

Abstract
Background
Clostridium difficile infection affects IBD patients. This study’s aim was to compare humoral response to C. difficile toxins in IBD and control outpatients.
Methods
We prospectively followed adult IBD and control subjects with serum and stool tested by PCR obtained at enrollment and during periods of CDI. Semi-quantitative serum levels of IgM, IgG and IgA to C. difficile toxins A and B were measured.
Results
119 stool and 117 serum samples were obtained from 150 subjects. Different levels of IgA to toxin A (p=0.0016) and toxin B (p=0.0468) were noted between different IBD groups. Toxin A IgA levels were higher in the CD group (p=0.0321) and IPAA group (p=0.001) compared to the UC group and toxin B IgA levels were higher in the IPAA group compared to the UC group (p=0.0309). There were lower levels of toxin A IgA in IBD patients with compared with those in subjects without new CDI (P=0.0488) and higher levels in IBD patients with compared with those in subjects without CDI history before enrollment (P=0.016). There were nonsignificant lower toxin A IgG levels in IBD patients with compared to without prior CDI (p=0.095) and higher levels in controls with history of prior CDI compared to IBD patients with prior CDI (p=0.049).
Conclusions
UC patients have lower IgA levels to C. difficile toxins compared to Crohn’s and IPAA patients. IBD patients with prior CDI failed to demonstrate any increase in anti-toxin IgG. Our findings suggest that IBD patients may benefit from immunization strategies targeting C. difficile toxins.
Keywords: Clostridium difficile, Inflammatory Bowel Disease, Ileal Pouch Anal Anastomosis, Antitoxin Antibodies
Introduction
Clostridium difficile infection (CDI) is a serious problem worldwide and in the U.S. comprises approximately 17.1% of hospital-associated infections affecting 500,000 individuals annually with an incidence that is climbing yearly.1,2 CDI leads to worse clinical outcomes, more frequent readmissions, longer length of stays and costs billions of dollars each year in health care expenses.3 The most well-known risk factor for infection is antibiotic use,4 but other risk factors include advanced age5, hospitalization within preceding 2 months6, proton-pump inhibitor use,6–8 and severity of underlying illness.4,6 Inflammatory bowel disease (IBD), which includes Crohn’s disease (CD), Ulcerative Colitis (UC), and patients after ileal pouch anal anastomosis (IPAA), is known to be an independent risk factor for CDI.4,9,10 In this population, traditional risk factors do not have the same correlation with symptomatic infection. Proton-pump inhibitors are a less common risk11 and in children colonization is more common over symptomatic disease.12 Patients with IBD and CDI are less likely to have preceding antibiotic use,13,14 more commonly have community-acquired infection,9,15 and are more likely to be on immunosuppression, which further increases the risk of CDI.11,16
After exposure to the anaerobic Gram-positive spore-forming rod, patients can have varied presentations. Some continue as asymptomatic carriers while others develop clinical infection ranging from mild diarrhea and abdominal pain to pseudomembranous colitis and toxic megacolon.17 IBD patients have a significantly higher rate of CDI compared to the general population,12,18 with UC patients having highest rates when compared to CD or patients after IPAA. 9,18 They also have higher colonization rates compared to the general population.15,19 The presentation of CDI varies in patients with IBD and can be confused with a IBD flare,11 which is important to differentiate as IBD patients with CDI also have increased inhospital mortality, longer hospital stays, and higher complication rates compared to the general population.14,20,21 Humoral immunity is an increasing area of interest in understanding the varied clinical presentation of CDI both in IBD and non-IBD patients and also to investigate why IBD patient may be more susceptible to clinical disease. This study was conducted to determine if IBD patients have any alteration in humoral response to Clostridium difficile toxins A or B when compared to a group of control patients enrolled from outpatient clinics at the same medical center.
Methods
Trial Registry
This trial was registered on clinicaltrials.gov, a site maintained by National Library of Medicine at the National Institutes of Health (CT01813500).
Study Population
English speaking subjects at least eighteen years of age and with an established diagnosis of inflammatory bowel disease based on clinical, radiographic, histologic and endoscopic data were recruited from patients seen at the Center for Digestive Disorders at Boston Medical Center in Boston, MA. At time of enrollment. IBD specific data was collected including anatomic distribution of disease, type of therapy, antibiotics, corticosteroids and smoking status. Diarrhea was defined as a change in bowel habits with 3 or more unformed bowel movements a day for at least 2 days. Control subjects were non-IBD patients also followed at the Center for Digestive Disorders or Adult Primary Care Clinics at Boston Medical Center. Demographic data was collected on all subjects including age, sex, race, and comorbidities by calculating the Charlson comorbidity index. CDI-related risk factors were also collected and included antibiotic use prior C diff infection, recent hospitalization or institutionalization. At time of enrollment and during periods of diarrhea subjects from both IBD and control groups submitted both stool and blood for analysis. Those with clinically significant diarrhea and positive stool studies were treated by their gastroenterologist as per standard of care. Serum samples were collected for anti-toxin antibody measurements. Subjects were followed prospectively for at least 12 month period.
