

MRIs from 18 patients with confirmed tubulin mutations and 15 patients with mutations of the genes encoding microtubule-associated proteins were evaluated. The patients with tubulin gene mutations were found to have cortical and subcortical abnormalities such as microcephaly, ventriculomegaly, abnormal gyral and sulcal patterns, small or absent corpus callosum, and a small pons. Fewer subcortical abnormalities were seen in the microtubule-associated protein mutations group.
Abstract
BACKGROUND AND PURPOSE:
A number of recent studies have described malformations of cortical development with mutations of components of microtubules and microtubule-associated proteins. Despite examinations of a large number of MRIs, good phenotype-genotype correlations have been elusive. Additionally, most of these studies focused exclusively on cerebral cortical findings. The purpose of this study was to characterize imaging findings associated with disorders of microtubule function.
MATERIALS AND METHODS:
MRIs from 18 patients with confirmed tubulin mutations (8 TUBA1A, 5 TUBB2B, and 5 TUBB3) and 15 patients with known mutations of the genes encoding microtubule-associated proteins (5 LIS1, 4 DCX, and 6 DYNC1H1) were carefully visually analyzed and compared. Specific note was made of the cortical gyral pattern, basal ganglia, and white matter to assess internal capsular size, cortical thickness, ventricular and cisternal size, and the size and contours of the brain stem, cerebellar hemispheres and vermis, and the corpus callosum of patients with tubulin and microtubule-associated protein gene mutations. Results were determined by unanimous consensus of the authors.
RESULTS:
All patients had abnormal findings on MR imaging. A large number of patients with tubulin gene mutations were found to have multiple cortical and subcortical abnormalities, including microcephaly, ventriculomegaly, abnormal gyral and sulcal patterns (termed “dysgyria”), a small or absent corpus callosum, and a small pons. All patients with microtubule-associated protein mutations also had abnormal cerebral cortices (predominantly pachygyria and agyria), but fewer subcortical abnormalities were noted.
CONCLUSIONS:
Comparison of MRIs from patients with known mutations of tubulin genes and microtubule-associated proteins allows the establishment of some early correlations of phenotype with genotype and may assist in identification and diagnosis of these rare disorders.
The relationship of lissencephaly and pachygyria to mutations of specific genes, such as LIS1,1 DCX,2 and ARX,3 has been known for >20 years, with some mutations resulting in milder forms known as double cortex2 or band heterotopia.4 Many lissencephalies, in particular lissencephalies associated with significant microcephaly (head circumference of ≥3 SDs below the mean for age, also called “microlissencephalies”5), were not associated with mutations of any of these genes. In the past decade, studies have shown that many lissencephalies (and microlissencephalies) are associated with mutations of genes coding for tubulins, proteins that form the components of microtubules (MTs),6,7 and microtubule-associated proteins (MAPs)8 which, interestingly, include LIS1 and DCX. The recent literature has revealed that mutations of genes encoding tubulin and MAPs cause severe developmental delay, epilepsy, and severe anomalies of brain development. Although most mutations of both tubulin and MAP genes cause abnormal sulcation, tubulin gene mutations are associated with severe microcephaly and white matter anomalies, while patients with mutations of MAP genes have small normal brains or mild microcephaly with normal or nearly normal white matter structures. Specific MR imaging phenotype-genotype correlations have been elusive, despite examinations of large numbers of MR images.8–10 In this report, we describe the imaging appearances of 18 patients with brain malformations secondary to tubulin mutations and 15 patients with malformations secondary to tubulin-associated protein mutations, including cortical malformations, white matter disorders (corpus callosum, white matter volume, brain stem anomalies), and head size. These data allow establishment of some early correlations of phenotype with genotype.
Materials and Methods
Imaging
MRIs were retrieved from a large file of imaging studies of >2500 patients with malformations of cortical development that is part of ongoing studies of the genetics of epilepsy and cerebral malformations. The file includes MRIs of patients of all ages who were imaged during the past 30 years, mainly for developmental disorders or epilepsy, and were referred to one of the authors for evaluation; all consented to the use of their studies for research purposes. All patients have mutations identified in specific genes that are thought to be the cause of the malformation. Because these studies were obtained in many different facilities in multiple countries during >25 years, both the MR imaging techniques and the techniques for identifying mutations differed considerably. Most MRIs were obtained at 1.5T, though some recent scans were obtained at 3T. T1WI and T2WI (spin-echo or spoiled gradient-echo in more recent volumetric acquisitions) with section thicknesses ranging from 1 to 3 mm were analyzed. For inclusion, MRIs required 3 orthogonal planes with adequate contrast between the cerebral cortex and (myelinated or unmyelinated) white matter to determine the cortical gyral pattern, basal ganglia, and white matter to visually assess internal capsular presence or absence, cortical thickness (normal, <4 mm), ventricular and cisternal size, and the size and contours of the brain stem, cerebellar hemispheres and vermis, and the corpus callosum.
