

Abstract
Cross-presentation is the process by which professional antigen presenting cells (APCs) (B cells, dendritic cells (DCs) and macrophages) present endocytosed antigens (Ags) via MHC-I to CD8+ T cells. This process is crucial for induction of adaptive immune responses against tumors and infected cells. The pathways and cellular compartments involved in cross-presentation are unresolved and controversial. Among the cells with cross-presenting capacity, DCs are the most efficient, which was proposed to depend on prevention of endosomal acidification to block degradation of the epitopes. Contrary to this view, we show in this report that some cargoes induce strong endosomal acidification following uptake by human DCs, while others not. Moreover, processing of the tumor-associated antigen HER2/neu delivered in nanoparticles (NP) for cross-presentation of the epitope HER2/neu369–377 on HLA-A2 depended on endosomal acidification and cathepsin activity as well as proteasomes, and newly synthesized HLA class I. However, the HLA-A*0201/HER2/neu369–377 complexes were not found in the endoplasmic reticulum (ER) nor in endolysosomes but in hitherto not described vesicles. The data thus indicate spatial separation of antigen processing and loading of MHC-I for cross-presentation: antigen processing occurs in the uptake compartment and the cytosol whereas MHC-I loading with peptide takes place in a distinct subcellular compartment. The findings further elucidate the cellular pathways involved in the cross-presentation of a full-length, clinically relevant tumor-associated antigen by human DCs, and the impact of the vaccine formulation on antigen processing and CD8+ T cell induction.
Keywords: cross-presentation, cancer, dendritic cells, HER2/neu, nanoparticles, tumor-associated antigens, vaccine
Abbreviations
- Ag
Antigen
- APC
Antigen presenting cell
- CTL
Cytotoxic T lymphocyte
- DC
Dendritic cell
- ER
Endoplasmic reticulum
- ERGIC
Endoplasmic reticulum Golgi intermediate compartment
- HLA
Human leukocyte antigen
- MHC
Major histocompatibility complex
- NP
Nanoparticle
- TAP
transporter associated with antigen processing
Introduction
The induction of CD8+ cytotoxic T lymphocytes (CTLs) that specifically recognize and kill tumor cells is the main goal of cancer vaccination. For the induction of primary and secondary immune responses with the generation of CD8+ cytotoxic T cells, exogenous Ags must be taken up by DCs, and be processed into peptides for loading onto major histocompatibility (MHC) class I molecules, a process termed cross-presentation.1,2 DCs are considered the most potent cross-presenting cells, highly efficient in the priming of CD8+ T cells and, therefore, the most important targets for tumor Ag vaccination.3-5
Cross-presentation was first proposed by Bevan in the 1970s.1 He immunized mice with allogeneic cells and found cytotoxic CD8+ T cell-mediated responses specific for Ags from the graft presented by MHC class I molecules of the host. This suggested that Ags from the transplanted cells were internalized and processed by APCs of the host for presentation by the host MHC class I molecules. Since then and despite intensive study, the cellular and molecular mechanisms of cross-presentation have not been clarified.
By current understanding, cross-presentation of Ags by DCs can occur via at least three distinct mechanisms. Following uptake, Ags can be translocated from the endocytic/phagocytic compartment into the cytosol6,7 where they are broken down into peptides by proteasome and transported via transporter associated with antigen processing (TAP) into the ER for loading onto nascent MHC class I molecules.6 This route is known as “phagosome-to-cytosol pathway”. An alternative route, termed “vacuolar pathway” was shown to be used for certain soluble proteins. In this model, Ags are processed in endosomal compartments by cathepsins where peptides are generated, loaded onto recycling MHC class I molecules, and transported to the cell surface for presentation.8,9 The third proposed pathway involves export of Ag from the endosome to the cytosol, followed by proteasome processing and reimport into the endosomal compartment for loading onto MHC class I molecules. This model is often referred to as “ER-phagosome fusion pathway” and involves fusion of the ER and/or ER-Golgi intermediate compartment (ERGIC) with the endosome/phagosome, leading to the formation of a compartment named “ERgosome” characterized by the presence of ER components such as Sec22b, TAP, and components of the peptide loading complex (PLC) as well as components of endosomes/phagosomes.10-12
Regardless of the mechanisms that lead to cross-presentation, it is accepted that DCs are the most efficient cells at that function. It has recently been proposed that one of the reasons for the superior efficiency of DCs in cross-presentation is that they possess a mechanism to prevent endosomal/phagosomal acidification.13,14 A mild or alkaline endosomal pH would prevent degradation of the epitopes, favoring their cross-presentation. This phenomenon seems to be more evident in the murine DC subset expressing CD8α,13 but with little evidences of whether it would occur also in human DCs.15-17 The understanding of the mechanisms that lead to cross-presentation of tumor Ags by human DCs is essential for rational strategies to induce tumor-specific CTLs.
The mode of delivery as well as the choice of tumor Ag are key parameters for the success of the cancer vaccine. The tumor-associated Ag, HER2/neu, is a clinical target of high interest.18 It is overexpressed by a wide range of tumors, such as breast, ovary and renal cell carcinomas, and colorectal and pancreatic adenocarcinomas,19-21 and it is the target for a humanized monoclonal antibody in clinical use and capable of impacting tumor growth.19 As humoral and cellular responses against HER2/neu are detected in almost all patients, it is targeted by immune responses of the patients.20,22 The epitope HER2/neu369–377, which binds HLA-A2, was found to be one of the immunodominant epitopes of the protein.21,23 HER2/neu369–377 is generated and presented by HLA-A2+ breast cancer cells and triggers cytolysis by epitope-specific CTLs.23
The mode of Ag delivery to DCs may affect its processing and, as a result, the selection of cross-presented peptides.17 This would impact specificity and quality of the immune responses. Understanding the Ag processing mechanisms of cross-presentation is crucial for the development of effector T cell-directed vaccines. Soluble proteins are usually poorly cross-presented by DCs.24,25 Therefore, advanced delivery systems are required to optimize and enhance this process. To elucidate the pathways of Ag processing by human DCs, we investigated the processing and cross-presentation of the tumor-associated Ag HER2/neu by human DCs, delivered in the soluble form or in different NP formulations.
Results
The composition of NPs determines their routing inside DCs
Immature DCs were incubated with the different NPs and harvested at different time points to determine their intracellular fate. Interestingly, none of the NPs co-localized with EEA1 (Fig. S1), a protein expressed on the outer leaflet of early endosomes.26 Endosomes containing PLGA-based NPs were acidified already 1 h after their addition to DCs, as indicated by LysoTracker Red staining, a lysosomotropic probe that accumulates in acidic compartments (Fig. S2). On the other hand, chitosan and silica NPs were routed into and stayed in nonacidic compartments over a 24 h incubation period (Fig. 1A). Furthermore, chitosan, PLGA, and PEG-PAGE-PLGA NPs induced the expression of DC-LAMP in the late endosomes/lysosomes while silica and mannose PEG-PAGE-PLGA did not (Fig. 1B). DC-LAMP is a glycoprotein expressed in the lysosomes of mature DCs.27 It is interesting that its expression was induced by PLGA and PP-PLGA without induction of DC maturation (Baleeiro et al., submitted). Also, PLGA and PEG-PAGE-PLGA co-localized with the DC-LAMP whereas the chitosan NPs did not. The appearance of the compartments was also different for the NPs routed into acidic vs. those routed into nonacidic compartments. Chitosan and silica NPs were found in vacuole-like structures, while all PLGA-based NPs were inside well-defined endosome-like compartments (Fig. 1C). Importantly, all NPs were contained within membrane-bounded compartments (Fig. 1C). Collectively, these data indicate that the routing of particulate cargoes into acidic or non-acidic compartments in human DCs is determined by their composition rather than a constitutive process, as suggested by some previous studies.13,16
Figure 1.