Duration of participation
Mean follow up for patients was for a period of 24 months. During that time patients were followed via chart review and follow up visits for the duration of the enrollment period from 10/2011–10/2013 and the dataset was closed 4/2014. The subset of patients that developed CDI was analyzed and incorporated into the manuscript as new CDI. This patient subset will be followed prospectively for CDI for potential future studies. The minimum duration of sample collection during symptomatic disease was 1 month and maximum was 12 months. CDI were treated per standard of care and repeat blood samples for antitoxin titers were collected to determine any change in magnitude of anti-toxin antibody response post infection. All patients were seen routinely by their gastroenterologist and treated with standard of care for any flare or infectious diarrhea.
Stool tests
Stool was analyzed for Clostridium difficile based on published methodologies using PCR based assays. Samples were stored in a minus 80 degree freezer. All samples were coded by personnel who had no knowledge of clinical characteristics.
Blood tests
Serum samples were collected and stored for future anti-toxin antibody measurements based on published methodologies. Samples were stored in a minus 80 degree freezer. All samples were coded by personnel who had no knowledge of clinical characteristics.
Serum C. difficile anti-toxin antibody assay
Serum samples were analyzed for antibody titers to toxin A and B by standardized ELISA and cell-based toxin neutralization assay. Samples were processed by methods that were previously reported.22,23 C. difficile toxins A and toxin B were purified from the culture supernatant of strain VPI 10463 (American Type Culture Collection 43255-FZ, Manassas, VA) as previously described23,24. Levels of antibody against C. difficile toxin A and toxin B were measured by an enzyme-linked immunosorbent assay (ELISA).
Stool Clostridium difficile analysis
Clostridium difficile testing was provided by the Microbiology Section at BMC. All specimens were tested for C. difficile using a PCR-based test (GeneXpert). Clostridium difficile infection was defined as diarrhea coupled with a positive stool C. difficile assay and not attributed to other causes. Clostridium difficile colonization was defined as positive stool C. difficile assay without any signs or symptoms of disease including pain, diarrhea, bloating, fever. If subjects developed CDI, treatment was per standard of care (detailed below). Repeat blood samples for anti-toxin titers will be collected during routine clinic visit or during hospitalization to determine any change in magnitude of anti-toxin antibody response post infection.
Standard of Care
Subjects with diarrhea were evaluated by their gastroenterologist and depending upon the clinical situation may have undergone blood or stools tests and possible colonoscopy per the clinical judgment of the medical provider. Treatment options included antibiotics, immunosuppressants, anti-diarrheal agents, other standard medication or possible surgery as needed.
Statistics
Continuous variables were compared between groups using analysis of variance (ANOVA) or the non-parametric Kruskal-Wallis test while categorical variables were compared between groups using the chi-square test or Fisher’s exact test, as appropriate. Toxin A and toxin B IgA, IgG and IgM levels were compared between CD, UC, IPAA (combined IPAA/CD and IPAA/UC) and control subjects using ANOVA. If the overall ANOVA was significant at the 0.05 level, then all 6 pairwise comparisons were examined using the Tukey-Kramer method to control for multiple comparisons. The overall ANOVA results were confirmed using the Kruskal-Wallis test. Antibody levels for IBD and control subjects with and without a prior CDI and with and without new CDIs were compared using similar methods. Statistical significance was assessed at the 0.05 level and all analyses were performed using SAS v9.3.
Ethical Considerations
The study was approved by the institutional review board at Boston Medical Center in Boston, MA (BMC IRB Number: H-31226). Informed consent was obtained from all participants using an IRB-approved research consent document. Participants were given $10 to cover travel and parking expenses if they provided a stool sample. No other reimbursement was provided.
Results
We enrolled 150 subjects; 4 withdrew from the study prior to sample submission. Of the 146 participants who remained in the study, 118 had IBD (CD (n=50), UC (n=45), IPAA (n=23)) and 28 were controls. As illustrated in Figure 1, 119 submitted stool samples, 118 patients submitted serum samples and 89 submitted both serum and stool. All stool samples were processed using PCR to detect C. difficile and serum samples were processed to measure IgM, IgG, and IgA levels to both toxin A and toxin B. Of the stool samples collected, 13 had CDI during the time of enrollment or during the study period and eleven of those had submitted serum. This group was processed to measure IgM, IgG, and IgA levels to both toxin A and B and levels were compared prior to and at the time of infection.
Figure 1. Inclusion of Patients, Rates of Sample Collection and Sample Processing.

* Inflammatory Bowel Disease
± Clostridium difficile infection
Demographics
The characteristics of all patients are shown in Table 1. Notably of the 118 who submitted serum, 44 had CD, 30 UC, 15 IPAA with UC, 6 IPAA with CD and 23 were controls. Nine IBD patients (7.6%), and 2 control patients (7.1%), had CDI in the month prior to study enrollment. The Charlson Comorbidity Index Score was lower at 0.7 in the IBD group, when compared to 1.88 in controls.