Patients
All patients were referred for seizures and developmental anomalies, often associated with microcephaly that was variable in severity. Genetic analyses required that the candidate gene have a logarithm of odds score of ≥3 or microarray demonstration of abnormal gene expression. Targeted genetic analyses during the past few years revealed 18 patients with confirmed diagnoses of tubulin mutations: 8 with TUBA1A mutations, 5 with TUBB2B mutations, and 5 with TUBB3 mutations. No TUBA8, TUBB5, or TUBG1 mutations were identified in any of our patients. The MR imaging findings of these patients were carefully analyzed and compared with those of 15 patients with known mutations of the MAP genes LIS1 (5 patients), DCX (4 patients), and DYNC1H1 (6 patients). Included patients ranged in age from 2 days to 33 years. Although the scans had already been reviewed and findings recorded, all were rereviewed by the authors. Results were then analyzed to determine which imaging features best defined mutations of tubulin and MAP genes, which features overlapped, and which features or combinations of features, if any, allowed specific diagnoses. Recorded results were determined by unanimous consensus of the authors.
Results
All patients had abnormal findings on brain MR imaging. In particular, abnormalities of white matter structures/pathways (corpus callosum, internal capsules, and portions of the brain stem), the cerebral cortex (gyral pattern), and the cerebellum (vermis more frequently than hemispheres) were seen in essentially all patients. When the olfactory sulci and bulbs were adequately visualized with coronal images, the sulcal depth and appearance of the bulbs were assessed. Abnormalities of the midbrain tectum were recorded when present.
Tubulin Mutations
All 18 patients with mutations of tubulin genes had diminished white matter volume, ranging from moderate to severe, associated, in all cases, with microcephaly (by provided clinical history). Ventriculomegaly was present in 8/8 patients with TUBA1A mutations, 3/5 patients with TUBB2B mutations, and 2/5 patients with TUBB3 mutations (Table). The degree of enlargement varied considerably, from mild to severe; in more severely affected patients, ventricular enlargement was diffuse (Figs 1 and 2), but in 2 TUBA1A mutations, ventriculomegaly was asymmetric (1 left > right, the other right > left). A third patient had colpocephaly with enlarged trigones and occipital horns but normal temporal horns, frontal horns, and ventricular bodies.
Features of tubulin and microtubule-associated gene mutations
| Tubulin Mutations |
MAP Mutations |
|||||
|---|---|---|---|---|---|---|
| TUBA1A | TUBB2B | TUBB3 | LIS1 | DCX | DYNC1H1 | |
| Dysmorphic/absent corpus callosum | 8/8a | 5/5 | 2/5 | 5/5b | 1/4 | 6/6c |
| Thin corpus callosum | 4/5 | 4/5 | 4/5 | 0/5 | 2/4 | 1/6d |
| Dysgyric cerebral cortex | 4/7e | 4/5 | 5/5 | 0/5 | 0/4 | 0/6 |
| Pachygyric/agyric cerebral cortex | 4/7e | 0/5 | 3/5 | 5/5f | 4/4g | 6/6h |
| Small cerebellar vermis | 8/8 | 5/5i | 4/5 | 2/5i | 0/4 | 3/6i |
| Small cerebellar hemisphere | 5/8 | 1/5 | 1/5j | 0/5 | 0/4 | 0/6 |
| Small brain stem | 7/8k | 5/5l | 5/5m | 3/5n | 0/3o | 3/6l |
| Large tectum | 4/8 | 1/5 | 1/5 | 0/5 | 0/4 | 0/6 |
| Mild ventriculomegaly | 1/8 | 2/5 | 0/5 | 0/5 | 0/4 | 2/6 |
| Moderate ventriculomegaly | 3/8 | 1/5 | 1/5 | 0/5 | 0/4 | 0/6 |
| Severe ventriculomegaly | 4/8 | 0/5 | 1/5 | 0/5 | 0/4 | 0/6 |
Corpus callosum absent 3/8.
L-shaped corpus callosum 4/5.
L-shaped corpus callosum 3/6.
Thick corpus callosum 5/6.
In 1 patient, poor gray-white differentiation precludes accurate cortical assessment.
Complete agyria 1/5; posterior agyria and anterior pachygyria 4/5.
Complete agyria 1/4; anterior pachygyria 3/4.
Posterior pachygyria 6/6.
Small anterior vermis in all cases.
Dysmorphic vermis.
Asymmetric 3/8.
Small pons in all cases.
Asymmetric in all cases.
Diffusely thin brain stem 2/5, small pons 1/5.
Brain stem not well-visualized, 1 case.
Fig 1.

A 5-year-old child with a TUBB3 mutation. Midline sagittal T1WI (A) shows a fully formed but thin corpus callosum, an enlarged third ventricle, a dysmorphic cerebellar vermis with a disproportionately small anterior vermis, and a thin pons. Axial T1WI (B) shows asymmetry of the basal ganglia with an unseparated left striatum (white arrowhead). Note that the gyral pattern is abnormal, with sulci coursing deeply at many different angles and in unusual locations (white arrows). This abnormal pattern is classified as “dysgyria.” Axial T1WI at a slightly higher level of the same patient (C) shows again the dysmorphic, asymmetric basal ganglia with an enlarged left caudate head (black arrows) and marked ventriculomegaly. Cortical dysgyria is again noted (white arrows).
Fig 2.

