

Abstract
Estrogens can initiate cancer by reacting with DNA. Specific metabolites of endogenous estrogens, the catechol estrogen-3,4-quinones, react with DNA to form depurinating estrogen-DNA adducts. Loss of these adducts leaves apurinic sites in the DNA, generating mutations that can lead to the initiation of cancer. A variety of endogenous and exogenous factors can disrupt estrogen homeostasis, which is the normal balance between estrogen activating and protective enzymes. In fact, if estrogen metabolism becomes unbalanced and generates excessive catechol estrogen 3,4-quinones, formation of depurinating estrogen-DNA adducts increases and the risk of initiating cancer is greater. The levels of depurinating estrogen-DNA adducts are high in women diagnosed with breast cancer and those at high risk for the disease. High levels of depurinating estrogen-DNA adducts before the presence of breast cancer indicates that adduct formation is a critical factor in breast cancer initiation. Women with thyroid or ovarian cancer also have high levels of estrogen-DNA adducts, as do men with prostate cancer or non-Hodgkin lymphoma. Depurinating estrogen-DNA adducts are initiators of many prevalent types of human cancer. These findings and other discoveries led to the recognition that reducing the levels of estrogen-DNA adducts could prevent the initiation of human cancer. The dietary supplements N-acetylcysteine and resveratrol inhibit formation of estrogen-DNA adducts in cultured human breast cells and in women. These results suggest that the two supplements offer an approach to reducing the risk of developing various prevalent types of human cancer.
Graphical abstract.
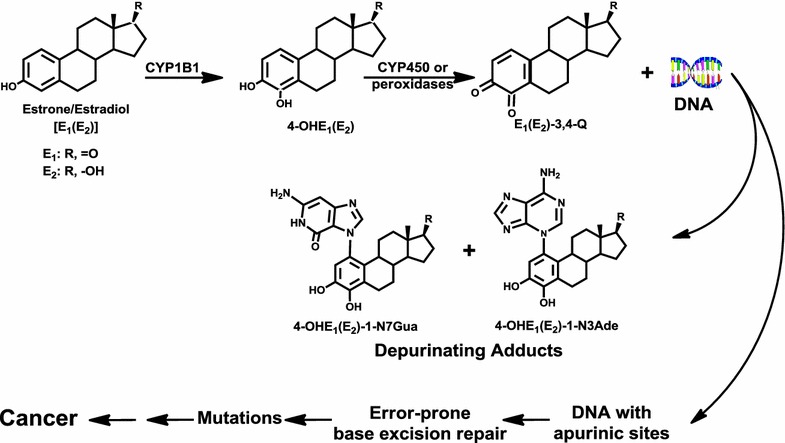
Major metabolic pathway in cancer initiation by estrogens.
Keywords: Estrogen metabolism; Catechol estrogen-3,4-quinones; Depurinating estrogen-DNA adducts; Estrogen genotoxicity; Estrogen carcinogenesis; Cancer prevention; N-acetylcysteine; Resveratrol
Mechanism of metabolic activation of estrogens to initiate cancer
One of the major obstacles in cancer research is related to the concept that cancer is a problem of many diseases. This viewpoint has kept researchers from investigating the etiology of cancer because a search for numerous causes would be prohibitively complex. While the expression of various cancers coincides with the concept of numerous diseases, we think many types of prevalent cancers have a common etiology. The understanding of this common mechanism of cancer initiation can lead to cancer prevention.
Exposure to estrogens is a known risk factor for developing cancer. The scientific community predominantly considers estrogens to be epigenetic carcinogens because these compounds do not induce mutations in standard bacterial and mammalian test systems. This presumably occurs because the reactive catechol estrogen quinone metabolites are not formed or cannot reach the target DNA [1–5]. These results have led scientists to classify estrone (E1) and estradiol (E2) as epigenetic carcinogens that function by stimulating abnormal cell proliferation via estrogen receptor (ER)-mediated processes [5–10]. These latter events can accelerate the process of carcinogenesis, but do not play the critical role in cancer initiation because the hypothetical mutations obtained are random.
Unbalanced estrogen metabolism is a critical factor in cancer initiation. The discovery that specific oxidative metabolites of estrogens, the catechol estrogen quinones, react with DNA supports the hypothesis that estrogens can become endogenous carcinogens by generating the mutations leading to the initiation of cancer [11–14]. This paradigm indicates that specific, critical mutations produce abnormal cell proliferation leading to cancer, rather than ER-mediated abnormal cell proliferation that generates random mutations [1, 6–10]. The specificity of the critical mutations is the result of the preliminary intercalating physical complex between estrogen and DNA that occurs before formation of the covalent bond between them. This intercalating mechanism has been demonstrated for the human carcinogen diethylstilbestrol (DES) [15].
Benzene and naphthalene
Natural and synthetic estrogens contain a benzene ring in their structure. For compounds containing one or two benzene rings, there is a common mechanism of metabolic activation, which produces extremely weak ultimate carcinogens. This mechanism of activation (Fig. 1) has been demonstrated to occur with benzene [16, 17], naphthalene [18, 19], the natural estrogens E1 and E2 [20–26], and the synthetic estrogens DES [15, 27] and hexestrol (HES) [23, 28, 29].
Fig. 1.
