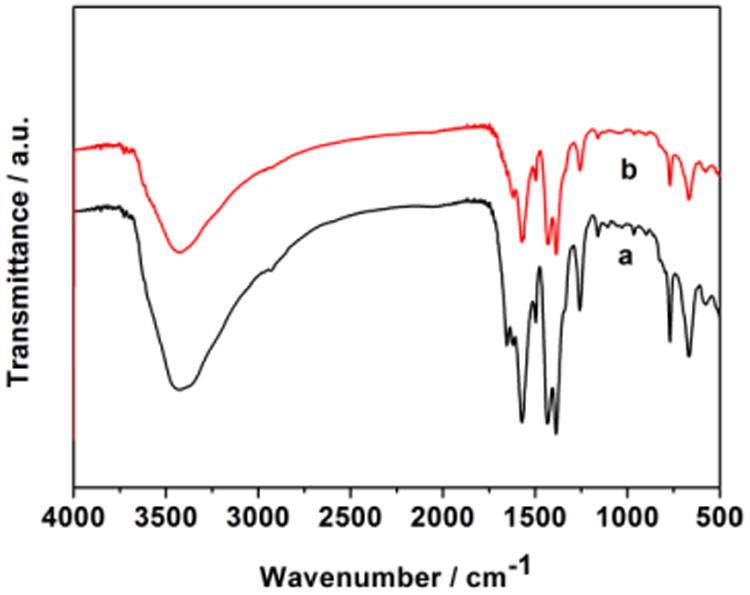
FTIR spectra of (a) UiO-66-NH2 and (b) HAuCl4@UiO-66-NH2.
Inductively coupled plasma-atomic emission spectroscopy (ICP-AES) was carried out to measure the gold content of HAuCl4@UiO-66-NH2, which was determined to be 2.3 wt%. This result is used to calculate the catalyst loading for organic transformation, since HAuCl4 is the catalytically active species. The Brunauer–Emmett–Teller (BET) area of the samples was measured by nitrogen adsorption/desorption measurement at 77 K. The result shows that the specific surface area of HAuCl4@UiO-66-NH2 only decreases from 1264 m2g-1 to 1149 m2g-1 after loading (Fig. S2 and 3). The size of the UiO-66-NH2 pore is narrowly distributed at around 0.58 nm according to nitrogen adsorption/desorption measurement. Thermal gravimetric analysis (TGA) was used to examine the thermal and structural stability of UiO-66-NH2 and HAuCl4@UiO-66-NH2 materials. The HAuCl4@UiO-66-NH2 catalyst showed thermal stabilities slight less to that of UiO-66-NH2 with a decomposition temperature of approximately 300 °C in air (Figure S4).
The catalytic activity of HAuCl4@UiO-66-NH2 was evaluated to show its great activity towards multi-step organic transformation. HAuCl4@UiO-66-NH2 can act as an efficient catalyst in the generation of dihydrochalcone 2a (Table 1). A solvent screen indicates that CH2Cl2 is the best solvent among other common organic solvents when HAuCl4@UiO-66-NH2 was used as the catalyst (Table 1, entries 1-4). Aromatic solvents such as toluene gave much lower yield, presumably due to low compatibility with the transitional metal catalyst (Table 1, entry 2). Methanol provided a much lower yield than chlorinated solvents due to the coordination to the Au(III) catalyst (Table 1, entry 3). In a similar way as methanol, THF also gave low yield of the desired dihydrochalcone 2a (Table 1, entry 4). Interestingly, the HAuCl4@UiO-66-NH2 performed poorly in the absence of water. Under anhydrous conditions, only 41% yield was achieved, which is significantly lower than the yield in Table 1, entry 1. The important role of water is discussed under the mechanism section. Furthermore, very low yield was achieved with HPdCl4@UiO-66-NH2 as the catalyst because of the decrease in efficient olefin activation when compared with Au(III) (Table 1, entry 6). It is worth mentioning that this reaction was not able to be promoted by aqueous HCl, which indicates the crucial role of the transition metal during the catalytic process (Table 1, entry 7).
Table 1.
Reaction optimization for a hydride shift/hydration sequencea.
