

NEDDylation controls the ubiquitination and proteasomal degradation of proteins that are critical for cell survival, oncogenic transformation, and therapeutic sensitivity including p27, CDT1, IκBα, NRF-2, cyclin E, c-Myc, p53, and hypoxia-inducible factor-1α.1 Aberrant NEDDylation has been implicated in malignant pathogenesis and drug resistance, thus providing strong rationale to develop strategies to disrupt this specific mechanism of protein turnover for cancer therapy. MLN4924 (Pevonedistat) is a first-in-class inhibitor of NEDDylation that has been evaluated in multiple phase I trials.2 Despite its robust effects in preclinical models of acute myeloid leukemia (AML) and preliminary efficacy in patients with relapsed/refractory AML and high-risk myelodysplastic syndromes (MDS), the specific pharmacodynamic (PD) effects that mediate the anti-leukemic activity of MLN4924 have not been completely defined and no predictive biomarkers of clinical sensitivity to MLN4924 have been validated.3,4, 5
Advanced proteomic profiling methods represent a powerful tool to determine specific protein signatures of individual cancers. In solid tumors, shotgun proteomics has rapidly become a validated approach to monitor changes in protein makeup for early detection and treatment guidance.6 We conducted comprehensive protein profiling of MV4-11 FLT3 ITD+ cells to determine the global impact of inhibiting NEDDylation with MLN4924 on the AML proteome. Cells were treated with MLN4924 (1 μM) for 24 hours and samples were processed for proteome quantification as previously described (Supplementary methods).7 3,812 unique proteins were detected in vehicle and MLN4924-treated MV4-11 cells (Figure 1A). Using a 2-fold PD change cut-off and a ratio of 1 unique peptide, 47 proteins were significantly upregulated following MLN4924 treatment (P< 0.05, Table 1). The effects of MLN4924 treatment on the levels of selected proteins [NEDDylated cullins, free NEDD8, CDT1, CDKN1B (p27), WEE1, HMOX1, NQO1, RRM2, BRD2, and TXNRD1] were confirmed by immunoblotting (Figure 1B). As expected, MLN4924 triggered increased levels of many established cullin-dependent substrates. Known cullin-RING ligase (CRL) substrate proteins that were upregulated 2 or more fold in response to drug treatment included WEE1, CLSPN, CDT1, CDKN1B (p27), MORF4L2, MRFAP1, MORF4L1, KEAP1, NUSAP1, and MLX. Other notable factors whose levels were modulated by MLN4924 treatment included the cell cycle progression 1 CCPG1, the electron transport chain component