A 7-week-old infant with a TUBB2B mutation. Midline sagittal T1WI (A) shows a short, thin corpus callosum; a small cerebellar vermis; and a thin pons with a disproportionately large tectum (white arrow). Axial T2WI (B–D from superior to inferior) reveals severe ventriculomegaly. There is diffuse cortical dysgyria with bilateral frontal lobe band heterotopia (B). More inferiorly, there are abnormally enlarged caudates with fused striata (C) and an asymmetric, small pons (D), smaller on the right (black arrowhead) with a central cleft.
All patients had abnormalities of the corpus callosum, dysmorphic or absent in all patients with TUBA1A or TUBB2B mutations and either dysmorphic (2/5) or abnormally thin (4/5) in patients with TUBB3 mutations (Figs 1–3 and On-line Fig 3). All patients with tubulin gene mutations also had abnormalities of the basal ganglia, including an enlarged caudate and an absent or diminutive internal capsule dividing the caudate from the putamen, resulting in a fused striatum (Figs 1–3). Additionally, nearly all patients with tubulin gene mutations had a small, often asymmetric brain stem (Table, Fig 2D, and On-line Fig 2) and a small cerebellar vermis; and a number had large midbrain tecta (Figs 2 and 3).
Fig 3.

Patients with TUBA1A mutations. Midline sagittal T1WI in a 2-day-old patient (A) shows a thick, dysmorphic corpus callosum; a small cerebellar vermis; and a thin pons with a disproportionately large tectum. Axial T2WI in the same patient (B) shows cortical lissencephaly with a cell sparse zone, enlarged caudate heads with fused striata, and mild ventriculomegaly. Midline sagittal (C) and axial (D) T1WI from a different 3-year-old patient with TUBA1A mutation reveals contrasting dysgyric cortex (D) and a thin corpus callosum with an absent genu and rostrum (C). Again note a thin pons and moderate ventriculomegaly.
Abnormalities of the cerebral cortex were seen in essentially all patients in this group, ranging from complete agyria to mixtures of agyria and pachygyria to complete “dysgyria” (a term intended to describe a cortex of normal thickness but with an abnormal gyral pattern characterized by abnormalities of sulcal depth or orientation: obliquely oriented sulci directed radially toward the center of the cerebrum and narrow gyri separated by abnormally deep or shallow sulci (Fig 1). Other patients had regions of pachygyria mixed with regions of dysgyria. Of the 5 patients with TUBB3 mutations, 3 had both dysgyria and pachygyria, with pachygyria located in the Sylvian and supra-Sylvian areas. The cortex in patients with TUBA1A mutations ranged from lissencephalic to pachygyric to dysgyric, with pachygyria usually present in the parietal and occipital lobes (Fig 4). Differentiation of regional pachygyria (with a cell sparse zone) from localized band heterotopia was based on the widths of the gyri and depths of the sulci: For classification, where sulci were deeper than the breadth of the gyri, the diagnosis of band heterotopia was made; and if the sulci were shallower than the breadth of the gyri, pachygyria was diagnosed. Four of the 5 patients with TUBB3 mutations exhibited dysgyria of varying severity, including 1 patient with both diffuse dysgyria and bifrontal band heterotopia (Fig 2). The fifth patient had nearly normal cerebrocortical morphology. Hypoplastic or absent olfactory bulbs or sulci were noted in 11/16 patients with tubulin mutations (4/6 TUBA1A, 3/5 TUBB2B, 4/5 TUBB3) for which they were adequately evaluated (On-line Fig 1).
Fig 4.

Patients with microtubule-associated protein mutations. Midline sagittal T1WI (A), axial T1WI (B), and T2WI (C) in an 8-month-old patient with a DYNC1H1 mutation. An abnormal corpus callosum with an absent inferior genu and rostrum and an L-shaped angulation of the body and splenium with small pons (A) are noted. Axial images (B and C) reveal parietal pachygyria with a cell sparse zone. Midline sagittal T1WI in a 7-year-old patient with a DCX mutation shows normal midline structures, including the corpus callosum, pons, cerebellar vermis, and tectum (D). Axial images from the same patient with a DCX mutation (E and F) reveal diffuse shallow sulci consistent with pachygyria and moderate band heterotopia. Midline sagittal T1WI from a 33-year-old patient with a LIS1 mutation (G) demonstrates a small anterior vermis and pons, with a mildly dysmorphic corpus callosum (small inferior genu and short splenium). Axial T2WI (H and I) from the same patient with a LIS1 mutation shows a typical cortical anteroposterior gradient with frontal pachygyria progressing to agyria in the parietal and occipital lobes. A thin cell sparse zone is present in the parietal and occipital lobes (H, white arrows). The basal ganglia are normal (I).
MAP Mutations
Of the 15 patients with mutations of MAPs, the imaging findings were more consistent within groups (Table). Patients with LIS1 mutations (5 total) had cortical malformations ranging from complete agyria to frontal pachygyria or shallow sulci with nearly normal cortical thickness associated with parietal and occipital pachygyria or agyria. They had anomalies of the corpus callosum, with a small or absent rostrum and inferior genu, normal body, and an unusual, sharp angle at the junction of the body and splenium (Fig 4), with the splenium coursing more caudal than usual (4/5) or a short splenium (1/5). These findings were very similar to the callosal anomaly seen in patients with DYNC1H1 mutations (6 total), who also had posterior pachygyria (less severe, overall, than that seen in the patients with LIS1 mutations, but some cases were indistinguishable by imaging).
