

Abstract
In the absence of extracellular stimulation the adaptor protein Grb2 and the phospholipase Plcγ1 compete for the same binding site on FGFR2. Reducing cellular Grb2 results in up-regulation of Plcγ1 and depletion of the phospholipid PI(4,5)P2. The functional consequence of this event on signaling pathways are unknown. We show that the decrease in PI(4,5)P2 level under non-stimulated conditions inhibits PTEN activity leading to the aberrant activation of the oncoprotein Akt. This results in excessive cell proliferation and tumor progression in a xenograft mouse model. As well as defining a novel mechanism of Akt phosphorylation with important therapeutic consequences, we also demonstrate that differential expression levels of FGFR2, Plcγ1 and Grb2 correlate with patient survival. Oncogenesis through fluctuation in the expression levels of these proteins negates extracellular stimulation or mutation and defines them as novel prognostic markers in ovarian cancer.
Introduction
Prolonged growth factor deprivation during tumor growth can paradoxically contribute to the progression of the oncogenic process1,2. Indeed, depletion of mitogenic signals through serum starvation has been shown to induce excessive proliferation in cancer cells2-7. Under these ‘basal’ conditions, non-homeostatic signaling has been linked to tyrosine kinases including fibroblast growth factor receptor 2 (FGFR2) even in the absence of autocrine/paracrine response8-10. However, the exact mechanism and underlying pathways for this oncogenic process remain elusive.
FGFR2 is vital for normal cellular processes but can also induce mitogenesis, angiogenesis and chemo-resistance in several types of cancer11-14. We previously reported that, in the absence of extracellular stimulation, growth factor receptor bound protein 2 (Grb2) and phospholipase C gamma 1 (Plcγ1) compete for the same binding site on FGFR2 to regulate cellular homeostasis and the membrane lipid pool. Under conditions of Grb2 depletion, Plcγ1 binds to FGFR2 and is aberrantly activated in a tyrosyl phosphorylation-independent manner leading to the excessive hydrolysis of its plasma membrane-localized phosphatidylinositol 4,5, bisphosphate (PI(4,5)P2) substrate10. PI(4,5)P2 is critical for recruitment of effector molecules and normal cellular processes15,16 but the mechanisms of aberrant signaling resulting from fluctuation in the membrane pool of PI(4,5)P2 remain poorly understood. Thus, whether variations in the PI(4,5)P2 level contribute to FGFR2-mediated oncogenic outcome remains an open question.
One of the main signaling pathways linked to PI(4,5)P2 concentration is the PI3K-Akt pathway which mediates survival and proliferation and can be aberrantly activated in an array of cancers17. This pathway is driven by the activity of class I phosphatidylinositol 3-kinases (PI3Ks) which phosphorylate PI(4,5)P2 to produce phosphatidylinositol 3,4,5-triphosphate (PI(3,4,5)P3)18 which in turn recruits a discrete set of pleckstrin homology (PH) domain-containing proteins to the membrane where they become activated. The main effector of this pathway is the serine/threonine protein kinase Akt which is activated on recruitment to the membrane localised PI(3,4,5)P319,20. The activity of PI3K is antagonized by phosphatase and tensin homologue deleted on chromosome 10 (PTEN) which dephosphorylates PI(3,4,5)P3 to PI(4,5)P2 (Supplementary Figure 1a) and can be conformationally affected by its lipid product PI(4,5)P221,22.
Hyperactivation or loss-of-function of the key proteins in the PI3K/PTEN/Akt pathway contribute to cancer development and they are frequently deregulated in gynecologic malignancies23-27. For example, the phosphorylation level of the oncoprotein Akt is an indicator of the response or resistance to therapy in ovarian cancer26. Interestingly FGFR2 expression is also implicated in the progression of this gynecological cancer responsible for the highest mortality rate of all types of female reproductive cancer13. However the connection between FGFR2 expression and Akt activation remain poorly understood. These observations raise the question as to whether it is possible for Akt to be activated via FGFR2 in the absence of mitogens through the previously observed perturbation of levels of phospholipids. If so what are the underlying mechanism and functional effects? And how does the expression of FGFR2, Grb2 and Plcγ1 correlate with level of Akt phosphorylation?
In this study we show that Grb2 depletion in non-stimulated cells leads to the inhibition of PTEN by drastically decreasing the levels of PI(4,5)P2 which results in the phosphorylation and activation of Akt. These findings support the notion that mitogens are not the sole regulators of the Akt pathway28,29 but that fluctuation in the expression levels of FGFR2, Grb2 and Plcγ1 are also competent in this role. Our findings demonstrate that variations in the levels of these proteins that influence membrane lipid concentration can modulate proliferation in cell lines and tumor formation in a xenograft mouse model. This novel mechanism occurring in our model cell line translates to ovarian cancer cell lines and to patient samples in which FGFR2, Grb2 and Plcγ1 concentrations correlate with Akt phosphorylation and clinical outcome. Therefore respective concentrations of these proteins provide novel prognostic markers of patient survival.
Results
Grb2-depletion in serum starved, FGFR2-expressing cells increases colony formation
Human embryonic kidney cells (HEK293T) represent an ideal initial model system to test receptor specificity because they lack endogenous FGFR2 (see parental HEK293T cells; Supplementary Figure 2a). We used these cells either untransfected (parental HEK293T cells: negative controls), or transfected with GFP-FGFR2 (Supplementary Figure 2a and b show the efficiency of transfection and knockdown for all cells adopted in this work) at an expression level that mimics that of FGFR2-dependent cancer cells9. By knocking down Grb2 in FGFR2-expressing cells (G2i cells) anchorage-independent colony formation was dramatically increased (in number and size imaged under the same scale) compared to control cells with scrambled shRNA (Ci cells). This effect was completely abrogated in FGFR2-expressing cells with Plcγ1 knocked down (Pγi cells) and in FGFR2-expressing cells with Grb2/Plcγ1 double knockdown (DKD) (Figure 1a and b). The observed proliferative outcome was shown to be FGFR2-dependent since in the parental HEK293T cells (PCi) and parental HEK293T cells with Grb2 or Plcγ1 knocked down (PG2i, PPγi respectively) colony formation was negligible compared to FGFR2-expressing cells (Figure 1a and b). The lack of involvement of the kinase activity of FGFR2 was confirmed after treating the cells with SU5402 (FGFR2 kinase inhibitor). We observed that in the absence of receptor kinase activity the level colony formation was unaffected in G2i cells (Figure 1c and d and Supplementary Figure 2c for inhibition efficiency). Thus a link between Grb2 and Plcγ1 concentrations (in the presence of FGFR2) and colony formation appears to exist under basal conditions. Colony formation after FGF9 stimulation of Ci and G2i cells resulted in a comparable number of colonies between the two cell lines where phosphorylation of extracellular signal-regulated kinase (Erk; aka. MAPK) was used to detect efficient stimulation (Supplementary Figure 2d and e).
Figure 1. Akt pathway-dependent colony formation is enhanced in Grb2 knockdown cells.

(a) HEK293T cells with and without FGFR2 were transfected with scrambled shRNA, Grb2 shRNA and/or Plcγ1 shRNA to generate stable knockdowns: PCi, PG2i and PPγi for parental cells, and Ci, G2i, Pγi and Grb2/Pγi (double knockdown, DKD) for cells transfected with GFP-FGFR2. 0.6 × 104 cells were cast as a single cell suspension in serum-deprived DMEM/agar in the upper layer with DMEM/agar with 1% FBS in the lower layer. Cells were left to grow for 8 days. DMEM with 1% FBS was added to the wells once every seven days. Pictures of the entire wells were taken. Pictures of colonies made from Ci or G2i cells transfected with GFP-FGFR2 were taken at 20X magnification to show the difference in colony size formed.