Table 1. Baseline Characteristics of Study Participants and Sample Submission Rates.
Description of study participant characteristics reported in percentages, with the exception of Charlson Comorbidity Index Score which is reported as mean. Submission rates of stool and serum samples, including initial enrollment and 12-month observation period, also reported.
| IBD | Control | Total | p-value* | ||||
|---|---|---|---|---|---|---|---|
| CD | UC | IPAA | IBD Group | ||||
| 50 | 45 | 23 | 118 | 28 | 146 | ||
| Age (years) | 41.8 +/− 15.1 | 38.6 +/− 14.3 | 48.9 +/− 14.5 | 42.0 +/− 15.0 | 45.6 +/−15.6 | 42.7 +/−15.1 | 0.0382 |
| Male | 36.0% | 51.1% | 34.8% | 41.5% | 50.0% | 43.2% | 0.3344 |
| Shelter or Nursing home resident | 4.0% | 0.0% | 0.0% | 1.7% | 7.1% | 2.7% | 0.2852 |
| Current tobacco use | 18.0% | 4.4% | 17.4% | 12.7% | 35.7% | 17.1% | 0.0056 |
| Tobacco use ever | 32.0% | 24.4% | 39.1% | 30.5% | 42.9% | 32.9% | 0.3707 |
| CDI in the past 1 month | 8.0% | 8.9% | 4.4% | 7.6% | 7.1% | 7.5% | 0.9714 |
| Flare or diarrhea at time of enrollment | 72.0% | 82.2% | 78.3% | 77.1% | 53.9% | 72.9% | 0.0683 |
| Abdominal pain | 32.0% | 35.6% | 34.8% | 33.9% | 60.7% | 39.0% | 0.0728 |
| Hospitalizations in the past 6 months | 30.0% | 2.2% | 8.7% | 15.3% | 29.6% | 17.9% | 0.0003 |
| Antibiotic use in the past 6 months | 32.0% | 33.3% | 47.8% | 35.6% | 50.0% | 38.4% | 0.2824 |
| PPI use in the past 6 months | 24.0% | 20.0% | 13.0% | 20.3% | 42.9% | 24.7% | 0.0658 |
| 5-ASA use in the past 6 months | 42.0% | 73.3% | 8.7% | 47.5% | 0.0% | 38.6% | <.0001 |
| Immunomodulator use in the past 6 months | 38.0% | 26.7% | 17.4% | 29.7% | 0.0% | 24.1% | 0.0022 |
| Anti-TNF use in the past 6 months | 22.0% | 17.8% | 4.4% | 16.9% | 0.0% | 13.8% | 0.0130 |
| Charlson Comorbidity Index Score (mean) | 1.5 +/− 1.1 | 1.5 +/− 0.8 | 0.9 +/− 1.1 | 1.4 +/− 1.0 | 0.8 +/− 1.0 | 1.3 +/− 1.0 | 0.0016 |
| Submitted blood samples | 88.0% | 66.7% | 91.3% | 80.5% | 78.6% | 80.1% | |
| Submitted stool samples | 84.0% | 75.6 | 95.7% | 83.1% | 75.0% | 81.5% | |
| # c. difficile infection | 12.0% | 6.7% | 4.3% | 8.5% | 10.7% | 8.9% | 0.7475 |
| # c. difficile colonization | 2.0% | 2.2% | 0% | 1.7% | 0% | 1.4% | |
| # NAP1/027 hypervirulent strain | 0% | 0% | 0% | 0% | 0% | 0% | |
| Submitted both blood and stool samples | 72.0% | 42.2% | 82.6% | 62.7% | 53.6% | 6.0% | |
Based on ANOVA for continuous variables and Chi-Square or Fisher’s Exact Test for categorical predictors, as appropriate.
Comparison of baseline anti-toxin antibody levels
IBD groups were divided into CD, UC and IPAA (including both IPAA/UC and IPAA/CD) for analysis as shown in Figure 2. There was a statistically significant difference in serum toxin A IgA between IBD diagnosis groups (p=0.0016) with higher values in the CD group and combined IPAA groups when compared to the UC group (p=0.0321 and p=0.001, respectively). The control group also tended to have higher values compared to the UC group but did not reach statistical significance (p=0.0963). There were no significant differences in serum toxin A IgM, or toxin A IgG between IBD groups or compared to controls. A significant difference was also found in toxin B IgA between IBD groups (p=0.0468). There were higher values in the combined IPAA group compared to the UC group (p=0.0309). There were no significant differences between IBD diagnosis groups or when compared to controls in toxin B IgM or toxin B IgG levels. There was also no significant difference between IBD diagnosis groups when groupings were examined with CD+IPAA/CD vs. UC+IPAA/UC for any of the antibody levels based on the overall ANOVA.
Figure 2.
