

Abstract
Model evaluation and selection is an important step and a big challenge in template-based protein structure prediction. Individual model quality assessment methods designed for recognizing some specific properties of protein structures often fail to consistently select good models from a model pool because of their limitations. Therefore, combining multiple complimentary quality assessment methods is useful for improving model ranking and consequently tertiary structure prediction. Here, we report the performance and analysis of our human tertiary structure predictor (MULTICOM) based on the massive integration of 14 diverse complementary quality assessment methods that was successfully benchmarked in the 11th Critical Assessment of Techniques of Protein Structure prediction (CASP11). The predictions of MULTICOM for 39 template-based domains were rigorously assessed by six scoring metrics covering global topology of Cα trace, local all-atom fitness, side chain quality, and physical reasonableness of the model. The results show that the massive integration of complementary, diverse single-model and multi-model quality assessment methods can effectively leverage the strength of single-model methods in distinguishing quality variation among similar good models and the advantage of multi-model quality assessment methods of identifying reasonable average-quality models. The overall excellent performance of the MULTICOM predictor demonstrates that integrating a large number of model quality assessment methods in conjunction with model clustering is a useful approach to improve the accuracy, diversity, and consequently robustness of template-based protein structure prediction.
Keywords: Protein structure prediction, model quality assessment, integration, template-based modeling, CASP
1. INTRODUCTION
In the genomic era, high-throughput genome or transcriptome sequencing technologies have generated a large amount (~100 million) of protein sequences. It is important to obtain the tertiary structures of these protein sequences in order to understand their biochemical, biological and cellular functions1-3. Experimental techniques (e.g. X-ray crystallography or NMR spectroscopy) can determine protein structures. However, these techniques cannot solve the structures of all proteins because they are relatively expensive and time consuming. Thus far, only a small portion of proteins (~99,000) have experimentally verified structures. Therefore, cheaper and faster computer-assisted prediction of protein tertiary structures is becoming increasingly popular and important4-8.
Computational prediction methods of protein tertiary structures generally fall into two categories: template-based modeling and template-free modeling. Template-based modeling methods generate the tertiary structure for a target protein by identifying its homologous structure templates and transferring the template structures to the structure of the target for further refinement9-11. These methods are the most widely used protein modeling methods, and their predictions are relatively accurate and usable if good homologous templates could be found. If no homologous templates could be found for a target protein, template-free modeling methods are employed to construct structural models for the target protein from scratch or from the combination of small structural fragments9,12. Since 1994, every two years both template-based and template free modeling methods (e.g. 13,14,15,16,17,18,19) were blindly and rigorously evaluated in the Critical Assessment of Protein Structure Prediction (CASP) experiments. In this work, we report our findings and analyses regarding the template-based predictions of our MULTICOM predictor based on massive integration of diverse and complementary protein model quality assessment methods in the CASP11 experiment held in 2014.
Evaluating the quality of predicted models and selecting the most accurate ones from them is an important step and a big challenge in protein structure prediction. There are two typical kinds of protein model quality assessment (QA) methods: single-model quality assessment method and multi-model quality assessment method 20. Single-model quality assessment methods12,18-27 evaluate the quality of a single model without referring to other models and assigned it a global quality score. Multi-model quality assessment methods 28-34 (also called clustering based methods) evaluate the predicted models for a target protein based on their pairwise structural similarity. For instance, some multi-model quality assessment methods 33,34 employ clustering techniques to cluster models into different groups according to their structural similarities, and then select the center model in each group as the presumably best model most similar to the native structure.
Because of the difficulty of predicting the real quality of a predicted protein model and the limitation of current techniques, one individual QA method generally cannot select the best model from the model pool. For example, single model QA methods may not be sensitive enough to rate a largely correct topology with significant local structural flaws higher than a native like but incorrect topology. Multiple model QA methods often fail when the majority of the predicted models is of bad qualities and is structurally similar to each other 20. The model selected by the clustering-based methods usually is not the best model if models in the largest cluster are of bad quality.
Therefore, some protein tertiary structure prediction methods in recent CASP experiments tried to use the consensus of QA methods to evaluate the predicted models. For example, Zhang-Server 28 evaluated the predicted models using the consensus score of seven MQAP methods (e.g. the I-TASSER C-score 14, structural consensus measured by pair-wise TM-score22, RW35, RWplus35, Dfire36, Dope37, verify3D27). MUFOLD38 used three single-model QA methods (e.g. OPUS-CA39, Dfire36, ModelEvaluator40) to filter out poor models and then used consensus QA method (e.g. clustering) to evaluate the remaining models. Pcons41 combined structural consensus42 with a single model machine learning-based QA method ProQ224 to evaluate the predicted models. Combining multiple quality assessment methods appeared to be an important approach to improve model evaluation as demonstrated in the CASP experiments. However, more extensive and sophisticated methods of integrating a large number of diverse and complementary QA methods need to be developed and analyzed.
Here we conduct a thorough analysis of our recently developed tertiary structure prediction methods based on a large-scale protein model quality assessment method – MULTICOM43 on its template-based model predictions in 2014 CASP11 experiment in order to investigate the strengths and weaknesses of massive quality assessment methods. Unlike other tertiary structure prediction methods using only one or several model quality assessment methods, MULTICOM integrated 14 complementary QA methods, which included both single-model QA methods and multi-model QA methods. Our tertiary structure prediction method participated in the CASP11 experiment as a human predictor and was ranked as one of top few methods for template-based protein structure modeling. The results indicate that the combination of the array of QA methods in conjunction with good model sampling and clustering is a promising direction for improving protein tertiary structure prediction.
2. METHODS
Our MULTICOM method (human group MULTICOM in CASP11 experiment), although categorized as MULTICOM human predictor, is largely an automated method. MULTICOM’s success, primarily, is because of exploiting appropriate use and combination of existing QA methods some of which we developed in house to complement existing methods, and not because of human intervention. Although the method has been discussed briefly in43, here we discuss it comprehensively with an emphasis on the details of the method and an extensive evaluation strategy.
2.1 Massive protein model quality assessment for ranking protein structural models
Figure 1 provides an overview of the entire workflow of MUTLICOM. MULTICOM takes a pool of structural models predicted by a variety of available protein structure prediction tools as input. This pool of models is supplied in parallel to both individual QA ranking methods and a model clustering tool - MUFOLD-CL44. The rankings generated by all QA methods are combined to obtain two consensus rankings. Since the consensus rankings may put similar models in the top ranks, in order to increase diversity in the top five selected models, the model clustering information is used to replace some similar top-ranked models with structurally different models from other model clusters if necessary. The final selected models are further refined by a model combination approach45.
Figure 1. Workflow of MULTICOM large-scale model quality assessment method.
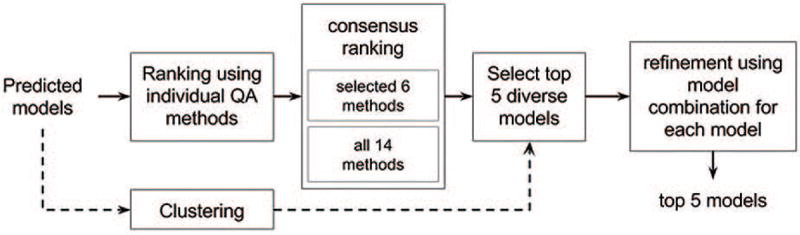
Predicted models are ranked by different QA methods followed by a consensus ranking and at the same time models are clustered based on structural similarity into groups. For diversity, top 5 ranks of consensus results are updated using clustering information and the corresponding models further refined using model combination approach.
