

Abstract
Astrocytes are one of the major cell types to combat cellular stress and protect neighboring neurons from injury. In order to fulfill this important role, astrocytes must sense and respond to toxic stimuli, perhaps including stimuli that are severely stressful and kill some of the astrocytes. The present study demonstrates that primary astrocytes that managed to survive severe proteotoxic stress were protected against subsequent challenges. These findings suggest that the phenomenon of preconditioning or tolerance can be extended from mild to severe stress for this cell type. Astrocytic stress adaptation lasted at least 96 hours, the longest interval tested. Heat shock protein 70 (Hsp70) was raised in stressed astrocytes, but inhibition of neither Hsp70 nor Hsp32 activity abolished their resistance against a second proteotoxic challenge. Only inhibition of glutathione synthesis abolished astrocytic stress adaptation, consistent with our previous report. Primary neurons were plated upon previously stressed astrocytes and the co-cultures were then exposed to another proteotoxic challenge. Severely stressed astrocytes were still able to protect neighboring neurons against this injury and the protection was unexpectedly independent of glutathione synthesis. Stressed astrocytes were even able to protect neurons after simultaneous application of proteasome and Hsp70 inhibitors, which otherwise elicited synergistic, severe loss of neurons when applied together. Astrocyte-induced neuroprotection against proteotoxicity was not elicited with astrocyte-conditioned media, suggesting that physical cell-to-cell contacts may be essential. These findings suggest that astrocytes may adapt to severe stress so that they can continue to protect neighboring cell types from profound injury.
Keywords: glia, adaptation, preconditioning, stress response, neurodegeneration
Introduction
Astrocytes are one of the most resistant cell types in the brain, providing trophic support and building scar tissue in areas of severe neuronal injury. As with many other cell types, astrocytes can be preconditioned by sublethal stress against subsequent insults [1–10]. However, we recently showed that astrocytes surviving severely toxic stress are also resistant to subsequent insults [11]. In other words, the astrocytes remaining after a lethal hit were able to survive subsequent challenges better than stress-naïve cells. This was one of the first studies to show that the phenomenon of preconditioning applies not only to sublethal stress but can be extended to toxic, severe stress for this cell type. Here we further characterize that stress model with neuron/astrocyte co-cultures to examine the interactions between severely stressed glia and neurons. Using this new model we test the hypothesis that severely stressed astrocytes are still able to protect neighboring neurons from injury. Although many previous studies have shown that astrocytes provide trophic, metabolic, and antioxidant support to surrounding neurons [12, 13], it is not clear if severely stressed astrocytes can continue to fulfill such neurosupportive roles. Indeed, some studies suggest that changes in astrocyte function in neurodegenerative disorders actually play detrimental roles [14–19]. It is therefore important to determine whether astrocytes that are subjected to severe stress mitigate or exacerbate neuronal injury.
Stress from protein misfolding leads to loss of protein homeostasis or proteotoxicity and is the major hallmark of neurodegenerative disorders such as Parkinson’s and Alzheimer’s disease [20–29]. For example, proteasome activity is reduced in several neurodegenerative conditions [30–32]. Therefore, in the present study, severe proteotoxic stress was elicited with the proteasome inhibitors MG132 and lactacystin. Proteasome inhibitors increase the burden of misfolded proteins by inhibiting protein degradation and thereby elicit cell death [33–38]. Although neurons are more vulnerable to loss of protein homeostasis than glia [14], many studies have shown that astrocytes are also exposed to proteotoxicity in neurodegenerative conditions, as shown by the presence of protein inclusions [39–43] and increased heat shock protein expression in this cell type [44]. For example, astrocytes in the amygdala, septum, cortex, thalamus, and striatum exhibit α-synuclein inclusions in Parkinson’s disease [43, 45]. Astrocytes are thought to remove α-synuclein from the extracellular space through endocytosis [46]. In addition, astrocytes may phagocytose β-amyloid from the extracellular space [47, 48] and are labeled by anti-Aβ antibodies in Alzheimer’s disease [49–51]. In amyotrophic lateral sclerosis, astrocytes in the cingulate gyrus exhibit tau deposits [52]. Furthermore, the frontal and temporal cortices exhibit astrocytic plaques and hyperphosphorylated astrocytic tau in amyotrophic lateral sclerosis [53, 54]. Tau-containing astrocytic inclusions are also found in supranuclear palsy, corticobasal degeneration, and Pick’s disease [55]. Finally, the frontal and entorhinal cortices also accumulate astrocytic tau with aging [56]. These studies are all in agreement that glia exhibit signs of protein misfolding stress in aging and in age-related neurological conditions, thereby supporting the use of proteasome inhibitors to model astrocytic proteotoxicity. Previous studies have shown that exposure to low, sublethal concentrations of proteasome inhibitors elicits resistance against subsequent insults in tumor cell lines [57, 58]. However, potentially beneficial effects of severe, lethal stress on the subpopulation of surviving cells are less well understood, especially in primary astrocytes.
One of the major endogenous mechanisms to protect against proteotoxicity is the heat shock protein family. Heat shock proteins are chaperones that help refold misfolded proteins or help degrade irreparably damaged proteins in conjunction with the proteasome or lysosome. Two of the best-studied chaperones are heat shock protein 70 (Hsp70) and heat shock cognate 70 (Hsc70). Hsp70 is thought to modulate the assembly of the proteasome under conditions of oxidative challenge [59]. Our previous study revealed a significant increase in Hsp70 levels in severely stressed astrocytes exposed to the proteasome inhibitor MG132 [11]. Thus, we tested the hypothesis that Hsp70 mediates the protection of severely stressed astrocytes against a second proteotoxic hit, perhaps in conjunction with the antioxidant glutathione. The results show that inhibition of Hsp70/Hsc70 ATPase activity does not abolish astrocytic stress adaptation. As shown previously, inhibition of glutathione synthesis renders stressed astrocytes highly susceptible to the second MG132 hit [11], supporting the notion that glutathione but not Hsp70 defenses mediate astrocytic stress adaptation. Furthermore, the astrocytes that survive severe stress are still able to protect neighboring neurons against proteasome inhibition in neuron/glia co-cultures. This protection remains significant even under conditions of glutathione loss (i.e., during mild oxidative stress) and appears to be dependent upon cell-to-cell contacts. Remarkably, severely stressed astrocyte survivors can protect neurons from the severe, synergistic proteotoxicity of simultaneous proteasome and Hsp70 inhibition. These results demonstrate the general robustness of this cell type and support the view that astrocytes have evolved to support surrounding cells even under conditions of oxidative stress and proteotoxic injury.
Materials and Methods
Astrocyte cultures
Astrocytes were harvested from postnatal day 1–2 Sprague-Dawley rats (Charles River, Wilmington, MA), because this cell type peaks around birth [60, 61] and survives well when harvested from neonatal tissue [62]. Tissue was dissected from the cerebral cortex after the primary sensorimotor neocortex was removed for neuronal cultures (see below). All procedures were in accordance with the NIH Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee. Briefly, cortical tissue was microdissected and dissociated by mechanical force through a 5 mL serological pipet following incubation in 0.25% Trypsin-EDTA (Cat. no. 25200, Gibco Life Technologies, Grand Island, NY) at 37 °C for 7 min. Tissue was then washed and triturated in cell culture medium [Dulbecco’s Modified Eagle medium (DMEM, Cat. no. 12100061, Gibco Life Technologies) with 10% Fetal Clone III (Cat. no. SH30109.03, Thermo Scientific Hyclone, Logan UT), penicillin, and streptomycin (50 U/mL and 50 μg/mL, respectively; Cat. no. 15070, Gibco Life Technologies)]. Dissociated cells were plated at a density of 1 × 106 cells/mL in 24 mL on T175 flasks coated with poly-D-lysine (10 μg/mL; Cat. no. P0899, Sigma-Aldrich, St. Louis, MO). After 7–10 days at 37°C and 5% CO2, the flask was rotated overnight at 37°C at 260 revolutions per minute to shake off non-astrocytic cells. The media was then refreshed. Following an additional 2–3 day incubation period, the cells were trypsinized and plated for experimental procedures. Cells were plated at a density of 20,000 cells/well in 96-well plates for viability experiments and 6.8 × 105 cells/well in 6-well plates for Western blotting (Costar, Corning Incorporated, Corning, NY). Primary astrocyte cultures were used for no longer than one month after dissection.
Neuron-astrocyte co-cultures
For contact co-cultures, primary neocortical neurons were harvested from the primary sensorimotor neocortex of postnatal day 1 Sprague Dawley rats (Charles River) as previously described [63, 64] and plated on top of purified primary cortical astrocytes (described above). Briefly, neocortical tissue was incubated in 10 Units/mL papain (Sigma-Aldrich, Cat. no. P3125) for 30 min and then in 10% type II-O trypsin inhibitor (Sigma-Aldrich, Cat. no. T9253). Tissue was washed and triturated in Basal Medium Eagle (Sigma-Aldrich, Cat. no. B1522) containing 10% bovine calf serum (BME/BCS, HyClone Thermo Scientific, Logan, UT, Cat. no. 2151,) supplemented with 35 mM glucose (Sigma-Aldrich, Cat. no. G8769), 1 mM L-glutamine (Gibco, Life Technologies, Cat. no. 25030-081), 50 Units/mL penicillin, and 50 μg/mL streptomycin (Gibco, Life Technologies, Cat. no. 15140-122). Dissociated cells were then seeded in Opti-MEM (Gibco, Life Technologies, Cat. no. 51985-034) supplemented with 20 mM glucose and incubated for 2 hours before switching to Neurobasal-A medium (Cat. no. 10888-022, Gibco Life Technologies) supplemented with 2% v/v B27 (Cat. no. 17504-044, Invitrogen Life Technologies) and L-glutamine (1:50, Cat. no. 25030-081, Gibco Life Technologies). Co-cultures were incubated in the same Neurobasal-A medium. For astrocyte-conditioned media experiments, the conditioned medium (DMEM supplemented as described above) was transferred to neocortical neurons in a 1:1 dilution in Neurobasal-A medium (described above).
Antibodies
Primary antibodies included the following, in alphabetical order: mouse anti-α-tubulin (1:200,000, Cat. no. T5168, lot no. 078K4781, Sigma-Aldrich), mouse anti-β-actin (1:50,000, Cat. no. A5441, lot no. 030M4788, Sigma-Aldrich), rabbit anti-DJ-1 (1:5000, Cat. no. RA19006, lot no. 400250, Neuromics, Edina, MN), rabbit anti-GAPDH (1:5000, Cat. no. 2118S, lot no. 8, Cell Signaling Technology, Danvers, MA), rabbit anti-glial fibrillary acidic protein (GFAP, 1:1000, Cat. no. Z0334, lot no. 20001046, Dako, Carpinteria, CA), rabbit anti-glutamate cysteine ligase C subunit (1:1000, Cat. no. AV54576, lot no. QC22671, Sigma-Aldrich), rabbit anti-glutamate cysteine ligase M subunit (1:1000, Cat. no. SAB2100907, lot no. QC23487, Sigma-Aldrich), rabbit anti-glutathione (1:300, Cat. no. AB5010, lot no. 2379274, Millipore, Billerica, MA), goat anti-glutathione S-transferase μ (1:3000, Cat. no. ab53942, lot no. GR157774-1, Abcam, Cambridge, MA), mouse anti-glutathione S-transferase π (1:2000, Cat. no. 610719, lot no. 3277736, BD Biosciences, Franklin Lakes, NJ), rabbit anti-heme oxygenase 1 (HO1, 1:200, Cat. no. H4535, lot no. 081M1122, Sigma-Aldrich), rabbit anti-Hip (1:1000, Cat. no. 2723, lot no. 1, Cell Signaling Technology), rat anti-Hsc70 (1:5000, Cat. no. Adi-Spa-815-D, lot no. 04231339, Enzo Life Sciences, Farmingdale, NY), goat anti-Hsp27 (1:1000, Cat. no. sc-1048, Santa Cruz, Santa Cruz, CA), rabbit anti-phospho-Hsp27 (1:1000, Cat. no. 2723, lot no. 11, Cell Signaling Technology), rabbit anti-Hsp40 (1:2000, Cat. no. 4868, lot no. 2, Cell Signaling Technology), rabbit anti-Hsp70 (1:1000, Cat. no. AB9920, lot no. 2278555, Millipore), rabbit anti-Hsp90 (1:2000, Cat. no. 4877, lot no. 3, Cell Signaling Technology), mouse anti-MAP2 (1:2000, Cat. no. M9942, 069K4770, Sigma-Aldrich), mouse anti-S100β (1:1000, Cat. no. S2532, lot no. 070M4767, Sigma-Aldrich), and mouse anti-ubiquitinated proteins (1:500, Cat. no. sc-8017, lot no. D0412, Santa Cruz). Secondary antibodies from Life Technologies (1:1000, Carlsbad, CA), LI-COR (1:20,000, Lincoln, NE) or Jackson ImmunoResearch (1:30,000, West Grove, PA) included the following: goat anti-mouse 488 nm (Cat. no. A11029, Life Technologies), goat anti-rabbit 555 nm (Cat. no. A21429, Life Technologies), goat anti-mouse 800 nm (Cat. no. 926-32210, LI-COR), donkey anti-rabbit 680 nm (Cat. no. 926-32223, LI-COR), donkey anti-rabbit 800 nm (LI-COR), donkey anti-mouse 680 nm (Cat. no. 926-32222, LI-COR), donkey anti-goat 800 nm (Cat. no. 926-32214, LI-COR), goat anti-rat 800 nm (Cat. no. 926-32219, LI-COR), donkey anti-rabbit 800 nm (Cat. no. 711-655-152, Jackson ImmunoResearch).
Inhibitors
We used the glutathione synthesis inhibitor buthionine sulfoximine (25 μM, Cat. no. 309475000, Acros Organics, Fair Lawn, New Jersey), the proteasome inhibitor MG132 (Cat. no. 474790, Calbiochem, EMD Chemicals, San Diego, CA or Cat no. F1100, Ubiquitin-Proteasome Biotechnologies, Aurora, CO), the proteasome inhibtor lactacystin (Cat. no. AG-CN2-0104, AdipoGen, San Diego, CA), the Hsp70/Hsc70 activity inhibitor VER155008 (12.5 μM, Cat. No. 3803, R&D Systems, Inc, Minneapolis, MN), and the heme oxygenase 1 (HO1) inhibitor tin (IV) protoporphyrin IX dichloride (SnPPx, 20 μM, Cat. no. Sn749-9, Frontier Scientific, Logan, Utah).
Two-hit protocol
Twenty-four hours after plating purified primary astrocytes, cells were treated with MG132 by adding a 10× concentration to the existing media at a 1:10 dilution. This treatment is referred to as the 1st MG132 hit. Twenty-four hours after the 1st MG132 hit, astrocytes were challenged with a 2nd MG132 hit, delivered at a 1× concentration in a full media exchange without dilution. The full media exchange allowed us to remove all the previously delivered MG132. Twenty-four hours after the 2nd hit, astrocytes were fixed in 3% paraformaldehyde in 75 mM phosphate buffer and 3% sucrose or lysed for ATP measurements as described below. A 24 hr interval between the 1st and 2nd hit was used in all experiments except those described in Figure 2D–I and the co-culture experiments in Figures 6 – 8. For lactacystin experiments, astrocytes were treated on the fifth and sixth days after plating with the 1st and 2nd hits, respectively, and fixed with paraformaldehyde on the seventh day. This delayed protocol was chosen for the sake of future studies employing RNA interference, which will necessitate 3–4 days of protein knockdown prior to the 1st hit.