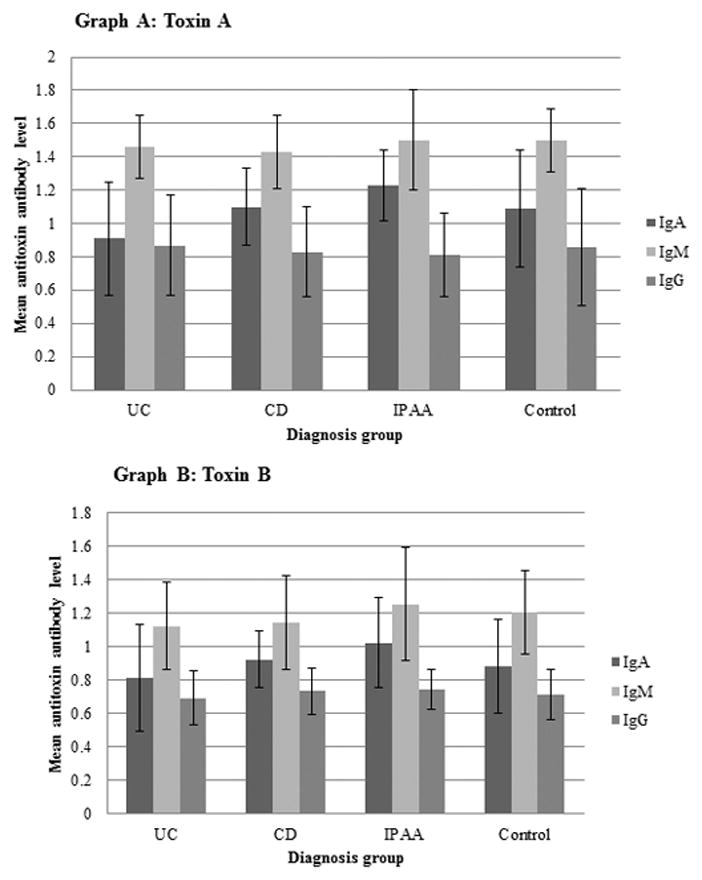
Comparison of baseline antitoxin antibodies to Clostridium difficile toxins among study participants
Clostridium difficile infection and colonization
At enrollment, 2 patients in the IBD group were found to be colonized with C. difficile and no colonization was found in the control group. No NAP1/027 hypervirulent strains were detected among any samples. Over the course of the study, 3 of the 28, or 10.7%, controls and 10 of 118, or 8.5%, IBD participants went on to develop CDI. Serum was available for 22 controls and 95 IBD patients and post-infection antibody levels from sera were available for 3 controls and 8 IBD patients.
Antibody levels in both IBD subjects and controls that developed a new CDI were compared with those without a new CDI, as shown in Figure 3. Levels were also compared between IBD subjects and controls with and without a prior CDI within the month prior to study enrollment, as shown in Figure 4. IBD subjects with a new CDI had significantly lower toxin A IgA levels compared to subjects without a new CDI (p = 0.0488). There were no other significant differences between IBD subjects with and without a new CDI and no significant differences between control subjects with and without a new CDI. The toxin A IgG levels tended to be lower in those with a prior CDI (p=0.0954) while toxin A IgA level among IBD subjects with a prior CDI was significantly higher compared to those without a prior CDI (p=0.0160). In controls, toxin A IgG was significantly higher in those with a prior CDI compared to those subjects without a prior CDI (p=0.0495). Toxin B IgM levels in controls with a prior CDI were also significantly lower than in those subjects without a prior CDI (p=0.0495).
Figure 3.

Comparison of mean antitoxin antibody titers to Clostridium difficile toxin A and toxin B among study participants with and without new Clostridium difficile infection.
Figure 4.

Comparison of mean antitoxin antibody titers to Clostridium difficile toxin A and toxin B among study participants with and without prior Clostridium difficile infection.
Discussion
IBD is an independent risk factor for CDI and is associated with higher mortality, longer hospital stays and worse outcomes. The reason for this is not completely understood but likely represents alterations in the colon microbiome as well as defects in both innate and humoral immunity. The body relies on intact mucous membranes25 with epithelial cell secretion containing mucins acting as a barrier, phosphatidylcholine providing local anti-inflammatory effects,26 and secretory IgA acting synergistically with the mucus layer to protect the luminal surface from bacteria.27 Normal gut flora is needed to compete with C. difficile for nutrients and regulate transformation of bile salts, particularly taucholate, which is needed for C. difficile germination.10,25 In its vegetative state C. difficile produces toxins which enter the cell and damage the intestinal epithelial cytoskeleton,17,26 act as a chemoattractant for neutrophils and lead to inflammation. It is this inflammatory response, not the degree of bacterial burden, which leads to cell injury and determines the clinical severity of CDI.28,29 Understanding the innate response to CDI helps highlight why IBD patients may have higher rates of infection and more severe, complicated clinical courses. Antibiotic use is the greatest and most well-known risk-factor for CDI because it leads to altered gut flora.17,30 IBD patients often have underlying dysbiosis even without preceding antibiotic exposure and do not have the normal flora to compete with C. difficile.31–34 IBD is also a condition that leads to chronic inflammation, tissue injury and altered T cell responses. Dysbiosis is both likely an effect of and an antigenic stimulus to cause this ongoing inflammation.31 In patients who already have chronically inflamed, damaged mucosa and may have a genetic predisposition to an overaggressive inflammatory response to enteric bacteria, it is not surprising there is an increased prevalence of infection and more severe clinical course. In addition, UC patients have decreased phosphatidylcholine levels and degradation of their mucosal barrier26 which may predispose them to even higher rates when compared to other types of IBD.