Intracellular routing of NPs after uptake by human iDCs. Confocal (A, B) and TEM (C) images of iDCs loaded with chitosan, Silica, PLGA, PEG-PAGE-PLGA, and Mannose PEG-PAGE-PLGA NPs. Confocal images (A, B) show DCs loaded with NPs (green) and stained with LysoTracker Red (red in A) for late/acidic endosomes, Alexa fluor 647-stained anti-MHC-I molecules (gray) to indicate plasma membrane and DAPI (blue) for nuclei – or DC-LAMP (red in B) for late endosomes. NPs inside late/acidic or DC-LAMP-positive compartments appear yellow in the merged images. Confocal micrographs were taken with a 63x oil objective lens. The TEM images (C) show chitosan and Silica NPs inside vacuole-like structures, whereas PLGA, PEG-PAGE-PLGA, and Mannose PEG-PAGE-PLGA NPs were inside well-defined vesicular compartments. Red arrows indicate NP localization. All NPs were within membrane-bounded compartments. Scale bar in (C)= 1 μm.
Kinetics of cross-presentation of HER2/neu369–377
To monitor the cross-presentation of antigen delivered to DCs in different carriers, we used an antibody specific for the HLA-A*0201/HER2/neu369–377 complex (Figs. S3–4). iDCs were incubated for 24 h with recombinant human HER2/neu protein in soluble form or encapsulated in different NPs. DCs loaded with HER2/neu delivered in the NPs presented higher levels of HLA-A*0201/HER2/neu369–377 complexes than DCs loaded with soluble HER2/neu (Fig. 2A). The amounts of HLA-A*0201/HER2/neu369–377 complexes increased for HER2/neu in PLGA and mannose PEG-PAGE-PLGA NPs to a plateau at and above 1 μg HER2/neu per mL, whereas for chitosan NPs the levels kept increasing with protein concentration (Fig. 2B). HER/neu in the soluble form yielded lower levels of specific complexes compared to the HER2/neu in the particulate formulations. Only at concentrations above 1 µg/mL soluble HER2/neu led to levels of the complex similar to NP-HER2/neu. In time-course experiments from 2 to 96 h after addition of the protein in soluble or particulate formulations to the DC cultures, presentation of the HLA-A*0201/HER2/neu369–377 complexes peaked at 48 h. Using the peptide HER2/neu369–377, the specific complexes were detected at the earliest time point already and declined thereafter to background levels within 12 h (Fig. 2C). To confirm the cross-presentation of HER2/neu to CD8+ T cells, DCs loaded with HER2/neu in the different formulations were incubated with purified autologous CD8+ T cells for 7 d. Expansion of peptide-specific CD8+ T cells was quantified by flow cytometry using HER2/neu369–377/HLA-A*0201 dextramers. DCs loaded with HER2/neu at 5 μg/mL in all NPs formulations showed similar immunostimulatory capacity (Fig. S5), thus confirming the data that we had obtained measuring the complexes HLA-peptide at the DCs surface.
Figure 2.
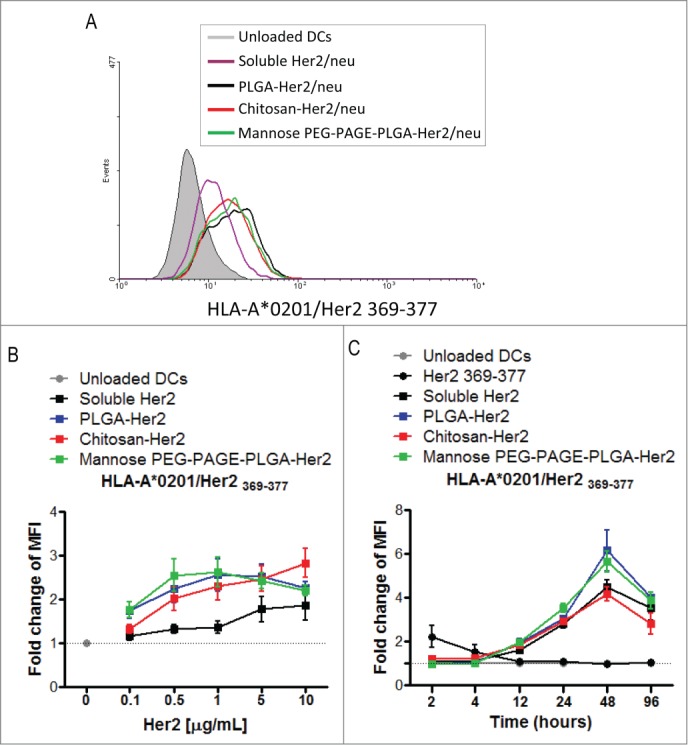
Kinetics of human DC-mediated cross-presentation of HER2/neu369–377. iDCs were loaded with 1µg/mL (A) or titrated doses of soluble or NP-encapsulated HER2/neu (B) and harvested 24 h post loading for assessment of the complexes on their surfaces by flow cytometry (A, B). For time course studies, iDCs were loaded with soluble HER2/neu (50 µg/mL), NP-HER2/neu (5 µg/mL) or soluble HER2/neu369–377 peptide (50 µg/mL), harvested at different time points between 2 and 96 h, washed, and stained for HLA-A2/HER2/neu369–377 complexes (C). Panel (A)shows one representative experiment of at least five; in (B) n = 5 and in (C) n = 3.
Cross-presentation of HER2/neuinvolves proteases of both the MHC class I and class II antigen-processing pathways, and de novo MHC class I biosynthesis
To assess the importance of endosomal processing, the cells were treated with inhibitors of acidification and endogenous proteases. Chloroquine, a reagent often used to block MHC class II antigen processing, inhibited cross-presentation of HER2/neu disregard of whether it was administered in soluble form or encapsulated in NPs (Fig. 3A). Ammonium chloride as a buffer and monensin as Na+/H+ antiporter are alternative inhibitors of endosomal acidification, and had the same effect on the cross-presentation (Data not shown). Impairment of cross-presentation by inhibition of acidification suggests endosomal processing of the antigen for cross-presentation. To identify the proteolytic enzymes involved in the antigen processing, we applied selective inhibitors for different endosomal proteases. The inhibition of serine, threonine and cysteine (e.g. cathepsin B) proteases by leupeptin abolished the cross-presentation of the soluble protein, but had no effect on the cross-presentation of the protein delivered within NPs (Fig. S6). Inhibition of cathepsin S with Z-FL-COCHO or Z-FA-FMK effectively inhibited cross-presentation of HER2/neu delivered in soluble or NP-bound form suggesting an essential role of this enzyme in the process (Figs. 3B–C). To test the possible involvement of the classical MHC class I antigen processing pathway we inhibited the proteasomes by treating the cells with lactacystin, which blocks the catalytic activity of the β subunits of the proteasome,28,29 or butabindide oxalate, an inhibitor of the tripeptidyl-peptidase II (TPP II), a cytosolic protease involved in processing some class I-restricted Ags.30 Lactacystin significantly reduced cross-presentation of HER2/neu delivered in all tested formulations (Fig. 3D). Butabindide oxalate, on the other hand, had only a slight effect (Fig. 3E). Moreover, pretreatment of iDCs with the inhibitor of protein biosynthesis cycloheximide abolished cross-presentation of HER2/neu delivered in the NPs but not in the soluble form (Fig 3F), which suggests that newly synthesized MHC class I molecules are required for cross-presentation of particulate Ags. Taken together, these results suggest that in addition to the MHC class II also the MHC class I antigen processing pathway is involved in antigen cross-presentation.
Figure 3.

Antigen processing pathways for cross-presentation of HER2/neu. iDCs were incubated for 24 h with HER2/neu in the soluble form or encapsulated in NPs. After incubation, cells were harvested and stained for the complexes HLA-A2/HER2/neu369–377 and analyzed by flow cytometry. To investigate the processing mechanisms of cross-presentation, cells were treated with inhibitors 30 min prior to adding the Ag. The inhibitors were: in (A) chloroquine at 50 μM, for acidification; (B) Z-FL-COCHO at 10 μM and 100 μM, for cathepsin S; (C) Z-FA-FMK at 10 μM and 100 μM for cathepsins B, H, L, and S; (D) lactacystin at 10 μM and 40 μM for proteasome; (E) butabindide oxalate at 200 μM for tripeptidyl peptidase II; (F) cycloheximide at 5 μg/mL for protein biosynthesis. In (A) n = 3; (B, C) n = 7; (D) n = 6; (E) n = 3; and (F) n = 4. Error bars = Standard error of the mean, n.s. non-significant, *P <0.05, **P <0.01 in student's t-test.