Half (3/6) of the patients with DYNC1H1 mutations had a slightly small anterior cerebellar vermis compared with published norms,11 compared with 1/5 patients with LIS1 mutations and no patient with DCX mutations. This vermian anomaly and the presence of large caudate heads (found in all 6/6 patients with DYNC1H1 mutations [Fig 4], but in no [0/5] patients with LIS1 mutations) were the only consistent differences between patients with LIS1 and DYNC1H1 mutations.
The patients with DCX mutations had different appearances of the corpus callosum, which were either normal (2/4) or diffusely thin (2/4). The cortex of patients with DCX mutations, as has been reported,12 varied from normal thickness to slightly thick, associated with shallow sulci and a layer of neurons (band heterotopia4) deep to the cortex; the cortical anomaly was more severe frontally than in the occipital cortex. No cerebellar or brain stem anomalies were identified; however, large caudate heads were identified in 3/4 patients with DCX mutations, and cerebral white matter volume was mildly or moderately diminished in 3/4.
Discussion
Microtubules are filamentous intracellular structures critical for long-range intracellular cargo transport in neurons and, therefore, important in brain formation and function.13 They have key roles in mitosis, organization of intracellular structure, axonal pathfinding, neuronal migration, protein transport, and ciliary motility.13–15 Not surprising, disorders of MT formation affect multiple aspects of brain development, resulting in multiple malformations, including microcephaly (presumably from impaired mitosis), lissencephaly, band heterotopia and other types of cortical dysgenesis (impaired neuronal migration), anomalies of white matter pathways (impaired axonal pathfinding), anomalies of the cranial nerves (impaired axonal pathfinding), and malformations of the midbrain and hindbrain (possibly impairment of both neuronal migration and axonal pathfinding); these malformations have some characteristic features that allow them to be recognized on routine MR imaging.9,15–23
Similar malformations had been previously reported in patients with mutations of genes encoding MAPs, such as LIS1 and DCX1,2,12 that bind to MTs and aid long-range intracellular transport into distal axons, dendrites, and leading processes (actin-based transport is more important for short-range transport to synapses and axonal growth cones24). Recent revelations on MT functions and their interactions with MAPs in so many developmental processes make it clear that disorders of MT and MAP formation and function cause many developmental brain disorders.8,13,25–29 Although some MR imaging findings of cortical malformations and cranial neuropathies associated with mutations of tubulins and MAPs have been described, the precise nature of the cortical anomalies (which have been nearly uniformly called “pachygyria” and “polymicrogyria”) and the other aspects of abnormal brain development associated with them, such as callosal anomalies, brain stem anomalies, and cerebellar anomalies, have not been previously compared and contrasted, to our knowledge. In making this comparison, we have discovered several MR imaging findings that may help narrow the search for genes responsible for malformations.
A brief description of how microtubules form and function may be useful in this discussion. MTs form by polymerization of tubulin heterodimers, which are composed of the 2 main tubulin proteins, α-tubulin (formed from proteins constructed by transcription and translation of TUBA genes) and β-tubulin (from TUBB genes), which spontaneously combine (dimerize) to form α-β-heterodimers. To initiate the formation of an MT, heterodimers bind to a γ-tubulin ring complex, composed of γ-tubulins (formed by transcription and translation of TUBG genes) bound to γ-tubulin complex proteins in the wall of a centrosome.25 The γ-tubulin ring complex functions as a scaffold, with exposed γ-tubulin on its periphery, to which α-β-heterodimers bind and begin to polymerize, ultimately forming the MT (Fig 5D).25 The advancing ends of the MTs, at the growth cone of a pathfinding axon or the leading process of a migrating neuron, follow actin and myosin scaffolding that interacts with chemical signals in the interstitium (Fig 5B, -C).24 Attractive signals cause rapid growth of the actin in that direction, which quickly stimulates MT polymerization in its wake, whereas repulsive signals cause actin breakdown, quickly followed by MT depolymerization.24,26
Fig 5.