Specifically, in CASP 11 experiment we used the hundreds of models for each target predicted by all CASP participants as input. Input models are first ranked by existing and our in-house developed single-model and multiple-model quality assessment (QA) methods - a total of 14 QA methods43 whose software were available. These include 8 single-model methods, see Table I, two in-house single-model QA methods: (a) MULTICOM-NOVEL, and (b) Modelcheck2 - an improved version of ModelEvaluator score40. We also use 4 multiple-model QA methods: (a) ModFOLDclust232, (b) APOLLO29, (c) Pcons41, and (d) QApro20.
Table I.
Publicly available single-model QA methods used in our MULTICOM method.
| Method | Description |
|---|---|
| OPUS-PSP46 | Method based on side-chain derived orientation-dependent all-atom statistical potential |
| ProQ224 | Uses support vector machines to predict local as well as global quality of protein models; features of ProQ combined with updated structural and predicted features |
| RWplus35 | Method based on a new pair-wise distance-dependent atomic statistical potential function (RW) and side-chain orientation-dependent energy term |
| ModelEvaluator40 | Uses only structural features with support vector machine regression; assigns absolute GDT-TS score to a model by comparing secondary structure, relative solvent accessibility, contact map, and beta sheet topology with prediction from sequence |
| RF_CB_SRS_OD50 | Uses residue-based pairwise distance dependent statistical potential at various spatial pair separations |
| SELECTpro58 | Structure-based energy function with energy terms that include predicted secondary structure, solvent accessibility, contact map, beta-strand pairing, and side-chain hydrogen bonding |
| Dope37 | Uses probability theory to derive an atomic distance-dependent statistical potential |
| DFIRE259 | Based on statistical energy function that uses orientation –dependent interaction from protein structures treating each polar atom as dipole |
The integration of both single-model QA methods and multi-model QA methods is to leverage the strengths of the two kinds of methods and alleviate their weaknesses in order to rank models better than any of the individual method. The single-model methods may distinguish quality variation among good models, but may mistakenly favor a physically appealing, but low-quality models over largely correct models with significant local flaws. In contrast, the multi-model methods can often select some good models of average quality, but fail to identify models of better-than-average quality.
The rankings obtained using these individual methods are combined in two ways: (a) a mean is computed for each model to produce an average ranking of all 14 methods, (b) rankings of only 6 selected methods are used to produce an average ranking. The selected 6 methods include 4 single-model QA methods, MULTICOM-NOVEL, Modelcheck2, Dope37, and OPUS_PSP46, and 2 multiple-model methods, QApro, and Pcons. Before the CASP11 experiment started, we tested all possible ways of combining the rankings on the data of 46 CASP10 targets, and found that combination of these selected 6 methods resulted in the lowest average loss of 0.037 GDT-TS score for the top one selected models in comparison with the best possible models, 0.02 GDT-TS score lower than combination of all 14 methods. However, since the benchmark testing was not comprehensive and may overfit the data, we retained both consensus approaches in our overall method for CASP11 experiment.
During CASP11, in order to choose between 6-methods based consensus and all 14-methods based consensus for our overall method, we predict the ‘difficulty’ of the target using the multi-model QA tool APOLLO in order to use separate methods for ‘hard’ and ‘easy’ cases43. APOLLO’s score of greater than 0.3 generally hints higher quality of models because of high pairwise similarity between them, for example, when matching templates are found for the target. Hence, if APOLLO’s similarity score for top ranked model is greater than 0.3, we compare this top model with the two top ranked models ranked by 6-method and all 14-method consensus, and finally select the ranking whose top model is more similar to APOLLO’s top model. Here, in addition to using APOLLO to break the ties between the two consensus methods, the other rational is to filter out models of an incorrect topology that the consensus methods may accidentally rank at the top due to their use of many single-model quality assessment methods, by taking advantage of APOLLO’s capability of selecting a good model in the case of easy prediction. According to our experiment on the CASP10 data, if the highest pairwise similarity score of the models measured by APOLLO is greater than 0.3, which often suggests the prediction is relatively easy, the top model selected by APOLLO generally has a good, but not necessarily the best topology. So, the idea of using the top model selected by APOLLO to re-rank the top models of the consensus methods here is to make sure the bad models with incorrect topologies selected by those methods will be completely ruled out. So, instead of directly using APOLLO’s ranking, APOLLO is only used to provide some auxiliary information to make sure one of the top models ranked by those methods would be correct when the prediction is relatively easy. Overall, the ranking of models is largely dominated by the two consensus methods, which performed better than using APOLLO score alone.
Furthermore, in order to further improve the reliability of the top one model, the top one models of the two consensus rankings and of the top server predictors (e.g., MULTICOM-CLUSTER and Zhang-Server) were compared with the top one model of APOLLO, and the model most similar to the top one model of APOLLO was used as the top one model in the final ranking without changing the ranking of all other models. However, if APOLLO’s pairwise score for the top ranked model is less than or equal to 0.3, we consider the target to be ‘hard’, and use ab initio biased decision to make the selection of consensus ranking. For this, we predict secondary structure of input target sequence using PSIPRED47,48 and compare this with secondary structure of top ranked models in both consensus rankings by computing accuracy. Again, we select the consensus ranking whose top model has higher secondary structure similarity with predicted secondary structure. Despite the seemingly complexity of the modeling ranking strategy used by MULTICOM, the selection of top one model was largely determined by the two consensus methods with some influence from the other factors such as APOLLO’s ranking scores, top server predictors’ top models, and predicted secondary structures.
After selection of the appropriate ranking, instead of simply using top 5 ranked models as final rank, we use model clustering information to increase diversity in the top 5 list of models which is important especially for hard targets whose real structure is often very uncertain. As top five models selected by the approach above may be similar, if one is incorrect, all of them will fail. Therefore, it useful to include different models in the top five list. As such, MULTICOM always keeps the top two ranked models. If the model ranked third belongs to any of the clusters that the previously selected models belong to, it will be removed from the complete rank and the remaining ranking below is lifted up repeatedly until we find a model in a different cluster. The process is repeated for fourth and fifth ranks ensuring diversity in the final top five models. In addition, we employed a model filtering technique to ensure that low quality models do not make their way up to the top 5 ranks. That is, during the re-ranking process, models that were ranked at bottom 10% by our in-house MULTICOM-NOVEL QA method were skipped because those models were mostly bad models such as largely unfolded models according to our experiment. Clustering is performed based on structural similarity of the models using MUFOLD-CL44, a model clustering method based on the comparison of protein distance matrices. Our comparison of MUFOLD-CL with other techniques based on structural distance like RMSD49, show similar accuracy but MUFOLD-CL runs much faster.
As the last step of MULTICOM method, a model combination approach is used to integrate each selected model with other similar models in the pool to obtain a refined model45. Basically, the Modeller is used to use each selected model and other similar models as templates to regenerate a number of combine models for a target. The model with minimum Modeller energy is selected as the refined model.
2.2 Summary of some individual QA methods used by MULTICOM
APOLLO, one of the 4 multiple-model methods we use, generates a pair-wise average GDT-TS score by performing a full pairwise comparison between all input models. The predicted GDT-TS score for a model is the average GDT-TS score between the model and all other models in the model pool. For models that are incomplete predictions (only parts of the target are predicted), the score is scaled down by the ratio of the models’ sequence length divided by the target length. ModFOLDclust2, another multiple model method, uses mean score of the global predicted model quality scores from the clustering based method ModFOLDclust and ModFOLDclustQ as its score to rank models. The Pcons protocol, on the other hand, analyzes input models looking for recurring three-dimensional structural patterns and assigns each model a score based on how common its three-dimensional structural patterns are in the whole model pool. Specifically, it estimates the quality of residues in a protein model by superimposing a model to all other models for the same target protein and calculating the S-score for each residue41, which positively correlates with the level of recurrence of local conformations. Pcons predicts the global quality of a model by assigning a score reflecting the average similarity to the entire ensemble of models. The principle of Pcons is that recurring patterns are more likely to be correct than patterns that only occur in one or just a few models. The multiple model method, QApro, combines the scores of ModelEvaluator and APOLLO by summing the product of APOLLO’s pairwise GDT-TS and ModelEvaluator score normalized by the sum of all ModelEvaluator scores.