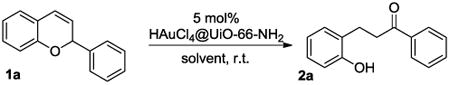
| |||
|---|---|---|---|
|
| |||
| Entry | Catalyst | Solvent | Yieldb |
| 1 | HAuCl4@UiO-66-NH2 | CH2Cl2 | 88% |
| 2 | HAuCl4@UiO-66-NH2 | PhCH3 | 56% |
| 3 | HAuCl4@UiO-66-NH2 | CH3OH | 28% |
| 4 | HAuCl4@UiO-66-NH2 | THF | 34% |
| 5c | HAuCl4@UiO-66-NH2 | CH2Cl2 | 41% |
| 6 | HPdCl4@UiO-66-NH2 | CH2Cl2 | 22% |
| 7 | HCl(aq) | CH2Cl2 | <10% |
Reaction conditions: 0.5 mmol of 1a, 5 mol% HAuCl4@UiO-66-NH2 or other catalyst, 0.2 M in the solvent for 24 h at room temperature
Isolated yield.
The HAuCl4@UiO-66-NH2 catalyst was dried under vacuum at 150 °C for 24 h before use and the reaction was conducted anhydrously.
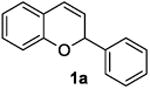
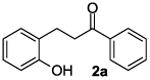
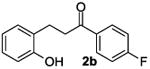
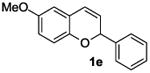
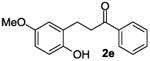
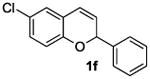
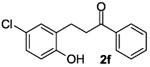
The ring opening reaction sequence of 2H-chromenes provides a powerful tool to gain rapid access to dihydrochalcone core structures. The optimized reaction using HAuCl4@UiO-66-NH2 proved to be general for a range of 2H-chromenes (Table 2). This reaction proceeded smoothly with phenyl substituted chromene 1a, furnishing dihydrochalcone 2a in good yield (Table 2, entry 1). Electron-deficient chromene 1b underwent this reaction in the same fashion, providing dihydrochalcone 2b in 85% yield at room temperature after 24 h (Table 2, entry 2). A decent yield of dihydrochalcone 2c was obtained when 2-(o-MeO-phenyl)-2H-chromene 1c was treated with a catalytic amount of HAuCl4@UiO-66-NH2 (Table 2, entry 3). These results prove that this HAuCl4@UiO-66-NH2-promoted ring opening reaction sequence approach is compatible with varying substitution at the C2 position. Furthermore, a substituted benzopyran ring was evaluated to show the good compatibility of our catalyst. Electron-rich benzopyran-derived 2H-chromenes 1d and 1e were compatible with the HAuCl4@UiO-66-NH2 catalyst, which gave the desired dihydrochalcone products in 82% and 87% yield, respectively (Table 2, entries 4 and 5). Lastly, the reaction with electron-deficient chromene 1f proceeded smoothly (Table 2, entry 6).
Table 2.
2H-chromene evaluation utilizing HAuCl4@UiO-66-NH2a.
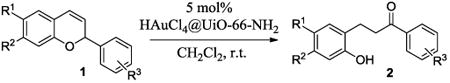
| |||
|---|---|---|---|
|
| |||
| Entry | 2H-chromene | Product | Yieldb |
| 1 |
|
|
88% |
| 2 |
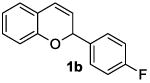
|
|
85% |
| 3 |
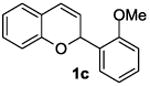
|
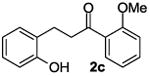
|
81% |
| 4 |
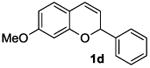
|
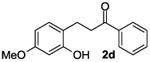
|
82% |
| 5 |
|
|
87% |
| 6 |
|
|
76% |
Reaction conditions: 0.5 mmol of 2H-chromene 1, 5 mol% catalyst, 0.2 M in CH2Cl2 for 6 h at room temperature.
Isolated yield.
To gain insight into the reaction mechanism, a deuterated analogue of 2-phenyl-2H-chromene 1a was synthesized.17 A reduction/cyclization reaction was carried out to furnish the C2 deuterated 2H-chromene 1a-D.18 Subjection of 1a-D to the HAuCl4@UiO-66-NH2-catalyzed reaction conditions afforded the deuterium label of dihydrochalcone 2a-D at the benzylic position adjacent to the phenol ring (Fig. 4). This observation strongly suggested a 1,5-hydride shift occurred during the dihydrochalcone formation.
Fig. 4.

Deuterium labeling study of deuterated 1a-D.
During our investigation, it was observed that water played a crucial role for promoting the generation of dihydrochalcones; the yield of dihydrochalcone decreased significantly if the reaction system was dried. To reveal the role of water in the dihydrochalcone formation process, a hydride shift/hydration sequence in the presence of D2O was conducted. The resulting deuterated dihydrochalcone 2a-2D was obtained with both the C3 and phenol positions deuterated (Fig.5). This observation further confirms the hypothesis that the water acts as a nucleophile for the benzopyran ring opening. The abstraction of two deuterium atoms leads to the formation of bisdeuterated dihydrochalcone 2a-2D.