Western blotting
Whole cell lysates were harvested 24 or 48 hr following treatments and sonicated in Cell Lysis Buffer (Cat. no. 9803, Cell Signaling, Danvers, MA) supplemented with 2% protease inhibitor cocktail (Cat. no. P8340, Sigma-Aldrich) and 10 mM sodium fluoride [65]. Equal amounts of protein (10 μg) were separated by gel electrophoresis on 10% polyacrylamide gels and transferred to Immobilon-FL polyvinylidene fluoride or nitrocellulose membranes (EMD Millipore). Membranes were washed three times in Tris-buffered saline (TBS) and blocked with 5% nonfat dry milk in TBS or Odyssey block (Cat. no. 927-40000, LI-COR) diluted 1:1 in TBS. Primary antibodies were diluted in TBS with 5% bovine serum albumin fraction V (Cat. no. A30075, Research Products International, Mount Prospect, IL) and 0.1% Tween-20 (Cat no. BP337, Fisher Scientific, Pittsburgh, PA) or Odyssey block diluted 1:1 in TBS with 0.1% Tween-20. Secondary antibodies were prepared in TBS with 5% nonfat dry milk and 0.1% Tween-20 or in Odyssey block diluted 1:1 in TBS with 0.1% Tween-20. Immunolabeled blots were then washed in TBS with 0.1% Tween-20 and fluorescent signal was measured on an Odyssey Imager and quantified using Image Studio Lite software (LI-COR).
Immunocytochemistry and cellular viability assays
Immunostaining was performed as previously described [11]. Primary astrocytes were immunolabeled for the astrocytic proteins glial fibrillary acidic protein (GFAP) and S100β. Primary neurons were immunolabeled for the specific neuronal marker microtubule associated protein 2 (MAP2) using an In-Cell Western assay as previously described [63, 66]. The MAP2 In-Cell Western assay has been shown to be in linear proportion to the number of neurons present [63, 66]. Nuclei were stained with Hoechst for viability assays (bisBenzidine or Hoechst 33258, 1:2000, Cat. no. B1155, Sigma-Aldrich) or with DRAQ5 (1:20,000, Cat. no. DR05500, Biostatus, United Kingdom) for the infrared In-Cell Western assays on neuron/astrocyte co-cultures. A blinded investigator used ImageJ software (NIH Image, Bethesda, MD) to generate cell counts of Hoechst-stained astrocyte nuclei and to measure nuclear size (reported as square microns) and staining intensity (reported as arbitrary units) with the “analyze particles” tool. DRAQ5 levels were imaged on the Odyssey Imager. Terminal deoxynucleotidyl transferease dUTP nick end labeling (TUNEL) was performed with the In Situ Cell Death Detection Kit (Cat. no. 11684795910, Roche Diagnostics, Indianapolis, IN). The adherent cells protocol was followed as per manufacturer’s instructions. TUNEL+ nuclei were counted by a blinded observer at 200× magnification.
ATP assay
ATP levels were measured using the CellTiter-Glo Assay for ATP (Cat. no. G7572, Promega, Madison, WI), with minor modifications to the manufacturer’s instructions, as previously published by us [11]. Briefly, 25 μL of the reconstituted CellTiter-Glo reagent was added to 50 μL media to lyse the astrocytes and generate a stable luminescent product. Luminescence was measured on a luminometer (VICTOR3 1420 multilabel counter; PerkinElmer, Waltham, MA) after a 15 min incubation period. This assay uses a thermostable form of luciferase to generate a luminescent signal that is in proportion to the amount of ATP present.
Total and reduced glutathione assays
Total glutathione levels were measured by an In-Cell Western assay, as described previously [11]. We have established that signal in this assay is reduced by application of the glutathione synthesis inhibitor buthionine sulfoximine and increased by the glutathione precursor N-acetyl cysteine [58, 63, 67, 68]. Reduced glutathione levels were specifically measured by the GSH-Glo glutathione assay according to the manufacturer’s instructions (Cat. no. V6911, Promega). This assay measures luminescence generated in the presence of glutathione by the action of glutathione S-transferase on a luciferin derivative. Glutathione levels were expressed as a function of Hoechst- or DRAQ5-stained nuclei on the same plate for the In-Cell Western assays or on parallel plates for the GSH-Glo assay, as the latter assay involved cell lysis.
Statistical analyses
Data are presented as the mean and standard error of the mean from a minimum of three independent experiments, each run in triplicate wells for all the viability assays. The average of these three wells represented an “n” of one experiment only. The Grubb’s outlier test was performed once on all the data. Western blotting experiments were run in one well per group, in at least three independent experiments. Statistical significance was determined the Student’s t test for two groups or by two- or three-way ANOVA for multiple groups, followed by the Bonferroni post hoc correction (IBM SPSS Statistics, Version 10.0, Armonk, NY or GraphPad Prism, Version 6, La Jolla, CA). In experiments where previous work led us to only expect increases in specific measurements, we performed the one-tailed t test. All other experiments with only two groups were analyzed by the two-tailed t test. Statistical differences were deemed significant only when p ≤ 0.05.
Results
We harvested astrocytes from postnatal rat cerebral cortices for the present study. An immunofluorescence analysis showed that the cultures were 89.6% GFAP+ and 91.8% S100β+ but that only 6.1% of cells expressed neither GFAP nor S100β (Figure 1A, B). This is somewhat higher purity than in our previous publication where we used different media [11]. In the present study, we counted Hoechst-stained nuclei to measure viability. In this analysis, fragmented or condensed nuclei under 350 pixels (53 μm2) in cross-sectional area were excluded by a blinded investigator using the threshold function in Image J. An example of these dying profiles is shown in Figure 1C and D.
Fig. 1.

Primary cortical astrocyte culture purity. A) Primary cortical astrocytes were stained with the nuclear marker Hoechst and immunolabeled for the glial markers glial fibrillary acid protein (GFAP) and S100β. B) The percentage of Hoechst+ cells that were immunolabeled for GFAP or S100β proteins or were immunonegative were quantified. C) Representative image of Hoechst-stained nuclei in untreated astrocytes. White arrow points to one cell with a condensed nucleus. D) The image in C was converted to a binary image using the threshold function in Image J. Nuclear cross-sectional areas appear next to each cell in square microns. Cells with nuclear areas under 350 pixels (53 μm2, see black arrow) were excluded from the counts of viable cells in subsequent figures
Stress-induced resistance against dual hits is long-lasting in nature
Astrocytes plated in DMEM media were more sensitive to the MG132 toxin than in our previous report [11]. For this reason, we used lower concentrations of MG132 to establish whether severely stressed astrocytes would be resistant against subsequent challenges (Figure 2A–C). As expected, astrocytes that survived a lethal challenge of 0.16 to 0.64 μM MG132 were resistant to a subsequent challenge of 3.125 μM MG132 administered on the following day. When we expressed all the data as a percentage of the 0 μM 2nd hit group (i.e., all gray bars were expressed as a percentage of the adjacent black bars; Figure 2B), a concentration-responsive increase in survival in previously stressed astrocytes was evident. The same stress-induced protection against additional cell loss was observed with the proteasome inhibitor lactacystin, which has a different mechanism of action from MG132 [69, 70] (Supplementary Figure 1). These results show that the astrocytes that manage to survive proteotoxic stress are indeed protected against a second proteotoxic challenge, somewhat akin to the phenomenon of preconditioning.
Fig. 2.

MG132-mediated severe proteotoxic stress preconditions astrocytes against subsequent insults. A) Primary cortical astrocytes were treated with indicated concentrations of the proteasome inhibitor MG132 one day after plating (1st hit) or an equal v/v of vehicle (dimethyl sulfoxide). Twenty-four hours later, astrocytes were challenged with a 2nd hit of MG132 (3.125 μM) or vehicle. Astrocyte viability was assessed 24h after the 2nd hit by counting viable Hoechst+ nuclei in blinded fashion. B) Data in panel A were expressed as a percentage of each 0 μM 2nd MG132 hit group (i.e., each gray bar was expressed as a percentage of the adjacent black bar). C) Representative images of Hoechst-stained astrocyte nuclei from the groups shown in A. The interval between the 1st hit and the 2nd hit was prolonged to 48h in D–F and to 96h in G–I. E and H illustrate data in D and G, respectively, expressed as a percentage of the 0 μM 2nd MG132 hit group. Representative Hoechst-stained nuclei are shown in F and I. J–K) The interval between the 2nd hit and the viability assay was extended to 72h. The data in panel J were expressed as a percentage of the 0 μM 2nd MG132 hit group and presented in panel K. L) Representative images of Hoechst-stained nuclei from groups shown in J. * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001 vs 0 μM 2nd MG132 hit; + p ≤ 0.05, ++ p ≤ 0.01, +++ p ≤ 0.001 vs 0 μM 1st MG132 hit, two-way ANOVA followed by Bonferroni post hoc correction
Our protocol in Figure 2A–C involved treating cells with two hits of MG132 that were 24h apart and then assaying them on the following day. In order to test the hypothesis that the effects of the 1st hit were longer lasting, we increased the interval between the two hits to 48h (Figure 2D–F) and to 96h (Figure 2G–I). For both sets of experiments, we also expressed the data as a percentage of the 0 μM 2nd hit group (see Figures 2E and H). These experiments reveal that the protective effects of the 1st hit lasted at least 96h, the longest between-hit interval tested. Furthermore, we increased the interval between the 2nd hit and the time of assay to 72h to verify that the protection lasted long after the dual hits had been administered (Figure 2J–L). Taken together, these results show that the protective effects of severe stress on astrocytes are not fleeting.
Most astrocytes surviving dual hits have viable nuclei
When cells are severely stressed, they often undergo nuclear condensation and fragmentation. Thus, we set out to determine whether there was any change in the size of the nucleus with the stress of MG132. Frequency histograms of nuclear area in μm2 in unstressed and MG132-treated astrocytes are illustrated in Figure 3. The histograms show that a distinct population of cells occupies sizes less than 350 pixels or approximately 50 μm2. The histogram in Figure 3A shows that there was a slight increase in median nuclear area after the 1st hit of MG132 relative to the vehicle-treated group. Figures 3B and 3C show that there was no change in median nuclear area after the 2nd hit by itself or after dual hits. When we examined average nuclear size, we observed a slight decrease in this measure in cells hit with dual hits relative to the cells hit with the 1st hit alone (Figure 3D, E). The data in these two figures are expressed both with (Figure 3D) and without (Figure 3E) the fragmented, small nuclei that occupy sizes less than ~50 μm2. Although this nuclear shrinkage may indicate an increase in cellular stress levels in the dual hit group, the difference was notably slight. Furthermore, the data demonstrate that the 1st hit elicits no decrease in average or median nuclear size.
Fig. 3.

Nuclear viability in stressed astrocytes. Astrocytes were treated with the indicated concentrations of MG132 24h (1st hit) and 48h (2nd hit) after plating. On the third day, astrocytes were stained with the nuclear marker Hoechst and analyzed by a blinded observer using ImageJ software. A–C) Frequency distributions of the number of nuclei corresponding to the indicated nuclear area. The vehicle-treated group is included on all three figures for comparison. D) Average nuclear size of all cellular profiles. E) Average size of all nuclei except those smaller than 53 μm2. F) Representative high-power image of Hoechst-stained nuclear profiles. G–I) Frequency distribution of the number of nuclei as a function of Hoechst nuclear staining intensity. The vehicle-treated group is included on all three figures for comparison. J) Average nuclear staining intensity of all cellular profiles. K) Average nuclear staining intensity of all profiles except cells with nuclei smaller than 53 μm2. L–O) Nuclear staining intensity as a function of nuclear size. P) Astrocytes were divided into two groups: those with small, bright nuclei (50–150 μm2; staining intensity greater than 20 arbitrary units for the mean gray value measurements) or large nuclei (greater than 150 μm2; all staining intensities). Raw data for this graph are shown in Supplementary Figure 2. Q) Number of TUNEL+ cells relative to the total number of Hoechst+ nuclei. R) Data presented in panel Q were expressed as a function of the 0 μM 2nd MG132 hit groups. S) Representative images of TUNEL staining. * p ≤ 0.05, ** p ≤ 0.01 vs 0 μM 2nd MG132 hit; + p ≤ 0.05, ++ p ≤ 0.01, +++ p ≤ 0.001 vs 0 μM 1st MG132 hit; ^ p ≤ 0.05, ^^ p ≤ 0.01 vs small, bright nuclei; two- or three-way ANOVA followed by Bonferroni post hoc correction
As dying cells often stain more intensely with the Hoechst nuclear stain because of chromatin condensation, we also measured the intensity of the Hoechst nuclear stain (defined in ImageJ as the gray value of all pixels in a nucleus divided by the number of pixels in that nucleus) and expressed the values as a frequency histogram (Figure 3G–I). The histograms reveal no change in median nuclear staining intensity with any MG132 treatment. Similarly, there was also no change in average nuclear staining intensity, whether or not we included the smallest, fragmented nuclei in this analysis (Figure 3J, K). Taken together, these results suggest that we are not counting dying, condensed nuclear profiles in the MG132-treated groups in our viability assays and that the vast majority of cells killed by MG132 have already detached from the plate at the time of fixation.
Next we determined the relationship between nuclear size and staining intensity (Figure 3L–O). According to the scatterplots, MG132 treatment eliminated a distinct population of cells with small, intensely-staining nuclei [nuclear sizes ranging between 50 and 150 μm2 and staining intensity greater than 20 arbitrary units (A.U.) for the mean gray value measurements]. In order to confirm this observation, we counted the number of cells with small, bright nuclei in each treatment group and compared it to the number of cells with large nuclei (nuclear size greater than 150 μm2). The raw data from this analysis are illustrated in Supplementary Figure 2 and show once again that severely stressed cells were less vulnerable to a 2nd hit, but that cells with small, bright nuclei appeared to be all the more vulnerable to MG132. To verify the latter observation statistically, we expressed the data as a percentage of the vehicle-treated groups in Figure 3P, which shows clearly that small, bright cells were indeed more vulnerable to proteotoxic stress than the cells with larger nuclei, consistent with the scatterplots. Nevertheless, both groups of small and large cells were less vulnerable to subsequent insults after exposure to the 1st hit.
Nuclei that are dying can be assessed by labeling the terminal end of nucleic acids in fragmented DNA [71, 72]. Therefore, we counted the number of TUNEL+ profiles in astrocytes hit once or twice with MG132 as a function of total cell numbers (Figure 3Q–S). Only a small fraction of nuclei were TUNEL+ even after MG132 treatment, suggesting that most dying cells wash off the plate by the time of fixation and that we are largely reporting live cells when presenting Hoechst+ cell numbers. Because of extremely low numbers of TUNEL+ cells, there was some variability in this measure in the raw data (Figure 3Q). Thus, we also present the data as a function of the 0 μM 2nd hit group in Figure 3R to control for inter-experimental variability. In the latter analysis, there was a significant increase in TUNEL+ profiles with the 2nd hit of MG132 whether or not the cells had been previously stressed. In other words, the 1st hit did not increase cellular vulnerability to DNA fragmentation when followed by the second challenge. These data are consistent with the analyses of nuclear size and intensity and demonstrate that the 1st hit does not increase vulnerability to the 2nd hit when measuring either live or dead cells.