Translocation of antigen from endosomes into the cytosol does not involve Sec61
Cytosolic processing of exogenous antigen requires its translocation from the endosomes into the cytosol. Sec61 is involved in retrotransport of proteins from the ER into the cytosol and was suggested to be involved in antigen cross-presentation as well.7 To test the possible involvement of Sec61 in the cross-presentation of HER2/neu we treated iDCs with the Sec61 inhibitor Pseudomonas exotoxin A (Exo A) and observed blockade of the cross-presentation (Fig. S7). However, Exo A is also an inhibitor of protein biosynthesis,31 and we had shown before (Fig. 3F) that protein de novo biosynthesis is required for cross-presentation. To clarify this issue we followed previous studies, which suggested that Sec61 is recruited from the ER to endosomes/phagosomes by vesicular transport involving Sec22b,10-12 and looked for co-localization of Sec22b and Sec61 with the NPs used to deliver HER2/neu. However, using iDCs loaded with fluorescent NPs and harvested at different time points after addition of the particles, and stained with antibodies against Sec22b and Sec61, we could detect the NPs inside the cells as well as Sec22b- and Sec61-positive subcellular compartments but no co-localization of the particles with Sec22b and/or Sec61 (Figs. 4A and B). Likewise, TAP, another ER protein proposed to be transported together with Sec61 to endosomes/phagosomes was not co-localized with the NPs (Fig. 4C). These observations exclude a role of Sec61 in the translocation of the antigen or fragments thereof from the NP-bearing uptake endosomes/endolysosomes into the cytosol. They also make it unlikely that other components directly derived from the ER via vesicular transport are involved in antigen processing for cross-presentation. However, endosomal trafficking is critical for antigen processing in this context as primaquine, an inhibitor of endosomal transport that does not affect endocytosis or vesicular export from ER via Golgi32 abrogates cross-presentation. This is obvious from experiments where iDCs incubated with primaquine prior to addition of HER2/neu either in soluble form or encapsulated in NPs were stained for the specific MHC-peptide complexes at the surface of the DC, and analyzed by flow cytometry (Fig. 5).
Figure 4.
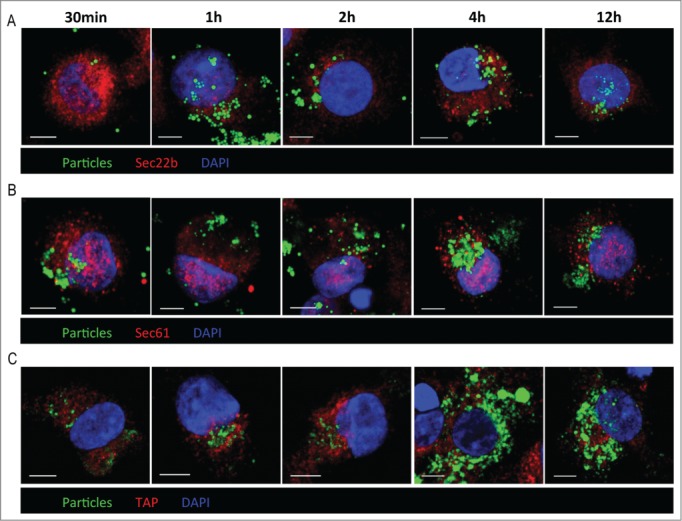
Sec22b, Sec61 or TAP in translocation of processed HER2/neu. iDCs were incubated with fluorescent PLGA NPs (green fluorescence), harvested at the indicated time points, and stained for Sec22b (A), Sec61 (B) or TAP (C) (red fluorescence), and counterstained with DAPI (blue fluorescence). The cells were examined by confocal microscopy. Scale bar = 5 μm.
Figure 5.
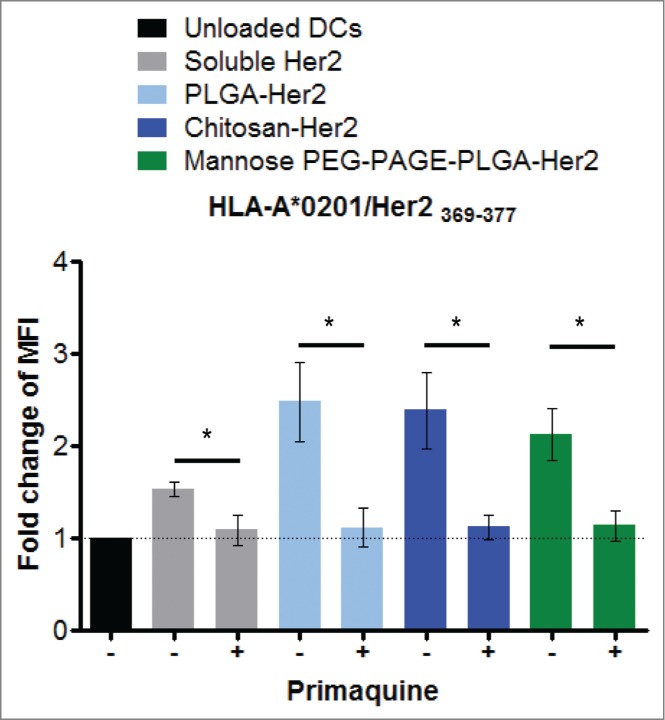
Endosomal transport in antigen cross-presentation. To investigate whether cross-presentation of HER2/neu involves endosomal transport, cells were treated with primaquine at 50 μM 30 min prior to adding the NPs. 24 h post loading, cells were harvested and surface-stained for HLA-A2/HER2/neu369–377 complexes, and analyzed by flow cytometry. n = 6; *P < 0.05 in student's t-test.
Antigen-loading compartment for cross-presentation
To identify the subcellular compartment where the MHC class I molecule is loaded with the processed antigen for cross-presentation we used the monoclonal antibody specific for the complex of the epitope HER2/neu369–377 with the human MHC class I molecule HLA-A*0201. This antibody stained well-defined intracellular vesicles of various sizes ranging up to about 1 µm in diameter (Fig. 6). The MHC-peptide complexes were detectable already after 4 h of incubation with the HER2/neu-loaded NPs, and roughly increased in numbers over the incubation period of 24 h. Using the specific MHC-peptide complex as a marker for the intracellular compartments where the MHC molecule are loaded with the processed antigen and intracellular compartments derived from this loading compartment, we investigated the different pathways that have been suggested to be implicated in antigen processing for direct or cross-presentation.33,34 The specific MHC-peptide complexes were not found in the canonical MHC class I processing pathway as we saw no co-localization with calnexin or TAP, two proteins of the ER, nor with TGN38 as marker for the trans-Golgi network (Fig. 6). Also the various implicated early endosomes (marker: EEA1), recycling endosomes (markers Rab 11 and CD71), late endosomes (markers: Rab 7, cathepsin S, Lysotracker) or storage endosomes (markers: Rab 14, IRAP) did not contain the specific MHC-peptide complexes, nor the MHC class II compartment (markers: HLA-DR, HLA-DM, the invariant chain CD74) or autophagosomes (marker: LC3). In contrast, for antigen processing of HER2/neu as an internal antigen (direct presentation) expressed by the breast cancer cell line MDA-MB-231 that also expresses HLA-A*0201 (Fig. S3), the specific HLA-A2/HER2/neu369–377 complexes were readily detected in the ER colocalized with calnexin (Fig. S8). Moreover, the presentation of the complexes at the surface of MDA-MB-231 cells was brefeldin A sensitive and primaquine insensitive (Fig. S9), which is compatible with its origin in the ER and the use of the classical secretory pathway for its expression at the cell surface. Altogether, these results exclude all known or proposed antigen-processing pathways for cross-presentation of HER2/neu by DCs. Thus, loading of MHC class I molecules with specific epitopes for cross-presentation of external Ags occurs in a novel subcellular compartment characterized by the presence of the specific MHC-peptide complexes.