Fig. 5.

The rearrangement of 2H-chromene in the presence of D2O.
At this stage, a flavone-type intermediate as the hydride shift outcome has been proposed. In order to support this mechanistic hypothesis, flavone 3 was synthesized and subjected into our HAuCl4@UiO-66-NH2-promoted reaction conditions.19 The desired dihydrochalcone, 2a, was isolated as the major product in 96% yield (Fig. 6). This observation indicates that the transformation flavone 3 to the desired product 2a is possible. A multi-step reaction mechanism involving a flavone 3 related intermediate is more plausible.
Fig. 6.

Treating flavone 3 with HAuCl4@UiO-66-NH2 catalyst.
The proposed mechanism for this transformation is shown in Figure 4, based on our experimental results (Figures 3-5). In the presence of a heterogeneous Au(III) catalyst, a 1,5-deuteride shift to the C4 position of intermediate 4 occurs. This type of hydride-shift is well studied in the literature.20 Intermediate 4 equilibrates to flavone 5 with an Au(III) attached. The formation of flavone 5 was elucidated as it was able to be trapped as a dienophile in the previous hetero-Diels-Alder reaction research.21 Lastly, the addition of H2O leads to ring opening product 6, which equilibrates to dihydrochalcone 2a-D through a tautomerization (Fig. 7).
Fig. 3.
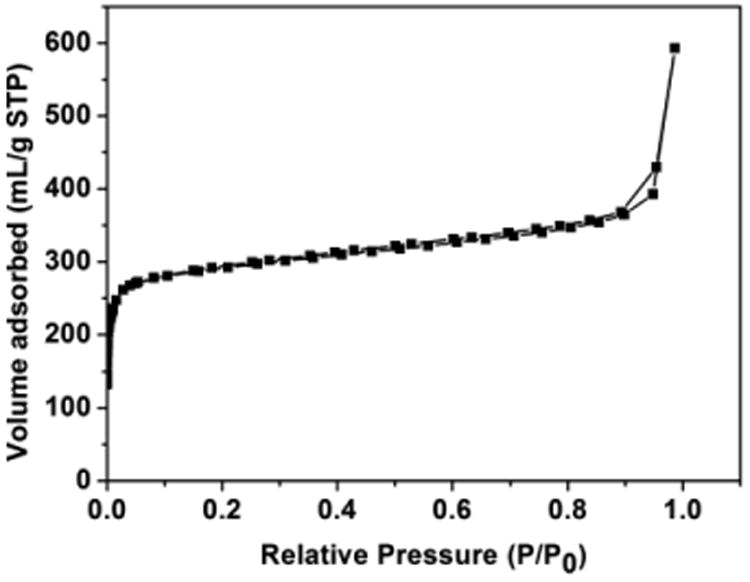
N2 adsorption/desorption isotherms of HAuCl4@UiO-66-NH2.
Fig. 7.

Proposed reaction mechanism.
The recyclability of the heterogeneous Au(III) catalyst was evaluated for the generation of 3-(2-hydroxyphenyl)-1-phenylpropan-1-one 2a. The recycled HAuCl4@UiO-66-NH2 catalyst showed no loss of dihydrochalcone yield for five runs (Fig. 8). The supernatant liquid of the CH2Cl2 suspension was collected after one catalytic cycle and the solid Au(III) catalyst was filtered. The liquid showed no reactivity towards to the 2H-chromene 1a, which suggests no leakage of the Au(III) catalyst. The catalyst before and after the hydride shift/hydration reaction sequence was assessed by PXRD to evaluate the crystalline structure of the HAuCl4@UiO-66-NH2 catalyst. The X-ray powder diffraction pattern of the HAuCl4@UiO-66-NH2 catalyst after five cycles of reuse was indistinguishable from those of the fresh catalyst (see ESI).
Fig. 8.