The humoral immune response in IBD patients, unlike innate immunity, is less well understood. Toxin-mediated pathogens, like Clostridium spp., produce an antibody-mediated response to exposure that aids in neutralization of toxins.35 A growing number of studies have examined the humoral response to CDI which have implications for novel therapies. Previous data show that up to 60% of older children and adults in the general population have antibodies to C. difficile toxin A and B.10 Early studies showed toxin antibody levels do not offer initial protection against colonization23 but are correlated with the occurrence of symptomatic disease. Kyne et al24 found that higher levels of Toxin A IgM on day 3 and IgG on day 12 after onset of c. difficile diarrhea were associated with single-episode CDI without recurrence. Katchar et al36 found similar evidence, showing patients with recurrent CDI had decreased levels of IgG2 and IgG3 to toxin A. While Kyne’s initial study showed elevated but not significant levels of IgG to Toxin B in asymptomatic carriers, later studies have shown a significant antibody response to Toxin B correlated with C. difficile colonization.6,22 A recent study by Wullt et al37 went one step further and measured sera at multiple time points preceding and after infection, finding that IgG levels at inclusion for both toxin A and B were lower in patients who later developed CDI when compared to controls and had a weak IgG response to infection. This study is of particular interest for IBD patients. Rai et al38 looked at total serum IgG levels in IBD patients without infection and found they had a lower level of IgG1 subgroup at baseline but overall IgG and IgG2 and IgG3 subgroups were normal compared to controls, whereas Seril et al39 found that in IPAA patients in particular, there was a significant lower level of IgG1. Though these studies are small, they support the idea that there may be an underlying humoral deficit not seen in the general population which predisposes IBD patients to CDI. In contrast, a study by Hourigan et al40 looked at CDI in pediatric IBD patients and found that they had higher antibody response compared to controls with a significant elevation specifically of IgA to toxin B but no difference in IgG anti-toxin.
Our paper examined serum levels of 117 samples (95 IBD, 22 controls) for IgM, IgG and IgA titers to C. difficile toxin A and B at initial enrollment of this study and at time of a new CDI within the surveillance period. We found that there were significant differences in initial IgA anti-toxin between IBD groups for both toxin A and toxin B. Ulcerative colitis patients had significantly lower levels of toxin A IgA when compared to CD and IPAA groups and tended towards lower levels compared to controls but did not reach statistical significance. Ulcerative colitis patients also had lower levels of toxin B IgA when compared to IPAA groups. In patients with new CDI, IBD patients with a new infection had significantly lower levels of IgA against toxin A when compared to patients without a new CDI. However, IBD patients with a history of prior CDI had relatively higher levels of IgA against toxin A when compared to IBD patients without such a history. IgA is an important area of focus for CDI immune response because serum IgA neutralizes the enterotoxic effect of C. difficile toxin A.26,41 Serum antitoxin antibodies bind luminal toxins in CDI by toxin-mediated paracellular transport of both serum IgA and IgG.42 While there is some risk in interpreting our data given the relatively small sample size, UC patients appear to have a deficit in humoral response with lower levels of toxin A and B IgA at baseline and IBD patients as a group have lowered IgA response against toxin A with new infection but have higher IgA levels against toxin A if previously exposed to CDI compared to their exposure-free counterparts. The significance of this is not yet fully understood but there is data to indicate the importance of IgA anti-toxin responses in improved clinical outcomes in CDI.43 Though there was no difference found when looking at the overall IgG levels at baseline to toxin A or B between IBD groups and controls, there was a difference between patients with previous CDI. IBD patients with prior CDI had lower IgG levels to toxin A, compared to controls which had higher IgG to toxin A with prior CDI. This is consistent with Kyne et al and Wullt et al, who showed a higher IgG response to infection correlated with colonization and avoidance of symptomatic disease in the future. There was also a significantly lower level of IgM to toxin B in controls with prior CDI compared with controls without prior CDI. The significance of this is not clear, and may be related to small sample size or an immune response favoring increased IgG production seen after initial infection.
Small sample size was a limitation to this study. One hundred fifty participants were recruited but not all submitted samples, and not all submitted both stool and sera so it is possible that active CDI was missed in serum-only patients as diarrhea at time of enrollment was reported in 51.7% of all participants and abdominal pain in 39%. There were multiple tests performed comparing different groups, exposures and infection and there is the risk that some of the significant differences found may be due to chance. Analysis of prior and new CDI, a much smaller grouping of subjects with 13 new infections and 11 having submitted sera, is at most risk of this. While some conclusions are consistent with previous data, all published information on humoral response to CDI in IBD patients are from small numbers of subjects and are at risk for the same source of error. The significance of humoral response in IBD patients and correlation with clinical severity of disease is not yet understood. There is also a lack of standardization of acceptable titer ranges so comparisons have been made within each study itself.