Figure 6.
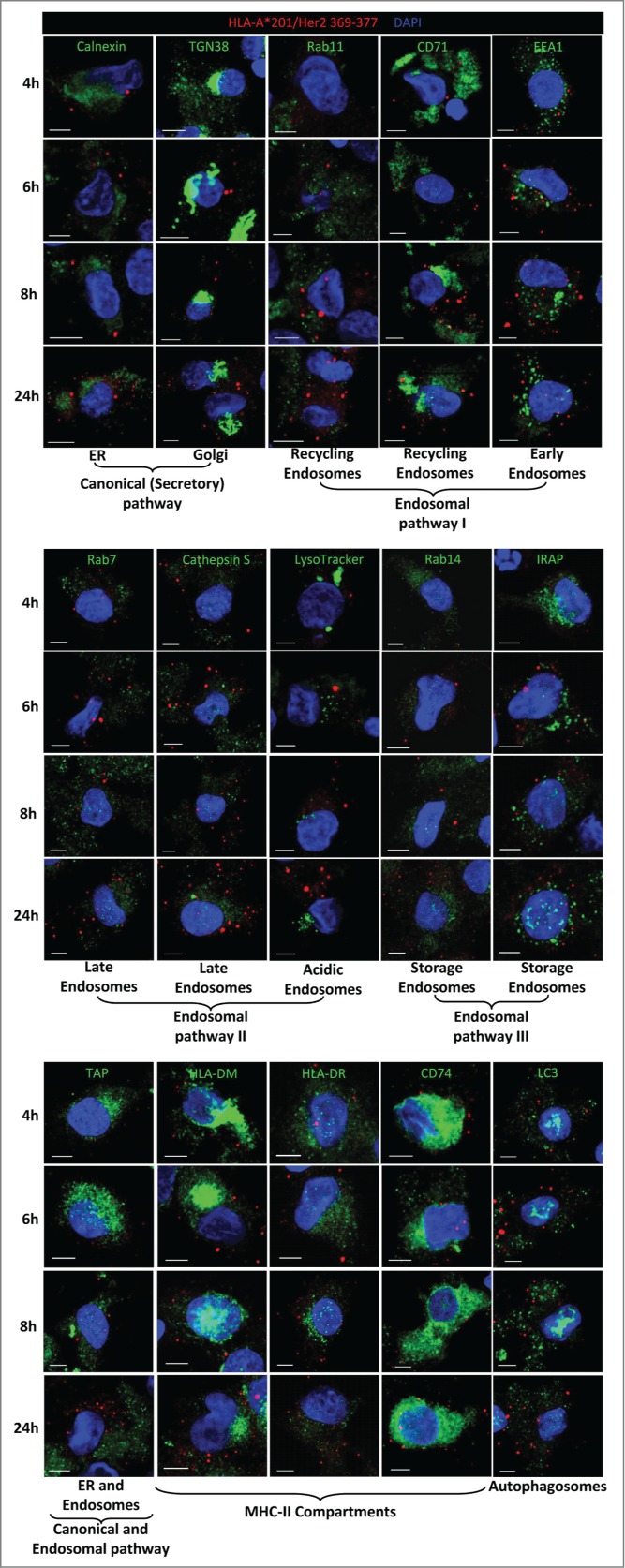
Cross-presenting compartment. To identify the loading compartment for cross-presentation, iDCs were incubated with PLGA-HER2/neu NPs, harvested at the indicated time points, and stained with the antibody for HLA-A2/HER2/neu369–377 (red fluorescence) and antibodies for specific subcellular compartments as indicated (green fluorescence). Cells were counterstained with DAPI (blue fluorescence) for nuclei visualization and examined by confocal microscopy. Scale bar = 5 μm.
Discussion
Based on the results of this study, we propose the following model for cross-presentation of external Ags by DCs. The Ag, here HER2/neu, is taken up by iDCs and routed into either strongly acidified (e.g., PLGA-based NPs) or non-acidified (e.g. Chitosan NPs) endosomes, where it is fragmented by cathepsins. The fragments are exported into the cytosol, further processed by proteasome, and the resulting epitopes are transported into endosomal vesicles distinct from the uptake compartments. In these vesicles they are loaded onto MHC class I molecules and transported to the cell surface for cross-presentation to cognate CD8+ T cells (Fig. S10). The source of the MHC class I molecule may vary as the particulate formulations of HER2/neu required de novo biosynthesized MHC class I molecules, whereas the soluble Ag did not. The latter is in agreement with studies with human8 and murine9 DCs that showed cross-presentation of soluble antigen by MHC class I molecules from the recycling endosomes and not from the ER.
Our model explains why endocytosed particulate HER2/neu and cross-presented HLA-A2/HER2/neu369–377 complexes were always in distinct endosomal compartments. Variations in the initial fate of the antigen, i.e. routing into strongly acidified endolysosomes or not, does not affect the efficiency of cross-presentation. Our model postulates the involvement of the proteolytic machineries of the canonical MHC class II and class I antigen processing pathways, antigen processing and loading of the epitope onto MHC class I molecules in separate compartments, and the requirement of de novo biosynthesized MHC class I molecules for cross-presentation of particulate Ags.
This model is in parts consistent with previous models for antigen cross-presentation but differs from these in important aspects. Processing of endocytosed Ags in endosomes/phagosomes by cathepsins for cross-presentation had been reported earlier.8,15 However, while earlier reports indicated the specific MHC-peptide complexes in the same processing compartment,8,9,11,15 we found the complexes in well-defined vesicles but never co-localized with the antigen-bearing NPs or with cathepsin S, the protease involved in the processing of HER2/neu. The sensitivity of antigen cross-presentation to proteasome inhibitors suggests that the antigen fragments are exported into the cytosol for proteasomal processing. The translocon was proposed to be Sec61, an ER-resident protein, responsible for the retrotranslocation of misfolded proteins from the ER into the cytosol.7,35 Treatment of DCs with Pseudomonas aeruginosa Exo A, an inhibitor of Sec61, indeed abolished cross-presentation of particulate HER2/neu369–377. However, Exo A also inhibits protein biosynthesis,31 and since we had shown that cross-presentation of particulate HER2/neu required newly synthesized MHC molecules, the effect of Exo A could also relate to its blocking protein biosynthesis. Recent reports suggested recruitment of Sec61 together with other ER-resident proteins such as TAP and components of the PLC to the endosomes/phagosomes via Sec22b.10-12 If that was the case in our experiments, Sec22b, Sec61, and TAP should be present on the membrane of endosomes containing NPs. However, staining of NP-loaded DCs for Sec22b, Sec61, and TAP revealed no co-localization of any of these proteins with the particles. Therefore, Sec61 can be excluded as the channel through which our Ag is transported into the cytosol. Alternatively, the other translocons such as ER-associated degradation machinery (ERAD)-related protein p97/VCP or Derlin-136 may be considered.
After proteasomal processing, peptides could follow the canonical MHC class I pathway and enter the ER via TAP to be loaded onto MHC class I molecules.36 That appears to be unlikely for HER2/neu, because we did not detect HLA-A2/HER2/neu369–377 complexes in the ER. For the OVA epitope OVA257–264 it has been shown that the peptides may be shuttled back into endosomes/phagosomes via endosomal TAP.9 In this study TAP was demonstrated in endosomes containing endocytosed OVA as well as H-2Kb/OVA257–264 complexes. Using a TAP inhibitor linked transferrin, which is delivered specifically to early/recycling endosomes, the authors saw complete inhibition of cross-presentation of OVA epitope. In contrast, in our study the HLA-A2/HER2/neu369–377 complexes and TAP were localized in distinct compartments, which rules out the contribution of TAP to peptide loading onto MHC class I molecules for HER2/neu. After proteasomal processing some peptides require further trimming by aminopeptidases such as the cytosolic tri-peptidil peptidase II (TPPII)30 and ER aminopeptidases (ERAPs).37,38 TPPII may have a slight impact on the presentation of HER2/neu369–377, as its inhibition caused a slight decrease in the HLA-A*201/HER2/neu369–377 complexes at the DCs surface. ERAPs however, appear to play no role in the processing of HER2/neu for cross-presentation, as we found no HLA-A2/HER2/neu369-377 complexes in the ER. Finally, an insulin-regulated aminopeptidase (IRAP), which is present on Rab14-positive endosomes, was shown to be recruited to phagosomes and to trim imported peptides for cross-presentation.39,40 However, in the case of HER2/neu the lack of co-localization of HLA-A2/HER2/neu369–377 complexes with IRAP or Rab14 ruled out these pathways. Also loading of MHC class I molecules with cross-presented epitopes in the MHC class II loading compartment proposed by some authors41-43 seems unlikely because HLA-A2/HER2/neu369–377 complexes were not found together with HLA-DR, the chaperon HLA-DM or the invariant chain CD74 in the same compartment. Likewise, loading of the MHC class I molecules with the HER2/neu epitope in early or recycling endosomes proposed by other models8,9 could be ruled out because of the lack of co-localization of the specific MHC peptide complexes with the respective marker proteins EEA1, CD71 or Rab11. While we could exclude the previously postulated compartments as sites for MHC loading with the HER2/neu epitope, the inhibition of its cross-presentation by primaquine still indicates transport of the complexes by endosomes.