(b) Colonies formed in each case in (a) were counted and averaged in 4 microscopic fields (independent experiments n=3). Error bars on the graph represent the standard deviation of the calculated mean. Student's t-test indicates p≤ 0.01 which denotes a significant increase in the number of colonies formed upon Grb2 depletion in FGR2 expressing cells.
(c) Same as (a) but in this case DMEM with 1% FBS in the presence of the indicated inhibitors was added to the relevant wells once every seven days. Only cells transfected with GFP-FGFR2 (Ci and G2i cells) were utilized. See also Supplementary Figure 2c, f-I for efficiency of inhibition.
(d) Colonies formed in each case in (c) were counted and averaged in 4 microscopic fields (independent experiments n=3). Error bars on the graph represent the standard deviation of the calculated mean. Student's t-test indicates *p≤ 0.05 and **p≤ 0.01.
(e) Heat map was generated via the R console statistical package. It summarizes the normalized linear RPPA values of the results obtained using 3 independent samples from Ci and G2i cells (unstimulated or stimulated with FGF9) as explained in the Supplemental Experimental Procedures section. See also Supplementary Figure 2j and Supplementary Figure 3a-c.
(f) Results of Akt in vitro kinase assay utilizing immunoprecipitated Akt (with purified GSK3α/β as a substrate) are shown in a western blot. The experiment was carried out in the presence (+) or absence (−) of the indicated inhibitors. The membrane was probed with the indicated antibodies where anti-Total Akt was used as a control. Numbers on the blot represent densitometric analysis results in arbitrary units. See also Supplementary Figure 3d.
Akt activation is responsible for basal state colony formation
Cell proliferation in oncogenesis is broadly attributable to aberrant signaling in either the mitogen-activated protein kinase (MAPK) pathway or the PI3K/Akt pathway. Phosphorylation of Erk is a downstream marker of MAPK signal activation. Under conditions of serum starvation Erk phosphorylation was negligible in Ci and G2i cells31 (also see Supplementary Figure 2e) confirming the absence of stimulation from growth factors in our culture medium. Furthermore colony formation under serum starvation is independent of the MAPK pathway, since addition of the potent Mek (aka. MAPKK) inhibitor U0126 had no effect on colony formation (Figure 1c and d and Supplementary Figure 2f (for U0126 inhibition efficiency)). It has been previously reported that cross-talk exists between the MAPK pathway and the PI3K pathway which affects Akt phosphorylation and activity32. This experiment rules out such an effect, since U0126 had no effect on Grb2-dependent Akt phosphorylation (Supplementary Figure 2f).
In the absence of evidence for MAPK signaling as a mediator of the effects of knockdown of Grb2, we focused our attention on the PI3K/Akt pathway. Treatment with Wortmannin (PI3K inhibitor) or PH domain-dependent Akt inhibitor VIII drastically impeded colony formation in both Ci and G2i cells. This effect was opposite to the one obtained through treatment with the inhibitor of the pathway antagonist PTEN, SF1670 (Figure 1c and d and Supplementary Figure 2g-i for inhibition efficiency). Interestingly, in all of these cases Akt phosphorylation was higher in G2i cells versus Ci cells under basal conditions. Also normalized reverse phase protein array (RPPA) results (Figure 1e, Supplementary Figure 2j (before normalization) and Supplementary Table 1) show that the phosphorylation level of Akt (on serine 473 and threonine 308) is high in the basal state compared to the stimulated state in G2i versus Ci cells. This was reflected in the phosphorylation level of the downstream effectors of Akt like glycogen synthase kinase 3 (GSK3), p70 S6 kinase (p70S6K) and mammalian target rapamacyin (mTOR) (Figure 1e, Supplementary Figure 2j and Supplementary Table 1). The RPPA results also validate the previous observation that Erk phosphorylation is not the driver of colony formation in G2i cells. Thus, Akt phosphorylation is dependent on Grb2 concentration. This is further validated through a rescue study knocking in Grb2 (Supplementary Figure 3a). In order to detect whether Akt phosphorylation negatively correlates with Grb2 expression levels in other cell lines, we investigated two additional FGFR2-expressing cancer cell lines (i.e. human colon adenocarcinoma cell line (LoVo) with an endogenously low level of Grb2 and a rat osteosarcoma-derived cell line in which we depleted the Grb2 concentration (ROS17/2.8, Ci or G2i)). These cells showed the same Grb2-dependent Akt phosphorylation trend as our model cell line (Supplementary Figure 3b). Interestingly knocking down Plcγ1 decreased pS473 (Supplementary Figure 3c) indicating that the concentration of Plcγ1 is also a contributor to the phosphorylation event observed.
By performing an in vitro kinase assay using a GSK3 fusion protein as a substrate for the enzyme, we observed that Akt activity was higher in G2i cells versus Ci cells and was abolished in Pγi cells (Figure 1f and Supplementary Figure 3d). Wortmannin and U73122 (Plcγ1 inhibitor) unlike SF1670 treatment decreased Akt activity while U0126 had no significant effect. Therefore Akt is phosphorylated and activated in the absence of extracellular stimulation and is responsible for anchorage independent growth in this model. This event is enhanced by knocking down Grb2 or inhibiting PTEN and diminished by knocking down Plcγ1, inhibiting Plcγ1 or inhibiting PI3K. It is thus essential to test whether the previously reported concentration-dependent competition between Grb2 and Plcγ1 for FGFR2 binding10 is affecting the activity of PI3K and/or PTEN leading to the observed effect.
Up-regulation of PI3K activity is not required for Akt phosphorylation, activation and subsequent colony formation
Upon stimulation PI3K is activated through binding to receptors either directly or via Grb233 and other linker molecules. To test whether the Grb2 knockdown results in a generalized or membrane associated increase in PI3K activity in the basal, non-stimulated state, we first established that the expression level of PI3K in both Ci and G2i cells was similar (Figure 2a, upper western blot). We then investigated PI3K activity in Ci and G2i cells using a competitive ELISA assay to measure the PI3K-mediated production of PI(3,4,5)P3 from PI(4,5)P2 (Figure 2b and Supplementary Figure 3e-h) in which the absorbance measured at 450nm is inversely proportional to the amount of lipid produced. A standard curve was generated utilizing serial dilutions of PI(4,5)P3 to determine the concentration of the lipid produced in each sample (Supplementary Figure 3e and f). The PI3K used in the assay was immunoprecipitated from FGFR2 transfected HEK293T cell lysates or membrane fractions to limit the experiment to the membrane localized pool of the enzyme. Wortmannin was used as an additional control for the specificity of the assay where Akt phosphorylation was the readout for the inhibition efficiency (Figure 2a, lower western blot). The amount of PI(3,4,5)P3 produced in non-stimulated Ci and G2i total cell lysates, or the membrane fraction specifically was similar and hence independent of the level of Grb2 expression (Figure 2b, and Supplementary Figure 3g and h). Another method was used to test PI3K activity in Ci and G2i cell lysates via [32P]PI(3,4,5)P3 production from unlabeled, ‘cold’ PI(4,5)P2 and [γ-32P]ATP. The level of the product generated after background subtraction was similar in both cell lines (Figure 2c). Furthermore the product was depleted upon addition of Wortmannin, demonstrating that the results obtained are specific to PI3K activity. We can therefore conclude that PI3K has a basal level of activation in non-stimulated cells, but an increase in membrane associated PI3K activity is not required for the increased colony formation observed in the absence of Grb2.
