

Abstract
Objective
Inhibition of matrix metalloproteinase-8 (MMP8) improves survival following cecal ligation and puncture (CLP) in mice, making it a potential therapeutic target. In the current study we expand our understanding of the role of MMP8 in sepsis by employing an adoptive transfer approach and alternative sepsis models.
Methods
We used three different sepsis models: CLP, cecal slurry, and intestinal implantation. In our first model, adoptive transfer experiments were followed by CLP to test the hypothesis that MMP8 containing myeloid cells are a critical factor in sepsis following CLP. Our second model, cecal slurry, used intraperitoneal injections of cecal contents to induce polymicrobial peritonitis without tissue compromise in the recipient. Our third model, intestinal implantation, involved ligating and puncturing a cecum from a donor, and then removing the cecum and placing it into the recipient’s peritoneal cavity. Clinically, blood samples were drawn from pediatric patients within 24 hours of meeting criteria for septic shock.
Measurements and Main Results
In our adoptive transfer experiments, MMP8 null mice receiving WT marrow had a survival advantage compared to WT mice receiving MMP8 null marrow, suggesting that MMP8 containing myeloid cells are not a critical factor in sepsis following CLP. In our cecal slurry model, no survival advantage was seen among MMP8 null mice. Our third model, intestinal implantation, found that mice receiving MMP8 null intestine had a survival advantage compared to mice receiving WT intestine, regardless of recipient genotype. Clinically, median MMP8 serum concentrations were higher in patients with sepsis and primary intestinal pathology compared to septic patients without primary intestinal pathology.
Conclusion
Intestine-derived MMP8 is a critical component of septic peritonitis secondary to intestinal compromise.
Keywords: sepsis, MMP-8, peritonitis, intestinal injury
INTRODUCTION
Despite advances in modern medicine such as antibiotics and vaccines, the number of cases of severe sepsis in the United States continues to rise [1]. This continued increase underscores the need for novel therapeutic targets and an improved understanding of the complex processes involved in sepsis. We previously demonstrated that increased matrix metalloproteinase-8 (MMP8) mRNA expression and activity correlate with decreased survival and increased organ failure in pediatric patients with septic shock [2]. We corroborated these observations in a murine model by showing that genetic ablation or pharmacologic inhibition of MMP8 improves survival in sepsis induced by cecal ligation and puncture (CLP), thereby propagating interest in MMP8 as a therapeutic target for sepsis [2, 3].
MMP8 was originally known as a neutrophil product involved in collagen degradation (neutrophil collagenase), but has since been found to also play a role in neutrophil cell migration and chemotaxis, chemokine modulation, and cleavage of non-matrix proteins [4–6]. MMP8 is found in numerous cell types including activated macrophages, smooth muscle cells, and endothelial cells [5].
In follow up to our previous studies, we investigated the source of MMP8, first by conducting adoptive transfer experiments to test the hypothesis that neutrophils are the key source of MMP8 in sepsis. Surprisingly, we found that following CLP, MMP8 null mice that received wild-type (WT) marrow had a survival advantage compared to WT mice that received MMP8 null marrow. These findings indicate that MMP8 containing myeloid cells are not a critical factor in sepsis following CLP and we conducted additional experiments to test the hypothesis that the intestine is an important source of MMP8 in sepsis following CLP.
METHODS
Murine models
All aspects of this study complied with the Guide for the Care and Use of Laboratory Animals published by the U.S. National Institutes of Health (NIH Publication No. 85-23 revised 1996) and met approval of the Institutional Animal Care and Use Committee. MMP8 null mice on a C57BL/6 background were provided by Dr. Steven Shapiro, University of Pittsburgh. Wild type (WT) C57BL/6 mice were obtained from Charles Rivers Laboratories (Wilmington, MA). All mice were fed standard rodent chow and maintained on 12 hour light-dark cycles.
Adoptive transfer
To generate chimeric mice via adoptive transfer, standard protocols from the Cincinnati Children’s Research Foundation Comprehensive Mouse and Cancer Core (CMCC) facility were used for transfer of either WT (C57BL/6) bone marrow or MMP8 null bone marrow into irradiated WT or MMP8 null mice. Briefly, the protocol consisted of lethal irradiation of the recipient mice (split dose, 7 and 4.75 Gy, 4 hours apart), harvesting of bone marrow cells via flushing of femurs and tibias from donor mice, and transfer of unfractionated bone marrow (107 cells per transfer) to recipient mice via tail vein injections.
Four weeks after recovery from the adoptive transfer procedure, these mice underwent CLP as previously described [7]. Briefly, mice were anesthetized, a midline laparotomy was performed, and the cecum was identified. A 6-0 silk suture was used to ligate the cecum to 30% of its original size and two punctures were made through-and-through using a 21-gauge needle. A small amount of fecal content was expressed from each puncture site and the cecum was returned to the abdominal cavity, which was closed in an interrupted fashion. The skin was closed using Gluture Tissue Adhesive (Abbott Laboratories) and mice received a 1 mL subcutaneous injection of normal saline for resuscitation. Animals were monitored for survival for up to 7 days.