Besides the four multi-model QA methods and some publicly available single-model QA methods (see their description in Table I), we developed a new in-house single-model QA method, MULTICOM-NOVEL, which uses features extracted from the structure and sequence to predict model quality. To assess the global quality we used following features, (1) amino acids encoded by a 20-digit vector of 0 and 1, (2) difference between secondary structure and solvent accessibility of the model (parsed using DSSP) and the prediction by Spine X (and also SSpro4) from the protein sequence, (3) physical-chemical features (pairwise Euclidean distance score, surface polar score, weighted exposed score, total surface area score), (4) normalized quality score generated by ModelEvaluator40, RWplus score35, dope score37, and RF_CB_SRS_OD score50. Performing statistical analysis for all global features on PISCES51 database, we obtain feature density maps, i.e. the distribution of the difference between the feature and GDT-TS score. For a model whose true quality is unknown, MULTICOM-NOVEL calculates the score for each feature, and combines these scores with the feature density maps to predict the model’s GDT-TS score. For local quality assessment, however, MULTICOM-NOVEL uses support vector machine with environment scores in different Euclidean distance ranges (8, 10, 12, 14, 16, 18, 20, and 30 angstrom) for each amino acid as input features. These environment scores extracted from a 15-residue sliding window that include secondary structure, solvent accessibility, and amino acid types, capture environmental information within a spatial sphere of a residue.
2.3 Evaluation
Together with 142 human and server predictors, our MULTICOM method was blindly tested on 42 human targets during CASP11 experiment. For the 39 TBM human domains of these 42 human targets, we downloaded native structures from CASP’s website (http://www.predictioncenter.org/casp11/index.cgi) for evaluation of the predicted structural models. We also downloaded the top 5 predictions by other server predictors to compare our results. All our evaluations use 6 different evaluation metrics GDT-HA52,53, SphereGrinder (SG)54, RMSD, Local Distance Difference Test (LDDT)55, GDC-all53, Molprobity score53. GDT-HA is a high accuracy version of global distance test (GDT) measure, which has half the size of distance cut off comparing with GDT measure. SG (SphereGrinder) score is an all-atom local structure fitness score, which was designed to complement and add value to GDT measure. Root-mean-square deviation (RMSD) is a measure for the superimposed proteins, which evaluates the average backbone atoms’ distance. It is not ideal for comparing cases when the structures are substantially different52. The Local Distance Difference Test (lDDT) is a superposition-free score that evaluates local distance differences of all atoms in a model. GDC-all score is global measures similar to GDT-HA, but it includes the positions of side-chain carbon atoms. Molprobity is a knowledge based metrics, which evaluates the physical reasonableness of molecular models. Besides the six evaluation metrics we also use various kinds of Z-scores. Z-score of a model is calculated as the model’s GDT-TS score minus the average GDT-TS score of all the models in the model pool of a target divided by the standard deviation of all GDT-TS scores.
3. RESULTS AND DISCUSSION
First, we systematically evaluate the performance of MULTICOM using global and local quality metrics to perform comparative analysis of MULTICOM against all the server predictors participating in CASP11 on 39 TBM human domains.
The distributions of accuracy for individual targets are subsequently explored along with specific case studies highlighting the importance of clustering in conjunction with model selection. Finally, we investigated the consistency and robustness of our massive model quality assessment method compared to any individual quality assessment method.
Table II shows the six quality scores of the first models submitted by MULTICOM and 25 top performing server predictors for 39 TBM human domains. According to the average scores of the first models, MULTICOM performs better than the overall best performing server predictor (Zhang-Server) in terms of GDC, LDDT and Sph-Gr score, and slightly worse than Zhang-Server in terms of GDT-HA, Mol, and RMSD. Table III reports the six quality scores of the best of top five models submitted by MULTICOM and the server predictors. According to the average score of the best of top five models, MULTICOM performs better than the overall best performing server predictor (Zhang-Server) in terms of GDT-HA, GDC, LDDT, and Sph-Gr score, and slightly worse than Zhang-Server in terms of Mol and RMSD score. The results show that, in addition to effectively selecting good top-one models, MULTICOM applies clustering technique to increase the diversity of top five models56 improves the quality of the best of five selected models.
Table II.
The average scores of the first models submitted by MULTICOM (bold) and top 25 performing server predictors.
| Gr. | Name | Num | GDT-HA | GDC | Mol | LDDT | RMSD | Sph-Gr |
|---|---|---|---|---|---|---|---|---|
| 277s | Zhang-Server | 39 | 38.18 | 28.06 | 7.19 | 3.01 | 0.50 | 50.96 |
| 290 | MULTICOM | 39 | 38.14 | 28.38 | 7.06 | 3.20 | 0.51 | 51.15 |
| 499s | QUARK | 39 | 37.59 | 27.65 | 7.76 | 2.96 | 0.49 | 48.95 |
| 038s | nns | 39 | 34.91 | 26.06 | 8.81 | 2.88 | 0.46 | 48.03 |
| 008s | MULTICOM-CONSTRUCT | 39 | 32.71 | 23.93 | 9.91 | 2.84 | 0.41 | 41.10 |
| 216s | myprotein-me | 39 | 31.64 | 23.86 | 10.06 | 2.49 | 0.41 | 42.38 |
| 346s | HHPredA | 39 | 29.78 | 21.34 | 10.77 | 4.28 | 0.36 | 36.01 |
| 420s | MULTICOM-CLUSTER | 39 | 32.49 | 23.96 | 10.42 | 2.90 | 0.42 | 39.49 |
| 279s | HHPredX | 39 | 31.86 | 23.16 | 11.69 | 4.27 | 0.38 | 38.90 |
| 050s | RaptorX | 39 | 32.23 | 23.42 | 8.97 | 2.47 | 0.44 | 41.05 |
| 184s | BAKER-ROSETTASERVER | 39 | 31.88 | 23.60 | 10.09 | 1.96 | 0.44 | 42.39 |
| 212s | FFAS-3D | 39 | 30.46 | 21.67 | 10.02 | 3.27 | 0.38 | 36.69 |
| 300s | PhyreX | 38 | 29.90 | 21.66 | 9.74 | 3.47 | 0.35 | 38.60 |
| 041s | MULTICOM-NOVEL | 39 | 30.60 | 22.40 | 11.77 | 3.33 | 0.40 | 38.97 |
| 251s | TASSER-VMT | 39 | 29.60 | 21.25 | 9.42 | 3.91 | 0.27 | 41.71 |
| 452s | FALCON_EnvFold | 39 | 28.02 | 19.78 | 11.14 | 3.35 | 0.40 | 34.31 |
| 335s | FALCON_TOPO | 39 | 27.92 | 19.56 | 11.19 | 3.43 | 0.39 | 34.11 |
| 381s | FALCON_MANUAL | 39 | 28.02 | 19.69 | 10.94 | 3.33 | 0.39 | 34.51 |
| 414s | FALCON_MANUAL_X | 39 | 27.86 | 19.61 | 11.26 | 3.37 | 0.39 | 34.18 |
| 479s | RBO_Aleph | 36 | 25.04 | 17.69 | 10.78 | 1.69 | 0.37 | 33.44 |
| 410s | Pcons-net | 39 | 27.66 | 19.83 | 14.88 | 2.97 | 0.37 | 32.84 |
| 022s | 3D-Jigsaw-V5_1 | 37 | 27.57 | 19.43 | 9.60 | 3.06 | 0.33 | 32.98 |
| 133s | IntFOLD3 | 39 | 28.60 | 19.88 | 17.02 | 3.35 | 0.39 | 35.68 |
| 117s | raghavagps-tsppred | 39 | 27.59 | 20.32 | 22.89 | 3.58 | 0.37 | 31.97 |
| 073s | SAM-T08-server | 25 | 18.94 | 13.33 | 6.99 | 1.89 | 0.23 | 21.82 |
Table III.