Despite initial routing of different antigen-loaded NPs into different compartments, the processing pathways for HER2/neu were the same, and leading to efficient cross-presentation. This is in contrast to other studies that found correlations between antigen routing and the efficiency of cross-presentation. According to some reports, antigen for cross-presentation needed to be routed into non-acidified compartments and acidification prevented or decreased cross-presentation.44,45 We found efficient cross-presentation in both situations. Three groups reported that Ags delivered via the mannose receptor were routed into early endosomes resulting in more efficient cross-presentation.9,44,46 In contrast, we observed that mannose NPs were not routed into early but rather into acidic endosomes, which did not interfere with cross-presentation of the epitope HER2/neu369–377. Actually, our studies showed that some degree of acidification was crucial for the generation of the peptide HER2/neu369–377, which is compatible with antigen processing by cathepsins that are activated at low pH. The failure to demonstrate strong acidification of the uptake compartments for some NPs despite the sensitivity of cross-presentation of the antigen to inhibitors of acidification might be due to the low sensitivity of the pH indicator used in our study. Our findings are in agreement with reports showing that processing of apoptotic monocytes infected with vaccinia virus expressing influenza A virus matrix protein (MP) 1 by human DCs required endosomal acidification and processing by cathepsins15 or that the generation of certain epitopes from proteins delivered to DCs in soluble form or in nanocarriers required endosomal acidification.17 The authors of the latter report showed that inhibition of endosomal acidification via the vacuolar-type H[+]-ATPase protein pump inhibitor ConB abolished cross-presentation of HLA-A2/NY-ESO-1157–165 and HLA-A2/Melan-A26–35 epitopes by human DCs loaded with full-length proteins.17 Other authors have demonstrated that inhibition of endosomal acidification either improved or had no effect on the cross-presentation.47-49 These discrepancies indicate that endosomal acidification plays a critical role for some epitopes, but might be detrimental for others, which may relate to destruction of some epitopes by proteases active in the different test situations.
Altogether, our findings suggest that different Ags in different formulations may be processed via different pathways which, however, converge into the same loading and export path for cross-presentation. This provides a framework for the rational design of vaccines meant to induce immunity based on CD8+ effector T cells as is required for therapeutic vaccines for treatment of cancer or vaccines against a number of intracellular infectious, in particular when vector-borne.
Material and Methods
Preparation of NPs and protein loading
The recombinant extracellular domain (ECD) of the human HER2/neu protein (amino acids 23–652; Accession Number P04626) was custom-produced by Biomatik (Biomatik, Wilmington, DE USA) and encapsulated into Chitosan, PLGA, PEG-PAGE-PLGA, Mannose-PEG-PAGE-PLGA NPs (PLGA from Evonik Industries AG, Darmstadt, Germany; PEG-PAGE-PLGA and Mannose-PEG-PAGE-PLGA were synthesized and characterized at the FSU Jena, Jena, Germany) as described by Baleeiro et al., (submitted). Silica NPs were produced as described elsewhere.50 The recombinant protein and all NP preparations were tested for endotoxin using a commercial limulus assay (Pierce LAL Chromogenic Endotoxin Quantitation Kit, Thermo Fisher Scientific, Rockford, IL USA) and proven endotoxin-free.
DC generation
DCs were generated in vitro from adherent peripheral blood mononuclear cells (PBMCs) of healthy donors isolated from leukoreduction filters or buffy coats by Ficoll gradient centrifugation. Adherent cells were cultured in RPMI 1640 GlutaMax culture medium (Invitrogen, Carlsbad, CA USA) with 10% foetal calf serum (FCS; Biochrom AG, Germany), recombinant human GM-CSF (50 ng/mL, Genzyme, Cambridge, MA USA), and IL-4 (50 ng/mL, PromoCell GmbH, Germany) added on day 0 and 4. After 5 d culture at 37°C in humidized atmosphere with 8% CO2 fluorochrome-labeled empty NPs or HER2/neu-loaded NPs were added, and the culture continued. The cells were harvested at different time points for transmission electron microscopy (TEM) and laser scanning confocal microscopy (LSCM). Where inhibitors were used, day-5 DCs were incubated for 30 min at 37°C with the respective inhibitor before adding soluble or NP-loaded HER2/neu. The inhibitors were chloroquine at 50 µM, lactacystin at 10 µM and 40 µM, leupeptin at 100 µM, Pseudomonas Exo A at 2 and 10 µg/mL (all Sigma-Aldrich, Steinheim, Germany), Z-FL-COCHO at 10 µM and 100 µM, cycloheximide at 5 μg/mL, and primaquine at 50 µM (all Calbiochem, Darmstadt, Germany), butabindide oxalate at 200 µM (Tocris Bioscience, Bristol, UK), and Z-FA-FMK (R&D Systems, Minneapolis, MN USA) at 10 µM and 100 µM. Cells were harvested on day 6 for flow cytometry. The use of human cells for the reported study was reviewed and approved by the institutional ethics committee of the Charité – Universitätsmedizin Berlin (EA1/148/08).
Cell lines
Human breast adenocarcinoma cell lines BT20, MDA-MB-231, and SKBR-3, and the human melanoma ChaMel84 and human TAP-deficient T2 cell line were cultured in RPMI-1640 medium with 10% heat-inactivated FCS, 100 U/mL penicillin and 0.1 mg/mL streptomycin. Human colon adenocarcinoma Caco-2 cells were cultured in minimum essential media (MEM) with 10% FCS, 1mM pyruvic acid, 0.1 mM non-essential amino acids (NEAA), 100 U/mL penicillin and 0.1 mg/mL streptomycin.
Loading of T2 cells with peptides
T2 cells were washed twice with RPMI 1640 medium and incubated with the peptides HER2/neu85–94 (LIAHNQVRQV), HER2/neu369–377 (KIFGSLAFL), HER2/neu435–443 (ILHNGAYSL), Flu M158–66 (GILGFVFTL), and CMV pp65495–503 (NLVPMVATV) (all EMC microcollections GmbH, Tübingen, Germany) at the indicated concentrations for 4 h at 37°C. Then, the cells were washed with PBS to remove free peptides and kept at 4°C until analysis.
CD8+ T cells expansion assay
CD8+ T lymphocytes were isolated from PBMCs by magnetosorting through positive selection using a commercial kit (Invitrogen, Carlsbad, CA USA) following the manufacturer's instruction. Sorted CD8+ cells were washed twice in PBS and cultured together with autologous HER2/neu-loaded DCs in 96-well plates of 2 × 104 DCs with 2 × 105 T lymphocytes for 7 d. On day 3 of the co-culture, cells were supplemented with 50 U/mL IL-2 (Novartis, Nuernberg, Germany). Then, cells were harvested and analyzed by flow cytometry.