Cecal slurry
We adapted the cecal slurry method from Wynn et al. and prepared a fresh slurry for each experiment using three C57BL/6 female donors, aged 6–9 weeks [8]. Following euthanasia with CO2, a midline laparotomy was made and the cecum excised from each donor. The distal portion of the appendix was removed and cecal contents were expressed from the most distal point and collected in a 50 ml sterile conical tube. Five percent dextrose (D5) was added to bring the stool concentration to 80 mg/ml. The solution was then vortexed and sonicated until all stool was in a homogeneous solution. The solution was then passed through a sterile 100 μm filter to capture any remaining large particles and ensure the solution passed through a 27 gauge needle.
Mice received an intraperitoneal injection of the cecal slurry at a dose of 0.8 mg/g via a 27 gauge needle. Control mice were injected with an equivalent volume of 5% dextrose. Following the cecal slurry injection, animals were monitored for survival (up to 7 days) or were sacrificed at 18 hours for procurement of biological specimens.
Intraperitoneal implantation of donor intestine
For each donor/recipient pair, the donor mouse was first anesthetized and a midline laparotomy made to adequately expose the cecum. Either cecum or distal small bowel was ligated with 6-0 silk suture proximally and distally and excised from the donor mouse. A lethal intracardiac injection of pentobarbital was then given to euthanize the donor. Two holes were then made with a 21 gauge needle in a through-and-through fashion in the excised intestine. The recipient mouse was then anesthetized and a midline laparotomy was made to allow for the donor intestine to be placed into the peritoneal cavity. The abdomen was then closed in an interrupted fashion and the skin closed with Gluture Tissue Adhesive. A 1 mL subcutaneous injection of saline was used for resuscitation. Animals were either followed for survival (up to 7 days) or sacrificed at 18 hours for procurement of biological specimens.
Ex vivo cecal decay
Donor mice were anesthetized and a midline laparotomy made to adequately expose the cecum. The cecum was ligated with 6-0 silk suture proximally and distally and removed from the donor mouse. A lethal intracardiac injection of pentobarbital was then given for euthanasia. Two holes were then made with a 21 gauge needle in a through-and-through fashion in the removed cecum. The cecums were placed into small tissue culture dishes with 3 mL of sterile PBS. The cecums were then placed in a 37°C incubator and removed at 0, 6 or 24 hours for processing.
Once removed from the PBS, excess stool was expressed from the cecums and discarded. Tissue was then processed using RIPA buffer and a rotator type tissue homogenizer. Protein levels were determined using Thermo Fisher Scientific Pierce Micro BCA Protein Assay Kit. MMP-8 levels were measured using a Luminex multiplex system (Luminex Corporation, Austin, TX) according to manufacturer instructions. Levels were standardized to the amount of protein within the peritoneal lavage samples.
Cytokines
Blood samples were also collected at 18 hours in serum separator tubes. Serum was analyzed for interleukins 1β, 6, 10 (IL-1 β, IL-6, IL-10), macrophage inflammatory protein-1α (MIP-1α), lipopolysaccharide induced CXC chemokine (LIX), tumor necrosis factor α (TNFα), and keratinocyte-derived chemokine (KC) using a Luminex multiplex system.
Patients and clinical samples
The clinical study protocol was approved by the Institutional Review Board (IRB) of each participating institution. The patient cohort (n=68) was derived from an ongoing multicenter pediatric septic shock database, which has previously been described in detail [9–11]. Briefly, children admitted to the pediatric intensive care unit (PICU) meeting pediatric-specific criteria for septic shock were eligible for enrollment [12]. After informed consent from parents or legal guardians was obtained, blood samples were obtained as close to the time of meeting criteria for septic shock as possible (<24 hours). Clinical data was collected through the first seven days of PICU admission and used to classify the patients as having “primary intestinal pathology” (e.g. ruptured appendicitis, volvulus, bowel perforation, bowel obstruction, or intussusception) as the etiology for their sepsis. These subjects were matched 2:1 to other children with septic shock and no evidence of primary intestinal pathology. Matching was based on age and PERSEVERE-based mortality risk [13]. Plasma MMP8 concentrations were measured from samples representing day 1 of septic shock using a Luminex multiplex system as according to instructions from the manufacturer [2].
Data Analysis
Statistical analyses were conducted using SigmaStat Software (Systat Software, Inc., San Jose, CA). For survival analysis, a Kaplan-Meier log rank survival analysis was used. Cytokine levels are described using means and error bars representing standard deviations (SD) of n observations, where n represents the number of subjects in each group. Comparisons between the study groups used ANOVA on ranks with Dunn’s test for pairwise comparisons. MMP expression levels are described relative to the levels at t=0 hours. Comparisons between groups at one time point were made using ANOVA on ranks. P values less than 0.05 were considered significant.