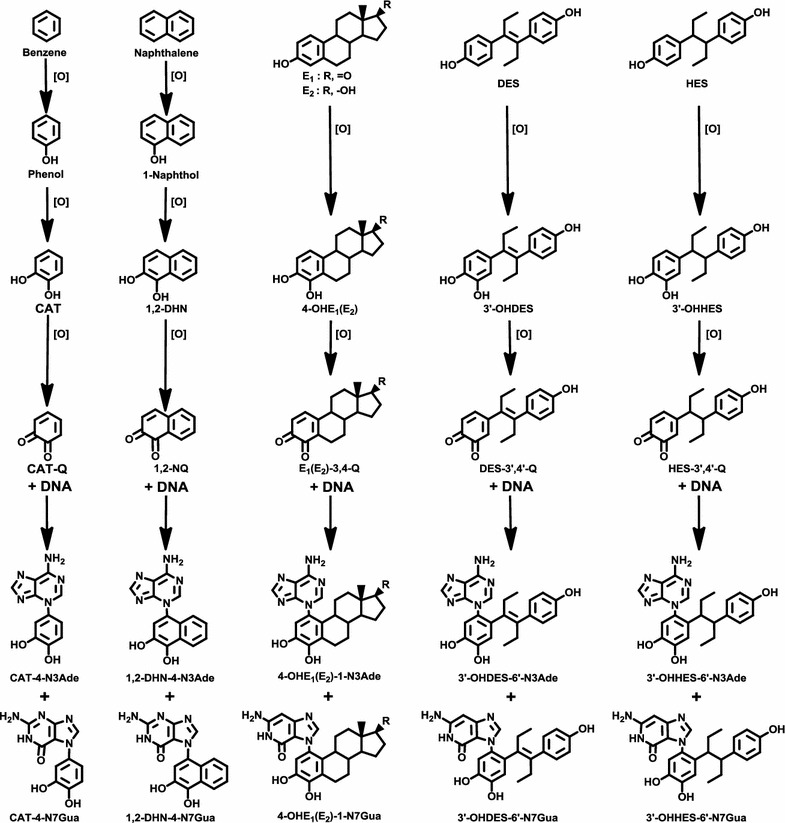
Common mechanism of metabolic activation and reaction with DNA to form the N3Ade and N7Gua depurinating adducts for benzene, naphthalene, estrone (E1), estradiol (E2), diethylstilbestrol (DES), and hexestrol (HES). The figure shows the progression from parent compound to hydroxy derivative, catechol, and then quinone, which reacts with DNA to form the depurinating adducts at the N3Ade and N7Gua
It has long been known that benzene causes acute myelogenous leukemia in humans [30, 31]. The benzene metabolites include catechol, (CAT, 1,2-dihydroxybenzene) and hydroquinone (1,4-dihydroxybenzene) [32, 33]. CAT and hydroquinone can accumulate in the bone marrow [34, 35], where they can be oxidized by peroxidases [36] to the corresponding quinones. The CAT-1,2 quinone reacts with DNA by 1,4 Michael addition to yield the depurinating adducts CAT-4-N7Gua and CAT-4-N3Ade (Fig. 1) [16, 17]. These results suggest that the ultimate carcinogenic metabolite of benzene is the benzene-1,2-quinone.
Inhalation of naphthalene by male and female rats for two years produced olfactory epithelial neuroblastomas in 5–10 % of the animals [37]. The logical mechanism of metabolic activation of naphthalene is analogous to the one described above for benzene. In fact, naphthalene-1,2-quinone reacts with DNA to produce the depurinating N3Ade and N7Gua adducts in vitro and in vivo (Fig. 1) [18, 19].
Natural and synthetic estrogens
One of the major metabolic pathways of the natural estrogens E1 and E2 is the formation of catechol estrogens. These metabolites are oxidized to semiquinones and then to quinones. Their reaction with DNA forms predominantly the depurinating adducts N3Ade and N7Gua that can initiate cancer (Fig. 1). Synthetic estrogens, such as the human carcinogen DES [38] and its hydrogenated derivative HES, display properties similar to the natural estrogens: (1) they are carcinogenic in the kidney of Syrian golden hamsters [39, 40]; (2) the major metabolites are their catechols [40–43]; (3) the catechol quinones of DES and HES have chemical and biochemical properties similar to those of the natural E1(E2)-3,4-quinones [E1(E2)-3,4-Q], namely they form N3Ade and N7Gua adducts after reaction with DNA (Fig. 1). Depurination of the N3Ade adduct is instantaneous, whereas depurination of the N7Gua adduct occurs rather slowly [15, 23, 27–29]. These data suggest that the catechol quinones of DES and HES are their cancer initiators.
Catechol estrogen metabolic pathway
Strong evidence from studies of estrogen metabolism, formation of DNA adducts, mutagenicity, cell transformation and carcinogenicity led to and supports the hypothesis that specific estrogen metabolites, the catechol estrogen quinones, can react with DNA to form estrogen-DNA adducts in critical genes that lead to the initiation of cancer [11, 12].