Figure 2. PI(3,4,5)P3 is generated equally by PI3K in Ci and G2i cells yet Akt membrane localization is enhanced in the latter.

(a) Upper western blot: Ci and G2i cells were serum starved and utilized for western blot analysis to detect the expression level of PI3K. Lower western blot: Untreated or Wortmannin treated cells (to ensure PI(3,4,5)P3 production is PI3K mediated) were used for western blot with pAkt as readout for inhibition efficiency.
(b) Left panel: PI3K was immuno-precipitated from untreated or Wortmannin treated cells on agarose beads using a PI3K specific antibody and used in a competitive ELISA assay. Absorbance at 450 nm was measured for samples run in triplicates (n=3) and used to extrapolate the amount of PI(3,4,5)P3 produced in each case (See Supplementary Figure 3e-h). Values were normalized against “untreated Ci” sample. Error bars denote standard deviation of the mean. Student's t-test indicates ***p≤ 0.001. Right panel: Same PI3K assay but in this case serum starved untreated Ci and G2i cells were subjected to fractionation first and only the membrane fraction was utilized in the experiment as an additional control to specifically quantify the membrane pool of PI(3,4,5)P3. The differences obtained were not statistically significant as determined by Student's t-test.
(c) Untreated or Wortmannin-treated cell lysates from Ci and G2i cells were used to detect the activity of intracellular PI3K by incubating fresh cell lysate with [32P]ATP and unlabeled PI(4,5)P2. The [32P]PI(3,4,5)P3 that was generated by PI3K was detected by TLC. Results of [32P] labeled lipids were visualized and quantified for adjusted volume as shown on the representative image.
(d) Ci and G2i cells transfected with an empty mCherry vector or mCherry-Akt-PH vector were serum starved and used for microscopy as explained in the Supplemental Experimental Procedures section. FGFR2-GFP was used as a membrane marker to show co-localization of the lipid binding PH domain on the membrane. Scale bar: 10 μm for empty mCherry vector and 25 μm for mCherry-Akt-PH vector. See also Supplementary Figure 4a for transfection efficiency.
(e) Top panel: serum starved Ci and G2i cells were fractionated into membrane and cytoplasmic portions and used for Western blot analysis then probed with the indicated antibodies followed by densitometric analysis. Bottom panel: graphical display of the quantified ratio of membrane total Akt to total FGFR2 (from 3 independent fractionation experiments) to show the increased Akt localization on the membrane. The red numbers on the blot presented in arbitrary units represent the ratio of total Akt to FGFR2 (the membrane marker).
Error bars denote standard deviation of the mean. Student's t-test was used to assess statistical significance *p≤ 0.05.
(f) Absorbance at 450 nm (assays are explained in the Supplementary Experimental Procedures section and shown in Supplementary Figure 4c-e) was used to extrapolate the amount of PI(3,4)P2, PI(4,5)P2 or PI(3,4,5)P3 produced. Values were averaged and normalized against Ci cells. Error bars denote standard deviation of the mean. ***p≤ 0.001 indicates a significant decrease in PI(4,5)P2 with a concomitant increase in PI(3,4,5)P3 in G2i cells compared to Ci cells.
Based on these observations, it might be assumed that the endogenous level of membrane localized PI(3,4,5)P3 which recruits and activates Akt 19,20 is not influenced by Grb2 depletion. However, this was not the case since after transfecting Ci and G2i cells with an empty mCherry or mCherry-Akt-PH vector (Supplementary Figure 4a), we observed a dramatic enhancement in the localization of the Akt PH domain on the membrane of G2i cells versus Ci cells (Figure 2d). In both cases GFP-FGFR2 was utilized as a membrane marker. We confirmed these results by performing a cell fractionation experiment in Ci and G2i cells where actin was used as a cytoplasmic marker and FGFR2 as a membrane marker (Figure 2e (western blot and histogram)). A larger population of Akt was found in the membrane of G2i cells compared to Ci cells. Accumulation of Akt on the membrane in Grb2 knockdown cells is driven by an increase in the pool of PI(3,4,5)P3, however we have shown that the increase in the PI(3,4,5)P3 pool is not dependent on differential PI3K activity. To demonstrate this we investigated phosphoinositide-dependent protein kinase 1 (PDK1), which is another protein that binds to membrane phospholipid PI(3,4,5)P3 in order to phosphorylate its substrate Akt. Upon binding to the membrane PDK1 is autophosphorylated on serine 241, which is required for its activation34. We found that PDK1 was indeed more highly phosphorylated in G2i cells compared to Ci cells (Figure 1e, Supplementary Figure 2j, Supplementary Figure 4b and Supplementary Table 1). This corroborated the notion that Grb2 depletion is causing an accumulation of PI(3,4,5)P3 in a PI3K-independent manner.
To validate this theory and also to determine the underlying causes of such an event, we quantified the cellular concentration of PI(3,4,5)P3 and the main phospholipids from which it is derived. Using mass ELISA, we measured PI(3,4)P2 (which is also a product of PI3K and an Akt membrane binding site), PI(4,5)P2 and PI(3,4,5)P3 levels in Ci and G2i cells following lipid extraction (Figure 2f and Supplementary Figure 4c-e for the controls and standard curves). We observed that the PI(3,4)P2 level is similar in the two cell lines unlike the PI(3,4,5)P3 level which was considerably higher in the absence of Grb2. The PI(4,5)P2 level however dropped significantly in G2i cells reflecting the previously reported Plcγ1 constitutive activation in Grb2 depleted cells10. Hence, in this work we show that the concentration of PI(3,4,5)P3 is affected by a Grb2-dependent mechanism under basal conditions. This perturbation has important consequences on the cellular localization of Akt and its concomitant phosphorylation and activation in the absence of extracellular stimulation.
Depletion of Grb2 leads to down-regulation of PTEN and accumulation of PI(3,4,5)P3 under non-stimulated conditions
Using the SF1670 inhibitor, we have shown that PTEN in Ci and G2i cells abrogates colony formation and decreases Akt phosphorylation and activity (Figure 1c, d and f, Supplementary Figure 3h). This led us to postulate that the cause of PI(3,4,5)P3 accumulation could lie in the decrease in the activity of its key phosphatase PTEN which is equally expressed in Ci and G2i cells as determined by western blot analysis (Figure 3a). To test this hypothesis and measure PTEN activity, we utilized TLC to validate the ELISA results obtained in Figure 2f and to measure the intracellular levels of membrane lipids in serum starved radiolabeled cells. This was the method of choice since other approaches like mass spectrometry are limited by the distribution and operational pools of phospholipids and can lead to lipid decomposition35. They also require spiking with a known concentration of lipids, which can affect the measured lipid phosphatase activity of proteins36. After extraction, separation and determination of retention value (Rf value)-based identification of labeled phospholipids by TLC, we observed accumulation of PI(3,4,5)P3 in non-stimulated G2i cells but not in Ci cells (Figure 3a and b).
Figure 3. PTEN activity is significantly reduced upon Grb2 depletion due to excessive PI(4,5)P2 hydrolysis via Plcγ1.

(a) Top panel: Western blot analysis was utilized to detect PTEN expression level in Ci and G2i cells. The blot was probed with the indicated antibodies. Bottom panel: Ci and G2i cells growing in serum free DMEM were labeled with myo-[2-3H(N)]inositol and used for lipid extraction followed by TLC. PI(4,5)P2 and PI(3,4,5)P3 were identified based on their Rf values reported in the literature 42.