RESULTS
MMP8 derived from myeloid cells does not play a role in CLP induced sepsis
In our first of three animal models, we sought to determine if the adoptive transfer procedure intrinsically affects the MMP8 null phenotype after CLP. WT and MMP8 null mice were transplanted with marrow from the same genotype and were monitored for survival following CLP. We observed a similar trend as that of our previous report in that MMP8 null mice transplanted with MMP8 null marrow have a survival advantage over WT mice transplanted with WT marrow following CLP (Figure 1A).
Figure 1. Adoptive transfer experiments followed by CLP indicate non-myeloid cells as the critical source of MMP8.
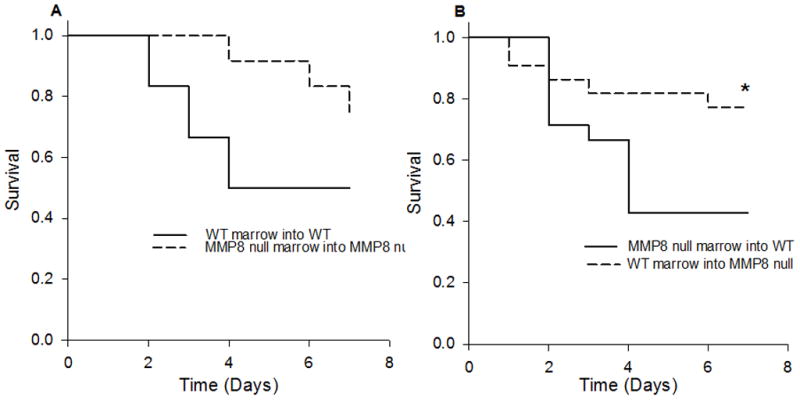
(Figure 1A) WT (n=12) and MMP8 null (n=11) mice were transplanted with bone marrow from the same genotype. Following CLP, MMP8 null mice transplanted with MMP8 null marrow showed a trend toward increased survival, consistent with our previous experiments. (Figure 1B) Following CLP, MMP8 null mice transplanted with WT marrow (n=22) had a survival advantage over WT mice transplanted with MMP8 null marrow (n=21, *p< 0.05).
WT marrow was then transplanted into MMP8 null mice and MMP8 null marrow was transplanted into WT mice. MMP8 null mice that received WT marrow had a survival advantage compared to WT mice that received MMP8 null marrow (Figure 1B). These findings suggest that MMP8 containing myeloid cells are not a critical factor in sepsis following CLP.
MMP8 does not contribute to sepsis in a peritonitis model without tissue injury
We then used a second model to test the hypothesis that injured cecum could be a source of MMP8 in CLP. To address this question, we employed an intraperitoneal injection of cecal slurry, an alternative model of polymicrobial peritonitis. This model mimics the polymicrobial aspect of the CLP model, but lacks the cecal injury. Figure 2 demonstrates the survival curves for WT and MMP8 null mice following intraperitoneal injections with cecal slurry. Survival times did not significantly differ between the two groups. Thus, unlike polymicrobial peritonitis secondary to CLP, genetic ablation of MMP8 does not confer a survival advantage during polymicrobial peritonitis in the absence of cecal injury. Coupled with our other data, these data suggest that cecal-derived MMP8 may play an important role in the CLP model.
Figure 2. Survival of WT (C57BL/6) and MMP8 null mice following intraperitoneal injections with cecal slurry.
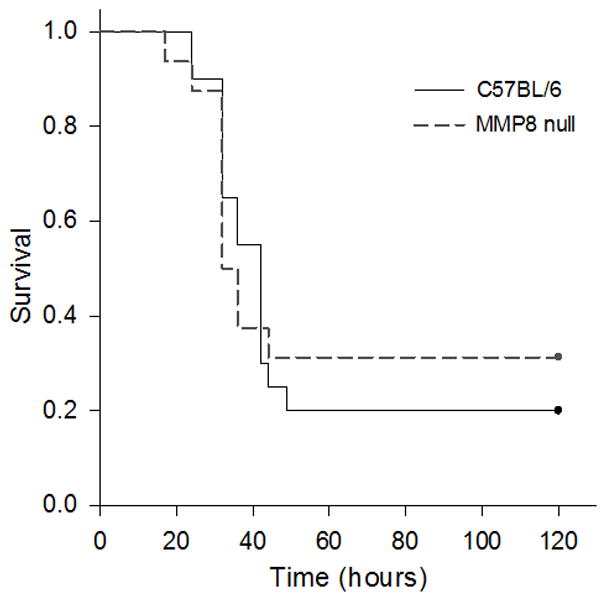
The mean survival time of WT mice (n=20) was 53.2 hours and did not differ significantly from the mean of 59.3 hours for MMP8 null mice (n=16).