Other possible limitations come from subject recruitment and selection of a control group. As IBD patients were only recruited from the Gastroenterology subspecialty clinic and primary care clinic from one medical center, there is possibility of sampling error. Known factors that decrease serum total Ig levels such as advanced age, malnutrition, and hypoalbuminemia38 were not controlled for in this study. Compared to the IBD group, the group of controls included in this study were overall sicker with higher rates of hospitalizations in the 6 months preceding enrollment in the study when compared to the IBD group (p=0.0003). The average Charlson Comorbidity Index score was 1.88 for the control group compared to 0.7 in the IBD patients, meaning the control group overall had more comorbidities and were a sicker group compared to the IBD group (p=0.0016). When comparing IBD patients to controls, the IBD patients were overall younger when compared to controls (p=0.0382) with a significant difference in active tobacco abuse (p0.0056). It is well documented that immunoglobulin levels are decreased in ill and elderly patients so this relatively older, sicker group of controls with a greater number of hospitalizations may have lower titers when compared to a group of younger, healthy controls.
Much is still unknown about the humoral immune response in IBD patients with CDI. This study reported data at one time point and serum antibody levels to toxin A and B in the 3 controls and 8 IBD patients who went on to develop CDI. Larger scale research is needed to determine if IBD patients do in fact have an altered humoral response to C. difficile toxin A and B, what is considered a normal antibody response to this infection, what this information means to future therapies for CDI and what anti-toxin antibody titers mean for clinical severity of disease. It is also not known if immunomodulator therapies, common in IBD populations, have clinically significant effects on humoral response in these individuals.
Our study showed that UC patients have lower IgA to C. difficile toxin A and B when compared to other IBD groups and IPAA groups, respectively. It also showed that IBD patients with active CDI have lower toxin A IgA levels but that these levels increased in IBD patients with prior CDI within 1 month of study enrollment. Toxin A IgG levels are overall lower in IBD patients with prior CDI and higher in controls with the same risk exposure. Toxin B IgM levels were lower in controls with prior CDI. Additional studies are needed to better understand baseline humoral immunity in IBD patients and the response to CDI in order to fully understand the increased prevalence of infection in this population. Further studies are also needed to better understand the clinical significance and potential treatment indications of these data.
Acknowledgments
Source of Funding: Funding from unrestricted grant to Boston Medical Center from Cubist Pharmaceuticals, now Merck Pharmaceuticals. Cepheid provided GeneXpert Epi tests for our study. Additional funding through a gift from Robin and Andrew Davis.
Supported through a gift from Robin and Andrew Davis
Footnotes
Conflict of Interest: Dr. Francis Farraye has previously served as a consultant for Cubist Pharmaceuticals. Drs. Ciaran Kelly and Xinhua Chen are receiving an NIH grant (RO1 AI 095256). The remaining authors have no sources of funding or conflicts of interest to disclose.
References
- 1.Magill SS, Edwards JR, Bamberg W, et al. Multistate point-prevalence survey of health care-associated infections. N Engl J Med. 2014;370:1198–1208. doi: 10.1056/NEJMoa1306801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lessa FC, Mu Y, Bamberg WM, et al. Burden of Clostridium difficile infection in the United States. N Engl J Med. 2015;372:825–834. doi: 10.1056/NEJMoa1408913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Centers for Disease Control and Prevention (CDC) Vital signs: preventing Clostridium difficile infections. MMWR Morb Mortal Wkly Rep. 2012;61:157–162. [PubMed] [Google Scholar]
- 4.Bignardi GE. Risk factors for Clostridium difficile infection. J Hosp Infect. 1998;40:1–15. doi: 10.1016/s0195-6701(98)90019-6. [DOI] [PubMed] [Google Scholar]
- 5.Garey KW, Sethi S, Yadav Y, et al. Meta-analysis to assess risk factors for recurrent Clostridium difficile infection. J Hosp Infect. 2008;70:298–304. doi: 10.1016/j.jhin.2008.08.012. [DOI] [PubMed] [Google Scholar]
- 6.Loo VG, Bourgault AM, Poirier L, et al. Host and pathogen factors for Clostridium difficile infection and colonization. N Engl J Med. 2011;365:1693–1703. doi: 10.1056/NEJMoa1012413. [DOI] [PubMed] [Google Scholar]