Stressed astrocytes are not completely refractory to the MG132 toxin and express higher Hsp70 levels
Misfolded proteins may be attached to ubiquitin molecules to target them for proteasomal degradation [73]. When proteasome function is inhibited, one expects to elicit an increase in ubiquitinated proteins because they can no longer be degraded. Thus, we examined ubiquitinated proteins by Western blot analysis (Figure 4A, B). As expected, the 1st hit elicited a significant increase in ubiquitinated proteins 24h later, at the time that the 2nd hit would normally have been applied (Figure 4A). This finding confirms that the 1st hit elicits protein misfolding stress. We also examined ubiquitinated proteins 24h after the 2nd hit, and found that ubiquitinated proteins were greatly increased by the 2nd hit regardless of whether the cells had been previously stressed (Figure 4B). When the MG132 concentrations are even higher, synergistic ubiquitinated protein responses are elicited in response to dual hits, as shown previously [11]. The current and previous findings strongly suggest that the astrocytes that survive the 1st hit are still somewhat responsive to the 2nd hit and not simply refractory to the MG132 poison, otherwise there would have not been any increase in this measure after dual hits. In Figure 4B, the 1st hit did not raise ubiquitinated protein levels 48h later, perhaps because the 1st hit of MG132 had to be removed from the media 24h earlier, at the time of the 2nd hit, in a full media exchange. This protocol was instituted so that we could rigorously compare the effects of the 1st hit alone (24h in duration) to that of the dual, sequential hits (each also 24h in duration) and because a fresh media exchange was essential for complete removal of the 1st hit of MG132.
Fig. 4.

MG132 increases protein misfolding stress and upregulates Hsp70 and HO1 levels. Astrocytes were treated with indicated concentrations of MG132 (1st hit) and probed for a number of stress-sensitive proteins 24h later (A, C–L). Astrocytes were treated with single or dual hits of MG132 and assayed for levels of ubiquitinated proteins and heat shock proteins Hsp70 and HO1 24h after the 2nd hit (B, M–N). *** p ≤ 0.001 vs 0 μM 2nd MG132 hit; + p ≤ 0.05, ++ p ≤ 0.01 vs 0 μM 1st MG132 hit, two-way ANOVA followed by Bonferroni post hoc correction for B, M, and N. The one-tailed, paired Student’s t-test was used for A, C–L
If exposure to the 1st proteotoxic hit elicits protection against the 2nd hit, then one might expect the 1st hit to raise levels of pro-survival proteins, such as the heat shock proteins and proteins involved in antioxidant defense. To test this hypothesis, we performed a series of Western blotting experiments for proteins involved in self-defense (Figure 4C–N). No significant change in the anti-apoptotic protein DJ-1, the heat shock proteins Hsp40 (p = 0.093), Hsp32 (also known as HO1, p = 0.058), Hsp25 (better known as Hsp27 in humans), Hsc70 (p = 0.070), Hsp90, or Hip was observed. However, phosphorylated Hsp25 was significantly higher in stressed astrocytes when expressed as a function of α-tubulin and there was a trend towards an increase in phosphorylated Hsp25 levels as a function of total Hsp25 (p = 0.0833). Furthermore, we observed a significant increase in Hsp70 after MG132 application (Figure 4L). When we examined Hsp70 and HO1 levels 24h after the 2nd hit, the patterns appeared similar to the ubiquitinated protein data (Figure 4M, N). Once again, these data confirm that stressed cells still do respond to the proteotoxicity of the 2nd hit, in that they still react with robust stress-induced increases in Hsp70 and HO1. Thus, we are not selecting for cells that are completely unresponsive to MG132 and, as a result, fail to respond to the 2nd hit with additional cell loss.
Finally, it is noteworthy that there is an increase in Hsp70 24h after the 1st hit alone (Figure 4L) that wanes within 48h (Figure 4M). These findings reveal that the increase in Hsp70 after the 1st hit is transient. The ubiquitin Western blots show a similar transient increase in protein ubiquitination after the 1st hit. The transient increase in Hsp70 is not consistent with the view the 1st hit leaves behind cells that exhibited higher expression of protective proteins to begin with and did not respond with any further adaptive changes. Instead, all the data–including the transient increases in Hsp70 and protein ubiquitination–suggest that stressed astrocytes respond actively to proteotoxicity and are not simply refractory to the MG132 poison from the beginning.
Loss of glutathione but not Hsp70 unmasks the toxic impact of the 2nd hit in previously stressed cells
Because of the significant increase in Hsp70 levels after the 1st MG132 hit, we tested the hypothesis that a decrease in Hsp70 activity would abolish stress resistance against the 2nd hit. For these experiments we used the Hsp70/Hsc70 activity inhibitor VER155008 (Figure 5A–C). The data are also expressed as a percentage of the 0 μM 2nd hit group in Figure 5B. As a positive control, we included the glutathione synthesis inhibitor buthionine sulfoximine, because our previous report had shown that stressed astrocytes resist a 2nd MG132 hit in a glutathione-dependent manner [11]. Guo and colleagues have shown that Hsp70 overexpression increases glutathione-related enzyme activities, such as glutathione peroxidase and glutathione reductase, after oxygen glucose deprivation, suggesting that there might also be a link between glutathione and Hsp70 in our model [74]. VER155008 failed to abolish the resistance of stressed astrocytes against a 2nd hit, although it significantly reduced basal viability. As expected, buthionine sulfoximine rendered stressed astrocytes vulnerable to a 2nd hit. Combining both inhibitors led to an overall reduction in viability (due to VER155008) and an increased sensitivity to the 2nd hit in previously stressed astrocytes (due to buthionine sulfoximine). In other words, the impact of VER155008 and buthionine sulfoximine appeared to be additive and not synergistic. In the dual hit group treated with VER155008 and buthionine sulfoximine, the survival rate was only 24.58%, approximately half of the survival rate in the 1st hit group. Furthermore, there was a trend towards reduced viability in the dual hit group in the presence of both VER155008 and buthionine sulfoximine relative to cells treated only with dual hits of MG132 (p = 0.064). Taken together, these data suggest that glutathione, but not Hsp70, is essential for stress-induced protection against a 2nd hit in astrocytes. In other words, forced glutathione loss unmasks the underlying toxic impact of the 2nd hit, suggesting again that astrocytes surviving the 1st hit of MG132 are simply refractory to the MG132 toxin but are quite vulnerable to proteasome inhibition when they are prevented from mounting active glutathione defenses.
Fig. 5.

Inhibition of glutathione synthesis but not Hsp70/Hsc70 activity abolishes stress-induced protection against the 2nd MG132 hit. A) Primary cortical astrocytes were treated with the indicated concentrations of MG132 in the absence or presence of the Hsp70/Hsc70 inhibitor VER155008 and glutathione synthesis inhibitor buthionine sulfoximine (BSO). Viable Hoechst+ nuclei were quantified 24h after the 2nd hit to measure viability. B) Data from panel A were expressed as a percentage of the 0 μM 2nd MG132 hit groups (i.e. all gray bars were expressed as a percentage of the adjacent black bars). C) Representative images of Hoechst-stained nuclei from data shown in panel A. D) Total glutathione levels were measured 24h after the 2nd MG132 hit by the In-Cell Western technique and expressed as a function of the corresponding number of Hoechst+ nuclei to control for differences in cell density. E) Representative image of total glutathione In-Cell Western. F–H) Reduced glutathione levels were measured intracellularly and in the extracellular media by the Glutathione-Glo assay and luminescence was expressed as a function of the number of Hoechst+ nuclei on parallel plates. All glutathione assays in F–H were performed 24h after the final MG132 hit. I–J) Western blot analysis of glutamate cysteine ligase modifier subunit (GCLM) and glutathione S-transferase μ (GST-μ) in lysates collected 24h after the 1st MG132 hit. K) Astrocytes were treated with single and dual hits of MG132 in the absence or presence of BSO and ATP levels were measured by the CellTiter-Glo assay 24h after the 2nd MG132 hit. ATP levels are expressed as a function of Hoechst+ cell numbers on parallel plates. * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001 vs 0 μM 2nd MG132 hit; + p ≤ 0.05, ++ p ≤ 0.01, +++ p ≤ 0.001 vs 0 μM 1st MG132 hit; ^ p ≤ 0.05, ^^ p ≤ 0.01 vs 0 μM VER155008 and/or 0 μM BSO, two- or three-way ANOVA followed by Bonferroni post hoc correction. For F–G and I–J, the two-tailed paired Student’s t test was employed
Similar to the Hsp70 data, inhibition of HO1 did not abolish stress-induced protection in astrocytes (Supplementary Figure 3), confirming the robust nature of the stress resistance in these cells. Notably, neither VER155008 nor the HO1 inhibitor SnPPx exacerbated the toxicity of single hits of MG132, suggesting that stressed astrocytes can rely on other molecules for protection against proteotoxic stress.
Next, we determined with the In-Cell Western technique whether MG132 raised glutathione levels and buthionine sulfoximine reduced glutathione levels (Figure 5D, E). We found that glutathione levels rose significantly with the 2nd hit and that there was a trend towards an increase in glutathione with the 1st hit (p=0.059). VER155008 greatly increased glutathione levels in cells stressed with the 1st and dual hits, perhaps explaining why stressed astrocytes were still able to resist a 2nd hit even after loss of Hsp70 activity.
The In-Cell Western assay shown in Figures 5D and E cannot distinguish between reduced (GSH) and oxidized (GSSG) glutathione. However, glutathione exists primarily in the reduced state [75], suggesting that total glutathione measures are likely to coincide with reduced glutathione levels. To confirm this, reduced glutathione levels were measured by the GSH-Glo assay, both intracellularly and in the extracellular media. A significant increase in intracellular but not extracellular reduced glutathione levels was observed 24h after the 1st hit (Figure 5F, G). We also measured reduced glutathione levels 24h after the 2nd hit and found similar trends as shown by the In-Cell Western assay (Figure 5H), supporting the view that most glutathione molecules are indeed in the reduced form. Thus, reduced glutathione levels were significantly higher after the 1st and 2nd hits compared to the untreated group. Furthermore, there was no additional change in glutathione levels in cells hit twice with MG132 relative to cells treated with the 1st hit alone.
In order to understand how glutathione defenses may be boosted in stressed cells, the rate-limiting enzyme in glutathione synthesis, glutamate cysteine ligase, was measured by Western blot analysis for the modifier and catalytic subunits of this protein. As the modifier subunit of glutamate cysteine ligase has been shown to increase the efficiency of the catalytic subunit [76], higher modifier subunit levels may be sufficient to permit an increase in glutathione synthesis. We also measured an enzyme responsible for the detoxification of xenobiotics in the presence of reduced glutathione, glutathione S-transferase. The family of cytosolic glutathione S-transferase enzymes are subdivided into classes based upon the composition of their N-terminal domain, such as π and μ [77]. Glutathione S-transferase μ is primarily found in astrocytes but glutathione S-transferase π, normally found in oligodendrocytes, has also been identified in reactive astrocytes [78–80]. Significant increases in the modifier subunit of glutamate cysteine ligase and glutathione S-transferase μ were observed 24h following the 1st hit of MG132, at the time the 2nd hit would normally have been applied (Figure 5I, J). No significant change in glutamate cysteine ligase catalytic subunit or glutathione S-transferase π was detected (Supplementary Figure 4).
ATP is important for fueling defensive responses in stressed cells and for supplying energy for glutathione synthesis and heat shock protein activity [76, 81–83]. Therefore, we examined ATP levels after MG132 and buthionine sulfoximine treatments to determine if there was a stress-induced increase in ATP and whether this was abolished when glutathione synthesis was disrupted to account for the loss of protection. There was a significant increase in ATP levels (expressed as a function of cell numbers) in cells hit with the 1st or 2nd hits alone (Figure 5K). However, no additional change in ATP was observed in cells hit twice with MG132, unless glutathione synthesis was inhibited with buthionine sulfoximine. That is, in the presence of buthionine sulfoximine, an unexpected rise in ATP output per cell was apparent after the dual hits. These findings suggest that in the absence of a stress responsive increase in glutathione, ATP levels are raised in a compensatory fashion to help the astrocytes survive. Without this ATP response to glutathione loss, astrocytes hit twice with MG132 might have been all the more vulnerable. Once again, these findings point to the general resilience of this cell type.
Stressed astrocytes can protect neighboring neurons from proteotoxicity in a glutathione-independent manner
Astrocytes are well known to provide support to neighboring cells, especially neurons. Thus, the major role of astrocytic adaptations to severe proteotoxic stress might be to continue to protect their injured neighbors. If this was true, severely stressed astrocytes should still be able to protect neurons from proteotoxic injury. To test this novel hypothesis, we developed a neuron/astrocyte co-culture model. First we treated astrocytes with MG132 or vehicle the day after plating. Twenty-four hours after this insult, we washed off the MG132 and plated primary cortical neurons on top of the astrocyte layer or in empty wells (neurons only group). As in our previous report [11], we have shown that the vulnerable astrocyte population has already died and detached from the plate by 24h after the 1st hit (data not shown), the time of addition of neurons in the present study. Two days following the plating of neurons, we treated the co-cultures with toxic concentrations of MG132, in this case a 2nd hit for the previously stressed astrocytes. Control primary neurons not plated on top of astrocytes were highly vulnerable to MG132 according to the In-Cell Western assay for the specific neuron marker MAP2 (Figure 6A, B). However, neurons plated on top of previously stressed astrocytes were significantly less vulnerable to MG132. Unstressed astrocytes were not significantly neuroprotective in these experiments, probably because the effect size was smaller. Surprisingly, this neuroprotection was not at all attenuated in the presence of buthionine sulfoximine. Thus, glutathione does not appear to be essential for astrocyte-mediated protection of neurons from proteotoxicity although it is essential for protection of previously stressed astrocytes from subsequent insults. In other words, the few astrocytes surviving dual hits of MG132 as well as glutathione loss can still robustly protect neighboring cells.
Fig. 6.

Severely stressed astrocytes can protect neurons from proteotoxic stress. A) Astrocytes were treated with the 1st MG132 hit or vehicle 24h prior to introduction of neocortical neurons, in the absence or presence of the glutathione synthesis inhibitor buthionine sulfoximine (BSO). Two days after the introduction of neurons, the neuron/astrocyte co-cultures were treated with MG132 as a 2nd hit or vehicle in the absence or presence of BSO. Two days after the 2nd hit, neuronal viability was measured by the In-Cell Western assay for the specific neuronal marker MAP2. B) Representative image of MAP2 In-Cell Western. C) Changes in glutathione levels were measured by In-Cell Western analyses and expressed as a function of the infrared nuclear stain DRAQ5 to control for changes in cell density. Representative images of glutathione immunostaining (D) and DRAQ5 levels (E) are included. F) The astrocytic marker GFAP was measured by In-Cell Western analysis in neuron/astrocyte co-cultures 48h after the 2nd MG132 hit or vehicle treatment in the absence or presence of BSO. G) Representative image of GFAP In-Cell Western. ** p ≤ 0.01, *** p ≤ 0.001 vs 0 μM 2nd MG132 hit; + p ≤ 0.05, ++ p ≤ 0.01, +++ p ≤ 0.001 vs neurons; ^ p ≤ 0.05, ^^ p ≤ 0.01, ^^^ p ≤ 0.001 vs 0 μM BSO, three-way ANOVA followed by Bonferroni post hoc correction
To ensure that buthionine sulfoximine successfully inhibited glutathione synthesis in the co-cultures, we also measured glutathione levels (Figure 6C–E). Buthionine sulfoximine significantly reduced glutathione levels in all neuron/astrocyte co-cultured groups. This analysis also revealed that glutathione levels were significantly increased by the presence of astrocytes in all the “neuron + astrocyte” groups and that high levels of proteotoxic stress from 1.68 μM MG132 further increased glutathione levels in all the neuron/astrocyte co-cultures. Taken together, these results reveal stress-induced increases in glutathione, astrocyte-induced increases in glutathione, and successful inhibition of glutathione synthesis by buthionine sulfoximine in neuron/astrocyte co-cultures.