Intestine derived MMP8 plays a role in peritonitis secondary to CLP
Our third animal model was designed to further test the possibility that intestine-derived MMP8 plays a role in peritonitis secondary to CLP. We developed a model in which a cecum was extracted from a donor mouse, punctured, and implanted into the peritoneum of a recipient mouse. First, recipient mice underwent cecal implantation from donors of the same genotype (i.e. WT cecums into WT mice and MMP8 null cecums into MMP8 null mice). We found that the survival curve mimicked our original CLP survival curve and that MMP8 null mice had a survival advantage compared to WT mice (Figure 3A).
Figure 3. Implantation of cecum into WT and MMP8 null recipients.
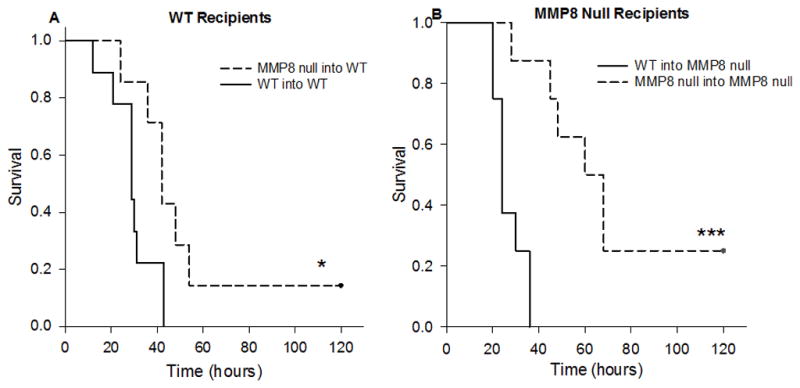
Figure 3A shows the survival of WT mice following intraperitoneal implantation of either a WT or MMP8 null cecum. WT mice that received a MMP8 null cecum (MMP8 null into WT, n=7) had a significant survival advantage over WT mice that received a WT cecum (WT into WT, n=9, *p<0.05). Figure 3B shows survival of MMP8 null mice following implantation of a WT or MMP8 null cecum. Mice that received a MMP8 null cecum (MMP8 null into MMP8 null, n=8) had a significant survival advantage compared to those that received a WT cecum (WT into MMP8 null, n=8, ***p<0.001).
Next, we implanted a cecum from the opposite genotype into the peritoneal cavity of the recipient mouse (i.e. WT cecums into MMP8 null mice and MMP8 null cecums into WT mice). WT mice that were implanted with an MMP8 null cecum had a survival advantage compared to MMP8 mice that were implanted with a WT cecum (Figure 3B). Similar results were seen when implanting distal small bowel into recipient mice (Figure 4).
Figure 4. Implantation of distal small bowel in WT and MMP8 null recipients.
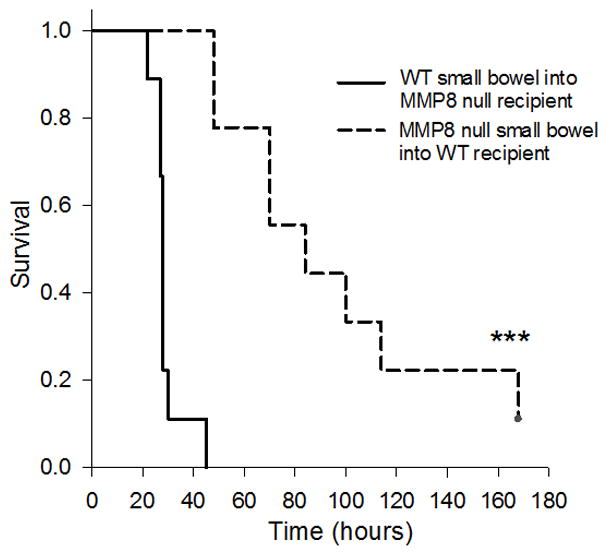
Figure 4 shows the survival of WT mice following intraperitoneal implantation of either WT or MMP8 distal small bowel. WT mice that received MMP8 null distal small bowel (n=9) had a significant survival advantage over MMP8 null mice that received WT distal small bowel (n=9, ***p<0.001).
Taken together, these data show that implantation of WT intestinal tissue is more lethal than implantation of MMP8 null intestinal tissue, regardless of the recipient’s genotype. These findings indicate that MMP8 derived from intestinal tissue may be a critical mediator of sepsis following CLP.
Cytokine expression after cecal implantation demonstrates greater inflammatory cytokine production following WT intestinal implantation
Serum cytokine levels were increased 18 hours after implantation with either a WT or MMP8 null cecum. As shown in Figure 5, serum IL-10, IL-1β, MIP-1α, and TNFα were significantly increased in mice that received WT cecums, compared to mice that received MMP8 null cecums, regardless of the recipient’s genotype. No significant differences were seen between serum levels of KC or LIX (not shown).
Figure 5. Intraperitoneal decay of WT cecum causes greater increase in recipient serum cytokines.

Serum levels of MIP 1α (4A), TNFα (4B), IL-1β (4C), IL-10 (4D), and IL-6 (not shown) were significantly higher in mice who received a WT cecum compared to those that received a MMP8 null cecum. Error bars are the standard deviation.