Flow Cytometry
To examine the presentation of HLA-A*0201/HER2/neu369–377 complexes by DCs, cell suspensions were labeled with the mouse IgG1 monoclonal antibody against HLA-A*0201/HER2/neu369–377 (clone RL1S; Pure Protein LLC, Oklahoma City, OK USA) and incubated for 25 min, followed by staining with FITC-labeled goat anti-mouse IgG1 (BD Bioscience, Chicago, IL USA) or Alexa Fluor 647-labeled rabbit anti-mouse IgG (Invitrogen, Carlsbad, CA USA) for another 30 min. The tumor cell lines BT20, MDA-MB-231, SKBR-3, ChaMel84, and peptide-pulsed TAP-deficient T2 cell line were stained with fluorochrome-labeled mouse IgG2b against HLA-A2 or mouse IgG1 against HER2/neu (all BioLegend, San Diego, CA USA) – or purified mouse IgG1 antibody against HLA-A*201/HER2/neu 369–377 complexes followed by staining with FITC-labeled goat anti-mouse IgG1 (BD Bioscience, Chicago, IL USA). For assessment of HER2/neu369–377-specific T lymphocytes, cells from the co-cultures (CD8+ T cells + DCs) were stained with fluorochrome-labeled antibody for CD8+ (BioLegend, San Diego, CA USA) and HLA-A*0201/HER2/neu 369–377 dextramers (Immudex, Copenhagen, Denmark). Expansion of the specific CD8+ T cells was determined by flow cytometry after incubation of these cells with DCs loaded with HER2/neu in different formulations. Stained cells were analyzed with a FACS Calibur flow cytometer (Becton Dickinson, San Jose, California), and data were processed using the CellQuest software (Becton Dickinson, San Jose, CA USA) or WinMDi (Purdue University, USA; www.purdue.edu).
Transmission Electron Microscopy (TEM)
DCs loaded with NPs were fixed with 2.5% glutaraldehyde in cacodylate buffer pH 7.5, followed by 100 mm cacodylate buffer with 2% osmium tetroxide at pH 7.5 and 4°C. The samples were washed thrice with buffer, dehydrated in increasing alcohol series and embedded in glycid ether 100 resin. Sections of about 70 nm (ultrathin sections) were prepared with a Leica Ultracut S and contrasted for observation with a Zeiss EM906 TEM.
Laser Scanning Confocal Microscopy (LSCM)
The kinetics of routing of NPs into early and late/acidic endosomes was examined by LSCM. DCs loaded with fluorochrome-labeled NPs were harvested at different time points and fixed for 20 min with 4% paraformaldehyde in PBS followed by permeabilization with 0.2% Triton X-100 in PBS for 10 min. Fixed and permeabilized cells were incubated for 45 min with 2% BSA in PBS to block non-specific binding, and stained for 1 h with a monoclonal mouse IgG1 antibody against human EEA1 (Santa Cruz Biotechnology, Santa Cruz, CA USA), mouse IgG1 against human DC-LAMP (Immunotech, Marseille, France), mouse IgG against human Sec22b, mouse IgG against human Sec61, or rabbit IgG against human TAP1 (All from Santa Cruz Biotechnology, Santa Cruz, CA USA) followed by 45 min with Alexa fluor 647 labeled rabbit anti-mouse or goat anti-rabbit secondary antibody (Invitrogen, Carlsbad, CA USA). To analyze the routing of NPs into acidic endosomes, unfixed NPs-loaded DCs were incubated for 30 min with 50–100 nm LysoTracker Red DND-99 (Invitrogen, Carlsbad, CA USA), then fixed with 4% paraformaldehyde for 20 min and stained with Alexa Fluor 647-labeled anti-HLA-A,B,C (BioLegend, San Diego, CA USA) for 25 min at 4°C. Cells were counterstained with 4′,6-Diamidino-2-Phenylindole, Dihydrochloride (DAPI; Roche Diagnostics GmbH, Mannheim, Germany) for nuclei visualization. To examine intracellular HLA-A*0201/HER2/neu369–377 complexes and their co-localization with intracellular marker, DCs loaded with HER2/neu bearing NPs were harvested, fixed and permeabilized as described above. Then, cells were stained with mouse IgG1 against HLA-A*201/HER2/neu369–377 complexes and rabbit IgG against calnexin, cathepsin S, CD71, CD74, EEA1, IRAP, LC3, Rab7, Rab11, Rab14, TAP1, or TGN38 (all Santa Cruz Biotechnology, Santa Cruz, CA USA), and HLA-DM and HLA-DR (all AbCam, Cambridge, UK). Alexa Fluor 488-labeled goat IgG anti-rabbit IgG and Alexa Fluor 594-labeled goat IgG anti-mouse IgG were used as secondary antibodies. Cells were then mounted on slides in Fluoromount-G (SouthernBiotech) and covered with coverslips. The intracellular localization and routing of the NPs was analyzed by LSCM (Leica TCS SP2). The images were processed with the Leica Confocal Software Version 2.5 Build 1227.
Statistical analysis
The statistical analyses were done with the Graphpad Software Prism 2.01 for Windows (GraphPad Software, La Jolla CA). Kolmogorov–Smirnov tests were used to check the data for normal distribution. Results from the independent experimental groups were compared by two-tailed student´s t-test. Differences were considered significant when P ≤ 0.05.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Armin Kübelbeck and Gregor Larbig (Merck KGaA) for the production of silica NP.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Funding
This study was supported by a grant from the Bundesministerium für Bildung und Forschung (BMBF, German Ministry for Education and Research): PeTra Project, Project Number 13N11455.
References
- 1.Bevan MJ. Minor H antigens introduced on H-2 different stimulating cells cross-react at the cytotoxic T cell level during in vivo priming. J Immunol 1976; 117:2233-2238; PMID:825578 [PubMed] [Google Scholar]
- 2.Berard F, Blanco P, Davoust J. Neidhart-Berard EM, Nouri-Shirazi M, Taquet N, Rimoldi D, Cerottini JC, Banchereau J, Palucka AK. Cross-priming of naive CD8+ T cells against melanoma antigens using dendritic cells loaded with killed allogeneic melanoma cells. J Exp Med 2000; 192:1535-1544; PMID:11104796; http://dx.doi.org/ 10.1084/jem.192.11.1535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Paglia P, Chiodoni C, Rodolfo M, Colombo MP. Murine dendritic cells loaded in vitro with soluble protein prime cytotoxic T lymphocytes against tumor antigen in vivo. J Exp Med 1996; 183:317-322; PMID:8551239; http://dx.doi.org/ 10.1084/jem.183.1.317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nussenzweig MC, Steinman RM, Gutchinov B, Cohn ZA. Dendritic cells are accessory cells for the development of anti-trinitrophenyl cytotoxic T lymphocytes. J Exp Med 1980; 152:1070-1084; PMID:6968335; http://dx.doi.org/ 10.1084/jem.152.4.1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Albert ML, Sauter B, Bhardwaj N. Dendritic cells acquire antigen from apoptotic cells and induce class I-restricted CTLs. Nature 1998; 392:86-89; PMID:9510252; http://dx.doi.org/ 10.1038/32183 [DOI] [PubMed] [Google Scholar]
- 6.Kovacsovics-Bankowski M, Rock KL. A phagosome-to-cytosol pathway for exogenous antigens presented on MHC class I molecules. Science 1995; 267:243-6; PMID:7809629; http://dx.doi.org/ 10.1126/science.7809629 [DOI] [PubMed] [Google Scholar]
- 7.Koopmann JO, Albring J, Hüter E, Bulbuc N, Spee P, Neefjes J, Hämmerling GJ, Momburg F. Export of antigenic peptides from the endoplasmic reticulum intersects with retrograde protein translocation through the Sec61p channel. Immunity 2000; 1:117-27; PMID:10933400; http://dx.doi.org/18376401 10.1016/S1074-7613(00)00013-3 [DOI] [PubMed] [Google Scholar]