NQO1, regulators of cellular redox status (GSR, GLUL, GCLM, TXNRD1, and HMOX1), the DNA helicase DNA2, the DNA replication factor ESCO2, the regulator of p53 stability KIAA0101 and heat shock proteins (HSBP1, DNAJB4). Several of these proteins such as CDT1 have been implicated as mediators of DNA re-replication and DNA damage-mediated apoptosis that are stimulated by MLN4924 treatment and responsible, at least in part, for the anti-neoplastic effects of this agent.8,9
Figure 1. Proteome profiling of MLN4924 provides rationale for its combination with azacitidine.
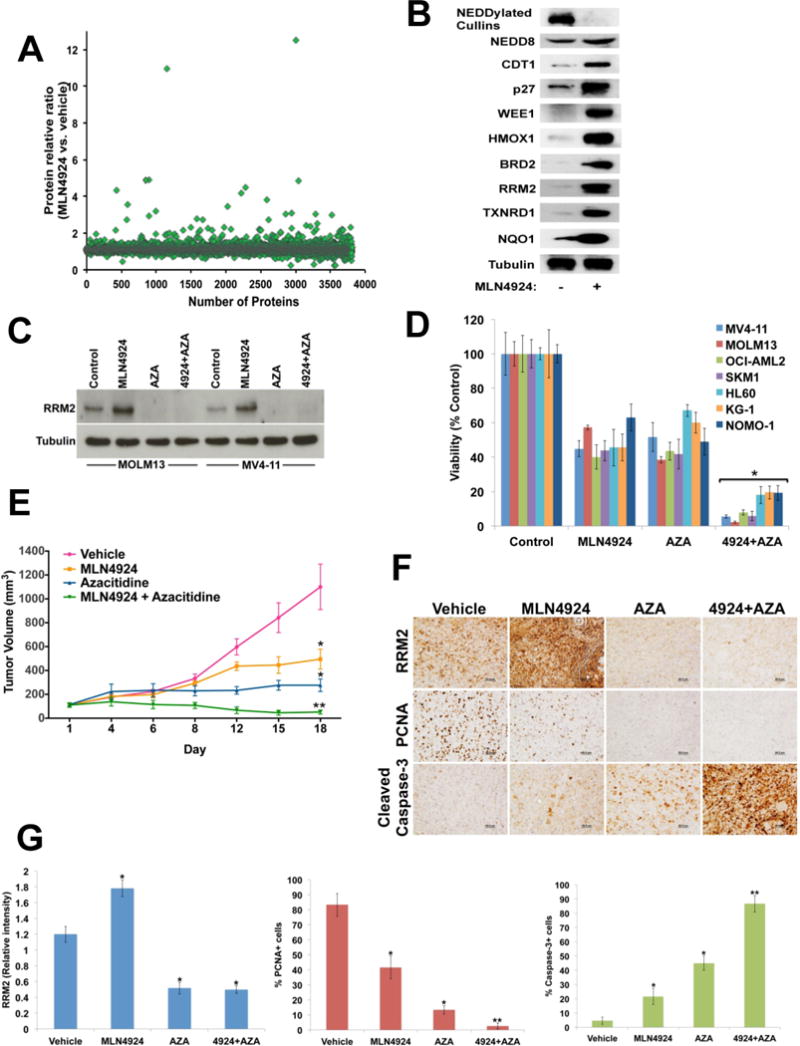
(A) Scatter plot illustrates the global protein expression of MV4-11 AML cells treated with MLN4924 (1 μM) for 24 hours. A total of 3,812 unique proteins were detected. Forty-seven proteins were found to increase in expression 2 or more fold following treatment with MLN4924. (B) MV4-11 cells were treated with MLN4924 (1 μM) for 24 hours. The expression level of selected proteins that were upregulated by MLN4924 treatment was validated by immunoblotting. Tubulin documented equal protein loading. (C) MV4-11 and MOLM13 cells were treated with MLN4924 in the presence or absence of azacitidine (AZA) for 24 hours. The effects of drug treatment on RRM2 levels were assessed by immunoblotting. Tubulin documented equal protein loading. (D) A panel of 7 human AML cell lines was treated with MLN4924, AZA, or the combination for 72 hours. The effects of drug treatment on cell viability were quantified by ATPLite assay. **P<0.05 based on comparison of combination versus single agent treatments. (E) MOLM13 human AML cells were implanted into immunodeficient nude mice (N=10 per group) to establish a xenograft model of AML. Mice were treated with vehicle control, 60 mg/kg MLN4924 (QDx5), 5 mg/kg AZA twice per week, or both agents. *P<0.05 based on comparison of single agent treatments versus vehicle control; **P<0.05 based on comparison of combination versus either single agent treatment. (F) Immunohistochemical analysis shows that administration of the MLN4924/AZA combination effectively diminishes RRM2 expression in-vivo, reduces proliferation (PCNA) and increases apoptosis (cleaved caspase-3). (G) Quantification of immunohistochemical analyses of RRM2 (relative expression), PCNA and cleaved caspase-3 (percentage of positive cells). *P<0.05 based on comparison of single agent treatments versus vehicle control;
**P<0.05 based on comparison of combination versus either single agent treatment.
Table 1.