One limitation of the co-culture experiments is the potential for incomplete wash-off of the MG132 toxin from the plate before neurons were added on top of the pretreated astrocytes. This could have inadvertently affected neuronal integrity in the group plated on top of MG132-pretreated astrocytes. To address this confound, we treated empty wells with vehicle or 0.16 μM MG132 and plated neurons in these wells immediately after wash-off of the MG132 toxin. Compared to vehicle treatment, the neurons in these previously treated wells exhibited no change in viability (data not shown), suggesting that there were no lingering effects of the previous MG132 treatment in the co-culture experiments.
It must be noted that our primary neuronal cultures have a small proportion of astrocytes (~9.5%) because they are harvested from postnatal brains [60, 61]. This presence of a small number of astrocytes in the control group was revealed by immunostaining for the astrocyte marker GFAP (Figure 6F, G). GFAP levels remained strikingly high in the presence or absence of MG132 in all the groups containing previously plated astrocytes. We have shown previously in multiple reports that the In-Cell Western assay for MAP2 is in linear proportion to the number of neurons [63, 66]. However, GFAP is a stress-responsive protein and, as a result, the In-Cell Westerns cannot be viewed as an approximation of astrocyte cell numbers. In other words, a lack of loss of GFAP+ signal does not signify a lack of loss of astrocyte numbers. Taken as a whole, these findings suggest that severely stressed astrocytes maintain or increase overall GFAP levels and can still protect neighboring neurons against MAP2 loss in response to severe proteotoxic stress.
Stressed astrocytes can protect neurons from the synergistic toxicity of proteasome and Hsp70 inhibitors
Although stressed astrocytes do not rely on Hsp70 activity for protection against subsequent proteotoxic insults, Hsp70 levels were shown to be higher in MG132 treated astrocytes in Figure 4 and may be released from these cells through exosomes in order to protect neighboring neurons from proteotoxicity [84, 85]. Thus, we tested the hypothesis that loss of Hsp70 activity would lead to loss of astrocytic neuroprotection and applied the Hsp70/Hsc70 inhibitor VER155008 to MG132-treated neuron/astrocyte co-cultures. As shown in our recent work in olfactory bulb and cortical neurons, we found strikingly synergistic effects of inhibitors of proteasomal activity and Hsp70/Hsc70 activity [86, 87]. Remarkably, severely stressed astrocytes protected neurons robustly against this synergistic, profound toxicity (Figure 7). That is, VER155008 greatly exacerbated the toxicity of MG132 in neurons, and astrocytes protected against the severe stress of simultaneous inhibition of proteasome and chaperone function. We have also presented the data as a function of 0 μM VER155008 in Figure 7B so that the synergy between MG132 and VER155008 can be fully appreciated. In these experiments, MG132 was less toxic to neurons than in Figure 6, probably due to variability either in the MG132 stock solutions or in the neuron culturing procedures. Because of lower toxicity of MG132, even unstressed astrocytes were able to protect neurons against MG132 toxicity. However, in the neurons treated with the higher concentration of MG132 (1.68 μM), only astrocytes that were previously stressed with MG132 were able to significantly protect against the synergy of MG132 and VER155008. Taken together, these data support the view that astrocytes are still highly neuroprotective even when they and the neurons have been exposed to severe proteotoxic stress, such as combined effects of proteasome and Hsp70/Hsc70 inhibitors.
Fig. 7.

Stressed astrocytes prevent the synergistic, severe toxicity of proteasome and Hsp70/Hsc70 inhibitors in stressed neurons. A) Astrocytes were treated with the 1st MG132 hit or vehicle 24h prior to introduction of neocortical neurons, in the absence or presence of the Hsp70/Hsc70 inhibitor VER155008. Two days after the introduction of neurons, the neuron/astrocyte co-cultures were treated with vehicle or MG132 as a 2nd hit in the absence or presence of VER155008. Two days after the 2nd hit, neuronal viability was measured by the In-Cell Western assay for MAP2. B) Data from panel A were expressed as a percentage of the 0 μM VER155008 groups to illustrate the exacerbation of MG132 toxicity by VER155008. C) Representative image of MAP2 In-Cell Western. D) The astrocytic marker glial fibrillary acidic protein (GFAP) was analyzed by In-Cell Western analyses 48h after the 2nd hit in the same neuron/glia co-cultures as shown in panels A–C. E) Representative image of GFAP In-Cell Western. * p ≤ 0.05, *** p ≤ 0.001 vs 0 μM 2nd MG132 hit; + p ≤ 0.05, ++ p ≤ 0.01, +++ p ≤ 0.001 vs neurons; ^ p ≤ 0.05, ^^ p ≤ 0.01, ^^^ p ≤ 0.001 vs 0 μM VER155008, three-way ANOVA followed by Bonferroni post hoc correction
In the VER155008 + MG132 experiments, an examination of GFAP levels showed a similar pattern as in the BSO experiments, with GFAP levels being very high in all groups containing pre-plated astrocytes (Figure 7D, E). VER155008 had no impact upon GFAP levels. When we treated co-cultures with VER155008 in combination with buthionine sulfoximine, no additional impact beyond that observed with VER155008 was apparent (data not shown). In other words, buthionine sulfoximine continued to have no impact on MAP2 levels in neuron/astrocyte co-cultures, even when applied together with an Hsp70/Hsc70 activity inhibitor.
Astrocyte-conditioned medium from stressed astrocytes fails to protect neurons against MG132 toxicity
Astrocytes are known to secrete diffusible substances that protect nearby cells from injury. We therefore examined whether astrocyte-conditioned medium from stressed or unstressed astrocytes would be sufficient to protect neurons from MG132 toxicity. The 1st hit of MG132 or an equivalent volume of vehicle was delivered for 24h to two groups: conditioned media (astrocytes present) and unconditioned media (astrocytes absent; empty wells with media only). Unconditioned media was used as a negative control and lacked any diffusible factors that might be released from astrocytes. Conditioned and unconditioned media–both still containing the 1st hit of MG132 or vehicle–were then transferred to neurons at a 1:1 dilution in Neurobasal media. The 2nd MG132 hit was administered to the neurons at the time of media transfer. As MG132 was still present from the 1st hit in transferred media, 2nd hit concentrations were reduced relative to the co-culture experiments described above. Neuronal viability was assessed 48h after the 2nd hit by the In-Cell Western assay for MAP2 levels.
We did not find that astrocyte-conditioned medium elicited neuroprotection against proteotoxic stress (Figure 8). Because the astrocyte media was diluted 1:1 in neuron media in these experiments, we also concentrated the factors in the astrocyte media by centrifugation and added them as a 10× stock to the neuron media. However, there was no protection with diffusible astrocyte factors using this alternative protocol either (data not shown). Therefore, the data support the view that stressed astrocytes must be in physical contact with stressed neurons to help prevent severe proteotoxic injury in the neurons.
Fig. 8.
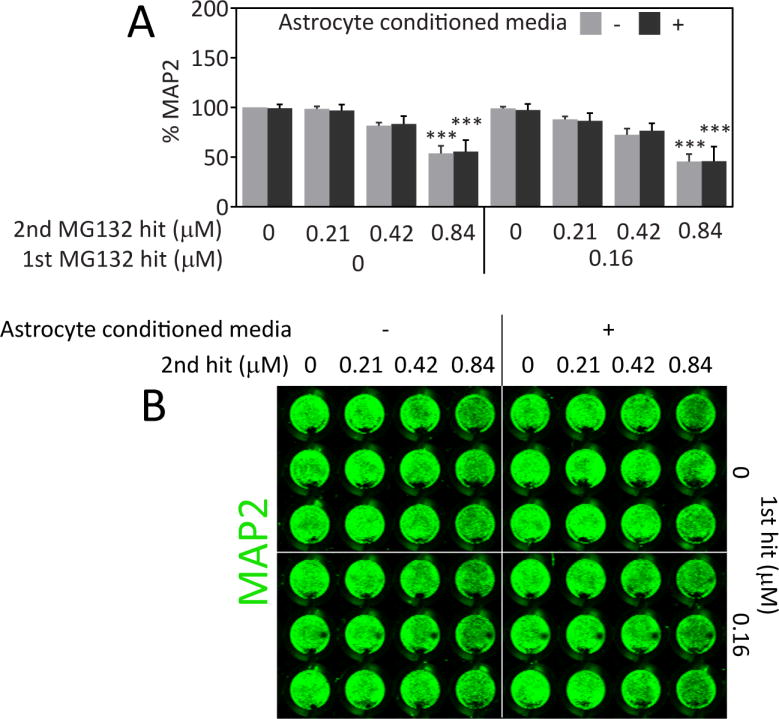
Stressed neurons are not protected from proteotoxicity by astrocyte-conditioned media. A) The 1st MG132 hit or an equivalent v/v of vehicle was added for 24h to wells in the presence of astrocytes (conditioned media) or in their absence (unconditioned media). Conditioned and unconditioned media (still containing 1st hit of MG132 or vehicle) were then delivered to neocortical neurons by diluting them 1:1 in neuron media. The 2nd MG132 hit or vehicle was administered at the time of media transfer. Neuronal viability was measured by the In-Cell Western assay for MAP2 48h after media transfer. B) Representative image of MAP2 In-Cell Western. *** p ≤ 0.001 vs 0 μM 2nd MG132 hit, three-way ANOVA followed by Bonferroni post hoc correction
Discussion
The findings of the present study support the novel view that astrocytes have the capacity to adapt to severe stress in order to retain their neurosupportive roles in the brain. This finding is somewhat reminiscent of the well-established preconditioning phenomenon, whereby cells that survive sublethal stress learn to tolerate subsequent challenges better than naïve cells. For the fraction of astrocytes that manage to survive high concentrations of MG132, the 1st hit was, by definition, also sublethal. Thus, our findings suggest that the definition of preconditioning in astrocytes should be expanded to include protective responses to severe stress, with severe stress being defined as stress that is lethal to some fraction of the cellular population under study. Recent findings in our lab further suggest that astrocytes surviving high concentrations of the oxidative toxin paraquat are also protected against subsequent challenges (Pant et al., in preparation), supporting the view that stress-induced protection in astrocytes is generalizable and can be achieved with multiple types of injury. Unlike what we observed in astrocytes, severe stress has been observed to weaken surviving neurons in previous studies, consistent with the dual hit hypothesis of neurodegeneration, whereby dual hits synergize in their toxic effects on neurons [68, 88–95]. In other words, the effect of stress is likely to be both injury and cell-type dependent and may also vary with age, among other variables [96]. Thus, in all likelihood, cell fate is determined by genetic determinants of susceptibility as well as a convergence of all previous stressors.
Astrocytes have been observed to upregulate heat shock proteins in a number of neurodegenerative disorders, supporting the view that they are exposed to some degree of proteotoxic injury in these conditions, because the major stimulus for heat shock protein induction is protein denaturation [97–107]. We discovered that severely stressed astrocyte survivors exhibit an increase in Hsp70 levels and a trend towards an increase in HO1 (p = 0.058) 24h after the first hit. However, the inhibitor experiments do not support the view that either Hsp70 or HO1 activity plays a role in mediating astrocytic resistance to proteotoxic stress. Furthermore, the rise in Hsp70 levels after the first hit appears to be transient in nature, in contrast to the stress-induced protection, which lasts for at least 96h after the first hit, further suggesting that Hsp70 molecules are not essential for stress adaptations in this model. On the other hand, the antioxidant thiol glutathione was essential for the protective effects of severe stress, as shown previously [11]. In the present study we established that stressed astrocytes raise glutathione levels by two independent assays for glutathione. These assays also verified that the glutathione synthesis inhibitor, buthionine sulfoximine, was indeed effective at reducing glutathione levels. Thus, the loss of a natural increase in glutathione helped to unmask the underlying, severely toxic effects of dual hits. This unmasking of vulnerability in the remaining astrocytes is inconsistent with the view that we were only selecting for “MG132 refractory” cells that do not take up the proteasome inhibitor in the first place.
Many studies have shown that glutathione levels are lowered in vulnerable brain regions in Parkinson’s disease [108–111]. However, one study from Mythri and colleagues suggests that resistant brain regions have higher glutathione levels in Parkinson’s patients than in control subjects [112]. Furthermore, there is evidence supporting a decrease in glutathione levels in Alzheimer’s patients [113, 114]. However, one study reported higher reduced and oxidized glutathione levels in the hippocampus of Alzheimer’s disease patients while another study reported age-related increases in glutathione levels in Alzheimer’s patients but not control subjects [115, 116]. If glutathione levels fall in vulnerable brain regions in neurodegenerative disorders, this loss may unmask the toxic impact of severe stressors, especially sequential, unremitting stress as in our dual hit model. If, on the other hand, glutathione levels are increased in some cells in neurodegenerative disorders, this might explain their lack of vulnerability to protein misfolding stress. Our data further support the view that the natural increase in glutathione in response to proteotoxic stress may depend on a slight increase in the modifier subunit of the enzyme glutamate cysteine ligase, an essential player in the rate-limiting step in glutathione synthesis, although RNA interference studies are needed to confirm this. Although one study did not find any changes in the activity levels of glutamate cysteine ligase in Parkinson’s patients [117], male Alzheimer’s patients exhibit lower glutamate cysteine ligase activity in erythrocytes [118].
Polymorphisms in glutathione S-transferase π have been associated with an increased risk for developing Parkinson’s disease in smokers [119] and glutathione S-transferase π loss is associated with increased susceptibility to experimental Parkinson’s disease in vivo [120]. Furthermore, patients lacking glutathione S-transferase μ 1 may experience an earlier onset of Parkinson’s disease [121]. A positive association has also been found between glutathione S-transferase μ 1 null genotypes and Alzheimer’s disease [122]. In the present study MG132-treated astrocytes expressed slightly higher levels of glutathione S-transferase μ, supporting the view that glutathione defenses are higher in astrocytes surviving severe proteotoxic stress than in stress-naïve astrocytes. Consistent with our findings on glutathione-mediated adaptations and the abovementioned clinical observations, many investigators view raising glutathione levels as a reasonable therapeutic strategy in neurodegenerative conditions [123–125].
In addition to an increase in glutathione levels, we also found an increase in ATP levels in stressed astrocytes. One major difference between our present report and our previous investigation [11] is that ATP levels here are expressed as function of cell numbers, reflecting ATP output per cell. In the absence of this type of normalization, one cannot be certain whether a loss or increase in ATP is due to changes in cell numbers, changes in ATP production, or both. The results of the present study therefore support the hypothesis that astrocytes fuel compensatory adaptations to stress with energy from ATP. However, when glutathione synthesis was inhibited in stressed astrocytes by buthionine sulfoximine, ATP levels did not fall in parallel with the loss of cell numbers. Instead, there was a striking increase in ATP output in cells hit twice with MG132 when treated with buthionine sulfoximine, the most vulnerable group of all. It is possible that these cells would be even more vulnerable to dual hits and loss of glutathione synthesis if ATP levels had not risen in this manner. Taken together, these findings are consistent with a robust ability of astrocytes to compensate against and survive injuries in the brain.