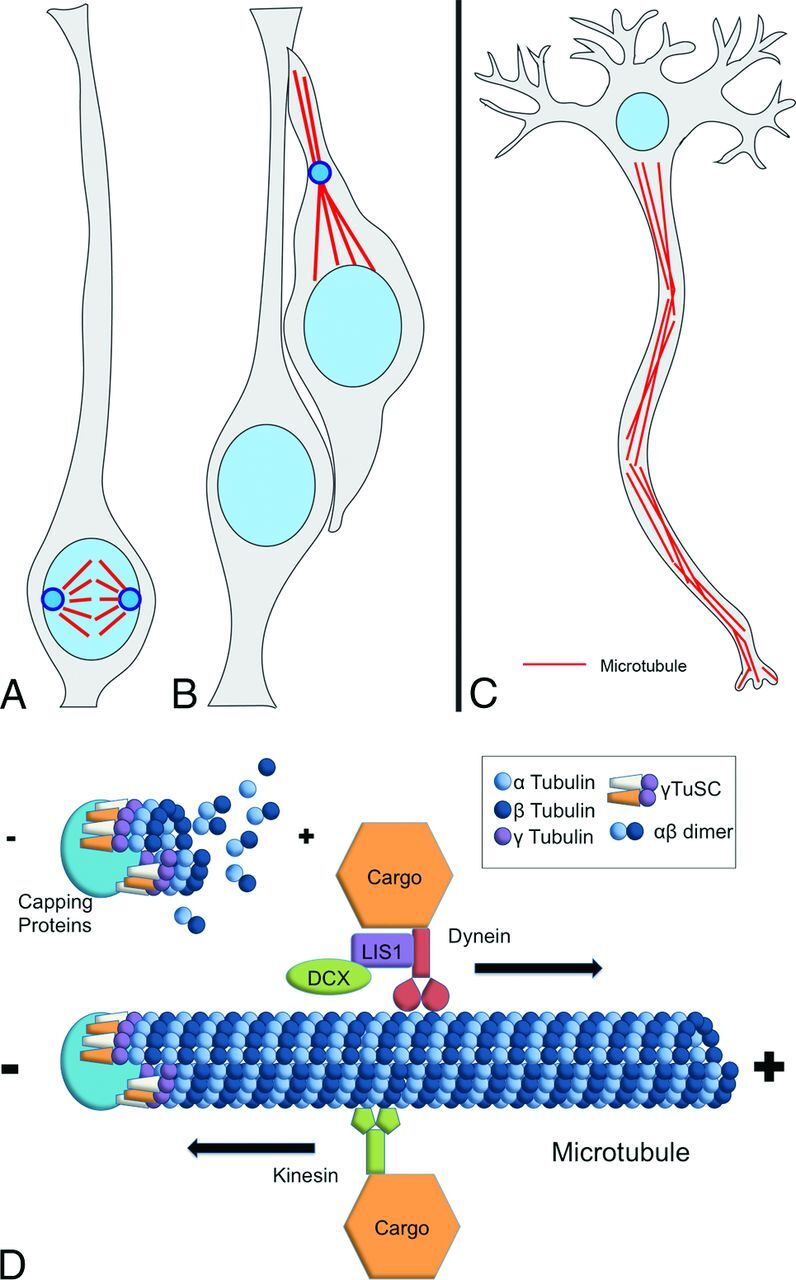
Multiple roles of microtubules in development. Schematic illustrates the importance of microtubules in key developmental functions, including mitosis/proliferation (A), migration (B), and axonal pathfinding/transport (C). Microtubules along with their associated proteins form attachments between chromosomes and centrosomes, establishing polarity in dividing neural progenitor cells, such as radial glia (A), during neurogenesis and providing the mechanical apparatus of mitosis and cell division. Following cell division, neuroblasts (immature neurons) migrate (B) from the germinal ventricular zone along the pial process of their mother radial glial cell into the developing cortical plate through the process of nucleokinesis, where the nucleolus are pulled along microtubules following the leading process of neuroblasts. Later in development, microtubules appear to play multiple important functional roles in projection neurons (C), including polarization, axonal pathfinding, synaptogenesis, and transport of intracellular materials along the axon. D, The prevailing “template model” for microtubule polymerization. γ-Tubulin molecules (usually 13) together with their associated scaffolding and capping proteins form the γ-tubulin ring complex, which serves as a template for polymerizing α-tubulin and β-tubulin dimers, forming a cylindric complex of filaments forming the microtubule. Microtubules are intrinsically polarized with distinct positive and negative ends. Microtubule-associated proteins include the molecular motor dynein (including DYNC1H1) and kinesis, which transport intracellular cargo along microtubules toward the positive and negative ends, respectively, as well as proteins such as LIS1 and DCX.
Thus, the MT plays a critical role in the navigation and formation of axons and neurons through the developing brain.13,27,28 The same process occurs during mitosis as the MTs migrate from centrosomes to attach to and separate chromosomes and begin cell division (Fig 5A). Thus, MTs are critical to the processes of mitosis, axon navigation, and neuron migration, all key factors in development. During neuronal migration, an important role is also played by MAPs (in particular LIS1, NudE, and Dynein), which are critical to the process of nucleokinesis, by which the nucleus of the migrating neuron is pulled forward after its leading process advances29; in the absence of nucleokinesis, the neuron does not migrate. The precise role of MAPs in axonal navigation is less well-established, but it is becoming clear that external axonal guidance cues in the brain interstitium couple with other molecules in the axonal growth cone (such as ROBO3 or DCC) to direct microtubule dynamics.30,31 It is entirely possible (and, indeed, likely) that MAPs participate in this process to some extent.
Knowledge of the developmental functions of MTs and MAPs helps in understanding imaging findings. MTs play a key role in mitosis; therefore, cell proliferation is diminished in patients who have mutations of MT genes. In addition, patients with tubulin mutations (TUBA1A, TUBB2B, TUBB3, TUBG, and so forth) almost universally have microcephaly6,8–10; the most severely affected do not survive gestation.32 A review of the recent literature indicates that patients with MT mutations very commonly have severe microcephaly, some profoundly so, and the severity of the microcephaly seems to be directly related to the severity of the brain malformation: most severe in microlissencephaly followed by lissencephaly and pachygyria and least severe with dysgyria (60% of these patients are normocephalic).9 The microcephaly seems likely to be related to abnormalities of mitosis. In contrast, patients with MAP mutations have much milder impairment of brain growth,33,34 with most normocephalic, and those with small heads usually mildly affected.