- 7.Dial S, Alrasadi K, Manoukian C, et al. Risk of Clostridium difficile diarrhea among hospital inpatients prescribed proton pump inhibitors: cohort and case-control studies. CMAJ. 2004;171:33–38. doi: 10.1503/cmaj.1040876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dial S, Delaney JA, Barkun AN, et al. Use of gastric acid-suppressive agents and the risk of community-acquired Clostridium difficile-associated disease. JAMA. 2005;294:2989–2995. doi: 10.1001/jama.294.23.2989. [DOI] [PubMed] [Google Scholar]
- 9.Rodemann JF, Dubberke ER, Reske KA, et al. Incidence of Clostridium difficile infection in inflammatory bowel disease. Clin Gastroenterol Hepatol. 2007;5:339–344. doi: 10.1016/j.cgh.2006.12.027. [DOI] [PubMed] [Google Scholar]
- 10.Sun X, Hirota SA. The roles of host and pathogen factors and the innate immune response in the pathogenesis of Clostridium difficile infection. Mol Immunol. 2015;63:193–202. doi: 10.1016/j.molimm.2014.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ananthakrishnan AN. Detecting and treating Clostridium difficile infections in patients with inflammatory bowel disease. Gastroenterol Clin North Am. 2012;41:339–353. doi: 10.1016/j.gtc.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 12.Hourigan SK, Oliva-Hemker M, Hutfless S. The prevalence of Clostridium difficile infection in pediatric and adult patients with inflammatory bowel disease. Dig Dis Sci. 2014;59:2222–2227. doi: 10.1007/s10620-014-3169-4. [DOI] [PubMed] [Google Scholar]
- 13.Bossuyt P, Verhaegen J, Van Assche G, et al. Increasing incidence of Clostridium difficile-associated diarrhea in inflammatory bowel disease. J Crohns Colitis. 2009;3:4–7. doi: 10.1016/j.crohns.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 14.Goodhand JR, Alazawi W, Rampton DS. Systematic review: Clostridium difficile and inflammatory bowel disease. Aliment Pharmacol Ther. 2011;33:428–441. doi: 10.1111/j.1365-2036.2010.04548.x. [DOI] [PubMed] [Google Scholar]
- 15.Clayton EM, Rea MC, Shanahan F, et al. The vexed relationship between Clostridium difficile and inflammatory bowel disease: an assessment of carriage in an outpatient setting among patients in remission. Am J Gastroenterol. 2009;104:1162–1169. doi: 10.1038/ajg.2009.4. [DOI] [PubMed] [Google Scholar]
- 16.Issa M, Vijayapal A, Graham MB, et al. Impact of Clostridium difficile on inflammatory bowel disease. Clin Gastroenterol Hepatol. 2007;5:345–351. doi: 10.1016/j.cgh.2006.12.028. [DOI] [PubMed] [Google Scholar]
- 17.Gerding DN, Johnson S. Clostridium Difficile Infection, Including Pseudomembranous Colitis. In: Longo DL, Fauci AS, Kasper DL, et al., editors. Harrison’s Principles of Internal Medicine. 18. New York, NY: McGraw-Hill; 2012. pp. 111–1111. [Google Scholar]
- 18.Nguyen GC, Kaplan GG, Harris ML, et al. A national survey of the prevalence and impact of Clostridium difficile infection among hospitalized inflammatory bowel disease patients. Am J Gastroenterol. 2008;103:1443–1450. doi: 10.1111/j.1572-0241.2007.01780.x. [DOI] [PubMed] [Google Scholar]
- 19.Bartlett JG, Gerding DN. Clinical recognition and diagnosis of Clostridium difficile infection. Clin Infect Dis. 2008;46(Suppl 1):S12–8. doi: 10.1086/521863. [DOI] [PubMed] [Google Scholar]
- 20.Ananthakrishnan AN, McGinley EL, Binion DG. Excess hospitalisation burden associated with Clostridium difficile in patients with inflammatory bowel disease. Gut. 2008;57:205–210. doi: 10.1136/gut.2007.128231. [DOI] [PubMed] [Google Scholar]
- 21.Trifan A, Stanciu C, Stoica O, et al. Impact of Clostridium difficile infection on inflammatory bowel disease outcome: a review. World J Gastroenterol. 2014;20:11736–11742. doi: 10.3748/wjg.v20.i33.11736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leav BA, Blair B, Leney M, et al. Serum anti-toxin B antibody correlates with protection from recurrent Clostridium difficile infection (CDI) Vaccine. 2010;28:965–969. doi: 10.1016/j.vaccine.2009.10.144. [DOI] [PubMed] [Google Scholar]
- 23.Kyne L, Warny M, Qamar A, et al. Asymptomatic carriage of Clostridium difficile and serum levels of IgG antibody against toxin A. N Engl J Med. 2000;342:390–397. doi: 10.1056/NEJM200002103420604. [DOI] [PubMed] [Google Scholar]
- 24.Kyne L, Warny M, Qamar A, et al. Association between antibody response to toxin A and protection against recurrent Clostridium difficile diarrhoea. Lancet. 2001;357:189–193. doi: 10.1016/S0140-6736(00)03592-3. [DOI] [PubMed] [Google Scholar]