In the present study, we performed a rigorous and extensive validation of our viability assays by measuring nuclear area and staining intensity in Hoechst+ MG132-treated cells. Furthermore, the stress of the first hit did not increase the number of TUNEL+ profiles following the second hit, a finding that supports the Hoechst nuclear count data. The examination of nuclear profiles and TUNEL staining further suggest that the vast majority of astrocytes remaining behind after MG132 treatment are indeed viable. Indeed, this may be the very reason they are still able to protect neighboring neurons, as discussed further below. As an additional positive control, we verified that a different proteasome inhibitor, lactacystin, has the same protective effects as MG132. The microbial metabolite lactacystin covalently and irreversibly binds the β-subunit of the proteasome and inhibits chymotrypsin, trypsin, and caspase activities [69, 126]. In contrast, the peptide aldehyde MG132 competitively and reversibly binds to the β-subunit of the proteasome to primarily inhibit chymotrypsin-like activities [70, 127, 128].
Severe proteotoxic stress upregulated the levels of ubiquitinated proteins in our model, as one would expect from a toxin that inhibits the degradation of misfolded proteins. The second hit elicited this robust response regardless of exposure to the first hit, supporting the view that we had not simply selected for astrocytes that did not take up the MG132 poison or were somehow refractory to its effects on protein misfolding. One caveat of our measure is that all ubiquitinated proteins are not necessarily misfolded, as protein ubiquitination also plays a role in normal turnover of a number of properly folded proteins [129]. Nevertheless, Hsp70 and HO1 expression patterns after dual MG132 hits were similar to the pattern of ubiquitinated proteins, further supporting the view that severely stressed astrocytes continue to respond to the stress of the second hit but that they do not die because of natural adaptations to this injury.
A recent study demonstrates that selective loss of GFAP+ astrocytes initiates neuronal loss [130], consistent with many previous studies showing that astrocytes can protect neurons, including in co-culture models [131–139]. Furthermore, astrocytes may protect neurons with the help of glutathione and Hsp70, the two molecules examined closely here [132, 134, 140–147]. Beta-amyloid has been shown to stimulate glutathione release from astrocytes [148] and astrocytes exposed to endogenous hydrogen peroxide can subsequently protect neurons from glutathione depletion [139]. Of special relevance to the present study are prior observations that astrocytes may mediate some of the protective effects of preconditioning stimuli [1, 149, 150]. Indeed, preconditioning with sublethal stress may render astrocytes all the more neuroprotective [151]. However, other studies have suggested that stressed astrocytes are neurotoxic, especially under conditions of severe stress [139, 152–155]. Thus, one of the major new findings of the present study is that severely stressed astrocytes can still protect neighboring neurons against proteotoxic injury, even when chaperone and proteasome functions are simultaneously compromised and when there is moderate glutathione loss. Considering that the stressed astrocytes have been exposed to an injury that is severe enough to kill a fraction of their population, the degree of neuroprotection by stressed astrocytes was unexpectedly robust. When astrocytes and neurons are injured, they may support neighboring neurons by releasing trophic factors, antioxidant support molecules, and metabolic precursors. Contrary to previous studies showing that glutathione is essential for astrocyte-mediated neuroprotection against oxidative stress (see above), our results with buthionine sulfoximine support the view that astrocytic protection of neurons against proteotoxicity is not necessarily dependent upon glutathione synthesis. Furthermore, astrocyte-induced neuroprotection against MG132 was also independent of Hsp70/Hsc70 because VER155008 did not attenuate the protective effects either. Instead, VER155008 exacerbated the toxicity of MG132 in neurons and stressed astrocytes greatly inhibited the synergistic toxic effects of the proteasome and Hsp70/Hsc70 inhibitors. This leads us to conclude that severely stressed astrocytes can protect neurons that are also severely injured. We did not gather support for the notion that stressed astrocytes protect neurons from proteotoxic injury through a diffusible signal. Instead, it seems more likely that the stressed astrocytes need to be in physical contact with injured neurons in the current co-culture model in order to elicit neuroprotective effects. However, they do not need to be in physical contact with neurons to protect themselves against second hits.
Some important limitations of our study must be conceded. First, our studies rely on very young cells to model the glial and neuronal pathology that characterize age-related proteotoxic disorders. It is quite possible that astrocytes from aged animals would not have the capacity to adapt to stress or protect neurons as observed in the present study. For example, astrocytes aged in culture are known to be less neuroprotective [156]. Although our cells were treated with MG132 to mimic the loss of protein homeostasis that is evident in neurodegenerative conditions, aged astrocytes might show different responses than young MG132-treated astrocytes and exacerbate proteotoxic injury in neighboring neurons. Second, the evolutionary divergence of humans and rats and the lack of any spontaneous model of proteinopathic disorders in aged rodents weaken interpretations of data in rodent models and negatively impact their predictive validity. Third, the present study relied on pharmacological inhibitors, all of which surely suffer from off-target effects. For example, MG132 also inhibits cathepsins and calpains in addition to the proteasome, thereby leading to further loss of protein homeostasis and exacerbation of proteotoxicity [157]. Fourth, it is not possible to verify loss of Hsp70 ATPase activity in whole cellular lysates after VER155008 treatment, because many cellular molecules besides Hsp70 also exhibit ATPase activity. Finally, our studies examine astrocytes in isolation from other glial cell types such as microglia, which are often found in close association with protein deposits and may either propel or prevent neurotoxicity in neurodegenerative disorders [158–160]. Indeed, astrocytes might have similar dualistic roles, particularly in the aged brain.
The novelty of the present study lies in showing for the first time that severely stressed astrocytes can still protect severely stressed neurons from proteotoxic injury, in contrast with traditional studies of the stress response, where adaptive defenses are engaged only with low-dose injury and inhibited with high-dose injury [4]. Our studies therefore extend the neuroprotective capacities of injured astrocytes and provide concrete evidence that stress does not need to be low in concentration to elicit adaptive defenses. Indeed, most authors would argue that severe or high-dose stress only weakens cells, consistent with the two-hit hypothesis of neurodegeneration [88, 90, 96, 161–164]. Furthermore, many authors have suggested that stressed astrocytes exacerbate injury in neighboring neurons [14–19] and that astrocytes have a strong potential for dysfunction despite their normal roles in cellular defense and homeostasis [165]. An additional novel finding is that astrocyte-mediated neuroprotection against proteotoxicity is not necessarily dependent upon glutathione as suggested previously for other insults [140]. Finally, it was also previously unknown that astrocytes can robustly protect neurons against the synergistic loss of chaperone and proteasome functions. Further studies to determine the mechanism underlying the impact of highly stressed astrocytes on highly stressed neurons are warranted, as stressed astrocytes might speed up or slow down the demise of neighboring neurons depending upon the cellular context and age of the organism. In addition, future studies to determine what distinguishes those astrocytes that survive the first hit from those that succumb and die might also be fruitful.
Supplementary Material
Lactacystin-mediated severe proteotoxic stress preconditions astrocytes against subsequent insults. A) Five days after plating, astrocytes were treated with various concentrations of lactacystin or an equivalent v/v of vehicle (phosphate buffered saline) as the 1st hit. On the following day, the astrocytes were treated with vehicle or a 2nd hit of lactacystin at the indicated concentrations. Viability was assayed 24h after the 2nd hit by counting Hoechst-stained nuclei. B) Representative images of Hoechst-stained nuclei for experiments shown in panel A. * p ≤ 0.05 vs 0 μM 2nd lactacystin hit; + p ≤ 0.05, ++ p ≤ 0.01, +++ p ≤ 0.001 vs 0 μM 1st lactacystin hit, two-way ANOVA followed by Bonferroni post hoc correction
{kind=link}
MG132 treatment is more lethal to small, bright nuclei than large nuclei. A) Hoechst-stained nuclei were categorized into two groups: those exhibiting small, brightly stained nuclei (50–150 μm2 in size and staining intensity greater than 20 arbitrary units for the mean gray value measurements) or large nuclei (greater than 150 μm2; all staining intensities). Raw counts of the numbers of nuclei per microscopic field of view (200× magnification) are shown here to supplement the transformed data in Figure 3P. * p ≤ 0.05, ** p ≤ 0.01 vs 0 μM 2nd MG132 hit; ++ p ≤ 0.01 vs vs 0 μM 1st MG132 hit; ^^ p ≤ 0.01, ^^^ p ≤ 0.001 vs small, bright nuclei, three-way ANOVA followed by Bonferroni post hoc correction
{kind=link}
Stressed astrocytes can resist dual hits despite inhibition of heme oxygenase 1 (HO1, also known as Hsp32). A) Astrocytes were treated with the 1st and 2nd MG132 hits or vehicle 24h apart in the absence or presence of the HO1 inhibitor SnPPx. Viability was assayed 24h after the 2nd hit. B) Representative images of Hoechst stained nuclei. * p ≤ 0.05, *** p ≤ 0.001 vs 0 μM 2nd MG132 hit; + p ≤ 0.05, +++ p ≤ 0.001 vs 0 μM 1st MG132 hit, two-way ANOVA followed by Bonferroni post hoc correction
{kind=link}
Impact of 1st hit on glutathione-related enzymes. Astrocytes were treated with the 1st MG132 hit or vehicle and the following glutathione-related proteins were measured in lysates collected 24h later: A) glutamate cysteine ligase catalytic subunit (GCLC), B) glutathione S-transferase π (GST-π). No significant difference was determined with the two-tailed paired Student’s t test.
{kind=link}
Acknowledgments
Wrote the paper: RKL and AMG. Designed the experiments: RKL. Conducted the experiments and analyzed the data: AMG, JMP, DP, MPH. Generated the figures: AMG. We are grateful to Mary Caruso, Deborah Willson, and Jackie Farrer for excellent administrative support and to Denise Butler-Bucilli and Christine Close for outstanding animal care. These studies were supported by a Hillman Foundation award (109033) and an R15 award from NIH (1R15NS093539) to RKL.
Footnotes
The authors have no conflicts to disclose.
References
- 1.Trendelenburg G, Dirnagl U. Neuroprotective role of astrocytes in cerebral ischemia: focus on ischemic preconditioning. Glia. 2005;50(4):307–20. doi: 10.1002/glia.20204. [DOI] [PubMed] [Google Scholar]
- 2.Gao C, Wang C, Liu B, Wu H, Yang Q, Jin J, Li H, Dong S, Gao G, Zhang H. Intermittent hypoxia preconditioning-induced epileptic tolerance by upregulation of monocarboxylate transporter 4 expression in rat hippocampal astrocytes. Neurochem Res. 2014;39(11):2160–9. doi: 10.1007/s11064-014-1411-2. [DOI] [PubMed] [Google Scholar]
- 3.Rajapakse N, Kis B, Horiguchi T, Snipes J, Busija D. Diazoxide pretreatment induces delayed preconditioning in astrocytes against oxygen glucose deprivation and hydrogen peroxide-induced toxicity. Journal of neuroscience research. 2003;73(2):206–14. doi: 10.1002/jnr.10657. [DOI] [PubMed] [Google Scholar]
- 4.Calabrese EJ. Astrocytes: adaptive responses to low doses of neurotoxins. Critical reviews in toxicology. 2008;38(5):463–71. doi: 10.1080/10408440802004023. [DOI] [PubMed] [Google Scholar]
- 5.Chu PW, Beart PM, Jones NM. Preconditioning protects against oxidative injury involving hypoxia-inducible factor-1 and vascular endothelial growth factor in cultured astrocytes. European journal of pharmacology. 2010;633(1–3):24–32. doi: 10.1016/j.ejphar.2010.02.008. [DOI] [PubMed] [Google Scholar]
- 6.Du F, Zhu L, Qian ZM, Wu XM, Yung WH, Ke Y. Hyperthermic preconditioning protects astrocytes from ischemia/reperfusion injury by up-regulation of HIF-1 alpha expression and binding activity. Biochimica et biophysica acta. 2010;1802(11):1048–53. doi: 10.1016/j.bbadis.2010.06.013. [DOI] [PubMed] [Google Scholar]
- 7.Du F, Qian ZM, Zhu L, Wu XM, Yung WH, Ke Y. A synergistic role of hyperthermic and pharmacological preconditioning to protect astrocytes against ischemia/reperfusion injury. Neurochemical research. 2011;36(2):312–8. doi: 10.1007/s11064-010-0327-8. [DOI] [PubMed] [Google Scholar]
- 8.Johnsen D, Murphy SJ. Isoflurane preconditioning protects astrocytes from oxygen and glucose deprivation independent of innate cell sex. Journal of neurosurgical anesthesiology. 2011;23(4):335–40. doi: 10.1097/ANA.0b013e3182161816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nikiforou M, Vlassaks E, Strackx E, Kramer BW, Vles JS, Gavilanes AW. Preconditioning by Oxygen-Glucose Deprivation Preserves Cell Proliferation and Reduces Cytotoxicity in Primary Astrocyte Cultures. CNS Neurol Disord Drug Targets. 2015 doi: 10.2174/1871527314666150116120518. [DOI] [PubMed] [Google Scholar]