(b) Pixel quantification results from the previous TLC were averaged from 3 independent experiments and normalized to generate a histogram. Error bars denote standard deviation of the mean with a calculated **p≤ 0.01.
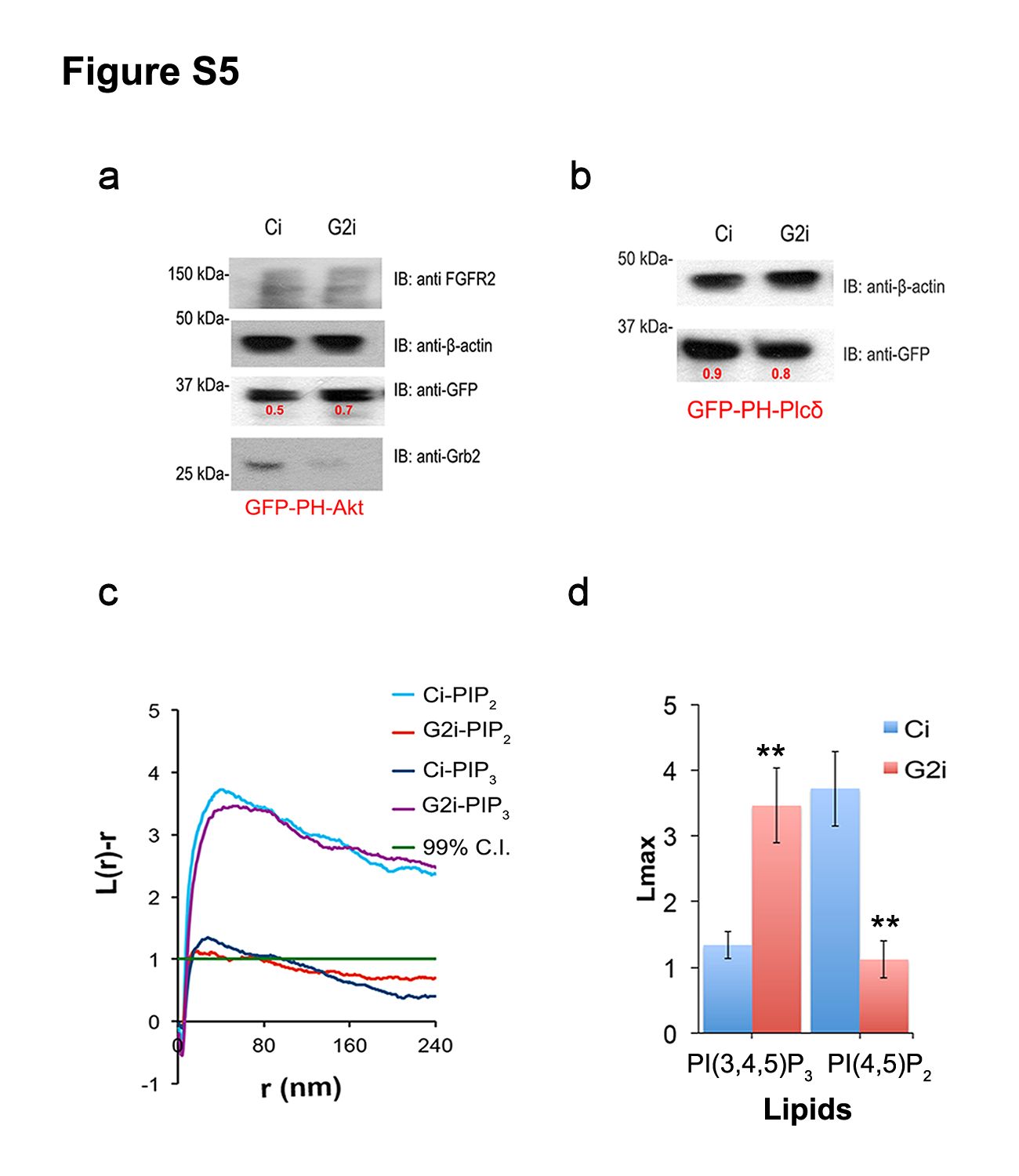
(c) Representative images of EM spatial mapping show the number of anti-GFP gold particles (and hence respective lipid concentrations) with a scale bar of 200 nm (for a 1μm × 1μm square). Ci and G2i cell lines were both transfected with GFP tagged lipid probes. See also Supplementary Figure 5a and b for transfection efficiency.
(d) At least 12 plasma membrane sheet images from (C) were acquired. The mean and standard error of the mean for the number of gold particles were calculated for each condition and Student's t-test was used to assess statistical significance which in both cases **p≤ 0.01. See also Figure S5C and D for clustering experiments performed as described in Supplementary Experimental Procedures section.
(e) Cell lysates from untreated cells or cells treated with the indicated inhibitors were incubated with [32P]PI(3,4,5)P3 as a PTEN substrate. TLC was used for the separation of extracted labeled lipids as shown on the blot.
(f) Pixel quantification data (n=3) was used to generate the bar graph showing the ratio of the product PI(4,5)P2 to the substrate PI(3,4,5)P3 following adjustment to the total amount of loaded lipid which reflects PTEN enzymatic activity. U73122 and SF1670 were used to depress Plcγ1 and PTEN activities respectively. The dramatic reduction in the PI(4,5)P2/PI(3,4,5)P3 ratio in the presence of SF1670 strongly suggests that the effect is specific to PTEN. Standard deviation of the mean is denoted by error bars. *p≤ 0.05
(g) Top panel: Ci cells were left untransfected or transfected with Plcγ1 followed by western blot and densitometric analysis of band intensity. Bottom panel: Lipids extracted from these cells were incubated with [32P]PI(3,4,5)P3 and subjected to TLC to measure PTEN activity. PI(4,5)P2/PI(3,4,5)P3 ratio quantification was obtained from duplicate experiments visualized and quantified for adjusted volume (pixel percentage).
(h) Top panel: Purified PTEN was incubated with Ptdser and [32P]PI(3,4,5)P3 in the presence or absence of 33 μM of cold PI(4,5)P2. TLC was run to separate [32P] labeled lipids followed by visualization and pixel quantification. Bottom panel: Results from three independent experiments were averaged following the calculation of the ratio of [32P]PI(4,5)P2 to [32P]PI(3,4,5)P3. Error bars represent the standard deviation of the mean. Student's t-test was used to calculate the statistical significance with ***p≤ 0.001.
Using EM spatial mapping37,38 (Figure 3c and d) also revealed PI(3,4,5)P3 accumulation and PI(4,5)P2 depletion in G2i cells compared to Ci cells expressing non-fluorescently tagged FGFR2. Images obtained are representative of Ci and G2i plasma membrane sheets with anti-GFP gold particles targeted against GFP-tagged lipid probes. The efficiency of transfection of GFP-PH-Plcδ (for specific labelling of PI(4,5)P2) or GFP-PH-Akt (for specific labelling of PI(3,4,5)P3) is shown in Supplementary Figure 5a and b). Gold particle spatial patterns were analyzed using univariant K functions to quantify the extent of clustering of the point pattern which reflects signaling intensity and hence in this case potential intracellular Akt activation. The results showed more clustering of PI(3,4,5)P3 in G2i cells compared with Ci cells. The opposite results were obtained for PI(4,5)P2 (Supplementary Figure 5c and d).