- 25.Ghose C, Kelly CP. The prospect for vaccines to prevent Clostridium difficile infection. Infect Dis Clin North Am. 2015;29:145–162. doi: 10.1016/j.idc.2014.11.013. [DOI] [PubMed] [Google Scholar]
- 26.Diebel LN, Liberati DM. Reinforcement of the intestinal mucus layer protects against Clostridium difficile intestinal injury in vitro. J Am Coll Surg. 2014;219:460–468. doi: 10.1016/j.jamcollsurg.2014.05.005. [DOI] [PubMed] [Google Scholar]
- 27.Olson A, Diebel LN, Liberati DM. Effect of host defenses on Clostridium difficile toxin-induced intestinal barrier injury. J Trauma Acute Care Surg. 2013;74:983–89. doi: 10.1097/TA.0b013e3182858477. discussion 989–90. [DOI] [PubMed] [Google Scholar]
- 28.Kelly CP, Kyne L. The host immune response to Clostridium difficile. J Med Microbiol. 2011;60:1070–1079. doi: 10.1099/jmm.0.030015-0. [DOI] [PubMed] [Google Scholar]
- 29.El Feghaly RE, Stauber JL, Deych E, et al. Markers of intestinal inflammation, not bacterial burden, correlate with clinical outcomes in Clostridium difficile infection. Clin Infect Dis. 2013;56:1713–1721. doi: 10.1093/cid/cit147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bassis CM, Theriot CM, Young VB. Alteration of the murine gastrointestinal microbiota by tigecycline leads to increased susceptibility to Clostridium difficile infection. Antimicrob Agents Chemother. 2014;58:2767–2774. doi: 10.1128/AAC.02262-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sartor RB. Microbial influences in inflammatory bowel diseases. Gastroenterology. 2008;134:577–594. doi: 10.1053/j.gastro.2007.11.059. [DOI] [PubMed] [Google Scholar]
- 32.Frank DN, St Amand AL, Feldman RA, et al. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci U S A. 2007;104:13780–13785. doi: 10.1073/pnas.0706625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Angriman I, Scarpa M, Castagliuolo I. Relationship between pouch microbiota and pouchitis following restorative proctocolectomy for ulcerative colitis. World J Gastroenterol. 2014;20:9665–9674. doi: 10.3748/wjg.v20.i29.9665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Komanduri S, Gillevet PM, Sikaroodi M, et al. Dysbiosis in pouchitis: evidence of unique microfloral patterns in pouch inflammation. Clin Gastroenterol Hepatol. 2007;5:352–360. doi: 10.1016/j.cgh.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 35.Brooks G, Carroll K, Butel J, et al. Chapter 8: Immunology. In: Brooks G, Carroll K, Butel J, et al., editors. Jawetz, Melnick, & Adelberg’s Medical Microbiology. 26. New York, NY: McGraw Hill; 2013. [Google Scholar]
- 36.Katchar K, Taylor CP, Tummala S, et al. Association between IgG2 and IgG3 subclass responses to toxin A and recurrent Clostridium difficile-associated disease. Clin Gastroenterol Hepatol. 2007;5:707–713. doi: 10.1016/j.cgh.2007.02.025. [DOI] [PubMed] [Google Scholar]
- 37.Wullt M, Noren T, Ljungh A, et al. IgG antibody response to toxins A and B in patients with Clostridium difficile infection. Clin Vaccine Immunol. 2012;19:1552–1554. doi: 10.1128/CVI.00210-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rai T, Wu X, Shen B. Frequency and risk factors of low immunoglobulin levels in patients with inflammatory bowel disease. Gastroenterol Rep (Oxf) 2015 doi: 10.1093/gastro/gou082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Seril DN, Ashburn JH, Lian L, et al. Risk factors and management of refractory or recurrent clostridium difficile infection in ileal pouch patients. Inflamm Bowel Dis. 2014;20:2226–2233. doi: 10.1097/MIB.0000000000000205. [DOI] [PubMed] [Google Scholar]
- 40.Hourigan SK, Chirumamilla SR, Ross T, et al. Clostridium difficile carriage and serum antitoxin responses in children with inflammatory bowel disease. Inflamm Bowel Dis. 2013;19:2744–2752. doi: 10.1097/01.MIB.0000435434.53871.36. [DOI] [PubMed] [Google Scholar]
- 41.Johnson S, Sypura WD, Gerding DN, et al. Selective neutralization of a bacterial enterotoxin by serum immunoglobulin A in response to mucosal disease. Infect Immun. 1995;63:3166–3173. doi: 10.1128/iai.63.8.3166-3173.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang Z, Chen X, Hernandez LD, et al. Toxin-mediated paracellular transport of antitoxin antibodies facilitates protection against Clostridium difficile infection. Infect Immun. 2015;83:405–416. doi: 10.1128/IAI.02550-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Warny M, Vaerman JP, Avesani V, et al. Human antibody response to Clostridium difficile toxin A in relation to clinical course of infection. Infect Immun. 1994;62:384–389. doi: 10.1128/iai.62.2.384-389.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]