- 10.Hirayama Y, Ikeda-Matsuo Y, Notomi S, Enaida H, Kinouchi H, Koizumi S. Astrocyte-Mediated Ischemic Tolerance. J Neurosci. 2015;35(9):3794–3805. doi: 10.1523/JNEUROSCI.4218-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Titler AM, Posimo JM, Leak RK. Astrocyte plasticity revealed by adaptations to severe proteotoxic stress. Cell and tissue research. 2013;352(3):427–43. doi: 10.1007/s00441-013-1571-4. [DOI] [PubMed] [Google Scholar]
- 12.Perea G, Sur M, Araque A. Neuron-glia networks: integral gear of brain function. Front Cell Neurosci. 2014;8:378. doi: 10.3389/fncel.2014.00378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Allen NJ. Astrocyte regulation of synaptic behavior. Annu Rev Cell Dev Biol. 2014;30:439–63. doi: 10.1146/annurev-cellbio-100913-013053. [DOI] [PubMed] [Google Scholar]
- 14.Jansen AH, Reits EA, Hol EM. The ubiquitin proteasome system in glia and its role in neurodegenerative diseases. Front Mol Neurosci. 2014;7:73. doi: 10.3389/fnmol.2014.00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Avila-Munoz E, Arias C. When astrocytes become harmful: Functional and inflammatory responses that contribute to Alzheimer’s disease. Ageing Res Rev. 2014;18C:29–40. doi: 10.1016/j.arr.2014.07.004. [DOI] [PubMed] [Google Scholar]
- 16.Jain P, Wadhwa PK, Jadhav HR. Reactive Astrogliosis: Role in Alzheimer’s Disease. CNS Neurol Disord Drug Targets. 2015 doi: 10.2174/1871527314666150713104738. [DOI] [PubMed] [Google Scholar]
- 17.Verkhratsky A, Parpura V. Astrogliopathology in neurological, neurodevelopmental and psychiatric disorders. Neurobiol Dis. 2015 doi: 10.1016/j.nbd.2015.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pekny M, Wilhelmsson U, Pekna M. The dual role of astrocyte activation and reactive gliosis. Neurosci Lett. 2014;565:30–8. doi: 10.1016/j.neulet.2013.12.071. [DOI] [PubMed] [Google Scholar]
- 19.Fu W, Jhamandas JH. Role of astrocytic glycolytic metabolism in Alzheimer’s disease pathogenesis. Biogerontology. 2014;15(6):579–86. doi: 10.1007/s10522-014-9525-0. [DOI] [PubMed] [Google Scholar]
- 20.Xilouri M, Stefanis L. Autophagy in the central nervous system: implications for neurodegenerative disorders. CNS & neurological disorders drug targets. 2010;9(6):701–19. doi: 10.2174/187152710793237421. [DOI] [PubMed] [Google Scholar]
- 21.Angot E, Steiner JA, Hansen C, Li JY, Brundin P. Are synucleinopathies prion-like disorders? Lancet neurology. 2010;9(11):1128–38. doi: 10.1016/S1474-4422(10)70213-1. [DOI] [PubMed] [Google Scholar]
- 22.Jellinger KA. Recent advances in our understanding of neurodegeneration. Journal of neural transmission. 2009;116(9):1111–62. doi: 10.1007/s00702-009-0240-y. [DOI] [PubMed] [Google Scholar]
- 23.Dickson DW. Neuropathology of non-Alzheimer degenerative disorders. International journal of clinical and experimental pathology. 2009;3(1):1–23. [PMC free article] [PubMed] [Google Scholar]
- 24.Uversky VN. Intrinsic disorder in proteins associated with neurodegenerative diseases. Frontiers in bioscience: a journal and virtual library. 2009;14:5188–238. doi: 10.2741/3594. [DOI] [PubMed] [Google Scholar]
- 25.Menzies FM, Ravikumar B, Rubinsztein DC. Protective roles for induction of autophagy in multiple proteinopathies. Autophagy. 2006;2(3):224–5. doi: 10.4161/auto.2696. [DOI] [PubMed] [Google Scholar]
- 26.Walker LC, Levine H, 3rd, Mattson MP, Jucker M. Inducible proteopathies. Trends in neurosciences. 2006;29(8):438–43. doi: 10.1016/j.tins.2006.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Walker LC, LeVine H. The cerebral proteopathies: neurodegenerative disorders of protein conformation and assembly. Molecular neurobiology. 2000;21(1–2):83–95. doi: 10.1385/MN:21:1-2:083. [DOI] [PubMed] [Google Scholar]
- 28.Morimoto RI. Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes & development. 2008;22(11):1427–38. doi: 10.1101/gad.1657108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gundersen V. Protein aggregation in Parkinson’s disease. Acta neurologica Scandinavica. Supplementum. 2010;(190):82–7. doi: 10.1111/j.1600-0404.2010.01382.x. [DOI] [PubMed] [Google Scholar]
- 30.McNaught KS, Belizaire R, Isacson O, Jenner P, Olanow CW. Altered proteasomal function in sporadic Parkinson’s disease. Exp Neurol. 2003;179(1):38–46. doi: 10.1006/exnr.2002.8050. [DOI] [PubMed] [Google Scholar]
- 31.McNaught KS. Proteolytic dysfunction in neurodegenerative disorders. Int Rev Neurobiol. 2004;62:95–119. doi: 10.1016/S0074-7742(04)62003-4. [DOI] [PubMed] [Google Scholar]
- 32.Keller JN, Hanni KB, Markesbery WR. Impaired proteasome function in Alzheimer’s disease. J Neurochem. 2000;75(1):436–9. doi: 10.1046/j.1471-4159.2000.0750436.x. [DOI] [PubMed] [Google Scholar]
- 33.Rideout HJ, Larsen KE, Sulzer D, Stefanis L. Proteasomal inhibition leads to formation of ubiquitin/alpha-synuclein-immunoreactive inclusions in PC12 cells. J Neurochem. 2001;78(4):899–908. doi: 10.1046/j.1471-4159.2001.00474.x. [DOI] [PubMed] [Google Scholar]
- 34.Rideout HJ, Stefanis L. Proteasomal inhibition-induced inclusion formation and death in cortical neurons require transcription and ubiquitination. Mol Cell Neurosci. 2002;21(2):223–38. doi: 10.1006/mcne.2002.1173. [DOI] [PubMed] [Google Scholar]
- 35.Rideout HJ, Lang-Rollin IC, Savalle M, Stefanis L. Dopaminergic neurons in rat ventral midbrain cultures undergo selective apoptosis and form inclusions, but do not up-regulate iHSP70, following proteasomal inhibition. J Neurochem. 2005;93(5):1304–13. doi: 10.1111/j.1471-4159.2005.03124.x. [DOI] [PubMed] [Google Scholar]
- 36.Sawada H, Kohno R, Kihara T, Izumi Y, Sakka N, Ibi M, Nakanishi M, Nakamizo T, Yamakawa K, Shibasaki H, Yamamoto N, Akaike A, Inden M, Kitamura Y, Taniguchi T, Shimohama S. Proteasome mediates dopaminergic neuronal degeneration, and its inhibition causes alpha-synuclein inclusions. J Biol Chem. 2004;279(11):10710–9. doi: 10.1074/jbc.M308434200. [DOI] [PubMed] [Google Scholar]
- 37.Sun F, Anantharam V, Zhang D, Latchoumycandane C, Kanthasamy A, Kanthasamy AG. Proteasome inhibitor MG-132 induces dopaminergic degeneration in cell culture and animal models. Neurotoxicology. 2006;27(5):807–15. doi: 10.1016/j.neuro.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 38.Xie W, Li X, Li C, Zhu W, Jankovic J, Le W. Proteasome inhibition modeling nigral neuron degeneration in Parkinson’s disease. Journal of neurochemistry. 2010;115(1):188–99. doi: 10.1111/j.1471-4159.2010.06914.x. [DOI] [PubMed] [Google Scholar]
- 39.Schultz C, Ghebremedhin E, Del Tredici K, Rub U, Braak H. High prevalence of thorn-shaped astrocytes in the aged human medial temporal lobe. Neurobiol Aging. 2004;25(3):397–405. doi: 10.1016/S0197-4580(03)00113-1. [DOI] [PubMed] [Google Scholar]
- 40.Rub U, Del Tredici K, Schultz C, de Vos RA, Jansen Steur EN, Arai K, Braak H. Progressive supranuclear palsy: neuronal and glial cytoskeletal pathology in the higher order processing autonomic nuclei of the lower brainstem. Neuropathol Appl Neurobiol. 2002;28(1):12–22. doi: 10.1046/j.0305-1846.2001.00374.x. [DOI] [PubMed] [Google Scholar]
- 41.Schultz C, Hubbard GB, Rub U, Braak E, Braak H. Age-related progression of tau pathology in brains of baboons. Neurobiol Aging. 2000;21(6):905–12. doi: 10.1016/s0197-4580(00)00176-7. [DOI] [PubMed] [Google Scholar]
- 42.Schultz C, Dehghani F, Hubbard GB, Thal DR, Struckhoff G, Braak E, Braak H. Filamentous tau pathology in nerve cells, astrocytes, and oligodendrocytes of aged baboons. J Neuropathol Exp Neurol. 2000;59(1):39–52. doi: 10.1093/jnen/59.1.39. [DOI] [PubMed] [Google Scholar]
- 43.Braak H, Sastre M, Del Tredici K. Development of alpha-synuclein immunoreactive astrocytes in the forebrain parallels stages of intraneuronal pathology in sporadic Parkinson’s disease. Acta neuropathologica. 2007;114(3):231–41. doi: 10.1007/s00401-007-0244-3. [DOI] [PubMed] [Google Scholar]
- 44.Willwohl D, Kettner M, Braak H, Hubbard GB, Dick EJ, Jr, Cox AB, Schultz C. Pallido-nigral spheroids in nonhuman primates: accumulation of heat shock proteins in astroglial processes. Acta Neuropathol (Berl) 2002;103(3):276–80. doi: 10.1007/s00401-001-0466-8. [DOI] [PubMed] [Google Scholar]
- 45.Wakabayashi K, Hayashi S, Yoshimoto M, Kudo H, Takahashi H. NACP/alpha-synuclein-positive filamentous inclusions in astrocytes and oligodendrocytes of Parkinson’s disease brains. Acta neuropathologica. 2000;99(1):14–20. doi: 10.1007/pl00007400. [DOI] [PubMed] [Google Scholar]
- 46.Lee HJ, Suk JE, Patrick C, Bae EJ, Cho JH, Rho S, Hwang D, Masliah E, Lee SJ. Direct transfer of alpha-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. The Journal of biological chemistry. 2010;285(12):9262–72. doi: 10.1074/jbc.M109.081125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wyss-Coray T, Loike JD, Brionne TC, Lu E, Anankov R, Yan F, Silverstein SC, Husemann J. Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat Med. 2003;9(4):453–7. doi: 10.1038/nm838. [DOI] [PubMed] [Google Scholar]
- 48.Thal DR, Schultz C, Dehghani F, Yamaguchi H, Braak H, Braak E. Amyloid beta-protein (Abeta)-containing astrocytes are located preferentially near N-terminal-truncated Abeta deposits in the human entorhinal cortex. Acta Neuropathol. 2000;100(6):608–17. doi: 10.1007/s004010000242. [DOI] [PubMed] [Google Scholar]
- 49.Akiyama H, Mori H, Saido T, Kondo H, Ikeda K, McGeer PL. Occurrence of the diffuse amyloid beta-protein (Abeta) deposits with numerous Abeta-containing glial cells in the cerebral cortex of patients with Alzheimer’s disease. Glia. 1999;25(4):324–31. doi: 10.1002/(sici)1098-1136(19990215)25:4<324::aid-glia2>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 50.Akiyama H, Schwab C, Kondo H, Mori H, Kametani F, Ikeda K, McGeer PL. Granules in glial cells of patients with Alzheimer’s disease are immunopositive for C-terminal sequences of beta-amyloid protein. Neurosci Lett. 1996;206(2–3):169–72. doi: 10.1016/s0304-3940(96)12474-5. [DOI] [PubMed] [Google Scholar]
- 51.Duyckaerts C, Delatour B, Potier MC. Classification and basic pathology of Alzheimer disease. Acta Neuropathol. 2009;118(1):5–36. doi: 10.1007/s00401-009-0532-1. [DOI] [PubMed] [Google Scholar]
- 52.Yang W, Sopper MM, Leystra-Lantz C, Strong MJ. Microtubule-associated tau protein positive neuronal and glial inclusions in ALS. Neurology. 2003;61(12):1766–73. doi: 10.1212/01.wnl.0000099372.75786.f8. [DOI] [PubMed] [Google Scholar]
- 53.Yang W, Strong MJ. Widespread neuronal and glial hyperphosphorylated tau deposition in ALS with cognitive impairment. Amyotrophic lateral sclerosis: official publication of the World Federation of Neurology Research Group on Motor Neuron Diseases. 2012;13(2):178–93. doi: 10.3109/17482968.2011.622405. [DOI] [PubMed] [Google Scholar]
- 54.Yokota O, Tsuchiya K, Oda T, Ishihara T, de Silva R, Lees AJ, Arai T, Uchihara T, Ishizu H, Kuroda S, Akiyama H. Amyotrophic lateral sclerosis with dementia: an autopsy case showing many Bunina bodies, tau-positive neuronal and astrocytic plaque-like pathologies, and pallido-nigral degeneration. Acta neuropathologica. 2006;112(5):633–45. doi: 10.1007/s00401-006-0141-1. [DOI] [PubMed] [Google Scholar]
- 55.Komori T. Tau-positive glial inclusions in progressive supranuclear palsy, corticobasal degeneration and Pick’s disease. Brain pathology. 1999;9(4):663–79. doi: 10.1111/j.1750-3639.1999.tb00549.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang W, Ang LC, Strong MJ. Tau protein aggregation in the frontal and entorhinal cortices as a function of aging. Brain research Developmental brain research. 2005;156(2):127–38. doi: 10.1016/j.devbrainres.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 57.Ding Q, Dimayuga E, Martin S, Bruce-Keller AJ, Nukala V, Cuervo AM, Keller JN. Characterization of chronic low-level proteasome inhibition on neural homeostasis. J Neurochem. 2003;86(2):489–97. doi: 10.1046/j.1471-4159.2003.01885.x. [DOI] [PubMed] [Google Scholar]
- 58.Leak RK, Zigmond MJ, Liou AK. Adaptation to chronic MG132 reduces oxidative toxicity by a CuZnSOD-dependent mechanism. J Neurochem. 2008;106(2):860–74. doi: 10.1111/j.1471-4159.2008.05459.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Grune T, Catalgol B, Licht A, Ermak G, Pickering AM, Ngo JK, Davies KJ. HSP70 mediates dissociation and reassociation of the 26S proteasome during adaptation to oxidative stress. Free Radic Biol Med. 2011;51(7):1355–64. doi: 10.1016/j.freeradbiomed.2011.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bayer SA, Altman J. Neocortical Development. New York: Raven Press; 1991. p. xiv.p. 255. [Google Scholar]
- 61.Miller FD, Gauthier AS. Timing is everything: making neurons versus glia in the developing cortex. Neuron. 2007;54(3):357–69. doi: 10.1016/j.neuron.2007.04.019. [DOI] [PubMed] [Google Scholar]
- 62.McCarthy KD, de Vellis J. Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol. 1980;85(3):890–902. doi: 10.1083/jcb.85.3.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Posimo JM, Titler AM, Choi HJ, Unnithan AS, Leak RK. Neocortex and allocortex respond differentially to cellular stress in vitro and aging in vivo. PLoS One. 2013;8(3):e58596. doi: 10.1371/journal.pone.0058596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Posimo JM, Weilnau JN, Gleixner AM, Broeren MT, Weiland NL, Brodsky JL, Wipf P, Leak RK. Heat shock protein defenses in the neocortex and allocortex of the telencephalon. Neurobiol Aging. 2015;36(5):1924–37. doi: 10.1016/j.neurobiolaging.2015.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Leak RK, Castro SL, Jaumotte JD, Smith AD, Zigmond MJ. Assaying multiple biochemical variables from the same tissue sample. J Neurosci Methods. 2010;191(2):234–8. doi: 10.1016/j.jneumeth.2010.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Posimo JM, Unnithan AS, Gleixner AM, Choi HJ, Jiang Y, Pulugulla SH, Leak RK. Viability assays for cells in culture. Journal of visualized experiments: JoVE. 2014;83(83):e50645. doi: 10.3791/50645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Unnithan AS, Jiang Y, Rumble JL, Pulugulla SH, Posimo JM, Gleixner AM, Leak RK. N-acetyl cysteine prevents synergistic, severe toxicity from two hits of oxidative stress. Neurosci Lett. 2014;560:71–6. doi: 10.1016/j.neulet.2013.12.023. [DOI] [PubMed] [Google Scholar]