The average scores of the best of top five models submitted by MULTICOM (bold) and top 25 performing server predictors.
| Gr. | Name | Num | GDT-HA | GDC | Mol | LDDT | RMSD | Sph-Gr |
|---|---|---|---|---|---|---|---|---|
| 290 | MULTICOM | 39 | 41.00 | 31.10 | 6.85 | 2.99 | 0.53 | 54.48 |
| 277s | Zhang-Server | 39 | 40.02 | 29.89 | 6.76 | 2.93 | 0.50 | 52.47 |
| 499s | QUARK | 39 | 39.69 | 29.38 | 6.92 | 2.99 | 0.49 | 52.88 |
| 184s | BAKER-ROSETTASERVER | 39 | 37.69 | 28.56 | 8.38 | 1.93 | 0.49 | 50.44 |
| 038s | nns | 39 | 37.56 | 28.07 | 8.00 | 2.91 | 0.49 | 50.29 |
| 420s | MULTICOM-CLUSTER | 39 | 34.98 | 26.10 | 10.08 | 2.91 | 0.44 | 43.26 |
| 216s | myprotein-me | 39 | 34.05 | 25.89 | 10.26 | 2.45 | 0.42 | 44.00 |
| 041s | MULTICOM-NOVEL | 39 | 34.76 | 25.93 | 9.96 | 3.24 | 0.43 | 43.34 |
| 008s | MULTICOM-CONSTRUCT | 39 | 34.38 | 25.84 | 10.67 | 2.90 | 0.42 | 41.33 |
| 251s | TASSER-VMT | 39 | 32.67 | 24.07 | 8.87 | 3.89 | 0.29 | 44.01 |
| 050s | RaptorX | 39 | 32.74 | 23.59 | 8.78 | 2.42 | 0.44 | 41.55 |
| 346s | HHPredA | 39 | 29.78 | 21.34 | 10.77 | 4.28 | 0.36 | 36.01 |
| 212s | FFAS-3D | 39 | 32.09 | 22.93 | 9.87 | 3.30 | 0.39 | 39.70 |
| 279s | HHPredX | 39 | 31.86 | 23.16 | 11.69 | 4.27 | 0.38 | 38.90 |
| 300s | PhyreX | 39 | 31.17 | 22.55 | 10.09 | 3.53 | 0.37 | 39.43 |
| 454s | eThread | 39 | 29.68 | 20.48 | 10.98 | 3.65 | 0.37 | 37.58 |
| 479s | RBO_Aleph | 36 | 27.41 | 19.89 | 10.31 | 1.65 | 0.38 | 34.95 |
| 452s | FALCON_EnvFold | 39 | 29.97 | 21.19 | 10.65 | 3.37 | 0.40 | 35.48 |
| 335s | FALCON_TOPO | 39 | 29.85 | 20.63 | 10.72 | 3.42 | 0.40 | 35.54 |
| 381s | FALCON_MANUAL | 39 | 29.83 | 21.04 | 10.69 | 3.35 | 0.40 | 35.72 |
| 073s | SAM-T08-server | 27 | 21.18 | 15.17 | 7.27 | 2.00 | 0.24 | 25.43 |
| 414s | FALCON_MANUAL_X | 39 | 29.80 | 21.14 | 10.50 | 3.35 | 0.41 | 35.61 |
| 237s | chuo-fams-server | 39 | 30.11 | 21.96 | 14.46 | 3.96 | 0.36 | 32.79 |
| 410s | Pcons-net | 39 | 29.46 | 21.00 | 14.53 | 2.91 | 0.38 | 34.66 |
| 466s | RaptorX-FM | 14 | 8.47 | 5.17 | 3.71 | 1.25 | 0.07 | 9.11 |
To evaluate the overall performance of MULTICOM in CASP11 TBM human targets relative to other server predictors and to explore any possible relationship between target difficulty and accuracy, we first investigated the median accuracy of first models submitted by MULTICOM and other server predictors against the number of residues in domain. Figure 2 shows the evaluation as judged by six different quality metrics. The lack of correlation between target length and accuracy might indicate the presence reliable template(s) irrespective of sequence length and the predictors’ ability to select them accordingly.
Figure 2. Performance of MULTICOM and server predictors with respect to number of residues in domain.
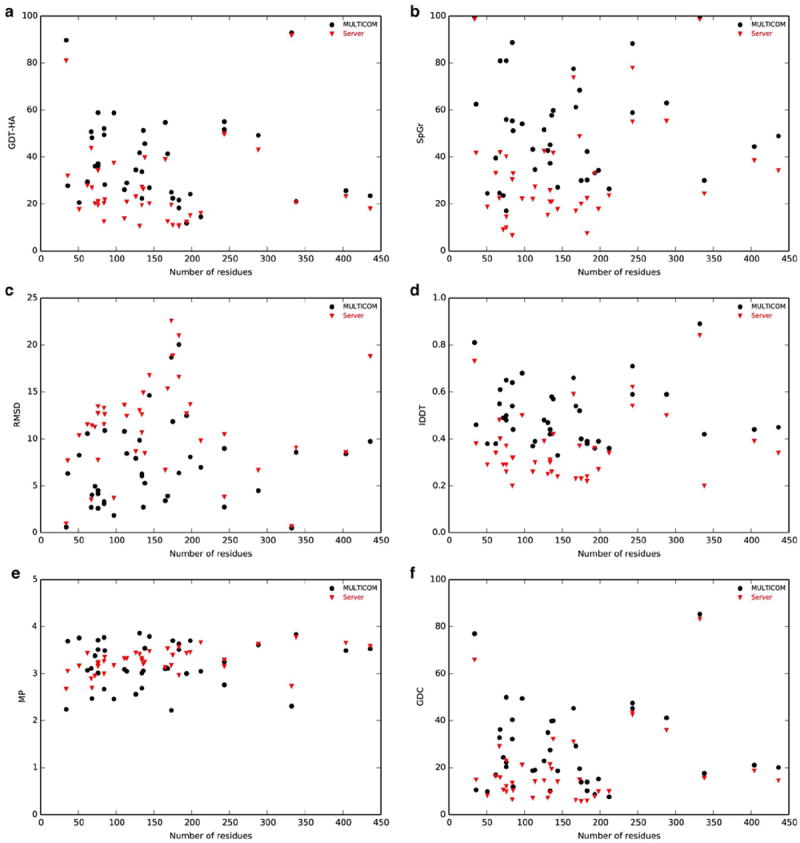
Relationships between number of residues in domain and the median accuracies are shown for these metrics: (a) GDT-HA, (b) SphereGrinder, (c) RMSD, (d) lDDT, (e) MolProbity and (f) GDC. MULTICOM and server predictors are represented by different style and color with the corresponding legends shown on the top-right.
To gain additional insight in target difficulty, we examined the percentage of sequence identity between the target and best template present in Protein Data Bank after optimal structural superposition (as provided by CASP11 assessors at http://www.predictioncenter.org/download_area/CASP11/templates/). In Figure 3, we report the accuracy of first models submitted by MULTICOM and the median performance of server predictors against the percentage of sequence identity for each of the six quality metrics. Once again, no systematic pattern can be observed from between the target difficulty and performance.
Figure 3. Performance of MULTICOM and server predictors with respect to difficulty of target.