* p<0.05 vs. MMP8 null mice implanted with WT cecum.
#p<0.05 vs. WT mice implanted with WT cecum.
Ex vivo cecal decay finds MMP8 expression is sustained at higher rate compared to other metalloproteinases
To further determine if the intestine is a source of MMP8, we removed cecums from WT mice and placed them in an incubator at 37°C for 24 hours and measured MMP8 expression within the tissue at 0, 6, and 24 hours. Ex vivo experiments demonstrated that as the cecal tissue decays, MMP8 expression is sustained at a much higher rate than other metalloproteinases (Figure 6).
Figure 6. Ratio of MMP protein expression relative to t=0 hours.
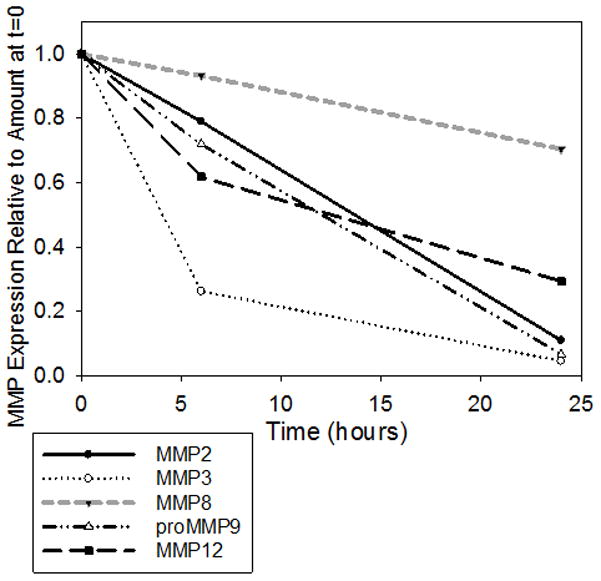
The expression of MMPs was measured using immuno-assays and expressed as a percentage of the respective baseline concentrations. Ex vivo experiments demonstrated that as the cecal tissue decays, MMP8 protein expression is sustained at a much higher rate than other metalloproteinases (n=12, ANOVA on ranks p=0.006).
MMP8 serum concentrations are elevated in patients with septic shock secondary to intestinal pathology
Twenty four pediatric subjects with septic shock were identified from the database as having primary intestinal pathology. These subjects were matched (1:2) based on age and PERSEVERE-based mortality risk to 48 children with septic shock and no evidence of primary intestinal pathology [13]. Figure 7 shows that children with septic shock and primary intestinal pathology had significantly higher MMP8 serum concentrations compared to those without primary intestinal pathology.
Figure 7. Log transformed serum MMP8 protein concentrations in children with septic shock, with and without primary intestinal pathology.
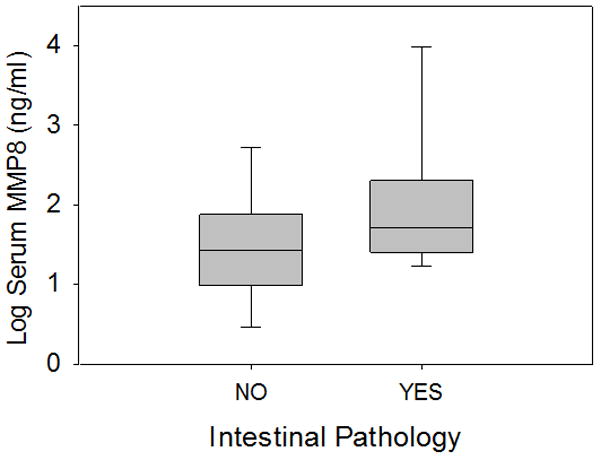
MMP8 serum protein concentrations were significantly higher in patients with primary intestinal pathology (n = 24), compared to age- and illness severity- matched patients without primary intestinal pathology (n=48, p<0.05).
DISCUSSION
The anatomic site of infection and associated tissue damage can influence the pathobiology of sepsis, with one recent report indicating that peritonitis associated with ischemic bowel has the highest mortality risk among patients with septic shock, compared to those having other anatomic sites of infection [14]. Previous experimental data by Ayala et al. demonstrated the importance of necrotic cecum in murine sepsis [15]. Our adoptive transfer experiments corroborate that MMP8 plays an important role in the pathobiology of sepsis secondary to CLP-induced polymicrobial peritonitis, but also indicate that myeloid cells are not the critical source of MMP8 in this model. Experiments involving intraperitoneal injection of a cecal slurry demonstrate that MMP8 does not play an important role in polymicrobial peritonitis without concomitant cecal injury.