Metabolic formation of estrogens derives from aromatization of testosterone and 4-androstene-3,17-dione, catalyzed by CYP19 (aromatase), to yield E2 and E1, respectively (Fig. 2). E2 and E1 are interconverted by 17β-hydroxysteroid dehydrogenase. If an excess of estrogen is obtained, it is stored as estrone sulfate. Estrogens are metabolized via two major pathways: formation of 16α-OHE1(E2) (not shown in Fig. 2) and formation of the catechol estrogens 2-OHE1(E2) and 4-OHE1(E2) (Fig. 2) [44]. Cytochrome P450 (CYP)1A and CYP3A catalyze the hydroxylation preferentially at the 2 position, whereas CYP1B1 catalyzes the hydroxylation almost exclusively at the 4 position [45–47]. The two catechol estrogens are inactivated by conjugation to glucuronides and sulfates especially in the liver (not shown in Fig. 2). In extrahepatic tissues, the most common path of conjugation of the catechol estrogens is O-methylation, catalyzed by catechol-O-methyltransferase (COMT) [48, 49]. A low activity of COMT renders more competitive oxidation of the catechol estrogens to E1(E2)-2,3-Q and E1(E2)-3,4-Q catalyzed by CYP or peroxidases (Fig. 2).
Fig. 2.
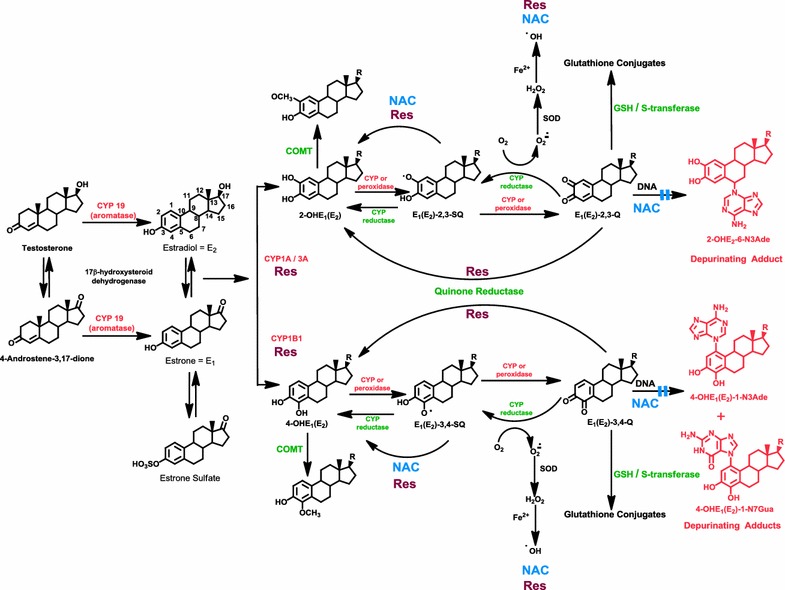
Formation of estrogens, catechol estrogen metabolic pathway and DNA adducts of estrogens. Activating enzymes and depurinating DNA adducts are in red and protective enzymes are in green. N-acetylcysteine (NAC, shown in blue) and resveratrol (Res, shown in burgundy) indicate the various steps where NAC and Res can improve unbalanced estrogen metabolism by reducing formation of depurinating estrogen-DNA adducts
Oxidation of semiquinones to quinones can also be obtained by molecular oxygen (Fig. 2). Reduction of estrogen quinones to semiquinones by CYP reductase completes the redox cycle. In this process, the molecular oxygen is reduced to superoxide anion radical, and then converted by superoxide dismutase to hydrogen peroxide. In the presence of Fe2+ the hydrogen peroxide is converted to hydroxyl radical. Reaction of the hydroxyl radical with lipids produces lipid hydroperoxides [50] (not shown in Fig. 2).
Following the formation of catechol estrogen quinones (Fig. 2), they can be inactivated by reacting with glutathione (GSH). A further inactivation pathway for the quinones is reduction to their respective catechols by quinone reductase [51, 52], a protective enzyme that can be induced by a variety of compounds [53].
If all the protective processes are insufficient, the catechol estrogen quinones can react with DNA to form predominantly the depurinating adducts 4-OHE1(E2)-1-N3Ade plus 4-OHE1(E2)-1-N7Gua (97 %) from E1(E2)-3,4-Q and 2-OHE1(E2)-6-N3Ade (3 %) from E1(E2)-2,3-Q (Fig. 2). The much larger amount of adducts formed by the E1(E2)-3,4-Q compared to those from the E1(E2)-2,3-Q results from the chemical properties of the quinones [26].
Depurinating estrogen-DNA adducts: generators of cancer initiation
Carcinogens react with DNA to form two types of adducts: stable adducts and depurinating adducts. In the Watson–Crick DNA model (Fig. 3), the backbone is constituted by deoxyribose and phosphate, the Gua is hydrogen-bonded to cytosine, and Ade is hydrogen-bonded to thymine. The Gua has one exocyclic NH2 group that can react with electrophiles to form a stable adduct (Fig. 3, hollow arrow). If reaction occurs at the N-7 and sometimes C-8 of Gua, depurinating adducts are formed (Fig. 3, solid arrows). In the case of Ade, reaction of an electrophile at the exocyclic NH2 group forms a stable adduct (Fig. 3, hollow arrow), whereas depurinating adducts are obtained after reaction at the N-3 and N-7 sites (Fig. 3, solid arrows). Following reaction at the N-3 of Ade, destabilization of the glycosyl bond occurs via formation of an intermediate oxocarbenium ion with subsequent depurination and generation of an apurinic site in the DNA [54].
Fig. 3.