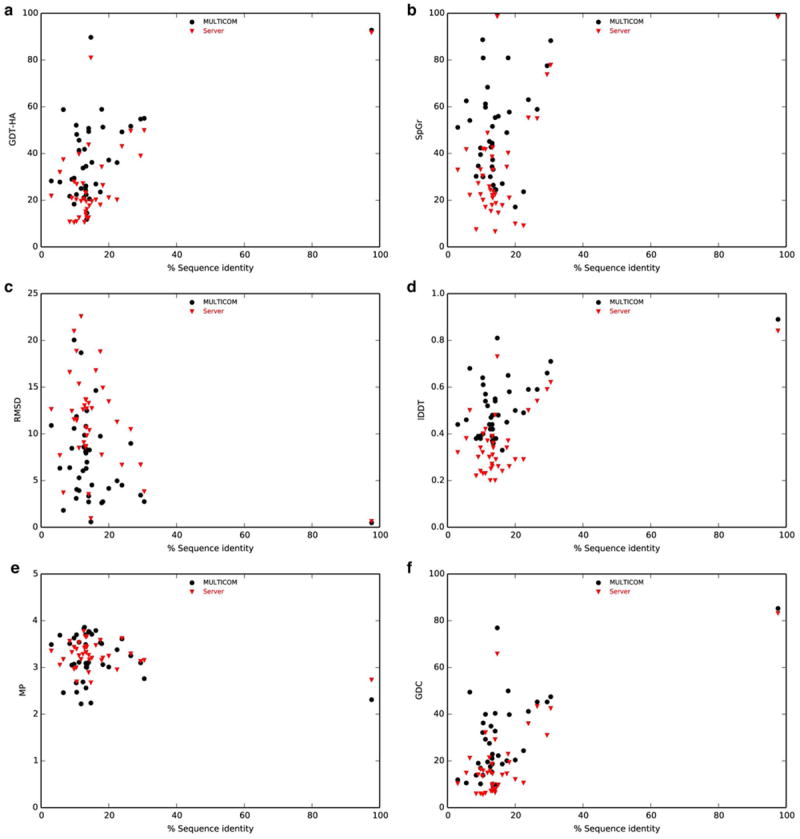
Relationships between the percentage of sequence identity between the target and best template present in Protein Data Bank after optimal structural superposition and the median accuracies are shown for these metrics: (a) GDT-HA, (b) SphereGrinder, (c) RMSD, (d) lDDT, (e) MolProbity and (f) GDC. MULTICOM and server predictors are represented by different style and color with the corresponding legends shown on the top.
In Figure 4, we examined the accuracy of the first models and the best of top five models submitted by MULTICOM and compared it with that of the server predictors. The comparison between the first models submitted by MULTICOM and the best server models (middle panels of Figure 4) indicates the ability of MULTICOM to often select good models from model pool. Furthermore, when the best of top five models submitted by MULTICOM are considered, MULTICOM’s performance of selecting some good models is even better (rightmost panels of Figure 4). This suggests that the massive integration of diverse protein quality assessment methods used in MULTICOM facilitates in selecting good models from the hundreds of alternative models generated by server predictors. MULTICOM’s performance in MolProbity was significantly worse than other quality metrics (Figure 4e), highlighting somewhat lack of physical reasonableness and enhanced stereochemistry in the submitted models. The problem may be caused by the poor quality of side chains and backbone atoms in the models, which could be corrected by using SCWRL57 to repack the side chains, and using a physically-realistic all-atom MD/Monte Carlo simulation to refine the model.
Figure 4. Accuracy of MULTICOM compared to other server predictors.
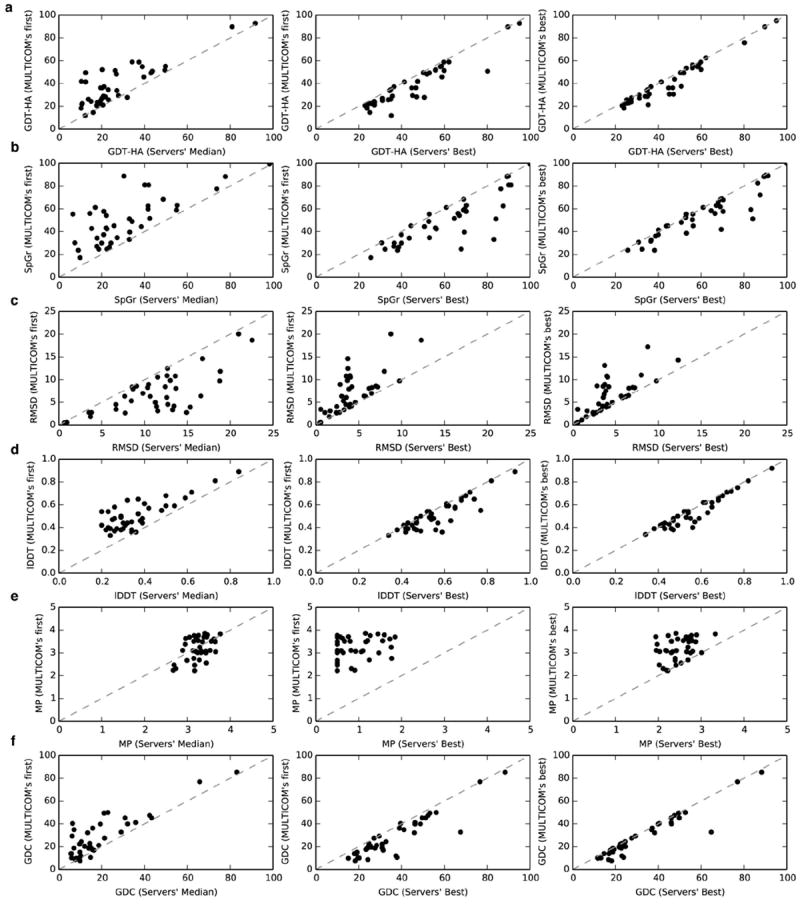
First models submitted by MULTICOM compared to median of the first models submitted by server predictors, first models submitted by MULTICOM compared to best models submitted by server predictors and best of five models submitted by MULTICOM compared to best models submitted by server predictors are shown for these metrics: (a) GDT-HA, (b) SphereGrinder, (c) RMSD, (d) lDDT, (e) MolProbity and (f) GDC. The dotted gray line represents the diagonal.
To study the distribution and degree of accuracy on a per target basis and to understand the diversity of MULTICOM’s five submitted models, we calculated Z-score for each of the six quality metrics considering all predictors and analyzed the quartile plots of Z-scores by highlighting the five models submitted by MULTICOM (see supplemental Figure S1). For several targets, MULTICOM’s performance was comparable with the best prediction submitted by any predictor. Moreover, the diversity between the five models submitted by MULTICOM indicates the effectiveness of using clustering together with model selection. Two representative examples are shown in Figure 5 for CASP11 targets T0853-D1 and T0830-D1. For target T0853-D1, the first submitted model (highlighted in red) proved to be the best as judged by GDT-HA while the five submitted models were quite diverse covering different aspects of model quality. A close resemblance can be observed between the experimental structure and prediction (Figure 5a). On the other hand, the fifth submitted model turned out to be the best in terms of GDT-HA for target T0830-D1 while having lesser diversity between five submitted models. In both the cases, the best out of five models by MULTICOM achieved accuracy close to the best-submitted model by any predictor.
Figure 5. Case study for CASP11 targets T0853-D1 and T0830-D1.