Because neuronal migration is disturbed in tubulinopathies, enlarged or asymmetric basal ganglia, heterotopias, and cortical dysgenesis are common. Pachygyria and agyria appear to be most common in TUBA1A and TUBG mutations. However, the MR imaging appearance of the cortex in the patients in our study and those described in the literature does not resemble true polymicrogyria. Pathologic analysis of the brains with tubulin mutations showed migration of neurons through gaps in the pial-limiting membrane into the subarachnoid space,32 which is not found in polymicrogyria. Moreover, the appearance of a smooth cortical surface with radially oriented sulci, even on high-resolution images, is not consistent with polymicrogyria. Because the appearance in these patients has not been previously described, we have coined the term “dysgyria” to describe a malformation in which sulci course at unusual angles and unusual depths, an appearance that has been illustrated in published MR imaging figures from patients with mutations of all the tubulin genes described at this time9,17,19,22,23 but not discussed in detail. We suggest that recognition of this characteristic feature aids in the diagnosis of tubulinopathy, especially when associated with microcephaly. In addition, the anterior cerebellar vermis is very commonly small, and the midbrain tectum is often large (which can be a result of abnormal migration of cerebellar granule cells35).
Finally, because axonal pathfinding is disturbed, patients with tubulin mutations may have congenital fibrosis of the extraocular muscles or hypoplastic olfactory nerves18; hypoplasia or absence of the corpus callosum; a small, often asymmetric brain stem; abnormal-appearing, fused striatum due to the absence of various parts of the internal capsule (most commonly the anterior limb); and diminished overall white matter volume.8,9 All of these findings were seen in some of our patients, and nearly all were seen in many of the patients with tubulin mutations.
Of particular interest in this study is the observation that abnormalities of axon pathfinding were much more common in patients with mutations of MT genes compared with those with MAP genes, while the abnormalities of neuronal migration were not. Therefore, these findings appear to be important in making the differentiation. Eighteen of 19 patients with MT gene mutations had a dysmorphic or absent corpus callosum (in the other, the corpus was uniformly thin). Most of the dysmorphic ones were thin and incompletely formed, with an absent rostrum and inferior genu and a thin, short splenium. One was of normal thickness but very short (rostrocaudally). Although only 2/15 patients with MAP gene mutations had a normal corpus callosum, 7 of them (4 with LIS1 mutations and 3 with DYNC1H1 mutations) had a very similar finding of a rather thick corpus callosum with a small inferior genu, absent rostrum, and a rather sharp angle in the posterior half of the callosal body, resulting in an overly vertical splenium situated within a few millimeters of the superior colliculus (Fig 4A). The brain stem was small in 17/18 patients with MT gene mutations and asymmetric in 8, but a small brain stem was seen in only 6/15 patients with MAP gene mutations and all brain stems were symmetric. Mutations of other axonal pathfinding genes, such as ROBO3,36 can cause similar (though usually symmetric) clefts in the pons and medulla, but they are not associated with cortical malformations. Therefore, a finding of a small, asymmetric brain stem with a midline cleft in the setting of cortical malformations should strongly suggest a tubulin mutation as the cause.
In terms of cortical malformations, areas of dysgyria were present in 13/17 patients with MT gene mutations, in whom the cortex was adequately visualized, while pachygyria was seen in 7. Peri-Sylvian pachygyria was seen in addition to dysgyria in 3/5 patients with TUBB3 mutations. In contrast, dysgyria was not seen in patients with MAP gene mutations, while pachygyria was present in all, mostly in the posterior parietal/occipital regions. Only 5/15 patients with MAP mutations (3/6 with DYNC1H1 mutations) had a small vermis (in contrast to 17/18 of patients with tubulin gene mutations); notably, anterior vermis atrophy predominated in both groups. No patients with predominantly posterior vermian atrophy were seen.
Conclusions
The human brain is formed in a complex and highly regulated process that includes neurogenesis (proliferation of progenitor cells and production of immature neurons), neuronal migration (migration of immature neurons from germinal zones into organized gray matter structures), and connectivity (formation of white matter tracts by extension of axons). Microtubules and their associated proteins are integral components of all of these processes in the developing brain; thus, their mutations can result in complex, overlapping imaging phenotypes. Diagnostically, these features can be useful because combinations of findings seen in other malformations of cortical development, when present together, can suggest a disorder of MT function. For example, in a patient with the combination of microcephaly, dysgyria, and a small corpus callosum and brain stem, the diagnosis of a tubulin mutation should be considered. It follows logically that while tubulin gene mutations often affect all categories (neurogenesis, migration and axonal pathfinding), mutations of MAPs often have more limited phenotypes, suggesting specialized or redundant functions. MAP gene mutations most notably caused errors in neuronal migration (particularly pachygyria and agyria), while axonal pathfinding abnormalities were much more common in the patients with tubulin mutations.