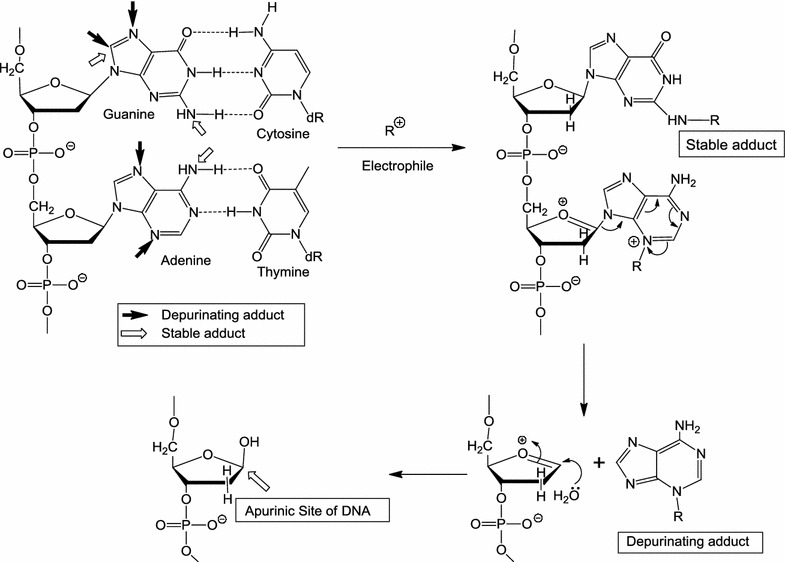
Formation of stable DNA adducts, and depurinating DNA adducts that generate apurinic sites
Cancer researchers have focused on stable adducts, which remain in DNA unless removed by repair. These adducts are routinely detected and quantified by the 32P-postlabeling technique, but their structure has not always been identified.
Stable adducts are formed when electrophilic carcinogenic compounds react with the exocyclic amino group of Ade or Gua [25]. If formation of adducts occurs at the N-3 or N-7 of Ade, or the N-7 of Gua, the most nucleophilic sites in Ade and Gua [55], destabilization of the glycosyl bond and subsequent depurination of the adduct from DNA takes place [20, 22, 25]. The critical relevance of these depurinating adducts is still not recognized by researchers 20 years after their discovery [56].
Evidence that depurinating DNA adducts play the predominant role in cancer initiation was first obtained from a correlation between the levels of depurinating polycyclic aromatic hydrocarbon-DNA adducts and oncogenic Harvey (H)-ras mutations in mouse skin papillomas [56, 57]. The very potent carcinogens 7,12-dimethylbenz[a]anthracene [58] and dibenzo[a,l]pyrene [59, 60] form predominantly depurinating Ade adducts and induce an A to T transversion in codon 61 of the H-ras oncogene. Instead, benzo[a]pyrene yields approximately twice as many Gua depurinating adducts as Ade depurinating adducts in mouse skin [61], and twice as many codon 13 G to T transversions as codon 61 A to T transversions [56, 61, 62].
A similar correlation between the sites of formation of depurinating DNA adducts and H-ras mutations was observed in mouse skin and rat mammary glands treated with E2-3,4-Q [63, 64].
E1(E2)-3,4-quinones and E1(E2)-2,3-quinones
The predominant cancer initiating pathway (97 %) derives from E1(E2)-3,4-Q and is shown in Fig. 4 [26]. E1 and E2 are metabolically converted to 4-OHE1(E2) by CYP1B1. Oxidation of the catechol estrogens leads to the corresponding E1(E2)-3,4-Q, which can react with DNA to form small amounts of stable adducts (1 %) remaining in the DNA and preponderant amounts of the depurinating adducts 4-OHE1(E2)-1-N3Ade and 4-OHE1(E2)-1-N7Gua (97 %), which detach from DNA leaving behind DNA with apurinic sites [26]. Possible errors in the repair of these sites can lead to the critical mutations initiating many common human cancers [63, 64].
Fig. 4.
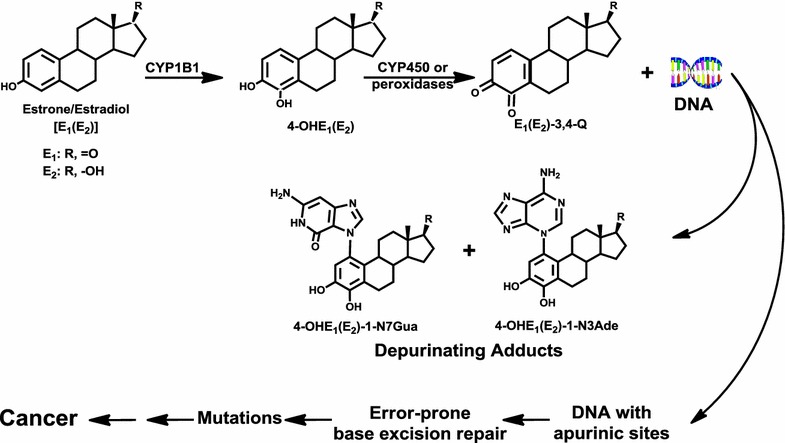
Major metabolic pathway in cancer initiation by estrogens
E1(E2)-2,3-Q form a much lower amount (2 %) of the depurinating adducts 2-OHE1(E2)-6-N3Ade by 1,6-Michael addition (Fig. 5) [26]. This product is obtained after tautomerization of the E1(E2)-2,3-Q to E1(E2)-2,3-Q methide [65]. The E1(E2)-2,3-Q form 10 to 50 times higher levels of stable DNA adducts than E1(E2)-3,4-Q [20, 24]. The level of stable adducts formed by E1(E2)-2,3-Q is still lower than the level of the depurinating adducts 2-OHE1(E2)-6-N3Ade [21, 26].