- 8.Di Pucchio T, Chatterjee B, Smed-Sörensen A, Clayton S, Palazzo A, Montes M, Xue Y, Mellman I, Banchereau J, Connolly JE. Direct proteasome-independent cross-presentation of viral antigen by plasmacytoid dendritic cells on major histocompatibility complex class I. Nat Immunol 2008; 9:551-7; PMID:18376401; http://dx.doi.org/ 10.1038/ni.1602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burgdorf S, Schölz C, Kautz A, Tampé R, Kurts C. Spatial and mechanistic separation of cross-presentation and endogenous antigen presentation. Nat Immunol 2008; 9:558-66; PMID:18376402; http://dx.doi.org/ 10.1038/ni.1601 [DOI] [PubMed] [Google Scholar]
- 10.Guermonprez P, Saveanu L, Kleijmeer M, Davoust J, Van Endert P, Amigorena S. ER-phagosome fusion defines an MHC class I cross-presentation compartment in dendritic cells. Nature 2003; 425:397-402; PMID:14508489; http://dx.doi.org/ 10.1038/nature01911 [DOI] [PubMed] [Google Scholar]
- 11.Houde M, Bertholet S, Gagnon E, Brunet S, Goyette G, Laplante A, Princiotta MF, Thibault P, Sacks D, Desjardins M. Phagosomes are competent organelles for antigen cross-presentation. Nature 2003; 425:402-6; PMID:14508490; http://dx.doi.org/ 10.1038/nature01912 [DOI] [PubMed] [Google Scholar]
- 12.Cebrian I, Visentin G, Blanchard N, Jouve M, Bobard A, Moita C, Enninga J, Moita LF, Amigorena S, Savina A. Sec22b regulates phagosomal maturation and antigen crosspresentation by dendritic cells. Cell 2011; 147:1355-68; PMID:22153078; http://dx.doi.org/ 10.1016/j.cell.2011.11.021 [DOI] [PubMed] [Google Scholar]
- 13.Savina A, Jancic C, Hugues S, Guermonprez P, Vargas P, Moura IC, Lennon-Duménil AM, Seabra MC, Raposo G, Amigorena S. NOX2 controls phagosomal pH to regulate antigen processing during crosspresentation by dendritic cells. Cell 2006; PMID:16839887 17351642126:205-18. 14; http://dx.doi.org/ 10.1016/j.cell.2006.05.035 [DOI] [PubMed] [Google Scholar]
- 14.Jancic C, Savina A, Wasmeier C, Tolmachova T, El-Benna J, Dang PM, Pascolo S, Gougerot-Pocidalo MA, Raposo G, Seabra MC. Rab27a regulates phagosomal pH and NADPH oxidase recruitment to dendritic cell phagosomes. Nat Cell Biol 2007; 9:367-78; PMID:17351642; http://dx.doi.org/ 10.1038/ncb1552 [DOI] [PubMed] [Google Scholar]
- 15.Fonteneau JF, Kavanagh DG, Lirvall M, Sanders C, Cover TL, Bhardwaj N, Larsson M. Characterization of the MHC class I cross-presentation pathway for cell-associated antigens by human dendritic cells. Blood 2003; 102:4448-55; PMID:12933572; http://dx.doi.org/ 10.1182/blood-2003-06-1801 [DOI] [PubMed] [Google Scholar]
- 16.Mantegazza AR, Savina A, Vermeulen M, Pérez L, Geffner J, Hermine O, Rosenzweig SD, Faure F, Amigorena S. NADPH oxidase controls phagosomal pH and antigen cross-presentation in human dendritic cells. Blood 2008; 112:4712-22; PMID:18682599; http://dx.doi.org/ 10.1182/blood-2008-01-134791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Robson NC, McAlpine T, Knights AJ, Schnurr M, Shin A, Chen W, Maraskovsky E, Cebon J. Processing and cross-presentation of individual HLA-A, -B, or -C epitopes from NY-ESO-1 or an HLA-A epitope for Melan-A differ according to the mode of antigen delivery. Blood 2010; 116:218-25; PMID:20430956; http://dx.doi.org/ 10.1182/blood-2009-10-249458 [DOI] [PubMed] [Google Scholar]
- 18.Baxevanis CN, Sotiropoulou PA, Sotiriadou NN, Papamichail M. Immunobiology of HER-2/neu oncoprotein and its potential application in cancer immunotherapy. Cancer Immunol Immunother 2004; 53:166-75; PMID:14685781; http://dx.doi.org/ 10.1007/s00262-003-0475-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yip YL, Ward RL. Anti-ErbB-2 monoclonal antibodies and ErbB-2-directed vaccines. Cancer Immunol Immunother 2002; 50:569-587; PMID:11807621; http://dx.doi.org/ 10.1007/s002620100226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kiessling R, Wie WZ, Herrmann F, Lindencrona JA, Choudhury A, Kono K, Seliger B. Cellular immunity to the Her-2/neu protooncogene. Adv Cancer Res 2002; 85:101-44; PMID:12374283; http://dx.doi.org/ 10.1016/S0065-230X(02)85004-7 [DOI] [PubMed] [Google Scholar]
- 21.Baxevanis CN, Sotiriadou NN, Gritzapis AD. Immunogenic HER-2/neu peptides as tumor vaccines. Cancer Immunol Immunother 2006; 55:85-95; PMID:15948002; http://dx.doi.org/ 10.1007/s00262-005-0692-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sotiropoulou PA, Perez SA, Voelter V, Echner H, Missitzis I, Tsavaris NB, Papamichail M, Baxevanis CN. Natural CD8+ T-cell responses against MHC class I epitopes of the HER-2/neu oncoprotein in patients with epithelial tumors. Cancer Immunol Immunother 2003; 52:771-9; PMID:13680193; http://dx.doi.org/ 10.1007/s00262-003-0420-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fisk B, Blevins TL, Wharton JT, Ioannides CG. Identification of an immunodominant peptide of HER-2/neu protooncogene recognized by ovarian tumor-specific cytotoxic T lymphocyte lines. J Exp Med 1995; 181:2109-17; PMID:7539040; http://dx.doi.org/ 10.1084/jem.181.6.2109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang H, Hong H, Li D, Ma S, Di Y, Stoten A, Haig N, Di Gleria K, Yu Z, Xu XN, McMichael A, Jiang S. Comparing pooled peptides with intact protein for accessing cross-presentation pathways for protective CD8+ and CD4+ T cells. J Biol Chem 2009; 284:9184-91; PMID:19193636; http://dx.doi.org/ 10.1074/jbc.M809456200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rosalia RA, Silva AL, Camps M, Allam A, Jiskoot W, van der Burg SH, Ossendorp F, Oostendorp J. Efficient ex vivo induction of T cells with potent anti-tumor activity by protein antigen encapsulated in nanoparticles. Cancer Immunol Immunother 2013; 62:1161-73; PMID:23613147; http://dx.doi.org/ 10.1007/s00262-013-1411-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vieira OV, Botelho RJ, Grinstein S. Phagosome maturation: aging gracefully. Biochem J 2002; 366:689-704; PMID:12061891; http://dx.doi.org/ 10.1042/BJ20020691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de Saint-Vis B, Vincent J, Vandenabeele S, Vanbervliet B, Pin JJ, Aït-Yahia S, Patel S, Mattei MG, Banchereau J, Zurawski S et al.. A novel lysosome-associated membrane glycoprotein, DC-LAMP, induced upon DC maturation, is transiently expressed in MHC class II compartment. Immunity 1998; 9:325-36; PMID:9768752; http://dx.doi.org/ 10.1016/S1074-7613(00)80615-9 [DOI] [PubMed] [Google Scholar]
- 28.Craiu A, Gaczynska M, Akopian T, Gramm CF, Fenteany G, Goldberg AL, Rock KL. Lactacystin and clasto-lactacystin beta-lactone modify multiple proteasome beta-subunits and inhibit intracellular protein degradation and major histocompatibility complex class I antigen presentation. J Biol Chem 1997; 272:13437-45; PMID:9148969; http://dx.doi.org/ 10.1074/jbc.272.20.13437 [DOI] [PubMed] [Google Scholar]
- 29.Lee DH, Goldberg AL. Proteasome inhibitors: valuable new tools for cell biologists. Trends Cell Biol 1998; 8:397-403; PMID:9789328; http://dx.doi.org/ 10.1016/S0962-8924(98)01346-4 [DOI] [PubMed] [Google Scholar]