After confirming the perturbation in the level of membrane lipids, we tested the level of activity of endogenous PTEN in Ci and G2i cells. The radiolabelled lipid substrate [32P]PI(3,4,5)P3 was prepared from ‘cold’ PI(4,5)P2 and [γ-32 P]ATP using purified active PI3K (starting material shown in the origin of TLC on Figure 3e). This experiment measures the basal rate of PI(3,4,5)P3 dephosphorylation to generate PI(4,5)P2 which, unlike the stimulation rate, is lipid concentration-dependent and not time-course (kinetics)-dependent. We noticed that in G2i cell lysates incubated with [32P]PI(3,4,5)P3 the ability of PTEN to restore the levels of PI(4,5)P2 was attenuated. This was reflected by the reduction in the PI(4,5)P2/PI(3,4,5)P3 ratio in untreated G2i cells compared to Ci cells (Figure 3e and f). This apparent increase in PI(3,4,5)P3 in G2i cells was shown to be dependent on PTEN by the addition of the PTEN selective inhibitor SF1670. The qualitative effect seen on the TLC plate was higher for G2i cells compared to Ci cells, which have more PTEN activity. Hence Ci cells were less sensitive to inhibitors than G2i cells. However after taking the ratio of product to substrate into consideration to correct for potential variability of total loaded material, the relative effect of the inhibitor was similar in both cell lines (Figure 3f).
How is PTEN activity down-regulated under serum starvation and Grb2 depletion? Is PI(4,5)P2, which has been previously reported to trigger a conformational change in PTEN22, the key regulator of its phosphatase activity under these conditions? We had previously shown that the reduction in Grb2 concentration elevates constitutive Plcγ1 activity and dramatically reduces cellular PI(4,5)P210. In order to test whether Plcγ1 is a regulator of PTEN activity in Grb2-depleted cells, we treated our cells with the phospholipase inhibitor U73122 (Figure 3f and g). The ratio of [32P]PI(4,5)P2/[32P]PI(3,4,5)P3 reflecting PTEN activity was drastically increased in G2i cells unlike Ci cells, possibly due to its initial elevated level in the latter. To confirm that the effect obtained is indeed due to Plcγ1 activity, we overexpressed it in Ci cells (Figure 3g, upper left panel). We then incubated the serum starved, untransfected and transfected cell lysates with radiolabeled [32P]PI(3,4,5)P3 (Figure 3g, lower left panel). The ratio of [32P]PI(4,5)P2/[32P]PI(3,4,5)P3 obtained via pixel quantification on TLC reflected more than a 50% decrease in PTEN activity upon Plcγ1 overexpression.
To test the direct effect of PI(4,5)P2 on PTEN activity, we incubated purified PTEN protein with labeled [32P]PI(3,4,5)P3 in the absence, or presence of non-labeled PI(4,5)P2. [32P]-labeled lipids were separated and visualized on TLC, followed by pixel quantification and statistical analysis (Figure 3h and i). In the absence of PI(4,5)P2, PTEN's ability to dephosphorylate [32P]PI(3,4,5)P3 into [32P]PI(4,5)P2 was weak. However, adding ‘cold’ PI(4,5)P2 at a physiologically effective concentration21 dramatically enhanced PTEN activity as it dephosphorylated the [32P]PI(3,4,5)P3 to produce [32P]PI(4,5)P2, i.e. the ratio of labeled [32P]PI(4,5)P2/[32P]PI(3,4,5)P3 was increased. Since the presence of PI(4,5)P2 activates PTEN, it follows that the PI(4,5)P2 concentration is a critical regulator of this tumor suppressor in the absence of extracellular stimulation.
Reducing cellular expression level of Grb2 enhances tumor formation
Tumor progression is known to be a multistep process in which an increase in tumor mass occurs concomitant with growth factor deprivation prior to invasion, angiogenesis and metastasis1. Our FGFR2-expressing, Grb2-depleted model cell line successfully formed colonies in an anchorage-independent manner via Akt activation. We subsequently investigated whether fluctuations in protein concentrations will modulate the potential to form tumors in a xenograft mouse model. To test this we subcutaneously injected PCi, PG2i, Ci and G2i cells in the right flank of adult female nude mice (4 mice per group). After 60 days the mice were subjected to MRI scanning (coronal and axial sections) to characterize tumor formation (Figure 4a). Mice injected with G2i cells showed higher tumor weight and volume with excessive multi-nodular burden compared to mice injected with Ci or PG2i cells. PCi injection failed to develop noticeable tumors (Figure 4a and b). Grb2 depletion results in the development of multiple tumors in the vicinity of the primary tumor indicating infiltration, invasion and/or local metastasis of tumor cells into the surrounding tissue. These results are consistent with our data from the colony formation assay performed under serum restriction where G2i cells were unique in their ability to proliferate into multiple colonies of increased size. Morphologically the G2i tumor is composed of similar types of neoplastic cells as Ci tumor but shows more anaplastic or higher degree of cellular pleomorphism (Figure 4c) characteristic of Akt pathway alteration39. Compared to Ci cells, G2i cells have lower amounts of cytoplasm and the nuclei have severely coarse, stippled or clumped chromatin with prominent multiple nucleoli and mitotic figures (as shown by the black arrows). Large areas of necrosis affected approximately 60-70% of the entire G2i tumor compared to 5-10% in Ci tumors (Figure 4c). The margins of the G2i tumor are indistinct and there is infiltration of tumor cells into the mammary tissues, fibroadipose tissue and skeletal muscles unlike Ci and PG2i tumors (as shown by the arrows in Figure 4c and d).
Figure 4. Knocking down Grb2 in FGFR2 expressing cells enhances their ability to form tumors with high levels of Akt phosphorylation.

(a) Top panel: Coronal MRI representative images of PCi, PG2i, Ci, G2i cell injected mice were taken following 60 days of subcutaneous injections. Arrows indicate the tumors formed. Middle panel: Axial MRI images of PCi, PG2i, Ci, G2i injected mice following subcutaneous injections. White arrows indicate the tumors formed. Bottom panel: A representative sample from each group (4 mice per group) showing both size and number of tumors formed.
(b) Upper panel (on the left): Tumor volumes were calculated in the axial sections obtained via MRI as explained in details in the Supplemental Experimental Procedures section. Results shown are the averages of the tumor volume in every group. Bottom panel (on the left): Tumors in every group were weighed (in grams) and averaged to generate a bar graph. Error bars denote standard deviation of the mean. Student's t-test was used to calculate the statistical significance with **p≤ 0.01. Right panel: Tumor nodules/mouse were counted and presented in a bar graph.
(c) Representative images of H & E staining of Ci and G2i tumors were taken at 40x and 600x to show infiltration to nearby tissues. Scale bars are 500 and 50 μm respectively. Images from 600x magnification were enlarged to show cellular morphology. Black arrows denote the nuclear/mitotic figures in G2i cells.
(d) Upper panels: Representative pictures of PG2i, Ci and G2i tumors (H & E staining) were taken at 400x magnification with a scale bar of 50 μm. Green arrows indicate the margins of PG2i and Ci tumors whereas black arrows (bottom panel) specify infiltration of G2i tumors into nearby tissues.
(e) Left panel: Representative images of pAkt staining of Ci and G2i tumors were taken at 40x, 200x and 400x. Scale bars are 500,100, 50 μm respectively. Black arrow is showing Akt phosphorylation in the perivascular region. Right panel: Western blot shows the level of Akt phosphorylation in tumor sections following sonication and protein extraction.