Supplementary Material
ABBREVIATIONS:
- MAP
microtubule-associated protein
- MT
microtubule
Footnotes
Disclosures: Annapurna Poduri—RELATED: Grant: National Institute of Neurological Disorders and Stroke K23 grant.* Mustafa Sahin—UNRELATED: Grants/Grants Pending: Novartis,* Shire,* Comments: I am the Principal Investigator of the site of grants from Novartis related to tuberous sclerosis. I am the Principal Investigator of a grant from Shire related to spinal muscular atrophy. Christopher A. Walsh—RELATED: Grant: National Institute of Neurological Disorders and Stroke,* National Institute of Mental Health,* Howard Hughes Medical Institute; UNRELATED: Consultancy: Hoffman–La Roche, Third Rock Ventures; Grants/Grants Pending: National Institute of Mental Health, National Institute of Neurological Disorders and Stroke,* Howard Hughes Medical Institute. Anthony James Barkovich—RELATED: Grant: National Institutes of Health/National Institute of Neurological Disorders and Stroke.* *Money paid to the institution.
This work was funded by the US Department of Health and Human Services–National Institutes of Health-National Institute of Biomedical Imaging and Bioengineering (2T32EB001631-11).
References
- 1. Dobyns W, Reiner O, Carrozzo R, et al. Lissencephaly: a human brain malformation associated with deletion of the LIS1 gene located at chromosome 17p13. JAMA 1993;270:2838–42 10.1001/jama.1993.03510230076039 [DOI] [PubMed] [Google Scholar]
- 2. Gleeson JG, Allen KA, Fox JW, et al. Doublecortin, a brain-specific gene mutated in human X-linked lissencephaly and double cortex syndrome, encodes a putative signaling protein. Cell 1998;92:63–72 10.1016/S0092-8674(00)80899-5 [DOI] [PubMed] [Google Scholar]
- 3. Kitamura K, Yanazawa M, Sugiyama N, et al. Mutation of ARX causes abnormal development of forebrain and testes in mice and X-linked lissencephaly with abnormal genitalia in humans. Nat Genet 2002;32:359–69 10.1038/ng1009 [DOI] [PubMed] [Google Scholar]
- 4. Barkovich AJ, Jackson DE Jr, Boyer RS. Band heterotopia: a newly recognized neuronal migration anomaly. Radiology 1989;171:455–58 10.1148/radiology.171.2.2468173 [DOI] [PubMed] [Google Scholar]
- 5. Barkovich AJ, Ferriero DM, Barr RM, et al. Microlissencephaly: a heterogeneous malformation of cortical development. Neuropediatrics 1998;29:113–19 10.1055/s-2007-973545 [DOI] [PubMed] [Google Scholar]
- 6. Poirier K, Keays DA, Francis F, et al. Large spectrum of lissencephaly and pachygyria phenotypes resulting from de novo missense mutations in tubulin alpha 1A (TUBA1A). Hum Mutat 2007;28:1055–64 10.1002/humu.20572 [DOI] [PubMed] [Google Scholar]
- 7. Bahi-Buisson N, Poirier K, Boddaert N, et al. Refinement of cortical dysgeneses spectrum associated with TUBA1A mutations. J Med Genet 2008;45:647–53 10.1136/jmg.2008.058073 [DOI] [PubMed] [Google Scholar]
- 8. Poirier K, Lebrun N, Broix L, et al. Mutations in TUBG1, DYNC1H1, KIF5C and KIF2A cause malformations of cortical development and microcephaly. Nat Genet 2013;45:639–47 10.1038/ng.2613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bahi-Buisson N, Poirier K, Fourniol F, et al. ; LIS-Tubulinopathies Consortium. The wide spectrum of tubulinopathies: what are the key features for the diagnosis? Brain 2014;137:1676–700 10.1093/brain/awu082 [DOI] [PubMed] [Google Scholar]
- 10. Cushion TD, Dobyns WB, Mullins JG, et al. Overlapping cortical malformations and mutations in TUBB2B and TUBA1A. Brain 2013;136:536–48 10.1093/brain/aws338 [DOI] [PubMed] [Google Scholar]
- 11. Doherty D, Millen KJ, Barkovich AJ. Midbrain and hindbrain malformations: advances in clinical diagnosis, imaging, and genetics. Lancet Neurol 2013;12:381–93 10.1016/S1474-4422(13)70024-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dobyns WB, Truwit CL, Ross ME, et al. Differences in the gyral pattern distinguish chromosome 17-linked and X-linked lissencephaly. Neurology 1999;53:270–77 10.1212/WNL.53.2.270 [DOI] [PubMed] [Google Scholar]
- 13. Franker MA, Hoogenraad CC. Microtubule-based transport: basic mechanisms, traffic rules and role in neurological pathogenesis. J Cell Sci 2013;126:2319–29 10.1242/jcs.115030 [DOI] [PubMed] [Google Scholar]
- 14. Kuijpers M, Hoogenraad CC. Centrosomes, microtubules and neuronal development. Mol Cell Neurosci 2011;48:349–58 10.1016/j.mcn.2011.05.004 [DOI] [PubMed] [Google Scholar]
- 15. Jaglin XH, Chelly J. Tubulin-related cortical dysgeneses: microtubule dysfunction underlying neuronal migration defects. Trends Genet 2009;25:555–66 10.1016/j.tig.2009.10.003 [DOI] [PubMed] [Google Scholar]