Fig. 5.
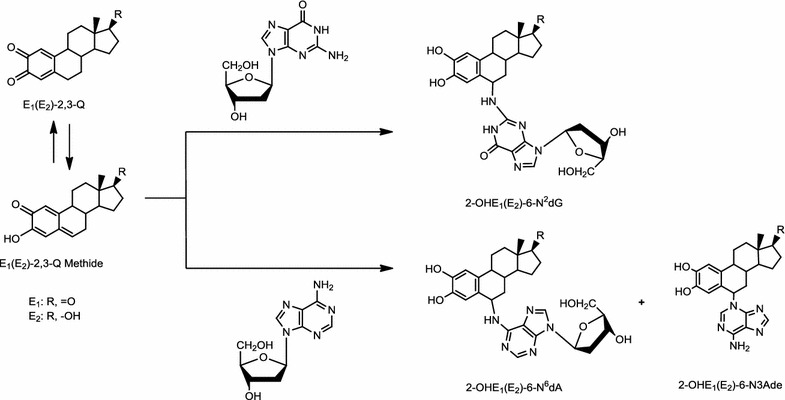
Reaction of E1(E2)-2,3-Q with dG or dA to form the stable 2-OHE1(E2)-6-N2dG or 2-OHE1(E2)-6-N6dA adducts (minor), respectively, and the depurinating 2-OHE1(E2)-6-N3Ade adducts (major)
The effectiveness of the E1(E2)-3,4-Q versus E1(E2)-2,3-Q to form depurinating adducts has been determined by reacting a mixture of E2-3,4-Q and E2-2,3-Q with DNA at different ratios. To achieve comparable levels of depurinating adducts, the mixture needs to contain 95 % E2-2,3-Q and 5 % E2-3,4-Q (Fig. 6a) [26].
Fig. 6.
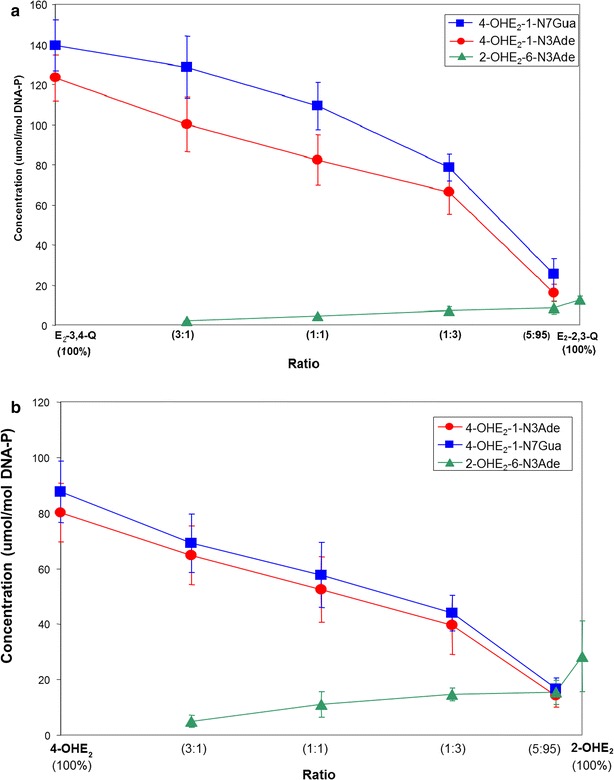
Depurinating adducts formed by mixtures of a E2-3,4-Q and E2-2,3-Q at different ratios after 10 h of reaction with DNA. The level of stable adducts formed in the mixtures ranged from 0.1 to 1 % of total adducts; and b 4-OHE2 and 2-OHE2 in the presence of tyrosinase at different ratios after 10 h of reaction with DNA. The level of stable adducts formed in the mixtures ranged from 0.1 to 0.7 % of total adducts [26]
Similar results are obtained from mixtures of 4-OHE2 and 2-OHE2 oxidized by tyrosinase in the presence of DNA (Fig. 6b). These results demonstrate the effectiveness of E2-3,4-Q to react with DNA in the formation of depurinating adducts compared to E2-2,3-Q.
The levels of depurinating DNA adducts formed by the catechol estrogen quinones [26] are in agreement with the greater carcinogenic activity of 4-OHE1(E2) compared with the borderline carcinogenic activity of 2-OHE1(E2) [66–68].
Imbalance of estrogen metabolism in cancer initiation
The metabolism of estrogens through the catechol estrogen pathway is characterized by homeostasis, a balanced set of activating and protective enzymes. Homeostasis minimizes the metabolic oxidation of catechol estrogens to catechol quinones and their reaction with DNA (Fig. 2). Disruption of homeostasis in the metabolism of estrogens, with excessive production of estrogen quinones and depurinating estrogen-DNA adducts, can lead to the initiation of cancer. A variety of endogenous and exogenous factors can disrupt estrogen homeostasis.