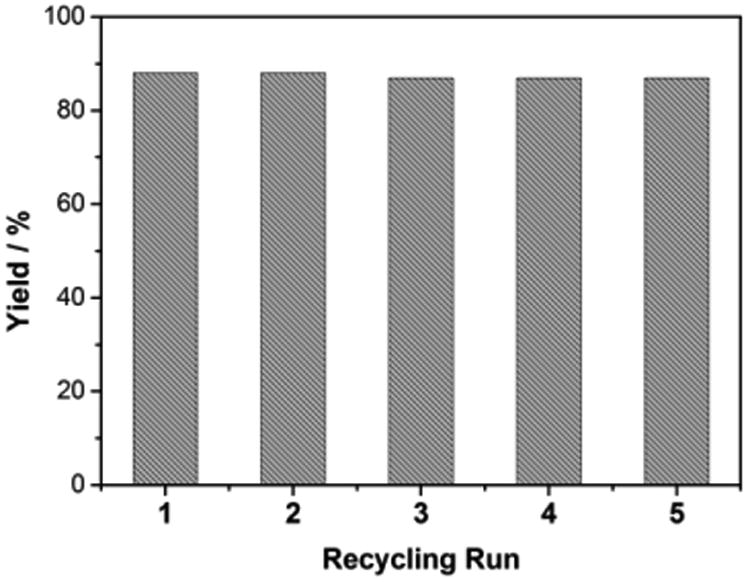
Catalyst recycling test of HAuCl4@UiO-66-NH2.
Conclusion
In conclusion, we have developed a rapid and facile method for the synthesis of a novel HAuCl4@UiO-66-NH2 material. This material was utilized as an efficient catalyst to access a variety of functionalized dihydrochalcone scaffolds from common 2H-chromenes. Furthermore, the HAuCl4@UiO-66-NH2 catalyst can be recycled 5 times without compromising the yield of the desired product. Deuterium-labeling studies have been performed in order to reveal the possible mechanism for dihydrochalcone formation, which suggest a tandem hydride shift/hydration reaction sequence via a flavone intermediate. Our synthetic approach provides a general method for the synthesis of a heterogeneous Au(III) catalyst and a mild synthetic route to a variety of dihydrochalcones.
Supplementary Material
Scheme 1.
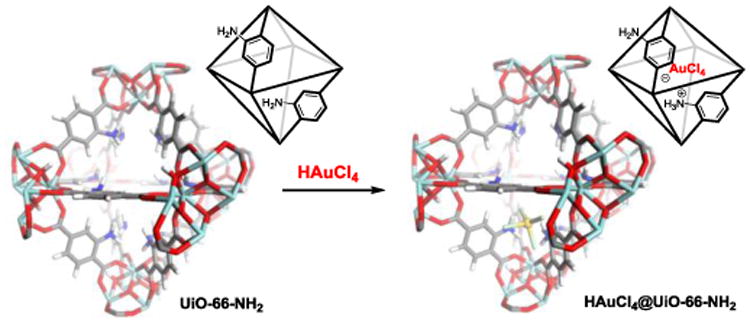
Schematic illustration of the synthesis of HAuCl4@MOF material.
Acknowledgments
The authors gratefully acknowledge Kathryn E. Summo for editorial review of the manuscript. Preliminary experiments were performed by Y.L. at Boston University. S.E.S. and Y.L. gratefully acknowledge the NIH for support (NIGMS R01 GM078240). Y.L., Y.Q., J.Y. and H.Y.G. thank Beijing Natural Science Foundation (Grant No. 2144052) and the China Postdoctoral Science Foundation (2014T70036) for support.
Footnotes
Electronic Supplementary Information (ESI) available: [Full experimental detail, 1H and 13C NMR spectra, and characterization data. This material can be found via the “Supplementary Content” section of this article's webpage].
Notes and references
- 1.Cook TR, Zheng YR, Stang PJ. Chem Rev. 2013;113:734. doi: 10.1021/cr3002824. [DOI] [PMC free article] [PubMed] [Google Scholar]; Song LF, Zhang J, Sun LX, Xu F, Li F, Zhang HZ, Si XL, Jiao CL, Li ZB, Liu S, Liu YL, Zhou HY, Sun DL, Du Y, Cao Z, Gabelica Z. Energy Environ Sci. 2012;5:7508. [Google Scholar]
- 2.Corma A, Carcia H, Xamena FXLI. Chem Rev. 2010;110:4606. doi: 10.1021/cr9003924. [DOI] [PubMed] [Google Scholar]; Dhakshinamoorthy A, Opanasenko M, Cejka J, Garcia H. Catal Sci Technol. 2013;3:2509. [Google Scholar]
- 3.Valtchev V, Majano G, Mintova S, Pérez-Ramírez J. Chem Soc Rev. 2013;42:263. doi: 10.1039/c2cs35196j. [DOI] [PubMed] [Google Scholar]
- 4.Dhakshinamoorthy A, Opanasenko M, Čejka J, Garcia H, Garcia H. Adv Synth Catal. 2013;355:247. [Google Scholar]
- 5.Liu L, Zhang X, Gao J, Xu C. Green Chem. 2012;14:1710. [Google Scholar]; Yang J, Li P, Wang L. Catal Commun. 2012;27:58. [Google Scholar]; Saha D, Sen R, Maity T, Koner S. Langmuir. 2013;29:3140. doi: 10.1021/la304147j. [DOI] [PubMed] [Google Scholar]