In contrast, MMP8 null or WT mice that undergo intraperitoneal implantation of a punctured MMP8 null cecum have a survival advantage compared to those that undergo intraperitoneal implantation of a punctured WT cecum. This effect seems generalizable to the intestine because intraperitoneal implantation of MMP8 null or WT distal small intestine leads to similar results. Ex vivo experiments indicate that MMP8 is expressed in the decaying cecum and that immuno-reactive MMP8 is conserved for a relatively greater time compared to other metalloproteinases. Taken together, these experiments indicate that MMP8 plays an important role in polymicrobial peritonitis with concomitant intestinal injury and that the intestine is an important source of MMP8. Clinical relevance is suggested by the observation that children with septic shock and primary intestinal pathology have higher serum concentrations of MMP8 compared to age- and illness severity-matched children with septic shock but no evidence of primary intestinal pathology.
The precise mechanism by which intestine-derived MMP8 contributes to sepsis remains to be determined. Our current data suggest a mechanism involving increased inflammation because regardless of recipient genotype, intraperitoneal implantation of a WT cecum induces greater levels of inflammatory cytokines when compared to intraperitoneal implantation of a MMP8 null cecum. These data are consistent with our previous report that MMP8 null mice, or WT mice treated with an MMP8 inhibitor, have reduced expression of inflammatory cytokines in association with a survival advantage. In addition, we previously demonstrated that in vitro MMP8 stimulation of macrophages leads to NF-κB activation and inflammatory cytokine expression, indicating that MMP8 has intrinsic pro-inflammatory signaling properties [2].
A mechanism linking MMP8 to increased inflammation and the pathobiology of sepsis is supported by previous studies indicating a pro-inflammatory role for MMP8. For example, in a murine model of TNFα-mediated hepatitis, MMP8 null animals have decreased neutrophil infiltration of the liver, reduced hepatocyte apoptosis, and are protected against liver failure, compared to WT mice [16]. Additionally, following injection of LPS into subcutaneous air pouches, MMP8 null mice had reduced PMN infiltration compared to WT mice [5]. Also, following intraperitoneal injections of LPS, administration of a MMP8 inhibitor results in decreased neuroinflammation compared to those animals that did not receive the inhibitor [17, 18].
Conversely, some experimental models indicate an anti-inflammatory role for MMP8. Murine studies have found that following pulmonary insults from intratracheal instillation of LPS, hypoxia, or bleomycin, resulted in increased pulmonary neutrophil infiltration and lung injury in MMP8 null mice compared to WT mice [4, 19, 20]. Similarly, following intraperitoneal injections of LPS, MMP8 deficient mice were found to have exacerbated lung injuries [21]. Thus, the pro- or anti-inflammatory role for MMP8 appears to depend on both the inflammatory stimulus as well as the anatomical compartment where the insult is initiated.
We note some limitations of our study. First, although animal models of sepsis are useful research tools, the data should be interpreted cautiously given that mice have different inflammatory responses compared to humans and may introduce artifacts [22, 23]. Second, it is possible that our data are confounded by adaptations associated with chronic genetic ablation of MMP8. For example, a previous study reported that in a wound healing model, MMP8 null mice had increased expression of MMP9 within the wound tissue, compared to wild-type mice [24].
MMP8 has been repeatedly identified as playing a role in inflammatory processes, which has led to interest in it as a therapeutic target [2, 11, 25]. MMP inhibition is currently being investigated as a therapy for pulmonary processes such as chronic obstructive pulmonary disease (COPD), acute lung injury (ALI), asthma, interstitial lung disease, and lung cancer [26, 27]. Previous failures with MMP inhibitors have been attributed to the lack of target specificity of the compounds and structural homology between MMPs [28, 29]. Our findings indicate that the septic or inflammatory source may also be relevant to the role of MMP8. As the design of clinical therapies are further considered, the source of inflammation and of MMP8 will also need to be considered while ensuring that the therapy is specific enough to target MMP8 and minimize off target adverse effects [29]. Our current study indicates that targeting MMP8 as a therapeutic strategy in sepsis may not be applicable to sepsis generically, but may be applicable for abdominal sepsis associated with intestinal injury.
Acknowledgments
Funding Source: Supported by NIH T32GM008478, R01GM096994, R01GM099773, and R01GM108025
Footnotes
Author Competing Interests: Dr. Wong and the Cincinnati Children’s Hospital Research Foundation have submitted a provisional patent application for PERSEVERE. The remaining authors have no competing interests to report.
Copyright form disclosures:
Dr. Wong received support for article research from the National Institutes of Health (NIH). His institution received funding from the NIH and has patents pending for PERSEVERE. Drs. Atkinson, Nolan, Lahni, and Klinbeil received support for article research from the National Institutes of Health (NIH). Their institutions received funding from the NIH (NIH T32GM008478, R01GM096994, R01GM099773, and R01GM108025). Dr. Harmon disclosed that she does not have any potential conflicts of interest. Dr. Zingarelli received support from the National Institutes of Health grants R01 GM67202 and R01GM27673 and received support for article research from the NIH. Her institution received funding from the National Institutes of Health grant R01 GM096994.