To determine if Akt activity is increased in the G2i subcutaneous tumors, we stained for pAkt. G2i cells stained positive consistent with the increases in mitotic progression and proliferation (Figure 4e, left panel). Tumor cells in the perivascular areas have increased pAkt staining in comparison with tumor cells that are further away from the blood vessels (depicted with a black arrow). Conversely the Ci tumor cells showed negligible staining with the pAkt (S473) antibody. Therefore, tumor tissues depleted for Grb2 exhibit the hallmarks of elevated Akt signaling. In western blot analysis of Ci and G2i tumors (Figure 4e, right panel), G2i tumors showed a higher level of Akt phosphorylation compared to Ci tumors. Total Akt and actin were used as loading controls. These results emphasize the dependence of tumor progression on respective G2i protein concentration.
FGFR2, Grb2 and Plcγ1 are modulators of Akt and predictive markers in ovarian cancer
Akt is highly phosphorylated and activated in ovarian cancer conversely dephosphorylation is known to limit tumor growth40. We performed IHC staining on an ovarian cancer patient tissue microarray (TMA) and observed an increase in Akt phosphorylation, which correlated with higher grade tumor and poorer prognosis (Figure 5a). Interestingly, database analysis of clinical information downloaded from Cancer Genome Atlas Project (TCGA; http://tcga-data.nci.nih.gov/) and Tothil et al., 200841 (Figure 5b and Supplementary Figure 6b-e) revealed that low levels of FGFR2 mRNA and Plcγ1 mRNA with a concomitantly high level of Grb2 mRNA is associated with a favorable prognosis as opposed to cases with high FGFR2, high Plcγ1 and low Grb2 mRNA levels. This mirrors the results obtained with cell lines and xenograft mouse models.
Figure 5. FGFR2, Grb2 and Plcγ1 modulate Akt phosphorylation in ovarian cancer and can act as prognostic markers for patient survival.

(a) Upper panel: H & E staining of representative TMA images taken at 400x with a scale bar of 50 μm. Lower panel: Representative images of pAkt staining were taken at 400x with a scale bar of 50 μm.
(b) Percentage survival of ovarian cancer patients is presented in a Kaplan-Meyer curve based on the downloaded and analyzed clinical data as explicitly explained in the Supplemental Experimental Procedure section. The calculated Log-rank test value is ****p≤ 0.0001 which indicates significant correlation between FGFR2, Grb2 and Plcγ1 (prognostic markers) with patient survival. The median of overall survival (OS) in months is shown in parentheses. See also Figure S6.
(c),(d) and (e) Western blot analysis was performed using OVCAR3 and OVCA433 cell lysates where the blots were probed with the indicated antibodies. The numbers in red are densitometric analysis results presented in arbitrary units on the blots.
(f) OVCAR3 cells were left un-transfected or transfected with Grb2 followed by western blot and densitometric analysis.
(g) Absorbance measured at 450 nm was used to extrapolate the amount of PI(3,4)P2, PI(4,5)P2 or PI(3,4,5)P3 produced. Values from 3 independent experiments were averaged and normalized against un-transfected OVCAR3 cells. Error bars denote standard deviation of the mean. **p≤ 0.01 indicates a significant increase in PI(4,5)P2 with a concomitant decrease in PI(3,4,5)P3 upon Grb2 transfection. See also Supplementary Figure 4c-e.
(h). Cells from (f) were utilized for 3D culture assay as described in the Materials and Methods section. The experiment was run in two chambers each containing a triplicate of each sample. Pictures of colonies (black arrows) formed in serum restricted DMEM were taken at 10X and 20X magnification.
(i) Cells from (f) were fixed and stained with the proliferation marker, Ki-67. scale bar is 50 μm.
(j) Left panel: Colonies formed in (h) were counted in 4 microscopic fields, averaged, normalized against un-transfected cells and utilized for bar graph generation. Error bars represent standard deviation of the calculated mean. Student's t-test was used to assess statistical significance (*p≤ 0.05). Right panel: Number of Ki-67+ cells were counted under same magnification, averaged and normalized against un-transfected cells. Error bars on the histogram generated denote the standard deviation of the calculated mean. Student's t-test was used to assess statistical significance (**p≤ 0.01).
(k) TMA of patients expressing FGFR2, Grb2, and Plcγ1 (IHC staining is described in the Supplemental Experimental Procedures) were scored and analyzed to generate a survival curve with log-rank test p-value <0.001.
(l),(m) Representative images of the TMAs stained for the proteins of interest. were taken at 20X magnification. The results obtained were used to correlate the expression level of the 3 biomarkers with pAkt level as explained in the Supplemental Experimental Procedures. Chi square test was used to detect statistical significance of the correlation. p-value <0.002.
To establish a better understanding of this we utilized 2 ovarian cancer cell lines, OVCAR3 and OVCA433. The latter expresses higher levels of Grb2, which in the background of lower levels of FGFR2 and Plcγ1 expression resulted in more Grb2 being immunoprecipitated by FGFR2 (i.e. enhanced Grb2-FGFR2 complex formation), than OVCAR3 cells (Figure 5c and d and Supplementary Figure 6a). Also, unstimulated OVCA433 showed lower levels of Akt phosphorylation compared to OVCAR3 (Figure 5e). Interestingly, overexpression of Grb2 in OVCAR3 cells resulted in an increase in PI(4,5)P2, a decrease in PI(3,4,5)P3 and a stable PI(3,4)P2 level (Figure 5f and g and Supplementary Figure 4c-e) consistent with the results obtained in Ci and G2i cells. The decrease in PI(3,4,5)P3 and pAkt were accompanied by a significant drop in colony formation in 3D culture under serum starvation conditions (Figure 5h and j). The 3D cultured cells were stained with Ki-67 (a marker of proliferation expressed in growth phase but absent in the resting phase), which was more prominent in the nucleus of untransfected cells (Figure 5i and j). This provides further evidence of Grb2-mediated cellular proliferation via Akt phosphorylation in the context of FGFR2.
Finally, we investigated the correlation between FGFR2, Grb2 and Plcγ1 protein expression (as one event) with patient survival using IHC staining of TMAs. Low FGFR2/low Plcγ1/high Grb2 correlated with favorable survival in contrast to high FGFR2/high Plcγ1/low Grb2 (Figure 5K and representative images of staining in Figure 5l). This confirms the results obtained via analysis of mRNA levels in the TCGA and in ovarian cancer cell lines. Interestingly, when TMAs were stained for pAkt (Figure 5l), low FGFR2/low Plcγ1/high Grb2 correlated with lower score of pAkt staining unlike high FGFR2/high Plcγ1/low Grb2 (Figure 5m).