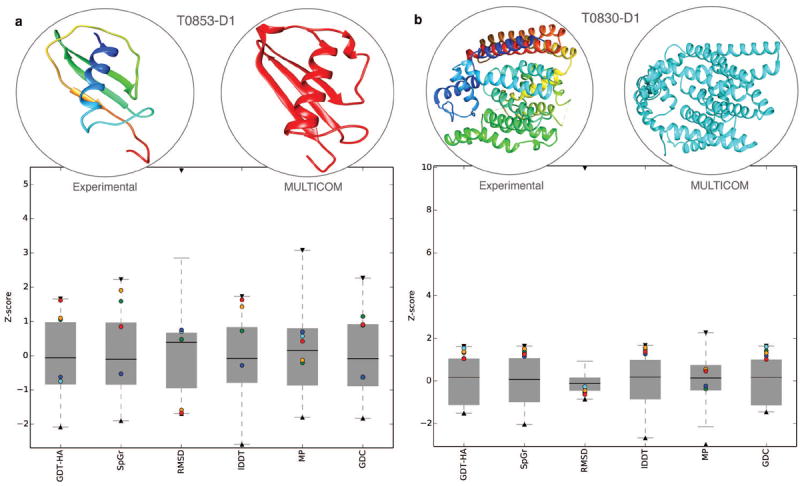
Quartile plots of Z-scores for all the submitted models are shown for six different quality metrics are shown for targets (a) T0853-D1 and (b) T0830-D1. The maximum and minimum Z-scores for each metric indicated by black down triangle and black up triangle respectively while five models submitted by MULTICOM are highlighted as red, orange, blue, green and cyan, in ascending order. For each target, the experimental structure is shown in the top left (rainbow colored from N terminal to C terminal) while the best prediction by MULTICOM (optimally superposed with experimental structure and translated) is shown in the top right.
In addition to assessing the overall performance, we specifically examined how massively integration of diverse protein quality assessment methods helps in improving the ranking of template-based models compared to any individual QA method and explored how average accuracy of the pool of model impacted model selection. Figure 6 presents the GDT-HA of the top model selected by each of the single QA and MULTICOM with respect to the median GDT-HA score of the ensemble of server predictors. The overall accuracy of MULTICOM is observed to be better than individual QA methods. Several additional interesting insights can be observed. For example, when the median GDT-HA scores are very high, several clustering-based methods display relatively poor performance compared to single model QA methods. One explanation for this could be that the presence of an easily identifiable template and relatively straightforward target-template alignment, causing almost all the server methods to perform similarly. This results in less diversity in the model ensemble and subsequently affects the performance of clustering-based QA techniques that favor average-quality models (i.e. the center of a model cluster).
Figure 6. Comparison of MULTICOM with individual QA methods.
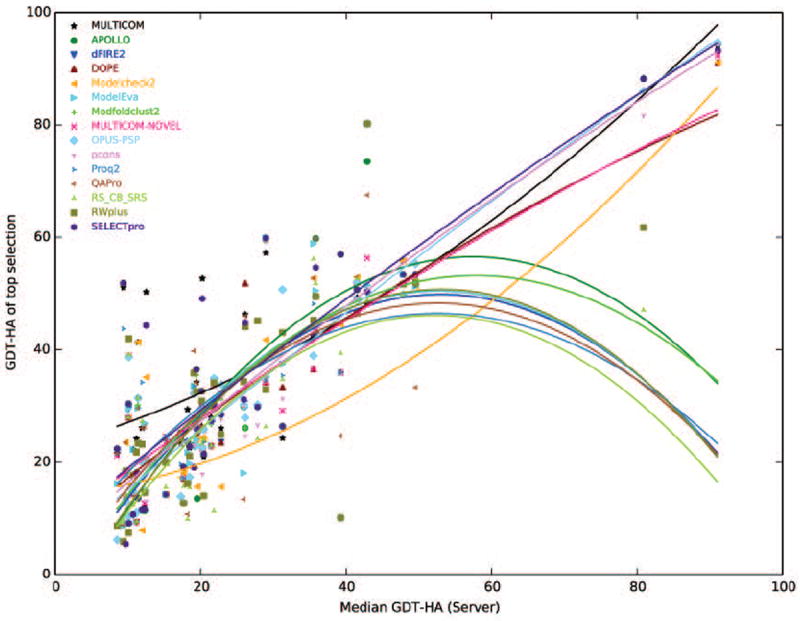
Relationships between median GDT-HA score of the server predictors and the GDT-HA of the top model selected by individual QA methods along with MULTICOM are shown. Individual QA methods are represented by different style and color while the curved lines are tendency lines constructed by fitting second-degree polynomial to the data. The corresponding legends are shown on the top left.
Table IV shows the comparison for the top 1 model selected by MULTICOM and each QA method based on GDT-HA score. As we can see from the table, in terms of average GDT-HA, and also Z-score on all targets, MULTICOM gets the best performance. In addition, we do a Wilcoxon signed ranked sum test on the top 1 model’s Z-score difference between MULTICOM and each QA method, and the p-value is shown in the table. The QA method QApro, ModelEva, and Proq2 actually perform very well on these TBM targets, and the difference between MULTICOM and them is not very significant given the confidence level 0.05. However, MULTICOM is significantly different with other QA methods based on the selected top 1 model’s Z score, suggesting Z-score is a more sensitive measure of the difference in model quality.
Table IV.
Comparison of MULTICOM with each QA method on the average GDT-HA score and Z-score of the top models selected, and the significant of each QA method.
| QA score name on all human targets | Ave. GDT-HA score on all | Ave. Z score on all | p-value of Z score diff. |
|---|---|---|---|
| MULTICOM | 36.3 | 1.417 | - |
| SELECTpro | 33.0 | 0.889 | 0.0159 |
| Proq2 | 31.8 | 1.158 | 0.0558 |
| Modelcheck2 | 31.8 | 0.959 | 0.0208 |
| MULTICOM-NOVEL | 31.4 | 0.936 | 0.0059 |
| Pcons | 31.1 | 0.681 | 0.0125 |
| ModelEva | 31.1 | 1.086 | 0.0829 |
| APOLLO | 30.9 | 0.830 | 0.0463 |
| Modfoldclust2 | 30.9 | 0.888 | 0.0425 |
| QApro | 30.9 | 1.117 | 0.1950 |
| Dope | 30.8 | 0.835 | 0.0061 |
| Dfire2 | 30.4 | 0.997 | 0.0224 |
| OPUS-PSP | 29.9 | 0.635 | 0.0016 |
| RWplus | 29.8 | 0.932 | 0.0161 |
| RF_CB_SRS | 27.6 | 0.489 | 0.0017 |
To investigate MULTICOM’s ability to rank the models, we studied the GDT-HA score of a model with respect to its ranking by MULTICOM on a per target basis. In supplemental Figure S2, we report the Gaussian kernel density estimates of MULTICOM’s ranking and GDT-HA score for all targets while highlighting the top model selected by each QA method. Strong convergence can be observed for several targets represented by inverted funnel shaped ranking landscape. In Figure 7, we present two typical example of MULTICOM’s ranking. For target T0822-D1, shown in Figure 7a, the majority of the models has GDT-HA score less than 0.15 GDT-HA score and was ranked low by MULTICOM, while few models have GDT-HA score more than 0.25 and were usually ranked higher. MULTICOM was able to select the better model compared to other QA methods, although it missed the best model myprotein-me_TS4 in the server model pool. In case of target T0838-D1, reported in Figure 7b, clear convergence to the optimal model can be observed as shown by distinct inverted funnel shaped ranking landscape. Even though in this case MULTICOM was neither able to pick the best model myprotein-me_TS1 in the server model pool, nor performed better than all the other QA methods. However, the performance of MULTICOM and the optimal QA methods (OPUS-PSP or DOPE) were comparable.
Figure 7. Landscape of MULTICOM’s ranking.