- 30.Reits E, Neijssen J, Herberts C, Benckhuijsen W, Janssen L, Drijfhout JW, Neefjes J. A major role for TPPII in trimming proteasomal degradation products for MHC class I antigen presentation. Immunity 2004; 20:495-506; PMID:15084277; http://dx.doi.org/ 10.1016/S1074-7613(04)00074-3 [DOI] [PubMed] [Google Scholar]
- 31.Beattie BK, Prentice GA, Merrill AR. Investigation into the catalytic role for the tryptophan residues within domain III ofPseudomonas aeruginosa exotoxin A. Biochemistry 1996; 35:15134-42; PMID:8952460; http://dx.doi.org/ 10.1021/bi961985t [DOI] [PubMed] [Google Scholar]
- 32.van Weert AW, Geuze HJ, Groothuis B, Stoorvogel W. Primaquine interferes with membrane recycling from endosomes to the plasma membrane through a direct interaction with endosomes which does not involve neutralisation of endosomal pH nor osmotic swelling of endosomes. Eur J Cell Biol 2000; 79:394-9; PMID:10928454; http://dx.doi.org/ 10.1078/0171-9335-00062 [DOI] [PubMed] [Google Scholar]
- 33.Li H, Li Y, Jiao J, Hu HM. Alpha-alumina nanoparticles induce efficient autophagy-dependent cross-presentation and potent antitumour response. Nat Nanotechnol 2011; 6:645-50; PMID:21926980; http://dx.doi.org/ 10.1038/nnano.2011.153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nair-Gupta P, Blander JM. An updated view of the intracellular mechanisms regulating cross-presentation. Front Immunol. 2013; 4:401; PMID:24319447; http://dx.doi.org/ 10.3389/fimmu.2013.00401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Imai J, Hasegawa H, Maruya M, Koyasu S, Yahara I. Exogenous antigens are processed through the endoplasmic reticulum-associated degradation (ERAD) in cross-presentation by dendritic cells. Int Immunol 2005; 17:45-53; PMID:15546887; http://dx.doi.org/ 10.1093/intimm/dxh184 [DOI] [PubMed] [Google Scholar]
- 36.Needham PG, Brodsky JL. How early studies on secreted and membrane protein quality control gave rise to the ER associated degradation (ERAD) pathway: the early history of ERAD. Biochim Biophys Acta 2013; 1833:2447-57; PMID:23557783; http://dx.doi.org/17277129 10.1016/j.bbamcr.2013.03.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ménager J, Ebstein F, Oger R, Hulin P, Nedellec S, Duverger E, Lehmann A, Kloetzel PM, Jotereau F, Guilloux Y. Cross-presentation of synthetic long peptides by human dendritic cells: a process dependent on ERAD component p97/VCP but Not sec61 and/or Derlin-1. PLoS One 2014; 9:e89897; PMID:24587108 http://dx.doi.org/17277129 10.1371/journal.pone.0089897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Joffre OP, Segura E, Savina A, Amigorena S. Cross-presentation by dendritic cells. Nat. Rev Immunol 2012; 12:557-69; PMID:22790179; http://dx.doi.org/17277129 10.1038/nri3254 [DOI] [PubMed] [Google Scholar]
- 39.Firat E, Saveanu L, Aichele P, Staeheli P, Huai J, Gaedicke S, Nil A, Besin G, Kanzler B, van Endert P et al.. The role of endoplasmic reticulum-associated aminopeptidase 1 in immunity to infection and in cross-presentation. J Immunol 2007; 178:2241-8; PMID:17277129; http://dx.doi.org/ 10.4049/jimmunol.178.4.2241 [DOI] [PubMed] [Google Scholar]
- 40.Saveanu L, Carroll O, Weimershaus M, Guermonprez P, Firat E, Lindo V, Greer F, Davoust J, Kratzer R, Keller SR et al.. IRAP identifies an endosomal compartment required for MHC class I cross-presentation. Science 2009; 325:213-7; PMID:19498108; http://dx.doi.org/22238454 10.1126/science.1172845 [DOI] [PubMed] [Google Scholar]
- 41.Weimershaus M, Maschalidi S, Sepulveda F, Manoury B, van Endert P, Saveanu L. Conventional dendritic cells require IRAP-Rab14 endosomes for efficient cross-presentation. J Immunol 2012; 188:1840-6; PMID:22238454; http://dx.doi.org/ 10.4049/jimmunol.1101504 [DOI] [PubMed] [Google Scholar]
- 42.Grommé M, Uytdehaag FG, Janssen H, Calafat J, van Binnendijk RS, Kenter MJ, Tulp A, Verwoerd D, Neefjes J. Recycling MHC class I molecules and endosomal peptide loading. Proc Natl Acad Sci USA 1999; 96:10326-31; PMID:10468607; http://dx.doi.org/11274420 10.1073/pnas.96.18.10326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.MacAry PA, Lindsay M, Scott MA, Craig JI, Luzio JP, Lehner PJ. Mobilization of MHC class I molecules from late endosomes to the cell surface following activation of CD34-derived human Langerhans cells. Proc Natl Acad Sci USA 2001; 98:3982-7; PMID:11274420; http://dx.doi.org/ 10.1073/pnas.071477498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Basha G, Omilusik K, Chavez-Steenbock A, Reinicke AT, Lack N, Choi KB, Jefferies WA. A CD74-dependent MHC class I endolysosomal cross-presentation pathway. Nat Immunol 2012; 13:237-45; PMID:22306692; http://dx.doi.org/ 10.1038/ni.2225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chatterjee B, Smed-Sörensen A, Cohn L, Chalouni C, Vandlen R, Lee BC, Widger J, Keler T, Delamarre L, Mellman I. Internalization and endosomal degradation of receptor-bound antigens regulate the efficiency of cross presentation by human dendritic cells. Blood 2012; 120:2011-20; PMID:22791285; http://dx.doi.org/ 10.1182/blood-2012-01-402370 [DOI] [PubMed] [Google Scholar]
- 46.Cohn L, Chatterjee B, Esselborn F, Smed-Sörensen A, Nakamura N, Chalouni C, Lee BC, Vandlen R, Keler T, Lauer P et al.. Antigen delivery to early endosomes eliminates the superiority of human blood BDCA3+ dendritic cells at cross presentation. J Exp Med 2013; 210:1049-63; PMID:23569326; http://dx.doi.org/ 10.1084/jem.20121251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tsuji T, Matsuzaki J, Kelly MP, Ramakrishna V, Vitale L, He LZ, Keler T, Odunsi K, Old LJ, Ritter G et al.. Antibody-targeted NY-ESO-1 to mannose receptor or DEC-205 in vitro elicits dual human CD8+ and CD4+ T cell responses with broad antigen specificity. J Immunol 2011; 186:1218-27; PMID:21149605; http://dx.doi.org/ 10.4049/jimmunol.1000808 [DOI] [PubMed] [Google Scholar]
- 48.Palliser D, Guillen E, Ju M, Eisen HN. Multiple intracellular routes in the cross-presentation of a soluble protein by murine dendritic cells. J Immunol 2005; 174:1879-87; PMID:15699114; http://dx.doi.org/ 10.4049/jimmunol.174.4.1879 [DOI] [PubMed] [Google Scholar]
- 49.Accapezzato D, Visco V, Francavilla V, Molette C, Donato T, Paroli M, Mondelli MU, Doria M, Torrisi MR, Barnaba V. Chloroquine enhances human CD8+ T cell responses against soluble antigens in vivo. J Exp Med 2005; 202:817-28; PMID:16157687; http://dx.doi.org/ 10.1084/jem.20051106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Garulli B, Stillitano MG, Barnaba V, Castrucci MR. Primary CD8+ T-cell response to soluble ovalbumin is improved by chloroquine treatment in vivo. Clin Vaccine Immunol 2008; 10:1497-504; PMID:18753338; http://dx.doi.org/ 10.1128/CVI.00166-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hermanson GT. Bioconjugates Techniques. 2nd Edition, Elsevier, 2008, 978-0-12-370-501-3, 620; ISBN-13: 978-0123705013 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.