Discussion
The role of growth factor deprivation in the modulation of signaling pathways is poorly understood or appreciated. Under these conditions, FGFR2 is capable of regulating cellular functions through competitive binding between Grb2 and Plcγ110. Here we investigated whether variations in FGFR2 levels and binding to Grb2 and Plcγ1 can activate other signaling pathways and induce additional oncogenic effects under growth factor deprivation via Grb2-Plcγ1 competition. In this study we show that proliferation is driven by a mechanism initiated in FGFR2-expressing cells independent of extracellular stimulation. This outcome is solely dependent on changes in protein concentrations and the resultant fluctuation in the levels of membrane phospholipids leading to Akt pathway regulation. To provide an appropriate pool of phospholipid for Akt recruitment a balance between lipid kinase and phosphatase activity is essential, thus perturbation of the activation of either of these enzymes can either promote or depress the onset of a proliferative signal. Here we also show that Grb2 depletion, by promoting Plcγ1 activity, limits PI(4,5)P2-dependent PTEN activity. As a result PI(3,4,5)P3 is not converted to PI(4,5)P2 and thus accumulates in the plasma membrane. This leads to the consequent recruitment and activation of Akt in the absence of extracellular stimulation (Supplementary Figure 1a and b). Thus, Grb2-Plcγ1 competition can negate the need for mitogen-dependent activation of signaling pathways by regulating the levels of phospholipids primarily through decreased PTEN activity. Under these conditions cells are highly effective in modulating anchorage-independent colony formation in culture. This is reflected in tumor progression in a xenograft mouse model and a correlation with survival in ovarian cancer patients. Thus, investigating signaling pathways and their functional effects under non-stimulatory conditions, which mimic early phases of cancer progression, is essential to understanding tumor development. In this case, the relative concentrations of Grb2 and Plcγ1 in FGFR2-expressing tumor cells provide a significant predictive marker for tumor staging and oncogenic outcome that could prove critical for therapeutic decision making.
Materials and methods
Cell culture. All cell lines with the exception of ROS17/2.8 osteosarcoma cells were cultured in Dulbecco's modified Eagle's high glucose medium (DMEM). The former was grown in phenol red-free DMEM. DMEM was supplemented with 10% (vol/vol) fetal bovine serum (FBS) and 1% antibiotic/antimycotic (Lonza) in a humidified incubator at 37°C with 10% CO2. Cells expressing GFP fused to the C-terminus of FGFR2 and the knockdowns were produced as previously described9,10,43.
PI(3,4,5)P3 phosphatase assays. Unilamellar liposomes containing radiolabeled substrate lipid in a background of phosphatidylserine were used to assay lipid phosphatase (PTEN) activity by modifying the described protocol44.
PI3K assay by thin layer chromatography. The activity of PI3K was measured via competitive ELISA (following the manufacturer's instructions) and through monitoring PI(4,5)P2 phosphorylation using γ[32P]ATP as co-substrate by modifying the described protocol45.
EM-spatial mapping was carried out as previously explained38.
Xenograft mouse model magnetic resonance imaging (MRI) and IHC staining were performed as required by MD Anderson small animal imaging facility. Clinical data analysis is detailed in the Supplementary Materials.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
This work was funded by the G. Harold and Leila Y. Mathers Charitable Foundation. This work was supported by NIH grants U54CA151668, P50 CA083639, P50 CA098258 and by the RGK Foundation and the Gilder Foundation.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
References
- 1.Storz P, Doppler H, Copland JA, Simpson KJ, Toker A. FOXO3a promotes tumor cell invasion through the induction of matrix metalloproteinases. Mol Cell Biol. 2009;29:4906–4917. doi: 10.1128/MCB.00077-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mahbub Hasan AK, Ijiri T, Sato K. Involvement of Src in the Adaptation of Cancer Cells under Microenvironmental Stresses. J Signal Transduct. 2012;2012:483796. doi: 10.1155/2012/483796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Risbud MV, Fertala J, Vresilovic EJ, Albert TJ, Shapiro IM. Nucleus pulposus cells upregulate PI3K/Akt and MEK/ERK signaling pathways under hypoxic conditions and resist apoptosis induced by serum withdrawal. Spine. 2005;30:882–889. doi: 10.1097/01.brs.0000159096.11248.6d. [DOI] [PubMed] [Google Scholar]
- 4.Yamamoto N, Mammadova G, Song RXD, Fukami Y, Sato K-i. Tyrosine phosphorylation of p145met mediated by EGFR and Src is required for serum- independent survival of human bladder carcinoma cells. J Cell Sci. 2006;119:4623–4633. doi: 10.1242/jcs.03236. [DOI] [PubMed] [Google Scholar]
- 5.Ching JK, Rajguru P, Marupudi N, Banerjee S, Fisher JS. A role for AMPK in increased insulin action after serum starvation. Am J Physiol Cell Physiol. 2010;299:1171–9. doi: 10.1152/ajpcell.00514.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pirkmajer S, Chibalin AV. Serum starvation: caveat emptor. Am J Physiol Cell Physiol. 2011;301:C272–279. doi: 10.1152/ajpcell.00091.2011. [DOI] [PubMed] [Google Scholar]
- 7.Troncoso R, Vicencio JM, Parra V, Nemchenko A, Kawashima Y, Del Campo A, et al. Energy-preserving effects of IGF-1 antagonize starvation- induced cardiac autophagy. Cardiovas Res. 2012;93:320–329. doi: 10.1093/cvr/cvr321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gauglhofer C, Sagmeister S, Schrottmaier W, Fischer C, Rodgarkia-Dara C, Mohr T, et al. Up-regulation of the fibroblast growth factor 8 subfamily in human hepatocellular carcinoma for cell survival and neoangiogenesis. Hepatology. 2011;53:854–864. doi: 10.1002/hep.24099. [DOI] [PubMed] [Google Scholar]
- 9.Ahmed Z, Lin CC, Suen KM, Melo FA, Levitt JA, Suhling K, et al. Grb2 controls phosphorylation of FGFR2 by inhibiting receptor kinase and Shp2 phosphatase activity. J Cell Biol. 2013;200:493–504. doi: 10.1083/jcb.201204106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Timsah Z, Ahmed Z, Lin CC, Melo FA, Stagg LJ, Leonard PG, et al. Competition between Grb2 and Plcgamma1 for FGFR2 regulates basal phospholipase activity and invasion. Nature Struct & Mol Biol. 2014;21:180–188. doi: 10.1038/nsmb.2752. [DOI] [PubMed] [Google Scholar]
- 11.Byron SA, Pollock PM. FGFR2 as a molecular target in endometrial cancer. Future Oncol. 2009;5:27–32. doi: 10.2217/14796694.5.1.27. [DOI] [PubMed] [Google Scholar]
- 12.Katoh Y, Katoh M. FGFR2-related pathogenesis and FGFR2-targeted therapeutics. Int J Mol Med. 2009;23:307–311. doi: 10.3892/ijmm_00000132. [DOI] [PubMed] [Google Scholar]