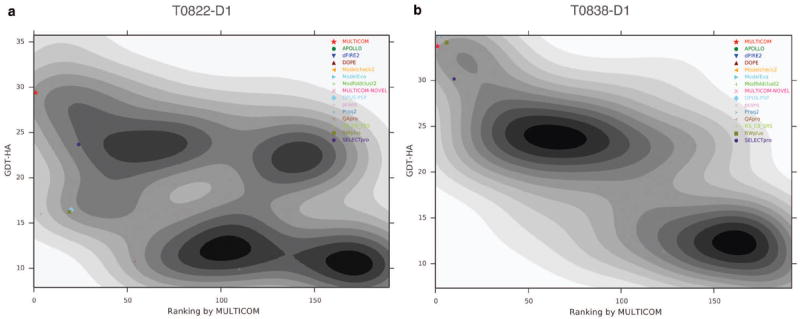
Gaussian kernel density estimates of GDT-HA score of models in the server pool and their ranking by MULTICOM are shown for targets (a) T0822-D1 and (b) T0838-D1 with lower rank indicating model predicted to be of higher quality. The top models selected by each of the QA methods are highlighted by different style and color. The corresponding legends are shown on the right.
4. CONCLUSION
We conducted a comprehensive analysis of our CASP11 human tertiary structure predictor MULTICOM on template-based targets. Our experiment demonstrates that the massive integration of diverse, complementary quality assessment methods is a promising approach to address the significant challenge of ranking protein models and improves the accuracy and reliability of template-based modeling. In order to further improve the template-based modeling, on one hand more accurate tertiary structure prediction methods need to be developed to generate a large portion of good structural models, and on the other hand more sensitive model quality assessment methods need to be included to reliably select good models from a pool of models that may only contain a few good models.
Supplementary Material
Acknowledgments
The work was partially supported by a NIH NIGMS grant (R01GM093123) to JC.
Footnotes
Conflict of Interest
The authors declare there is no conflict of interest.
References
- 1.Eisenhaber F, Persson B, Argos P. Protein structure prediction: recognition of primary, secondary, and tertiary structural features from amino acid sequence. Critical reviews in biochemistry and molecular biology. 1995;30(1):1–94. doi: 10.3109/10409239509085139. [DOI] [PubMed] [Google Scholar]
- 2.Rost B. Protein structure prediction in 1D, 2D, and 3D. The Encyclopaedia of Computational Chemistry. 1998;3:2242–2255. [Google Scholar]
- 3.Anfinsen CB, Haber E, Sela M, White F., Jr The kinetics of formation of native ribonuclease during oxidation of the reduced polypeptide chain. Proceedings of the National Academy of Sciences of the United States of America. 1961;47(9):1309. doi: 10.1073/pnas.47.9.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Floudas C. Computational methods in protein structure prediction. Biotechnology and bioengineering. 2007;97(2):207–213. doi: 10.1002/bit.21411. [DOI] [PubMed] [Google Scholar]
- 5.Shah M, Passovets S, Kim D, Ellrott K, Wang L, Vokler I, LoCascio P, Xu D, Xu Y. A computational pipeline for protein structure prediction and analysis at genome scale. Bioinformatics. 2003;19(15):1985. doi: 10.1093/bioinformatics/btg262. [DOI] [PubMed] [Google Scholar]
- 6.Fox BG, Goulding C, Malkowski MG, Stewart L, Deacon A. Structural genomics: from genes to structures with valuable materials and many questions in between. Nature methods. 2008;5(2):129–132. doi: 10.1038/nmeth0208-129. [DOI] [PubMed] [Google Scholar]
- 7.Lemer CMR, Rooman MJ, Wodak SJ. Protein structure prediction by threading methods: evaluation of current techniques. Proteins: Structure, Function, and Bioinformatics. 1995;23(3):337–355. doi: 10.1002/prot.340230308. [DOI] [PubMed] [Google Scholar]
- 8.Moult J, Pedersen JT, Judson R, Fidelis K. A large-scale experiment to assess protein structure prediction methods. Proteins: Structure, Function, and Bioinformatics. 1995;23(3):ii–iv. doi: 10.1002/prot.340230303. [DOI] [PubMed] [Google Scholar]
- 9.Zhang Y. Progress and challenges in protein structure prediction. Current opinion in structural biology. 2008;18(3):342–348. doi: 10.1016/j.sbi.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bowie JU, Luthy R, Eisenberg D. A method to identify protein sequences that fold into a known three-dimensional structure. Science. 1991;253(5016):164–170. doi: 10.1126/science.1853201. [DOI] [PubMed] [Google Scholar]
- 11.Jones DT, Taylort W, Thornton JM. A new approach to protein fold recognition. 1992 doi: 10.1038/358086a0. [DOI] [PubMed] [Google Scholar]
- 12.Simons K, Kooperberg C, Huang E, Baker D. Assembly of protein tertiary structures from fragments with similar local sequences using simulated annealing and Bayesian scoring functions. Journal of Molecular Biology. 1997;268(1):209–225. doi: 10.1006/jmbi.1997.0959. [DOI] [PubMed] [Google Scholar]
- 13.Zhang Y. Interplay of I-TASSER and QUARK for template-based and ab initio protein structure prediction in CASP10. Proteins: Structure, Function, and Bioinformatics. 2014;82(S2):175–187. doi: 10.1002/prot.24341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Y. I-TASSER server for protein 3D structure prediction. BMC bioinformatics. 2008;9(1):40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Källberg M, Wang H, Wang S, Peng J, Wang Z, Lu H, Xu J. Template-based protein structure modeling using the RaptorX web server. Nature protocols. 2012;7(8):1511–1522. doi: 10.1038/nprot.2012.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang J, Wang Q, Barz B, He Z, Kosztin I, Shang Y, Xu D. MUFOLD: A new solution for protein 3D structure prediction. Proteins: Structure, Function, and Bioinformatics. 2010;78(5):1137–1152. doi: 10.1002/prot.22634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Simons KT, Kooperberg C, Huang E, Baker D. Assembly of protein tertiary structures from fragments with similar local sequences using simulated annealing and Bayesian scoring functions. Journal of molecular biology. 1997;268(1):209–225. doi: 10.1006/jmbi.1997.0959. [DOI] [PubMed] [Google Scholar]
- 18.Xu D, Zhang Y. Ab initio protein structure assembly using continuous structure fragments and optimized knowledge-based force field. Proteins: Structure, Function, and Bioinformatics. 2012;80(7):1715–1735. doi: 10.1002/prot.24065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li SC, Bu D, Xu J, Li M. Fragment-HMM: A new approach to protein structure prediction. Protein Science. 2008;17(11):1925–1934. doi: 10.1110/ps.036442.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cao R, Wang Z, Cheng J. Designing and evaluating the MULTICOM protein local and global model quality prediction methods in the CASP10 experiment. BMC Structural Biology. 2014;14(1):13. doi: 10.1186/1472-6807-14-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cao R, Wang Z, Wang Y, Cheng J. SMOQ: a tool for predicting the absolute residue-specific quality of a single protein model with support vector machines. BMC bioinformatics. 2014;15(1):120. doi: 10.1186/1471-2105-15-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Y, Skolnick J. Scoring function for automated assessment of protein structure template quality. Proteins: Structure, Function, and Bioinformatics. 2004;57(4):702–710. doi: 10.1002/prot.20264. [DOI] [PubMed] [Google Scholar]
- 23.Wallner B, Elofsson A. Can correct protein models be identified? Protein Science. 2003;12(5):1073–1086. doi: 10.1110/ps.0236803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ray A, Lindahl E, Wallner B. Improved model quality assessment using ProQ2. BMC bioinformatics. 2012;13(1):224. doi: 10.1186/1471-2105-13-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Benkert P, Tosatto SC, Schomburg D. QMEAN: A comprehensive scoring function for model quality assessment. Proteins: Structure, Function, and Bioinformatics. 2008;71(1):261–277. doi: 10.1002/prot.21715. [DOI] [PubMed] [Google Scholar]
- 26.Benkert P, Künzli M, Schwede T. QMEAN server for protein model quality estimation. Nucleic acids research. 2009 doi: 10.1093/nar/gkp322. gkp322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eisenberg D, Luthy R, Bowie J. VERIFY3D: assessment of protein models with three-dimensional profiles. Methods Enzymol. 1997;277:396–404. doi: 10.1016/s0076-6879(97)77022-8. [DOI] [PubMed] [Google Scholar]