Proteins significantly elevated by MLN4924 treatment in MV4-11 AML cells
| Gene symbol | Description | Ratio (MLN4924 vs. vehicle) |
|---|---|---|
| WSB1 | WD repeat and SOCS box-containing protein 1 isoform 1 | 12.5 |
| HMOX1 | heme oxygenase 1 | 11.0 |
| GLUL | glutamine synthetase | 4.9 |
| LOC1005080 | predicted: aldo-keto reductase family 1 member C2-like isoform 1 | 4.9 |
| CDKN1B | cyclin-dependent kinase inhibitor 1B | 4.8 |
| MRFAP1 | MORF4 family-associated protein 1 | 4.5 |
| NQO1 | NAD(P)H dehydrogenase [quinone] 1 isoform c | 4.3 |
| MORF4L2 | mortality factor 4-like protein 2 | 4.2 |
| ESCO2 | N-acetyltransferase ESCO2 | 3.8 |
| GSR | glutathione reductase, mitochondrial isoform 3 precursor | 3.5 |
| MLX | max-like protein X isoform beta | 3.4 |
| GCLM | glutamate--cysteine ligase regulatory subunit | 3.2 |
| WEE1 | wee1-like protein kinase isoform 1 | 3.1 |
| CLSPN | claspin isoform 1 | 3 |
| CCPG1 | cell cycle progression protein 1 isoform 2 | 3 |
| APPBP2 | amyloid protein-binding protein 2 | 3 |
| MORF4L1 | mortality factor 4-like protein 1 isoform 1 | 3 |
| SLBP | histone RNA hairpin-binding protein | 2.8 |
| RRM2 | ribonucleoside-diphosphate reductase subunit M2 isoform 1 | 2.7 |
| DNAJB4 | dnaJ homolog subfamily B member 4 | 2.7 |
| NUSAP1 | nucleolar and spindle-associated protein 1 isoform 2 | 2.7 |
| NCF2 | neutrophil cytosol factor 2 isoform 1 | 2.5 |
| SHKBP1 | SH3KBP1-binding protein 1 | 2.4 |
| CLNS1A | methylosome subunit pICln | 2.3 |
| NAA30 | N-alpha-acetyltransferase 30, NatC catalytic subunit | 2.3 |
| KIAA0101 | PCNA-associated factor isoform 1 | 2.3 |
| TK1 | thymidine kinase, cytosolic | 2.3 |
| F5 | coagulation factor V precursor | 2.3 |
| SPIN4 | spindlin-4 | 2.2 |
| CHD3 | chromodomain-helicase-DNA-binding protein 3 isoform 3 | 2.2 |
| KCTD9 | BTB/POZ domain-containing protein KCTD9 | 2.2 |
| SKA3 | spindle and kinetochore-associated protein 3 isoform 1 | 2.2 |
| ETNK1 | ethanolamine kinase 1 isoform A | 2.2 |
| TXNRD1 | thioredoxin reductase 1, cytoplasmic isoform 1 | 2.2 |
| GMNN | geminin | 2.2 |
| CDT1 | DNA replication factor Cdt1 | 2.2 |
| C3orf37 | chromosome 3 open reading frame 37 | 2.2 |
| KEAP1 | kelch-like ECH-associated protein 1 | 2.1 |
| BRD2 | bromodomain-containing protein 2 isoform 1 | 2.1 |
| HSBP1 | heat shock factor-binding protein 1 | 2.1 |
| DIAPH3 | protein diaphanous homolog 3 isoform a | 2.1 |
| HNRNPR | heterogeneous nuclear ribonucleoprotein R isoform 1 | 2.1 |
| DNA2 | DNA2-like helicase | 2.1 |
| TACC3 | transforming acidic coiled-coil-containing protein 3 | 2.1 |
| UFD1L | ubiquitin fusion degradation protein 1 homolog isoform B | 2 |
| WRNIP1 | ATPase WRNIP1 isoform 1 | 2 |
| ATAD2 | ATPase family AAA domain-containing protein 2 | 2 |
Reactome network analysis of the 47 top proteins whose levels were increased 2 or more fold by MLN4924 treatment demonstrated that the affected proteins primarily clustered in the cell cycle, mitosis, and stress response pathways (Table S1). We also merged our proteomic profiling of MV4-11 AML cells with a comprehensive SILAC analysis performed in A375 melanoma cells similarly treated with MLN4924. We determined that 36% (17/47) of the proteins that were pharmacodynamically increased by MLN4924 were identical between the two analyses.9 The proteins that differed between these two studies are detailed in Table S2. Considering that the majority (nearly two-thirds) of the proteins that were modulated by drug treatment were distinct in this analysis, the repertoire of proteins that are modulated by this agent may be tumor-type dependent.7, 9 It is possible that the spectrum of affected targets in individual tumors may be important in determining sensitivity to MLN4924 as patients with MDS/AML have responded better to this agent than those with other malignancies based on initial phase I studies.2
We conducted an extensive literature search to identify potential genetic interactions and therapeutic implications of the proteins significantly affected by MLN4924 treatment in AML (Table S3). Notably, several of the proteins modulated by MLN4924 in our study could represent biomarkers for patient stratification. For example, the helicase DNA binding protein CHD3 (fold change: 2.23) was reported to be up-regulated in patients with MYST3-CREBBP AML or AML with a monocytic phenotype and high FLT3 expression that experienced short complete remissions following conventional therapy.10,11 Considering this, it would be worthwhile to investigate whether patients with high basal CHD3 levels derive less benefit from treatment with MLN4924. We also detected drug-induced changes in 27 members of the RNA helicase family including the newly discovered DDX41, which has been found to be mutated in patients with AML and MDS (Table S4).12 Out of these 27 helicases, DDX24 and DDX54 were most increased (1.74- and 1.51-fold, respectively). Notably, DDX24 was also upregulated (> 1.8-fold) in A375 melanoma cells following MLN4924 treatment.9 Although the impact of drug treatment on individual helicases fell below our set threshold of significance, the collective data suggest that MLN4924 may have a previously undefined class effect on RNA helicase function. Further investigation is required to assess whether NEDD8 plays a novel role in the regulation of RNA helicases and to determine how this may impact MLN4924 efficacy.