- 13.Cole C, Lau S, Backen A, Clamp A, Rushton G, Dive C, et al. Inhibition of FGFR2 and FGFR1 increases cisplatin sensitivity in ovarian cancer. Cancer Biol Ther. 2010;10:495–504. doi: 10.4161/cbt.10.5.12585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matsuda Y, Ueda J, Ishiwata T. Fibroblast growth factor receptor 2: expression, roles, and potential as a novel molecular target for colorectal cancer. Pathol Res Internat. 2012;2012:574768. doi: 10.1155/2012/574768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Srinivasan S, Wang F, Glavas S, Ott A, Hofmann F, Aktories K, et al. Rac and Cdc42 play distinct roles in regulating PI(3,4,5)P3 and polarity during neutrophil chemotaxis. J Cell Biol. 2003;160:375–385. doi: 10.1083/jcb.200208179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thapa N, Sun Y, Schramp M, Choi S, Ling K, Anderson RA. Phosphoinositide signaling regulates the exocyst complex and polarized integrin trafficking in directionally migrating cells. Dev Cell. 2012;22:116–30. doi: 10.1016/j.devcel.2011.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 18.Vanhaesebroeck B, Leevers SJ, Ahmadi K, Timms J, Katso R, Driscoll PC, et al. Synthesis and function of 3-phosphorylated inositol lipids. Annu Rev Biochem. 2001;70:535–602. doi: 10.1146/annurev.biochem.70.1.535. [DOI] [PubMed] [Google Scholar]
- 19.Franke TF, Kaplan DR, Cantley LC, Toker A. Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3,4-bisphosphate. Science. 1997;275:665–668. doi: 10.1126/science.275.5300.665. [DOI] [PubMed] [Google Scholar]
- 20.Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3- kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–19. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- 21.Campbell RB, Liu FH, Ross AH. Allosteric activation of PTEN phosphatase by phosphatidylinositol 4,5-bisphosphate. J Biol Chem. 2003;278:33617–33620. doi: 10.1074/jbc.C300296200. [DOI] [PubMed] [Google Scholar]
- 22.Redfern RE, Redfern D, Furgason ML, Munson M, Ross AH, Gericke A. PTEN phosphatase selectively binds phosphoinositides and undergoes structural changes. Biochemistry. 2008;47:2162–71. doi: 10.1021/bi702114w. [DOI] [PubMed] [Google Scholar]
- 23.Altomare DA, Testa JR. Perturbations of the AKT signaling pathway in human cancer. Oncogene. 2005;24:7455–7464. doi: 10.1038/sj.onc.1209085. [DOI] [PubMed] [Google Scholar]
- 24.Janku F, Wheler JJ, Westin SN, Moulder SL, Naing A, Tsimberidou AM, et al. PI3K/AKT/mTOR inhibitors in patients with breast and gynecologic malignancies harboring PIK3CA mutations. J Clin Oncol. 2012;30:777–782. doi: 10.1200/JCO.2011.36.1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Khan KH, Yap TA, Yan L, Cunningham D. Targeting the PI3K-AKT-mTOR signaling network in cancer. Chin J Cancer. 2013;32:253–265. doi: 10.5732/cjc.013.10057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mazzoletti M, Broggini M. PI3K/AKT/mTOR inhibitors in ovarian cancer. Curr Med Chem. 2010;17:4433–4447. doi: 10.2174/092986710794182999. [DOI] [PubMed] [Google Scholar]
- 27.Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol. 2012;13:283–296. doi: 10.1038/nrm3330. [DOI] [PubMed] [Google Scholar]
- 28.Ushio-Fukai M, Alexander RW, Akers M, Yin Q, Fujio Y, Walsh K, et al. Reactive oxygen species mediate the activation of Akt/protein kinase B by angiotensin II in vascular smooth muscle cells. J Biol Chem. 1999;274:22699–22704. doi: 10.1074/jbc.274.32.22699. [DOI] [PubMed] [Google Scholar]
- 29.Pan J, Chang Q, Wang X, Son Y, Zhang Z, Chen G, et al. Reactive oxygen species-activated Akt/ASK1/p38 signaling pathway in nickel compound- induced apoptosis in BEAS 2B cells. Chem Res Toxicol. 2010;23:568–577. doi: 10.1021/tx9003193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ahmed Z, Schuller AC, Suhling K, Tregidgo C, Ladbury JE. Extracellular point mutations in FGFR2 elicit unexpected changes in intracellular signalling. Biochem J. 2008;413:37–49. doi: 10.1042/BJ20071594. [DOI] [PubMed] [Google Scholar]
- 31.Lin C-C, Melo FA, Ghosh R, Suen KM, Stagg LJ, Kirkpatrick J, et al. Inhibition of basal FGF receptor signaling by dimeric Grb2. Cell. 2012;149:1514–1524. doi: 10.1016/j.cell.2012.04.033. [DOI] [PubMed] [Google Scholar]
- 32.Turke AB, Song Y, Costa C, Cook R, Arteaga CL, Asara JM, et al. MEK inhibition leads to PI3K/AKT activation by relieving a negative feedback on ERBB receptors. Cancer Res. 2012;72:3228–3237. doi: 10.1158/0008-5472.CAN-11-3747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cully M, You H, Levine AJ, Mak TW. Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat Rev Cancer. 2006;6:184–192. doi: 10.1038/nrc1819. [DOI] [PubMed] [Google Scholar]
- 34.Casamayor A, Morrice NA, Alessi DR. Phosphorylation of Ser-241 is essential for the activity of 3-phosphoinositide-dependent protein kinase-1: identification of five sites of phosphorylation in vivo. Biochemical J. 1999;342:287–292. [PMC free article] [PubMed] [Google Scholar]
- 35.Clark J, Anderson KE, Juvin V, Smith TS, Karpe F, Wakelam MJ, et al. Quantification of PtdInsP3 molecular species in cells and tissues by mass spectrometry. Nature Methods. 2011;8:267–272. doi: 10.1038/nmeth.1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang K, Han X. Accurate quantification of lipid species by electrospray ionization mass spectrometry - Meet a key challenge in lipidomics. Metabolites. 2011;1:21–40. doi: 10.3390/metabo1010021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rotblat B, Prior IA, Muncke C, Parton RG, Kloog Y, Henis YI, et al. Three separable domains regulate GTP-dependent association of H-ras with the plasma membrane. Mol Cell Biol. 2004;24:6799–6810. doi: 10.1128/MCB.24.15.6799-6810.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou Y, Liang H, Rodkey T, Ariotti N, Parton RG, Hancock JF. Signal integration by lipid-mediated spatial cross talk between Ras nanoclusters. Mol Cell Biol. 2014;34:862–786. doi: 10.1128/MCB.01227-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rodriguez EF, Scheithauer BW, Giannini C, Rynearson A, Cen L, Hoesley B, et al. PI3K/AKT pathway alterations are associated with clinically aggressive and histologically anaplastic subsets of pilocytic astrocytoma. Acta Neuropathologica. 2011;121:407–420. doi: 10.1007/s00401-010-0784-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Altomare DA, Wang HQ, Skele KL, De Rienzo A, Klein-Szanto AJ, Godwin AK, et al. AKT and mTOR phosphorylation is frequently detected in ovarian cancer and can be targeted to disrupt ovarian tumor cell growth. Oncogene. 2004;23:5853–5857. doi: 10.1038/sj.onc.1207721. [DOI] [PubMed] [Google Scholar]
- 41.Tothill RW, Tinker AV, George J, Brown R, Fox SB, Lade S, et al. Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin Cancer Res. 2008;14:5198–5208. doi: 10.1158/1078-0432.CCR-08-0196. [DOI] [PubMed] [Google Scholar]
- 42.Kȍnig S, Hoffmann M, Mosblech A, Heilmann I. Determination of content and fatty acid composition of unlabeled phosphoinositide species by thin-layer chromatography and gas chromatography. Analyt Biochem. 2008;378:197–201. doi: 10.1016/j.ab.2008.03.052. [DOI] [PubMed] [Google Scholar]
- 43.Ahmed Z, George R, Lin CC, Suen KM, Levitt JA, Suhling K, et al. Direct binding of Grb2 SH3 domain to FGFR2 regulates SHP2 function. Cellular Signal. 2010;22:23–33. doi: 10.1016/j.cellsig.2009.08.011. [DOI] [PubMed] [Google Scholar]
- 44.Ooms LM, Dyson JM, Kong AM, Mitchell CA. Analysis of phosphatidylinositol 3,4,5 trisphosphate 5-phosphatase activity by in vitro and in vivo assays. Methods Mol Biol. 2009;462:223–239. doi: 10.1007/978-1-60327-115-8_15. [DOI] [PubMed] [Google Scholar]
- 45.Fry MJ. Phosphoinositide (PI) 3-kinase assays. Methods Mol Biol. 2009;462:345–62. doi: 10.1007/978-1-60327-115-8_22. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.