- 68.Unnithan AS, Choi HJ, Titler AM, Posimo JM, Leak RK. Rescue from a two hit, high-throughput model of neurodegeneration with N-acetyl cysteine. Neurochemistry international. 2012;61(3):356–368. doi: 10.1016/j.neuint.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 69.Fenteany G, Standaert RF, Lane WS, Choi S, Corey EJ, Schreiber SL. Inhibition of proteasome activities and subunit-specific amino-terminal threonine modification by lactacystin. Science. 1995;268(5211):726–31. doi: 10.1126/science.7732382. [DOI] [PubMed] [Google Scholar]
- 70.Kisselev AF, Goldberg AL. Proteasome inhibitors: from research tools to drug candidates. Chem Biol. 2001;8(8):739–58. doi: 10.1016/s1074-5521(01)00056-4. [DOI] [PubMed] [Google Scholar]
- 71.Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119(3):493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35(4):495–516. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Finley D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu Rev Biochem. 2009;78:477–513. doi: 10.1146/annurev.biochem.78.081507.101607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Guo S, Wharton W, Moseley P, Shi H. Heat shock protein 70 regulates cellular redox status by modulating glutathione-related enzyme activities. Cell Stress Chaperones. 2007;12(3):245–54. doi: 10.1379/CSC-265.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Owen JB, Butterfield DA. Measurement of oxidized/reduced glutathione ratio. Methods Mol Biol. 2010;648:269–77. doi: 10.1007/978-1-60761-756-3_18. [DOI] [PubMed] [Google Scholar]
- 76.Franklin CC, Backos DS, Mohar I, White CC, Forman HJ, Kavanagh TJ. Structure, function, and post-translational regulation of the catalytic and modifier subunits of glutamate cysteine ligase. Mol Aspects Med. 2009;30(1–2):86–98. doi: 10.1016/j.mam.2008.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mazzetti AP, Fiorile MC, Primavera A, Lo Bello M. Glutathione transferases and neurodegenerative diseases. Neurochem Int. 2015;82:10–8. doi: 10.1016/j.neuint.2015.01.008. [DOI] [PubMed] [Google Scholar]
- 78.Johnson JA, el Barbary A, Kornguth SE, Brugge JF, Siegel FL. Glutathione S-transferase isoenzymes in rat brain neurons and glia. J Neurosci. 1993;13(5):2013–23. doi: 10.1523/JNEUROSCI.13-05-02013.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cammer W, Zhang H. Atypical localization of the oligodendrocytic isoform (PI) of glutathione-S-transferase in astrocytes during cuprizone intoxication. J Neurosci Res. 1993;36(2):183–90. doi: 10.1002/jnr.490360208. [DOI] [PubMed] [Google Scholar]
- 80.Abramovitz M, Homma H, Ishigaki S, Tansey F, Cammer W, Listowsky I. Characterization and localization of glutathione-S-transferases in rat brain and binding of hormones, neurotransmitters, and drugs. J Neurochem. 1988;50(1):50–7. doi: 10.1111/j.1471-4159.1988.tb13228.x. [DOI] [PubMed] [Google Scholar]
- 81.Lu SC. Regulation of glutathione synthesis. Mol Aspects Med. 2009;30(1–2):42–59. doi: 10.1016/j.mam.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lindquist S, Craig EA. The heat-shock proteins. Annu Rev Genet. 1988;22:631–77. doi: 10.1146/annurev.ge.22.120188.003215. [DOI] [PubMed] [Google Scholar]
- 83.Morimoto RI, Santoro MG. Stress-inducible responses and heat shock proteins: new pharmacologic targets for cytoprotection. Nat Biotechnol. 1998;16(9):833–8. doi: 10.1038/nbt0998-833. [DOI] [PubMed] [Google Scholar]
- 84.Lancaster GI, Febbraio MA. Exosome-dependent trafficking of HSP70: a novel secretory pathway for cellular stress proteins. J Biol Chem. 2005;280(24):23349–55. doi: 10.1074/jbc.M502017200. [DOI] [PubMed] [Google Scholar]
- 85.Zhan R, Leng X, Liu X, Wang X, Gong J, Yan L, Wang L, Wang Y, Wang X, Qian LJ. Heat shock protein 70 is secreted from endothelial cells by a non-classical pathway involving exosomes. Biochem Biophys Res Commun. 2009;387(2):229–33. doi: 10.1016/j.bbrc.2009.06.095. [DOI] [PubMed] [Google Scholar]
- 86.Crum TS, Gleixner AM, Posimo JM, Mason DM, Broeren MT, Heinemann SD, Wipf P, Brodsky JL, Leak RK. Heat shock protein responses to aging and proteotoxicity in the olfactory bulb. J Neurochem. 2015 doi: 10.1111/jnc.13041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Posimo JM, Weilnau JN, Gleixner AM, Broeren MT, Weiland NL, Brodsky JL, Wipf P, Leak RK. Heat shock protein defenses in the neocortex and allocortex of the telencephalon. Neurobiol Aging. 2015 doi: 10.1016/j.neurobiolaging.2015.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Boger HA, Granholm AC, McGinty JF, Middaugh LD. A dual-hit animal model for age-related parkinsonism. Progress in neurobiology. 2010;90(2):217–29. doi: 10.1016/j.pneurobio.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cory-Slechta DA, Thiruchelvam M, Barlow BK, Richfield EK. Developmental pesticide models of the Parkinson disease phenotype. Environmental health perspectives. 2005;113(9):1263–70. doi: 10.1289/ehp.7570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhu X, Lee HG, Perry G, Smith MA. Alzheimer disease, the two-hit hypothesis: an update. Biochimica et biophysica acta. 2007;1772(4):494–502. doi: 10.1016/j.bbadis.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 91.Carvey PM, Punati A, Newman MB. Progressive dopamine neuron loss in Parkinson’s disease: the multiple hit hypothesis. Cell Transplant. 2006;15(3):239–50. doi: 10.3727/000000006783981990. [DOI] [PubMed] [Google Scholar]
- 92.Gao HM, Hong JS. Gene-environment interactions: key to unraveling the mystery of Parkinson’s disease. Progress in neurobiology. 2011;94(1):1–19. doi: 10.1016/j.pneurobio.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Weidong L, Shen C, Jankovic J. Etiopathogenesis of Parkinson disease: a new beginning? The Neuroscientist: a review journal bringing neurobiology, neurology and psychiatry. 2009;15(1):28–35. doi: 10.1177/1073858408319974. [DOI] [PubMed] [Google Scholar]
- 94.Sulzer D. Multiple hit hypotheses for dopamine neuron loss in Parkinson’s disease. Trends in neurosciences. 2007;30(5):244–50. doi: 10.1016/j.tins.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 95.Manning-Bog AB, Langston JW. Model fusion, the next phase in developing animal models for Parkinson’s disease. Neurotoxicity research. 2007;11(3–4):219–40. doi: 10.1007/BF03033569. [DOI] [PubMed] [Google Scholar]
- 96.Leak RK. Adaptation and sensitization to proteotoxic stress. Dose-response: a publication of International Hormesis Society. 2014;12(1):24–56. doi: 10.2203/dose-response.13-016.Leak. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Seidel K, Vinet J, Dunnen WF, Brunt ER, Meister M, Boncoraglio A, Zijlstra MP, Boddeke HW, Rub U, Kampinga HH, Carra S. The HSPB8-BAG3 chaperone complex is upregulated in astrocytes in the human brain affected by protein aggregation diseases. Neuropathol Appl Neurobiol. 2012;38(1):39–53. doi: 10.1111/j.1365-2990.2011.01198.x. [DOI] [PubMed] [Google Scholar]
- 98.Wilhelmus MM, Otte-Holler I, Wesseling P, de Waal RM, Boelens WC, Verbeek MM. Specific association of small heat shock proteins with the pathological hallmarks of Alzheimer’s disease brains. Neuropathol Appl Neurobiol. 2006;32(2):119–30. doi: 10.1111/j.1365-2990.2006.00689.x. [DOI] [PubMed] [Google Scholar]
- 99.Dabir DV, Trojanowski JQ, Richter-Landsberg C, Lee VM, Forman MS. Expression of the small heat-shock protein alphaB-crystallin in tauopathies with glial pathology. The American journal of pathology. 2004;164(1):155–66. doi: 10.1016/s0002-9440(10)63106-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Renkawek K, Stege GJ, Bosman GJ. Dementia, gliosis and expression of the small heat shock proteins hsp27 and alpha B-crystallin in Parkinson’s disease. Neuroreport. 1999;10(11):2273–6. doi: 10.1097/00001756-199908020-00009. [DOI] [PubMed] [Google Scholar]
- 101.Renkawek K, Bosman GJ, Gaestel M. Increased expression of heat-shock protein 27 kDa in Alzheimer disease: a preliminary study. Neuroreport. 1993;5(1):14–6. doi: 10.1097/00001756-199310000-00003. [DOI] [PubMed] [Google Scholar]
- 102.Shinohara H, Inaguma Y, Goto S, Inagaki T, Kato K. Alpha B crystallin and HSP28 are enhanced in the cerebral cortex of patients with Alzheimer’s disease. Journal of the neurological sciences. 1993;119(2):203–8. doi: 10.1016/0022-510x(93)90135-l. [DOI] [PubMed] [Google Scholar]
- 103.Renkawek K, Bosman GJ, de Jong WW. Expression of small heat-shock protein hsp 27 in reactive gliosis in Alzheimer disease and other types of dementia. Acta neuropathologica. 1994;87(5):511–9. doi: 10.1007/BF00294178. [DOI] [PubMed] [Google Scholar]
- 104.Renkawek K, Voorter CE, Bosman GJ, van Workum FP, de Jong WW. Expression of alpha B-crystallin in Alzheimer’s disease. Acta neuropathologica. 1994;87(2):155–60. doi: 10.1007/BF00296185. [DOI] [PubMed] [Google Scholar]
- 105.Durrenberger PF, Filiou MD, Moran LB, Michael GJ, Novoselov S, Cheetham ME, Clark P, Pearce RK, Graeber MB. DnaJB6 is present in the core of Lewy bodies and is highly up-regulated in parkinsonian astrocytes. Journal of neuroscience research. 2009;87(1):238–45. doi: 10.1002/jnr.21819. [DOI] [PubMed] [Google Scholar]
- 106.Kawamoto Y, Akiguchi I, Shirakashi Y, Honjo Y, Tomimoto H, Takahashi R, Budka H. Accumulation of Hsc70 and Hsp70 in glial cytoplasmic inclusions in patients with multiple system atrophy. Brain Res Brain Res Protoc. 2007;1136(1):219–27. doi: 10.1016/j.brainres.2006.12.049. [DOI] [PubMed] [Google Scholar]
- 107.Lowe J, McDermott H, Pike I, Spendlove I, Landon M, Mayer RJ. alpha B crystallin expression in non-lenticular tissues and selective presence in ubiquitinated inclusion bodies in human disease. The Journal of pathology. 1992;166(1):61–8. doi: 10.1002/path.1711660110. [DOI] [PubMed] [Google Scholar]
- 108.Sofic E, Lange KW, Jellinger K, Riederer P. Reduced and oxidized glutathione in the substantia nigra of patients with Parkinson’s disease. Neurosci Lett. 1992;142(2):128–30. doi: 10.1016/0304-3940(92)90355-b. [DOI] [PubMed] [Google Scholar]
- 109.Sian J, Dexter DT, Lees AJ, Daniel S, Agid Y, Javoy-Agid F, Jenner P, Marsden CD. Alterations in glutathione levels in Parkinson’s disease and other neurodegenerative disorders affecting basal ganglia. Ann Neurol. 1994;36(3):348–55. doi: 10.1002/ana.410360305. [DOI] [PubMed] [Google Scholar]
- 110.Fitzmaurice PS, Ang L, Guttman M, Rajput AH, Furukawa Y, Kish SJ. Nigral glutathione deficiency is not specific for idiopathic Parkinson’s disease. Mov Disord. 2003;18(9):969–76. doi: 10.1002/mds.10486. [DOI] [PubMed] [Google Scholar]
- 111.Pearce RK, Owen A, Daniel S, Jenner P, Marsden CD. Alterations in the distribution of glutathione in the substantia nigra in Parkinson’s disease. J Neural Transm. 1997;104(6–7):661–77. doi: 10.1007/BF01291884. [DOI] [PubMed] [Google Scholar]
- 112.Mythri RB, Venkateshappa C, Harish G, Mahadevan A, Muthane UB, Yasha TC, Srinivas Bharath MM, Shankar SK. Evaluation of markers of oxidative stress, antioxidant function and astrocytic proliferation in the striatum and frontal cortex of Parkinson’s disease brains. Neurochemical research. 2011;36(8):1452–63. doi: 10.1007/s11064-011-0471-9. [DOI] [PubMed] [Google Scholar]
- 113.Saharan S, Mandal PK. The emerging role of glutathione in Alzheimer’s disease. J Alzheimers Dis. 2014;40(3):519–29. doi: 10.3233/JAD-132483. [DOI] [PubMed] [Google Scholar]
- 114.Johnson WM, Wilson-Delfosse AL, Mieyal JJ. Dysregulation of glutathione homeostasis in neurodegenerative diseases. Nutrients. 2012;4(10):1399–440. doi: 10.3390/nu4101399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Adams JD, Jr, Klaidman LK, Odunze IN, Shen HC, Miller CA. Alzheimer’s and Parkinson’s disease. Brain levels of glutathione, glutathione disulfide, and vitamin E. Mol Chem Neuropathol. 1991;14(3):213–26. doi: 10.1007/BF03159937. [DOI] [PubMed] [Google Scholar]
- 116.Makar TK, Cooper AJ, Tofel-Grehl B, Thaler HT, Blass JP. Carnitine, carnitine acetyltransferase, and glutathione in Alzheimer brain. Neurochem Res. 1995;20(6):705–11. doi: 10.1007/BF01705539. [DOI] [PubMed] [Google Scholar]
- 117.Sian J, Dexter DT, Lees AJ, Daniel S, Jenner P, Marsden CD. Glutathione-related enzymes in brain in Parkinson’s disease. Ann Neurol. 1994;36(3):356–61. doi: 10.1002/ana.410360306. [DOI] [PubMed] [Google Scholar]
- 118.Liu H, Harrell LE, Shenvi S, Hagen T, Liu RM. Gender differences in glutathione metabolism in Alzheimer’s disease. J Neurosci Res. 2005;79(6):861–7. doi: 10.1002/jnr.20424. [DOI] [PubMed] [Google Scholar]
- 119.Deng Y, Newman B, Dunne MP, Silburn PA, Mellick GD. Case-only study of interactions between genetic polymorphisms of GSTM1, P1, T1 and Z1 and smoking in Parkinson’s disease. Neurosci Lett. 2004;366(3):326–31. doi: 10.1016/j.neulet.2004.05.061. [DOI] [PubMed] [Google Scholar]
- 120.Smeyne M, Boyd J, Raviie Shepherd K, Jiao Y, Pond BB, Hatler M, Wolf R, Henderson C, Smeyne RJ. GSTpi expression mediates dopaminergic neuron sensitivity in experimental parkinsonism. Proc Natl Acad Sci U S A. 2007;104(6):1977–82. doi: 10.1073/pnas.0610978104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ahmadi A, Fredrikson M, Jerregard H, Akerback A, Fall PA, Rannug A, Axelson O, Soderkvist P. GSTM1 and mEPHX polymorphisms in Parkinson’s disease and age of onset. Biochem Biophys Res Commun. 2000;269(3):676–80. doi: 10.1006/bbrc.2000.2338. [DOI] [PubMed] [Google Scholar]