One factor that can imbalance estrogen metabolism is the excessive synthesis of estrogens by overexpression of CYP19 (aromatase) in target tissues (Fig. 2) [69–71]. A second factor that can imbalance estrogen homeostasis is the presence of unregulated sulfatase that converts excessive stored E1-sulfate into E1 (Fig. 2) [72, 73]. A third factor in imbalance is the production of high levels of 4-OHE1(E2), due to overexpression of CYP1B1, which converts E1(E2) predominantly to 4-OHE1(E2) (Fig. 2) [45–47, 74, 75]. Higher levels of 4-OHE1(E2) can give rise to higher levels of the strongest ultimate carcinogenic metabolites, E1(E2)-3,4-Q. An analogous effect can be produced by a lack or low level of COMT activity due to polymorphic variation [49, 76]. Insufficient activity of this enzyme would be translated into low levels of methylation of 4-OHE1(E2) and subsequent increase in the competitive oxidation of 4-OHE1(E2) to E1(E2)-3,4-Q (Fig. 2). Higher levels of E1(E2)-3,4-Q can also be obtained by polymorphism in quinone reductase (NQO1) which leads to decreased conversion of quinones into catechols (Fig. 2) [77]. Furthermore, low cellular levels of GSH, which reacts efficiently with the quinones, can leave higher levels of E1(E2)-3,4-Q available.
Imbalances in estrogen metabolism have also been observed in animal models for estrogen carcinogenicity: the prostate of Nobel rats [78], the kidney of male Syrian golden hamsters [79] and the mammary gland of ER-α knockout mice [80]. Imbalance of estrogen homeostasis can also be seen by comparing analyses of breast tissue from women with and without breast cancer [81]. In non-tumor breast tissue from women with breast carcinoma, the levels of 4-OHE1(E2) were nearly four-times higher than the levels in breast tissue from women without breast cancer. Further evidence of imbalance in estrogen homeostasis derives from the greater expression of estrogen-protective enzymes, COMT and NQO1 (Fig. 2), in women without breast cancer and greater expression of estrogen-activating enzymes, CYP19 and CYP1B1 (Fig. 2), in breast tissue of women with breast cancer [82].
Imbalance in estrogen metabolism can also be triggered by environmental factors. These factors include substances we ingest by mouth, skin and nose. It is logical to hypothesize that these environmental compounds are capable of changing estrogen metabolism, leading to increased formation of catechol estrogen quinones. Dioxin, for example, induces expression of the activating enzyme CYP1B1 [74, 75] (Fig. 2). This compound is not carcinogenic by itself, but makes the estrogens become carcinogenic by disrupting their metabolic homeostasis.
Depurinating estrogen-DNA adducts, the biomarkers of risk for women with and without breast cancer
Development of biomarkers for cancer risk has been a major goal in cancer research for decades. The ratio of the depurinating estrogen-DNA adducts 4-OHE1(E2)-1-N3Ade, 4-OHE1(E2)-1-N7Gua and 2-OHE1(E2)-6-N3Ade to their respective catechol estrogen metabolites and catechol estrogen conjugates provides a biomarker of risk that is related to the initiating step of breast and other prevalent types of human cancer.
Three case–control studies have been conducted in women diagnosed with breast cancer, as well as women at high risk or normal risk for the disease (Fig. 7) [83–85]. The high-risk women were identified by using the Gail model score to estimate a 5-year risk greater than 1.66 % [86]. Calculation of the Gail model score is based on age, age at menarche, age at first birth, prior breast biopsies or atypical hyperplasia, and number of first-degree relatives with breast cancer.
Fig. 7.
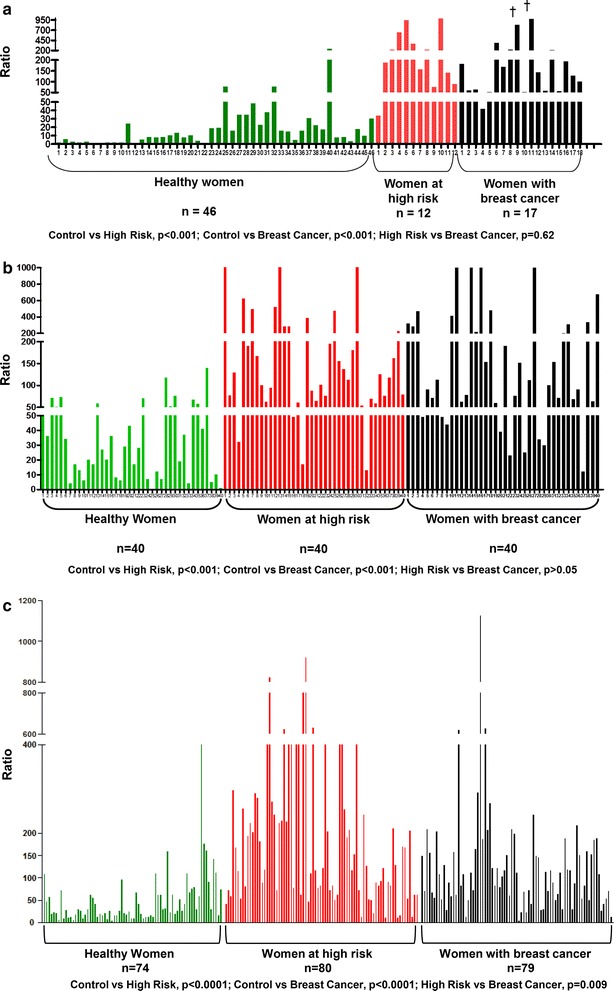
Ratios of depurinating estrogen-DNA adducts to catechol estrogen metabolites and catechol estrogen conjugates in a first study [83]: urine of healthy women, high risk women and women with breast cancer; b second study [84]: urine of healthy women, high risk women and women with breast cancer; c third study [85]: serum of healthy women, high risk women and women with breast cancer
In the first two studies [83, 84], a spot urine sample (~50 ml) was collected from each subject. An aliquot of the sample was partially purified by solid-phase extraction and analyzed for 38 catechol estrogen metabolites, catechol estrogen conjugates and depurinating estrogen-DNA adducts. The estrogen analytes were identified and quantified by using ultraperformance liquid chromatography/tandem mass spectrometry, and the ratio (see equation above) was calculated for each subject (Fig. 7a, b). In the first study of 46 normal-risk women, 12 high-risk women and 17 women diagnosed with breast cancer, the ratios in the high risk (p < 0.001) and breast cancer (p < 0.001) were significantly higher than the ratios in the normal-risk women (Fig. 7a) [83]. Similar differences were observed in the second study between 40 normal-risk women, 40 high-risk women and 40 women with breast cancer (both p < 0.001) (Fig. 7b) [84].