Author Contributions: Sarah J. Atkinson: Conducted the experiments and analysis, wrote the manuscript. Meghan Nolan: Assisted with experiments and analysis, edited the manuscript. Lindsey Klingbeil: Assisted with experiments and analysis, edited the manuscript. Kelli Harmon: Maintained animal colony and assisted with animal experiments. Patrick Lahni: Conducted cytokine measurements from all samples. Basilia Zingarelli: Assisted with study design and manuscript editing. Hector R. Wong: Conceived and developed the study, obtained funding for the study, conducted the analysis, and edited the manuscript.
References
- 1.Lagu T, Rothberg MB, Shieh MS, Pekow PS, Steingrub JS, Lindenauer PK. Hospitalizations, costs, and outcomes of severe sepsis in the United States 2003 to 2007. Crit Care Med. 2012;40(3):754–761. doi: 10.1097/CCM.0b013e318232db65. [DOI] [PubMed] [Google Scholar]
- 2.Solan PD, Dunsmore KE, Denenberg AG, Odoms K, Zingarelli B, Wong HR. A novel role for matrix metalloproteinase-8 in sepsis. Crit Care Med. 2012;40(2):379–387. doi: 10.1097/CCM.0b013e318232e404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fink MP. Matrix metalloproteinase-8 as a potential drug target for the therapy of sepsis. Crit Care Med. 2012;40(2):655–656. doi: 10.1097/CCM.0b013e31823b97d7. [DOI] [PubMed] [Google Scholar]
- 4.Quintero PA, Knolle MD, Cala LF, Zhuang Y, Owen CA. Matrix metalloproteinase-8 inactivates macrophage inflammatory protein-1 alpha to reduce acute lung inflammation and injury in mice. J Immunol. 2010;184(3):1575–1588. doi: 10.4049/jimmunol.0900290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tester AM, Cox JH, Connor AR, Starr AE, Dean RA, Puente XS, Lopez-Otin C, Overall CM. LPS responsiveness and neutrophil chemotaxis in vivo require PMN MMP-8 activity. PLoS One. 2007;2(3):e312. doi: 10.1371/journal.pone.0000312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Van Lint P, Libert C. Chemokine and cytokine processing by matrix metalloproteinases and its effect on leukocyte migration and inflammation. J Leukoc Biol. 2007;82(6):1375–1381. doi: 10.1189/jlb.0607338. [DOI] [PubMed] [Google Scholar]
- 7.Zingarelli B, Piraino G, Hake PW, O’Connor M, Denenberg A, Fan H, Cook JA. Peroxisome proliferator-activated receptor {delta} regulates inflammation via NF-{kappa}B signaling in polymicrobial sepsis. Am J Pathol. 2010;177(4):1834–1847. doi: 10.2353/ajpath.2010.091010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wynn JL, Scumpia PO, Delano MJ, O’Malley KA, Ungaro R, Abouhamze A, Moldawer LL. Increased mortality and altered immunity in neonatal sepsis produced by generalized peritonitis. Shock. 2007;28(6):675–683. doi: 10.1097/SHK.0b013e3180556d09. [DOI] [PubMed] [Google Scholar]
- 9.Wong HR. Genome-wide expression profiling in pediatric septic shock. Pediatr Res. 2013;73(4 Pt 2):564–569. doi: 10.1038/pr.2013.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cvijanovich N, Shanley TP, Lin R, Allen GL, Thomas NJ, Checchia P, Anas N, Freishtat RJ, Monaco M, Odoms K, et al. Validating the genomic signature of pediatric septic shock. Physiol Genomics. 2008;34(1):127–134. doi: 10.1152/physiolgenomics.00025.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shanley TP, Cvijanovich N, Lin R, Allen GL, Thomas NJ, Doctor A, Kalyanaraman M, Tofil NM, Penfil S, Monaco M, et al. Genome-level longitudinal expression of signaling pathways and gene networks in pediatric septic shock. Mol Med. 2007;13(9–10):495–508. doi: 10.2119/2007-00065.Shanley. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goldstein B, Giroir B, Randolph A. International pediatric sepsis consensus conference: definitions for sepsis and organ dysfunction in pediatrics. Pediatr Crit Care Med. 2005;6(1):2–8. doi: 10.1097/01.PCC.0000149131.72248.E6. [DOI] [PubMed] [Google Scholar]
- 13.Wong HR, Salisbury S, Xiao Q, Cvijanovich NZ, Hall M, Allen GL, Thomas NJ, Freishtat RJ, Anas N, Meyer K, et al. The pediatric sepsis biomarker risk model. Crit Care. 2012;16(5):R174. doi: 10.1186/cc11652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leligdowicz A, Dodek PM, Norena M, Wong H, Kumar A, Kumar A. Association between source of infection and hospital mortality in patients who have septic shock. Am J Respir Crit Care Med. 2014;189(10):1204–1213. doi: 10.1164/rccm.201310-1875OC. [DOI] [PubMed] [Google Scholar]