- 122.Piacentini S, Polimanti R, Squitti R, Ventriglia M, Cassetta E, Vernieri F, Rossini PM, Manfellotto D, Fuciarelli M. GSTM1 null genotype as risk factor for late-onset Alzheimer’s disease in Italian patients. J Neurol Sci. 2012;317(1–2):137–40. doi: 10.1016/j.jns.2012.01.026. [DOI] [PubMed] [Google Scholar]
- 123.Pocernich CB, Butterfield DA. Elevation of glutathione as a therapeutic strategy in Alzheimer disease. Biochimica et biophysica acta. 2012;1822(5):625–30. doi: 10.1016/j.bbadis.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Braidy N, Zarka M, Welch J, Bridge W. Therapeutic Approaches to Modulating Glutathione Levels as a Pharmacological strategy in Alzheimer’s Disease. Curr Alzheimer Res. 2015 doi: 10.2174/1567205012666150302160308. [DOI] [PubMed] [Google Scholar]
- 125.Bavarsad Shahripour R, Harrigan MR, Alexandrov AV. N-acetylcysteine (NAC) in neurological disorders: mechanisms of action and therapeutic opportunities. Brain Behav. 2014;4(2):108–22. doi: 10.1002/brb3.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Craiu A, Gaczynska M, Akopian T, Gramm CF, Fenteany G, Goldberg AL, Rock KL. Lactacystin and clasto-lactacystin beta-lactone modify multiple proteasome beta-subunits and inhibit intracellular protein degradation and major histocompatibility complex class I antigen presentation. J Biol Chem. 1997;272(20):13437–45. doi: 10.1074/jbc.272.20.13437. [DOI] [PubMed] [Google Scholar]
- 127.Alexandrova A, Petrov L, Georgieva A, Kirkova M, Kukan M. Effects of proteasome inhibitor, MG132, on proteasome activity and oxidative status of rat liver. Cell biochemistry and function. 2008;26(3):392–8. doi: 10.1002/cbf.1459. [DOI] [PubMed] [Google Scholar]
- 128.Braun HA, Umbreen S, Groll M, Kuckelkorn U, Mlynarczuk I, Wigand ME, Drung I, Kloetzel PM, Schmidt B. Tripeptide mimetics inhibit the 20 S proteasome by covalent bonding to the active threonines. J Biol Chem. 2005;280(31):28394–401. doi: 10.1074/jbc.M502453200. [DOI] [PubMed] [Google Scholar]
- 129.MacGurn JA, Hsu PC, Emr SD. Ubiquitin and membrane protein turnover: from cradle to grave. Annu Rev Biochem. 2012;81:231–59. doi: 10.1146/annurev-biochem-060210-093619. [DOI] [PubMed] [Google Scholar]
- 130.Schreiner B, Romanelli E, Liberski P, Ingold-Heppner B, Sobottka-Brillout B, Hartwig T, Chandrasekar V, Johannssen H, Zeilhofer HU, Aguzzi A, Heppner F, Kerschensteiner M, Becher B. Astrocyte Depletion Impairs Redox Homeostasis and Triggers Neuronal Loss in the Adult CNS. Cell Rep. 2015 doi: 10.1016/j.celrep.2015.07.051. [DOI] [PubMed] [Google Scholar]
- 131.Rosenberg PA, Aizenman E. Hundred-fold increase in neuronal vulnerability to glutamate toxicity in astrocyte-poor cultures of rat cerebral cortex. Neurosci Lett. 1989;103(2):162–8. doi: 10.1016/0304-3940(89)90569-7. [DOI] [PubMed] [Google Scholar]
- 132.Dringen R, Gutterer JM, Hirrlinger J. Glutathione metabolism in brain metabolic interaction between astrocytes and neurons in the defense against reactive oxygen species. Eur J Biochem. 2000;267(16):4912–6. doi: 10.1046/j.1432-1327.2000.01597.x. [DOI] [PubMed] [Google Scholar]
- 133.Wilson JX. Antioxidant defense of the brain: a role for astrocytes. Can J Physiol Pharmacol. 1997;75(10–11):1149–63. [PubMed] [Google Scholar]
- 134.Rathinam ML, Watts LT, Narasimhan M, Riar AK, Mahimainathan L, Henderson GI. Astrocyte mediated protection of fetal cerebral cortical neurons from rotenone and paraquat. Environ Toxicol Pharmacol. 2012;33(2):353–60. doi: 10.1016/j.etap.2011.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Mullett SJ, Di Maio R, Greenamyre JT, Hinkle DA. DJ-1 expression modulates astrocyte-mediated protection against neuronal oxidative stress. J Mol Neurosci. 2013;49(3):507–11. doi: 10.1007/s12031-012-9904-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Mullett SJ, Hinkle DA. DJ-1 knock-down in astrocytes impairs astrocyte-mediated neuroprotection against rotenone. Neurobiology of disease. 2009;33(1):28–36. doi: 10.1016/j.nbd.2008.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Mullett SJ, Hinkle DA. DJ-1 deficiency in astrocytes selectively enhances mitochondrial Complex I inhibitor-induced neurotoxicity. Journal of neurochemistry. 2011;117(3):375–87. doi: 10.1111/j.1471-4159.2011.07175.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Cipriani S, Desjardins CA, Burdett TC, Xu Y, Xu K, Schwarzschild MA. Protection of dopaminergic cells by urate requires its accumulation in astrocytes. J Neurochem. 2012;123(1):172–81. doi: 10.1111/j.1471-4159.2012.07820.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Haskew-Layton RE, Payappilly JB, Smirnova NA, Ma TC, Chan KK, Murphy TH, Guo H, Langley B, Sultana R, Butterfield DA, Santagata S, Alldred MJ, Gazaryan IG, Bell GW, Ginsberg SD, Ratan RR. Controlled enzymatic production of astrocytic hydrogen peroxide protects neurons from oxidative stress via an Nrf2-independent pathway. Proc Natl Acad Sci U S A. 2010;107(40):17385–90. doi: 10.1073/pnas.1003996107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Drukarch B, Schepens E, Jongenelen CA, Stoof JC, Langeveld CH. Astrocyte-mediated enhancement of neuronal survival is abolished by glutathione deficiency. Brain Res. 1997;770(1–2):123–30. doi: 10.1016/s0006-8993(97)00790-7. [DOI] [PubMed] [Google Scholar]
- 141.Xu L, Lee JE, Giffard RG. Overexpression of bcl-2, bcl-XL or hsp70 in murine cortical astrocytes reduces injury of co-cultured neurons. Neurosci Lett. 1999;277(3):193–7. doi: 10.1016/s0304-3940(99)00882-4. [DOI] [PubMed] [Google Scholar]
- 142.Narasimhan M, Rathinam M, Patel D, Henderson G, Mahimainathan L. Astrocytes Prevent Ethanol Induced Apoptosis of Nrf2 Depleted Neurons by Maintaining GSH Homeostasis. Open J Apoptosis. 2012;1(2) doi: 10.4236/ojapo.2012.12002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Miao Y, Qiu Y, Lin Y, Miao Z, Zhang J, Lu X. Protection by pyruvate against glutamate neurotoxicity is mediated by astrocytes through a glutathione-dependent mechanism. Mol Biol Rep. 2011;38(5):3235–42. doi: 10.1007/s11033-010-9998-0. [DOI] [PubMed] [Google Scholar]
- 144.Sandhu JK, Gardaneh M, Iwasiow R, Lanthier P, Gangaraju S, Ribecco-Lutkiewicz M, Tremblay R, Kiuchi K, Sikorska M. Astrocyte-secreted GDNF and glutathione antioxidant system protect neurons against 6OHDA cytotoxicity. Neurobiol Dis. 2009;33(3):405–14. doi: 10.1016/j.nbd.2008.11.016. [DOI] [PubMed] [Google Scholar]
- 145.Vargas MR, Johnson DA, Sirkis DW, Messing A, Johnson JA. Nrf2 activation in astrocytes protects against neurodegeneration in mouse models of familial amyotrophic lateral sclerosis. J Neurosci. 2008;28(50):13574–81. doi: 10.1523/JNEUROSCI.4099-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Vargas MR, Pehar M, Cassina P, Beckman JS, Barbeito L. Increased glutathione biosynthesis by Nrf2 activation in astrocytes prevents p75NTR-dependent motor neuron apoptosis. J Neurochem. 2006;97(3):687–96. doi: 10.1111/j.1471-4159.2006.03742.x. [DOI] [PubMed] [Google Scholar]
- 147.Shih AY, Johnson DA, Wong G, Kraft AD, Jiang L, Erb H, Johnson JA, Murphy TH. Coordinate regulation of glutathione biosynthesis and release by Nrf2-expressing glia potently protects neurons from oxidative stress. J Neurosci. 2003;23(8):3394–406. doi: 10.1523/JNEUROSCI.23-08-03394.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ye B, Shen H, Zhang J, Zhu YG, Ransom BR, Chen XC, Ye ZC. Dual pathways mediate beta-amyloid stimulated glutathione release from astrocytes. Glia. 2015 doi: 10.1002/glia.22886. [DOI] [PubMed] [Google Scholar]
- 149.Brucklacher RM, Vannucci RC, Vannucci SJ. Hypoxic preconditioning increases brain glycogen and delays energy depletion from hypoxia-ischemia in the immature rat. Dev Neurosci. 2002;24(5):411–7. doi: 10.1159/000069051. [DOI] [PubMed] [Google Scholar]
- 150.Jones NM, Bergeron M. Hypoxia-induced ischemic tolerance in neonatal rat brain involves enhanced ERK1/2 signaling. J Neurochem. 2004;89(1):157–67. doi: 10.1111/j.1471-4159.2004.02324.x. [DOI] [PubMed] [Google Scholar]
- 151.Sen E, Basu A, Willing LB, Uliasz TF, Myrkalo JL, Vannucci SJ, Hewett SJ, Levison SW. Preconditioning induces the precocious differentiation of neonatal astrocytes to enhance their neuroprotective properties. ASN neuro. 2011;3(3):e00062. doi: 10.1042/AN20100029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Gouix E, Buisson A, Nieoullon A, Kerkerian-Le Goff L, Tauskela JS, Blondeau N, Had-Aissouni L. Oxygen glucose deprivation-induced astrocyte dysfunction provokes neuronal death through oxidative stress. Pharmacol Res. 2014;87:8–17. doi: 10.1016/j.phrs.2014.06.002. [DOI] [PubMed] [Google Scholar]
- 153.Jana A, Pahan K. Fibrillar amyloid-beta-activated human astroglia kill primary human neurons via neutral sphingomyelinase: implications for Alzheimer’s disease. J Neurosci. 2010;30(38):12676–89. doi: 10.1523/JNEUROSCI.1243-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Fogal B, Li J, Lobner D, McCullough LD, Hewett SJ. System x(c)- activity and astrocytes are necessary for interleukin-1 beta-mediated hypoxic neuronal injury. J Neurosci. 2007;27(38):10094–105. doi: 10.1523/JNEUROSCI.2459-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Jackman NA, Melchior SE, Hewett JA, Hewett SJ. Non-cell autonomous influence of the astrocyte system xc- on hypoglycaemic neuronal cell death. ASN Neuro. 2012;4(1) doi: 10.1042/AN20110030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Pertusa M, Garcia-Matas S, Rodriguez-Farre E, Sanfeliu C, Cristofol R. Astrocytes aged in vitro show a decreased neuroprotective capacity. J Neurochem. 2007;101(3):794–805. doi: 10.1111/j.1471-4159.2006.04369.x. [DOI] [PubMed] [Google Scholar]
- 157.Lee DH, Goldberg AL. Proteasome inhibitors: valuable new tools for cell biologists. Trends Cell Biol. 1998;8(10):397–403. doi: 10.1016/s0962-8924(98)01346-4. [DOI] [PubMed] [Google Scholar]
- 158.Mandrekar-Colucci S, Landreth GE. Microglia and inflammation in Alzheimer’s disease. CNS Neurol Disord Drug Targets. 2010;9(2):156–67. doi: 10.2174/187152710791012071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Ojo JO, Rezaie P, Gabbott PL, Stewart MG. Impact of age-related neuroglial cell responses on hippocampal deterioration. Front Aging Neurosci. 2015;7:57. doi: 10.3389/fnagi.2015.00057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Theriault P, ElAli A, Rivest S. The dynamics of monocytes and microglia in Alzheimer’s disease. Alzheimers Res Ther. 2015;7(1):41. doi: 10.1186/s13195-015-0125-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Zhu X, Castellani RJ, Takeda A, Nunomura A, Atwood CS, Perry G, Smith MA. Differential activation of neuronal ERK, JNK/SAPK and p38 in Alzheimer disease: the ‘two hit’ hypothesis. Mech Ageing Dev. 2001;123(1):39–46. doi: 10.1016/s0047-6374(01)00342-6. [DOI] [PubMed] [Google Scholar]
- 162.Chan J, Jones NC, Bush AI, O’Brien TJ, Kwan P. A mouse model of Alzheimer’s disease displays increased susceptibility to kindling and seizure-associated death. Epilepsia. 2015;56(6):e73–7. doi: 10.1111/epi.12993. [DOI] [PubMed] [Google Scholar]
- 163.Zhu X, Raina AK, Perry G, Smith MA. Alzheimer’s disease: the two-hit hypothesis. Lancet neurology. 2004;3(4):219–26. doi: 10.1016/S1474-4422(04)00707-0. [DOI] [PubMed] [Google Scholar]
- 164.Hawkes CH, Del Tredici K, Braak H. Parkinson’s disease: a dual-hit hypothesis. Neuropathol Appl Neurobiol. 2007;33(6):599–614. doi: 10.1111/j.1365-2990.2007.00874.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Verkhratsky A, Parpura V, Pekna M, Pekny M, Sofroniew M. Glia in the pathogenesis of neurodegenerative diseases. Biochem Soc Trans. 2014;42(5):1291–301. doi: 10.1042/BST20140107. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Lactacystin-mediated severe proteotoxic stress preconditions astrocytes against subsequent insults. A) Five days after plating, astrocytes were treated with various concentrations of lactacystin or an equivalent v/v of vehicle (phosphate buffered saline) as the 1st hit. On the following day, the astrocytes were treated with vehicle or a 2nd hit of lactacystin at the indicated concentrations. Viability was assayed 24h after the 2nd hit by counting Hoechst-stained nuclei. B) Representative images of Hoechst-stained nuclei for experiments shown in panel A. * p ≤ 0.05 vs 0 μM 2nd lactacystin hit; + p ≤ 0.05, ++ p ≤ 0.01, +++ p ≤ 0.001 vs 0 μM 1st lactacystin hit, two-way ANOVA followed by Bonferroni post hoc correction
MG132 treatment is more lethal to small, bright nuclei than large nuclei. A) Hoechst-stained nuclei were categorized into two groups: those exhibiting small, brightly stained nuclei (50–150 μm2 in size and staining intensity greater than 20 arbitrary units for the mean gray value measurements) or large nuclei (greater than 150 μm2; all staining intensities). Raw counts of the numbers of nuclei per microscopic field of view (200× magnification) are shown here to supplement the transformed data in Figure 3P. * p ≤ 0.05, ** p ≤ 0.01 vs 0 μM 2nd MG132 hit; ++ p ≤ 0.01 vs vs 0 μM 1st MG132 hit; ^^ p ≤ 0.01, ^^^ p ≤ 0.001 vs small, bright nuclei, three-way ANOVA followed by Bonferroni post hoc correction
Stressed astrocytes can resist dual hits despite inhibition of heme oxygenase 1 (HO1, also known as Hsp32). A) Astrocytes were treated with the 1st and 2nd MG132 hits or vehicle 24h apart in the absence or presence of the HO1 inhibitor SnPPx. Viability was assayed 24h after the 2nd hit. B) Representative images of Hoechst stained nuclei. * p ≤ 0.05, *** p ≤ 0.001 vs 0 μM 2nd MG132 hit; + p ≤ 0.05, +++ p ≤ 0.001 vs 0 μM 1st MG132 hit, two-way ANOVA followed by Bonferroni post hoc correction
Impact of 1st hit on glutathione-related enzymes. Astrocytes were treated with the 1st MG132 hit or vehicle and the following glutathione-related proteins were measured in lysates collected 24h later: A) glutamate cysteine ligase catalytic subunit (GCLC), B) glutathione S-transferase π (GST-π). No significant difference was determined with the two-tailed paired Student’s t test.