In addition to its potential value in refining patient characteristics that are linked to drug sensitivity, the proteomic technology we utilized may also facilitate the selection of rational combination partners for optimal therapeutic benefit. Notably, a number of the PD targets that were elevated in response to treatment with MLN4924 are directly actionable with existing approved and investigational drugs (Table S3). We hypothesized that the ability of azacitidine (AZA) to antagonize ribonucleotide reductase (RRM2), a well-established mediator of resistance to many cytotoxic anticancer agents, may translate into increased sensitivity to MLN4924.13, 14 To investigate this possibility, we first tested the ability of AZA to antagonize MLN4924-mediated RRM2 induction. Immunoblotting analyses showed that AZA effectively countered the PD-related increase in RRM2 in both MOLM13 and MV4-11 AML cells (Figure 1C). Subsequent analysis of the effects of the AZA/MLN4924 combination on the viability of a panel of 7 AML cell lines demonstrated that the reduction in RRM2 expression was associated with a synergistic (CI less than 1.0) reduction in cell viability in all models evaluated (Figure 1D). The in vitro synergy we observed was recapitulated in a FLT3-ITD+ AML xenograft model where administration of the AZA/MLN4924 combination yielded significantly greater benefit than either monotherapy (P<0.05, Figure 1E). Analysis of specimens obtained from mice in each treatment group showed that the combination effectively suppressed RRM2 expression in vivo and led to a significantly greater reduction in AML cell proliferation (PCNA) and elevated levels of apoptosis compared to single agent treatments (P<0.05, Figure 1F–G). These findings demonstrate proof of concept that proteomic profiling is a valuable approach to identify rational promising drug combinations and also support the investigation of a potential relationship between RRM2 expression and clinical response in the ongoing trial of MLN4924 plus AZA in elderly patients with AML (NCT0181426).
In addition to AZA, our data also suggest a number of other potential combination partners for MLN4924. For example, the ability of MLN4924 to increase BRD2 levels may heighten the sensitivity of AML cells to BET inhibitors like OTX015.15 We are currently assessing this possibility in preclinical models of AML. Considering that OTX015 and other BET inhibitors are currently in clinical trials, novel combination approaches with MLN4924 could be seamlessly incorporated into existing studies. In summary, our study demonstrates that high-throughput proteomic technology is a powerful tool with potential applications in patient refinement and the identification of rational actionable targets for precision combination therapeutic strategies. These findings support the implementation of high-throughput proteomics as a synergistic complement to genomics in novel anticancer drug development.
Supplementary Material
Acknowledgments
This work was supported by the National Cancer Institute grant R01CA172443 (JSC).
Footnotes
Conflicts of interest
Anthony Possemato and Sean Beausoleil are employees of Cell Signaling Technology.
Author contributions
V.V. analyzed and interpreted data and wrote the manuscript; S.T.N. analyzed and interpreted data and participated in manuscript preparation; C.M.E. performed experiments, analyzed the data, and edited the manuscript; K.R.K. provided intellectual input and contributed to manuscript preparation; A.P. and S.A.B. performed proteome profiling and contributed to manuscript preparation; Y.H. participated in manuscript preparation; H.E.C., A.N., A.S.A., J.P.M., and M.A.S. contributed to data interpretation and manuscript preparation; J.S.C. designed the study, analyzed and interpreted data, and wrote the manuscript. All authors approved the manuscript before submission.