In the third study, serum was collected from each of the 74 normal-risk women, 80 high-risk women and 79 women diagnosed with breast cancer (Fig. 7c) [85]. Once again, the ratio of adducts to metabolites and conjugates was significantly lower in the women at normal risk, compared to the high-risk and breast cancer women (both p < 0.001).
In all three studies, the high ratios arose from high levels of adducts and low levels of metabolites and conjugates, although in some samples the levels of adducts were average, but the levels of metabolites and conjugates were very low [83–85], yielding a similar ratio in both cases. Overall, the high ratio of depurinating estrogen-DNA adducts to the catechol estrogen metabolites and catechol estrogen conjugates is a biomarker of risk for breast cancer.
Since estrogens initiate breast cancer by a genotoxic mechanism, the observation of higher levels of estrogen-DNA adducts in women at high risk for breast cancer suggests that formation of these adducts is a causative factor in the etiology of breast cancer and not a consequence of the cancer itself.
Similar case–control studies were conducted with women diagnosed with ovarian cancer and healthy women [87], and women with thyroid cancer and healthy women [88]. In both cases, the women diagnosed with the disease had much higher ratios of depurinating estrogen-DNA adducts to catechol estrogen metabolites and conjugates. Similar results were obtained in case–control studies of men with prostate cancer [89] or with non-Hodgkin lymphoma [90].
We think that other prevalent types of cancer, which have not yet been investigated for depurinating estrogen-DNA adduct formation, are also initiated by estrogens. These cancers include brain, colon, endometrium, kidney, leukemia, lung of non-smokers, melanoma, myeloma, pancreas and testis.
Prevention of cancer initiation by N-acetylcysteine and resveratrol acting as antioxidants, enzyme modulators and inhibitors of depurinating estrogen-DNA adduct formation
The metabolism of estrogens in the catechol estrogen pathway is regulated by homeostasis, a balanced set of activating and protective enzymes. Homeostasis can be maintained or re-established by the use of specific compounds, N-acetylcysteine (NAC) and resveratrol (Res), which are particularly effective in blocking formation of estrogen-DNA adducts [91]. NAC is the acetyl derivative of the amino acid cysteine (Fig. 8), which is one component of the tripeptide GSH. Res, which is the 3,5,4′-hydroxy stilbene (Fig. 8), is a natural antioxidant present in grapes, wine, peanuts and other plants. NAC and Res can prevent oxidative and/or electrophilic damage to DNA by inhibiting formation of the electrophilic catechol estrogen quinones and/or reacting with them.
Fig. 8.
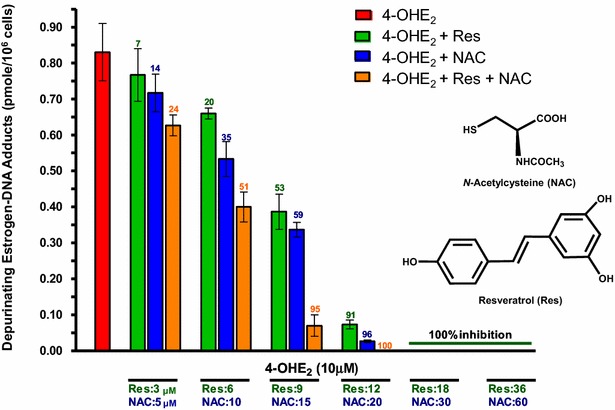
Ability of NAC, Res or their combination to block formation of depurinating estrogen-DNA adducts in MCF-10F human breast epithelial cells treated with 4-OHE2. The numbers on bars are the percentage of the inhibition of the depurinating estrogen-DNA adducts compared to treatment with 4-OHE2 alone [106]
The anticarcinogenic properties of NAC are attributed to multiple protective mechanisms, such as its nucleophilicity, antioxidant activity and inhibition of DNA adduct formation [92, 93]. Hydrolysis of NAC by acylase in the liver and gut yields cysteine, one of the precursors in the synthesis of intracellular GSH. The presence of cysteine guarantees replenishment of this crucial tripeptide. Changes in GSH homeostasis have been implicated in the etiology and progression of cancer and other human diseases [94]. GSH cannot be used as a preventive agent because it does not cross cell membranes. The use of cysteine as a preventive agent is limited by its toxicity. NAC, instead, has very low toxicity and it can cross the blood–brain barrier [92, 93]. NAC reacts efficiently with the electrophilic E1(E2)-3,4-Q [95, 96] to prevent their reaction with DNA to form adducts (Fig. 2). Furthermore, NAC reduces catechol estrogen semiquinones to catechol estrogens (Fig. 2) [97] and prevents malignant transformation of the human MCF-10F cells [98], as well as the mouse E6 mammary cells treated with 4-OHE2 [99].