- 6.Nakamura Y, Watanabe S, Miyake N, Kohno H, Osawa T. J Agric Food Chem. 2003;51:3309. doi: 10.1021/jf0341060. [DOI] [PubMed] [Google Scholar]; Dziedzic SZ, Hudson BJF, Barnes GJ. J Agric Food Chem. 1985;33:244. [Google Scholar]; Mathiesen L, Malterud KE, Sund RB. Free Radical Biol Med. 1997;22:307. doi: 10.1016/s0891-5849(96)00277-8. [DOI] [PubMed] [Google Scholar]
- 7.Yang WM, Liu JK, Qin XD, Wu WL, Chen ZH. Z Naturforsch C. 2004;59c:481. doi: 10.1515/znc-2004-7-805. [DOI] [PubMed] [Google Scholar]
- 8.Ichimaru M, Nakatani N, Takahashi T, Nishiyama Y, Moriyasu M, Kato A, Mathenge SG, Juma FD, Nganga JN. Chem Pharm Bull. 2004;52:138. doi: 10.1248/cpb.52.138. [DOI] [PubMed] [Google Scholar]
- 9.Uyanik M, Okamoto H, Yasui T, Ishihara K. Science. 2010;328:1376. doi: 10.1126/science.1188217. [DOI] [PubMed] [Google Scholar]
- 10.Cho CS, Shim SC. J Organomet Chem. 2006;691:4329. [Google Scholar]; Fuchs A, De Vries FW, Landheer CA, Veldhuizen AV. Phytochemistry. 1984;23:2199. [Google Scholar]
- 11.Ding BQ, Zhang ZF, Liu YG, Sugiya M, Imamoto T, Zhang WB. Org Lett. 2013;15:3690. doi: 10.1021/ol401560q. [DOI] [PubMed] [Google Scholar]; Lakshmi KM, Parsharamulu T, Manorama SV. J Mol Catal A: Chem. 2012;365:115. [Google Scholar]
- 12.Shakil NA, Singh MK, Sathiyendiran M, Kumar J, Padaria JC. Eur J Med Chem. 2013;59:120. doi: 10.1016/j.ejmech.2012.10.038. [DOI] [PubMed] [Google Scholar]; Dong XW, Liu T, Chen J, Ying HZ, Hu YZ. Synth Commun. 2009;39:733. [Google Scholar]
- 13.Briot A, Baehr C, Brouillard R, Wagner A, Mioskowski C. J Org Chem. 2004;69:1374. doi: 10.1021/jo034936c. [DOI] [PubMed] [Google Scholar]
- 14.Maiti G, Kayal U, Karmakar R, Bhattacharya RN. Tetrahedron Lett. 2012;53:6321. [Google Scholar]
- 15.Kandiah M, Ussegqlio S, Svelle S, Olsbye U, Lillerud KP, Tilset M. J Mater Chem. 2010;20:9848. [Google Scholar]
- 16.Ingleson MJ, Barrio JP, Guilbaud JB, Khimyak YZ, Rosseinsky MJ. Chem Commun. 2008:2680. doi: 10.1039/b718367d. [DOI] [PubMed] [Google Scholar]
- 17.Krohn K, Ahmed I, John M. Synthesis. 2009;5:779. [Google Scholar]
- 18.Ashihara Y, Nagata Y, Kurosawa K. Bull Chem Soc Jpn. 1977;50:3298. [Google Scholar]
- 19.Bird TGC, Brown BR, Stuart IA, Tyrrell AWR. J Chem Soc Perkin Trans. 1983;1:1831. [Google Scholar]
- 20.Pastine SJ, McQuaid KM, Sames D. J Am Chem Soc. 2005;127:12180. doi: 10.1021/ja053337f. [DOI] [PubMed] [Google Scholar]; Pastine SJ, Sames D. Org Lett. 2005;7:5429. doi: 10.1021/ol0522283. [DOI] [PubMed] [Google Scholar]; McQuaid KM, Sames D. J Am Chem Soc. 2009;131:402. doi: 10.1021/ja806068h. [DOI] [PMC free article] [PubMed] [Google Scholar]; McQuaid KM, Long JZ, Sames D. Org Lett. 2009;11:2972. doi: 10.1021/ol900915p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luan Y, Sun H, Schaus SE. Org Lett. 2011;13:6480. doi: 10.1021/ol202772k. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.