References
- 1.Podust VN, Brownell JE, Gladysheva TB, Luo RS, Wang C, Coggins MB, et al. A Nedd8 conjugation pathway is essential for proteolytic targeting of p27Kip1 by ubiquitination. Proc Natl Acad Sci U S A. 2000;97:4579–4584. doi: 10.1073/pnas.090465597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nawrocki ST, Griffin P, Kelly KR, Carew JS. MLN4924: a novel first-in-class inhibitor of NEDD8-activating enzyme for cancer therapy. Expert Opin Investig Drugs. 2012;21:1563–1573. doi: 10.1517/13543784.2012.707192. [DOI] [PubMed] [Google Scholar]
- 3.Swords RT, Kelly KR, Smith PG, Garnsey JJ, Mahalingam D, Medina E, et al. Inhibition of NEDD8-activating enzyme: a novel approach for the treatment of acute myeloid leukemia. Blood. 2010;115:3796–3800. doi: 10.1182/blood-2009-11-254862. [DOI] [PubMed] [Google Scholar]
- 4.Nawrocki ST, Kelly KR, Smith PG, Keaton M, Carraway H, Sekeres MA, et al. The NEDD8-activating enzyme inhibitor MLN4924 disrupts nucleotide metabolism and augments the efficacy of cytarabine. Clin Cancer Res. 2015;21:439–447. doi: 10.1158/1078-0432.CCR-14-1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Swords RT, Erba HP, DeAngelo DJ, Bixby DL, Altman JK, Maris M, et al. Pevonedistat (MLN4924), a First-in-Class NEDD8-activating enzyme inhibitor, in patients with acute myeloid leukaemia and myelodysplastic syndromes: a phase 1 study. Br J Haematol. 2015;169:534–543. doi: 10.1111/bjh.13323. [DOI] [PubMed] [Google Scholar]
- 6.Hayashi E, Kuramitsu Y, Okada F, Fujimoto M, Zhang X, Kobayashi M, et al. Proteomic profiling for cancer progression: Differential display analysis for the expression of intracellular proteins between regressive and progressive cancer cell lines. Proteomics. 2005;5:1024–1032. doi: 10.1002/pmic.200401132. [DOI] [PubMed] [Google Scholar]
- 7.Nawrocki ST, Kelly KR, Smith PG, Espitia CM, Possemato A, Beausoleil SA, et al. Disrupting protein NEDDylation with MLN4924 is a novel strategy to target cisplatin resistance in ovarian cancer. Clin Cancer Res. 2013;19:3577–90. doi: 10.1158/1078-0432.CCR-12-3212. [DOI] [PubMed] [Google Scholar]
- 8.Lin JJ, Milhollen MA, Smith PG, Narayanan U, Dutta A. NEDD8-targeting drug MLN4924 elicits DNA rereplication by stabilizing Cdt1 in S phase, triggering checkpoint activation, apoptosis, and senescence in cancer cells. Cancer Res. 2010;70:10310–10320. doi: 10.1158/0008-5472.CAN-10-2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liao H, Liu XJ, Blank JL, Bouck DC, Bernard H, Garcia K, et al. Quantitative proteomic analysis of cellular protein modulation upon inhibition of the NEDD8-activating enzyme by MLN4924. Mol Cell Proteomics. 2011;10:M111 009183. doi: 10.1074/mcp.M111.009183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Camos M, Esteve J, Jares P, Colomer D, Rozman M, Villamor N, et al. Gene expression profiling of acute myeloid leukemia with translocation t(8;16)(p11;p13) and MYST3-CREBBP rearrangement reveals a distinctive signature with a specific pattern of HOX gene expression. Cancer Res. 2006;66:6947–6954. doi: 10.1158/0008-5472.CAN-05-4601. [DOI] [PubMed] [Google Scholar]
- 11.Mills KI, Kohlmann A, Williams PM, Wieczorek L, Liu WM, Li R, et al. Microarray-based classifiers and prognosis models identify subgroups with distinct clinical outcomes and high risk of AML transformation of myelodysplastic syndrome. Blood. 2009;114:1063–1072. doi: 10.1182/blood-2008-10-187203. [DOI] [PubMed] [Google Scholar]
- 12.Polprasert C, Schulze I, Sekeres MA, Makishima H, Przychodzen B, Hosono N, et al. Inherited and Somatic Defects in DDX41 in Myeloid Neoplasms. Cancer Cell. 2015;27:658–670. doi: 10.1016/j.ccell.2015.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aimiuwu J, Wang H, Chen P, Xie Z, Wang J, Liu S, et al. RNA-dependent inhibition of ribonucleotide reductase is a major pathway for 5-azacytidine activity in acute myeloid leukemia. Blood. 2012;119:5229–5238. doi: 10.1182/blood-2011-11-382226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burton TR, Kashour T, Wright JA, Amara FM. Cellular signaling pathways affect the function of ribonucleotide reductase mRNA binding proteins: mRNA stabilization, drug resistance, and malignancy (Review) Int J Oncol. 2003;22:21–31. [PubMed] [Google Scholar]
- 15.Coude MM, Braun T, Berrou J, Dupont M, Bertrand S, Masse A, et al. BET inhibitor OTX015 targets BRD2 and BRD4 and decreases c-MYC in acute leukemia cells. Oncotarget. 2015;6:17698–17712. doi: 10.18632/oncotarget.4131. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.