- 15.Ayala A, Song GY, Chung CS, Redmond KM, Chaudry IH. Immune depression in polymicrobial sepsis: the role of necrotic (injured) tissue and endotoxin. Crit Care Med. 2000;28(8):2949–2955. doi: 10.1097/00003246-200008000-00044. [DOI] [PubMed] [Google Scholar]
- 16.Van Lint P, Wielockx B, Puimege L, Noel A, Lopez-Otin C, Libert C. Resistance of collagenase-2 (matrix metalloproteinase-8)-deficient mice to TNF-induced lethal hepatitis. J Immunol. 2005;175(11):7642–7649. doi: 10.4049/jimmunol.175.11.7642. [DOI] [PubMed] [Google Scholar]
- 17.Folgueras AR, Fueyo A, Garcia-Suarez O, Cox J, Astudillo A, Tortorella P, Campestre C, Gutierrez-Fernandez A, Fanjul-Fernandez M, Pennington CJ, et al. Collagenase-2 deficiency or inhibition impairs experimental autoimmune encephalomyelitis in mice. J Biol Chem. 2008;283(14):9465–9474. doi: 10.1074/jbc.M709522200. [DOI] [PubMed] [Google Scholar]
- 18.Lee EJ, Han JE, Woo MS, Shin JA, Park EM, Kang JL, Moon PG, Baek MC, Son WS, Ko YT, et al. Matrix metalloproteinase-8 plays a pivotal role in neuroinflammation by modulating TNF-alpha activation. J Immunol. 2014;193(5):2384–2393. doi: 10.4049/jimmunol.1303240. [DOI] [PubMed] [Google Scholar]
- 19.Owen CA, Hu Z, Lopez-Otin C, Shapiro SD. Membrane-bound matrix metalloproteinase-8 on activated polymorphonuclear cells is a potent, tissue inhibitor of metalloproteinase-resistant collagenase and serpinase. J Immunol. 2004;172(12):7791–7803. doi: 10.4049/jimmunol.172.12.7791. [DOI] [PubMed] [Google Scholar]
- 20.Craig VJ, Quintero PA, Fyfe SE, Patel AS, Knolle MD, Kobzik L, Owen CA. Profibrotic activities for matrix metalloproteinase-8 during bleomycin-mediated lung injury. J Immunol. 2013;190(8):4283–4296. doi: 10.4049/jimmunol.1201043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gonzalez-Lopez A, Aguirre A, Lopez-Alonso I, Amado L, Astudillo A, Fernandez-Garcia MS, Suarez MF, Batalla-Solis E, Colado E, Albaiceta GM. MMP-8 deficiency increases TLR/RAGE ligands S100A8 and S100A9 and exacerbates lung inflammation during endotoxemia. PLoS One. 2012;7(6):e39940. doi: 10.1371/journal.pone.0039940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, Richards DR, McDonald-Smith GP, Gao H, Hennessy L, et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci USA. 2013;110(9):3507–3512. doi: 10.1073/pnas.1222878110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Osuchowski MF, Remick DG, Lederer JA, Lang CH, Aasen AO, Aibiki M, Azevedo LC, Bahrami S, Boros M, Cooney R, et al. Abandon the mouse research ship? Not just yet! Shock. 2014;41(6):463–475. doi: 10.1097/SHK.0000000000000153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gutierrez-Fernandez A, Inada M, Balbin M, Fueyo A, Pitiot AS, Astudillo A, Hirose K, Hirata M, Shapiro SD, Noel A, et al. Increased inflammation delays wound healing in mice deficient in collagenase-2 (MMP-8) FASEB J. 2007;21(10):2580–2591. doi: 10.1096/fj.06-7860com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rella JM, Jilma B, Fabry A, Kaynar AM, Mayr FB. MMP-8 genotypes influence the inflammatory response in human endotoxemia. Inflammation. 2014;37(2):451–456. doi: 10.1007/s10753-013-9758-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fingleton B. MMPs as therapeutic targets--still a viable option? Semin Cell Dev Biol. 2008;19(1):61–68. doi: 10.1016/j.semcdb.2007.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jacobsen JA, Major Jourden JL, Miller MT, Cohen SM. To bind zinc or not to bind zinc: an examination of innovative approaches to improved metalloproteinase inhibition. Biochim Biophys Acta. 2010;1803(1):72–94. doi: 10.1016/j.bbamcr.2009.08.006. [DOI] [PubMed] [Google Scholar]
- 28.Vandenbroucke RE, Libert C. Is there new hope for therapeutic matrix metalloproteinase inhibition? Nat Rev Drug Discov. 2014;13(12):904–927. doi: 10.1038/nrd4390. [DOI] [PubMed] [Google Scholar]
- 29.Whittaker M, Floyd CD, Brown P, Gearing AJ. Design and therapeutic application of matrix metalloproteinase inhibitors. Chem Rev. 1999;99(9):2735–2776. doi: 10.1021/cr9804543. [DOI] [PubMed] [Google Scholar]