- 28.Zhang Y. Interplay of I-TASSER and QUARK for template-based and ab initio protein structure prediction in CASP10. Proteins: Structure, Function, and Bioinformatics. 2014;82(S2):175–187. doi: 10.1002/prot.24341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Z, Eickholt J, Cheng J. APOLLO: a quality assessment service for single and multiple protein models. Bioinformatics. 2011;27(12):1715–1716. doi: 10.1093/bioinformatics/btr268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McGuffin LJ. The ModFOLD server for the quality assessment of protein structural models. Bioinformatics. 2008;24(4):586–587. doi: 10.1093/bioinformatics/btn014. [DOI] [PubMed] [Google Scholar]
- 31.McGuffin LJ. Prediction of global and local model quality in CASP8 using the ModFOLD server. Proteins: Structure, Function, and Bioinformatics. 2009;77(S9):185–190. doi: 10.1002/prot.22491. [DOI] [PubMed] [Google Scholar]
- 32.McGuffin LJ, Roche DB. Rapid model quality assessment for protein structure predictions using the comparison of multiple models without structural alignments. Bioinformatics. 2010;26(2):182–188. doi: 10.1093/bioinformatics/btp629. [DOI] [PubMed] [Google Scholar]
- 33.Dobson CM, Šali A, Karplus M. Protein folding: a perspective from theory and experiment. Angewandte Chemie International Edition. 1998;37(7):868–893. doi: 10.1002/(SICI)1521-3773(19980420)37:7<868::AID-ANIE868>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 34.Shortle D, Simons KT, Baker D. Clustering of low-energy conformations near the native structures of small proteins. Proceedings of the National Academy of Sciences. 1998;95(19):11158–11162. doi: 10.1073/pnas.95.19.11158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang J, Zhang Y. A novel side-chain orientation dependent potential derived from random-walk reference state for protein fold selection and structure prediction. PloS one. 2010;5(10):e15386. doi: 10.1371/journal.pone.0015386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou H, Zhou Y. Distance-scaled, finite ideal-gas reference state improves structure-derived potentials of mean force for structure selection and stability prediction. Protein Science. 2002;11(11):2714–2726. doi: 10.1110/ps.0217002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shen My, Sali A. Statistical potential for assessment and prediction of protein structures. Protein Science. 2006;15(11):2507–2524. doi: 10.1110/ps.062416606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang J, He Z, Wang Q, Barz B, Kosztin I, Shang Y, Xu D. Functional Genomics. Springer; 2012. Prediction of protein tertiary structures using MUFOLD; pp. 3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu Y, Lu M, Chen M, Li J, Ma J. OPUS-Ca: A knowledge-based potential function requiring only Cα positions. Protein Science. 2007;16(7):1449–1463. doi: 10.1110/ps.072796107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Z, Tegge AN, Cheng J. Evaluating the absolute quality of a single protein model using structural features and support vector machines. Proteins: Structure, Function, and Bioinformatics. 2009;75(3):638–647. doi: 10.1002/prot.22275. [DOI] [PubMed] [Google Scholar]
- 41.Wallner B, Elofsson A. Identification of correct regions in protein models using structural, alignment, and consensus information. Protein Science. 2006;15(4):900–913. doi: 10.1110/ps.051799606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Larsson P, Skwark M, Wallner B, Elofsson A. Assessment of global and local model quality in CASP8 using Pcons and ProQ. Proteins. 2009;77(S9):167–172. doi: 10.1002/prot.22476. [DOI] [PubMed] [Google Scholar]
- 43.Cao R, Bhattacharya D, Adhikari B, Li J, Cheng J. Large-Scale Model Quality Asessment for Improving Protein Tertiary Structure Prediction. Bioinformatics (accepted); 23rd International Conference on Intelligent Systems for Molecular Biology (ISMB); 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang J, Xu D. Fast algorithm for population-based protein structural model analysis. Proteomics. 2013;13(2):221–229. doi: 10.1002/pmic.201200334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang Z, Eickholt J, Cheng J. MULTICOM: a multi-level combination approach to protein structure prediction and its assessments in CASP8. Bioinformatics. 2010;26(7):882–888. doi: 10.1093/bioinformatics/btq058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lu M, Dousis AD, Ma J. OPUS-PSP: an orientation-dependent statistical all-atom potential derived from side-chain packing. Journal of molecular biology. 2008;376(1):288–301. doi: 10.1016/j.jmb.2007.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jones DT. Protein secondary structure prediction based on position-specific scoring matrices. Journal of molecular biology. 1999;292(2):195–202. doi: 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
- 48.McGuffin LJ, Bryson K, Jones DT. The PSIPRED protein structure prediction server. Bioinformatics. 2000;16(4):404–405. doi: 10.1093/bioinformatics/16.4.404. [DOI] [PubMed] [Google Scholar]
- 49.Kabsch W. A solution for the best rotation to relate two sets of vectors. Acta Crystallographica Section A: Crystal Physics, Diffraction, Theoretical and General Crystallography. 1976;32(5):922–923. [Google Scholar]
- 50.Rykunov D, Fiser A. Effects of amino acid composition, finite size of proteins, and sparse statistics on distance-dependent statistical pair potentials. Proteins: Structure, Function, and Bioinformatics. 2007;67(3):559–568. doi: 10.1002/prot.21279. [DOI] [PubMed] [Google Scholar]
- 51.Wang G, Dunbrack RL. PISCES: a protein sequence culling server. Bioinformatics. 2003;19(12):1589–1591. doi: 10.1093/bioinformatics/btg224. [DOI] [PubMed] [Google Scholar]
- 52.Cozzetto D, Kryshtafovych A, Fidelis K, Moult J, Rost B, Tramontano A. Evaluation of template-based models in CASP8 with standard measures. Proteins: Structure, Function, and Bioinformatics. 2009;77(S9):18–28. doi: 10.1002/prot.22561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huang YJ, Mao B, Aramini JM, Montelione GT. Assessment of template-based protein structure predictions in CASP10. Proteins: Structure, Function, and Bioinformatics. 2014;82(S2):43–56. doi: 10.1002/prot.24488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kryshtafovych A, Monastyrskyy B, Fidelis K. CASP Prediction Center infrastructure and evaluation measures in CASP10 and CASP ROLL. Proteins: Structure, Function, and Bioinformatics. 2014;82(S2):7–13. doi: 10.1002/prot.24399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mariani V, Biasini M, Barbato A, Schwede T. lDDT: a local superposition-free score for comparing protein structures and models using distance difference tests. Bioinformatics. 2013;29(21):2722–2728. doi: 10.1093/bioinformatics/btt473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cao R, Bhattacharya D, Adhikari B, Li J, Cheng J. Large-scale model quality assessment for improving protein tertiary structure prediction. Bioinformatics. 2015;31(12):i116–i123. doi: 10.1093/bioinformatics/btv235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Krivov GG, Shapovalov MV, Dunbrack RL., Jr Improved prediction of protein side-chain conformations with SCWRL4. Proteins: Structure, Function, and Bioinformatics. 2009;77(4):778–795. doi: 10.1002/prot.22488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Randall A, Baldi P. SELECTpro: effective protein model selection using a structure-based energy function resistant to BLUNDERs. BMC structural biology. 2008;8(1):52. doi: 10.1186/1472-6807-8-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yang Y, Zhou Y. Specific interactions for ab initio folding of protein terminal regions with secondary structures. Proteins: Structure, Function, and Bioinformatics. 2008;72(2):793–803. doi: 10.1002/prot.21968. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.