- 16. Breuss M, Heng JI, Poirier K, et al. Mutations in the β-tubulin gene TUBB5 cause microcephaly with structural brain abnormalities. Cell Rep 2012;2:1554–62 10.1016/j.celrep.2012.11.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cushion TD, Paciorkowski AR, Pilz DT, et al. De novo mutations in the beta-tubulin gene TUBB2A cause simplified gyral patterning and infantile-onset epilepsy. Am J Hum Genet 2014;94:634–41 10.1016/j.ajhg.2014.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chew S, Balasubramanian R, Chan WM, et al. A novel syndrome caused by the E410K amino acid substitution in the neuronal β-tubulin isotype 3. Brain 2013;136:522–35 10.1093/brain/aws345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jaglin XH, Poirier K, Saillour Y, et al. Mutations in the beta-tubulin gene TUBB2B result in asymmetrical polymicrogyria. Nat Genet 2009;41:746–52 10.1038/ng.380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kumar RA, Pilz DT, Babatz TD, et al. TUBA1A mutations cause wide spectrum lissencephaly (smooth brain) and suggest that multiple neuronal migration pathways converge on alpha tubulins. Hum Mol Genet 2010;19:2817–27 10.1093/hmg/ddq182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Morris-Rosendahl DJ, Najm J, Lachmeijer AM, et al. Refining the phenotype of alpha-1a tubulin (TUBA1A) mutation in patients with classical lissencephaly. Clin Genet 2008;74:425–33 10.1111/j.1399-0004.2008.01093.x [DOI] [PubMed] [Google Scholar]
- 22. Poirier K, Saillour Y, Bahi-Buisson N, et al. Mutations in the neuronal β-tubulin subunit TUBB3 result in malformation of cortical development and neuronal migration defects. Hum Mol Genet 2010;19:4462–73 10.1093/hmg/ddq377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Romaniello R, Tonelli A, Arrigoni F, et al. A novel mutation in the β-tubulin gene TUBB2B associated with complex malformation of cortical development and deficits in axonal guidance. Dev Med Child Neurol 2012;54:765–69 10.1111/j.1469-8749.2012.04316.x [DOI] [PubMed] [Google Scholar]
- 24. Kneussel M, Wagner W. Myosin motors at neuronal synapses: drivers of membrane transport and actin dynamics. Nat Rev Neurosci 2013;14:233–47 10.1038/nrn3445 [DOI] [PubMed] [Google Scholar]
- 25. Kollman JM, Merdes A, Mourey L, et al. Microtubule nucleation by γ-tubulin complexes. Nat Rev Mol Cell Biol 2011;12:709–21 10.1038/nrm3209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Slováková J, Speicher S, Sánchez-Soriano N, et al. The actin-binding protein Canoe/AF-6 forms a complex with Robo and is required for Slit-Robo signaling during axon pathfinding at the CNS midline. J Neurosci 2012;32:10035–44 10.1523/JNEUROSCI.6342-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tanaka E, Ho T, Kirschner MW. The role of microtubule dynamics in growth cone motility and axonal growth. J Cell Biol 1995;128:139–55 10.1083/jcb.128.1.139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tanaka EM, Kirschner MW. Microtubule behavior in the growth cones of living neurons during axon elongation. J Cell Biol 1991;115:345–63 10.1083/jcb.115.2.345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Huang J, Roberts Anthony J, Leschziner Andres E, et al. Lis1 acts as a “clutch” between the ATPase and microtubule-binding domains of the dynein motor. Cell 2012;150:975–86 10.1016/j.cell.2012.07.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Qu C, Dwyer T, Shao Q, et al. Direct binding of TUBB3 with DCC couples netrin-1 signaling to intracellular microtubule dynamics in axon outgrowth and guidance. J Cell Sci 2013;126:3070–81 10.1242/jcs.122184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu G, Dwyer T. Microtubule dynamics in axon guidance. Neurosci Bull 2014;30:569–83 10.1007/s12264-014-1444-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fallet-Bianco C, Laquerriere A, Poirier K, et al. Mutations in tubulin genes are frequent causes of various foetal malformations of cortical development including microlissencephaly. Acta Neuropathol Commun 2014;2:69 10.1186/2051-5960-2-69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bahi-Buisson N, Souville I, Fourniol FJ, et al. ; SBH-LIS European Consortium. New insights into genotype-phenotype correlations for the doublecortin-related lissencephaly spectrum. Brain 2013;136(pt 1):223–44 10.1093/brain/aws323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Uyanik G, Morris-Rosendahl D, Stiegler J, et al. Location and type of mutation in the LIS1 gene do not predict phenotypic severity. Neurology 2007;69:442–47 10.1212/01.wnl.0000266629.98503.d0 [DOI] [PubMed] [Google Scholar]
- 35. Goldowitz D, Hamre K, Przyborski S, et al. Granule cells and cerebellar boundaries: analysis of Unc5h3 mutant chimeras. J Neurosci 2000;20:4129–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jen JC, Chan WM, Bosley TM, et al. Mutations in a human ROBO gene disrupt hindbrain axon pathway crossing and morphogenesis. Science 2004;304:1509–13 10.1126/science.1096437 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.