Res exerts chemopreventive effects in various in vitro and in vivo systems [100, 101]. These properties are attributed to the easy hydrogen abstraction from the 4′-OH bond with formation of an oxy radical [102]. The easy abstraction is due to the great resonance stabilization energy of the oxy radical intermediate. Res is a modulator of CYP1B1 [74, 75, 103] and an inducer of quinone reductase (Fig. 2) [75, 104]. Res also reduces estrogen semiquinones to catechol estrogens (Fig. 2) [75]. When MCF-10F cells are cultured in the presence of 4-OHE2 and Res, formation of depurinating estrogen-DNA adducts is inhibited in a dose-dependent manner [75, 105]. To investigate whether the inhibitory effects of NAC and Res on the formation of estrogen-DNA adducts are additive or synergistic, MCF-10F cells were cultured in the presence of 4-OHE2 plus NAC or Res or NAC and Res together (Fig. 8) [106]. It was found that the effects of NAC and Res combined were additive in inhibiting formation of the depurinating estrogen-DNA adducts (p < 0.0001) [106]. NAC and Res had similar inhibitory effects at low concentrations, but the effects of Res were about 50 % greater than those of NAC at high concentrations.
A Healthy Breast Protocol that included NAC and Res was administered to women [107]. A group of 21 women (ages 30–70), who had never been diagnosed with cancer, participated in a study of the Healthy Breast Protocol [107]. They followed the treatment daily for 3 months and provided a spot urine sample before starting the treatment and after the 3 month period. The urine samples were analyzed for catechol estrogen metabolites and conjugates, and depurinating estrogen-DNA adducts by ultraperformance liquid chromatography/tandem mass spectrometry, and the ratio of adducts to metabolites and conjugates was calculated for each sample (Fig. 9). Of the 21 women participants, 16 experienced a decrease (green bars) in the ratio of adducts to metabolites and conjugates, four remained the same (blue bars) and one had an increase (red bars). The decrease in the ratio after treatment was statistically significant (p < 0.03) [107]. These results indicate that a treatment including NAC and Res can reduce depurinating estrogen-DNA adduct levels in people. This preventive approach does not require knowledge of the genes involved or the complex series of events that follow cancer initiation.
Fig. 9.
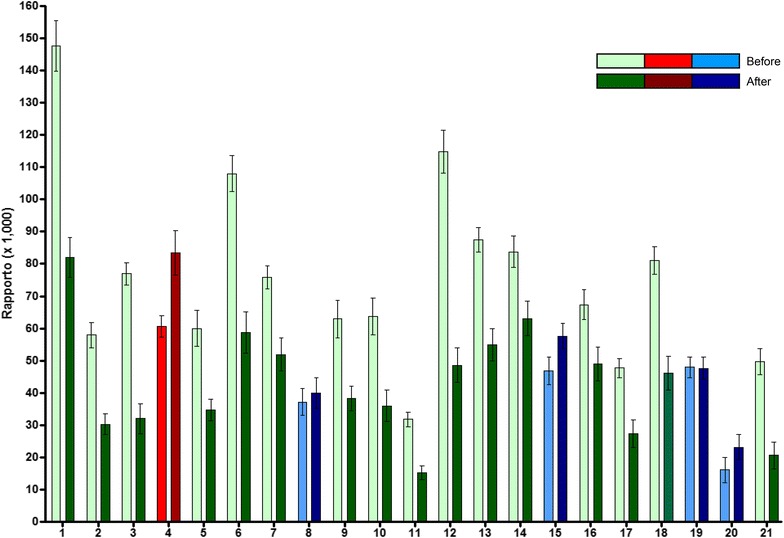
Assessment of depurinating estrogen-DNA adduct ratios before women began treatment with the Healthy Breast Protocol and after having been on the treatment for 3 months. Green bars represent women whose adduct ratios decreased, blue bars represent women whose adduct ratios remained the same and the red bars represent a woman whose adduct ratio increased over the course of the study [107]
In summary, NAC and Res are both able to reduce estrogen semiquinones to catechol estrogens [75, 97]. Furthermore, NAC keeps the cell replenished with GSH and reacts efficiently with the potential carcinogens, catechol estrogen quinones (Fig. 2). Res induces the enzyme quinone reductase and modulates the CYP1B1 activity (Fig. 2). Thus, NAC and Res, by inhibiting formation of depurinating estrogen-DNA adducts, maintain homeostasis in the metabolism of estrogens.
Conclusions
Metabolism of estrogens via the catechol estrogen pathway is characterized by homeostasis, a balanced set of activating and protective enzymes (Fig. 2). Under these conditions, formation of the catechol estrogen quinones, the ultimate carcinogenic metabolites of estrogens, is minimized. These compounds are not available to react with DNA; therefore, cancer cannot be initiated. When homeostasis is disrupted, however, excessive oxidation of catechol estrogens to quinones occurs. The quinones can react with DNA to form predominantly the depurinating adducts 4-OHE1(E2)-1-N3Ade and 4-OHE1(E2)-1-N7Gua. The apurinic sites derived from the loss of these adducts from DNA lead to the mutations that can initiate cancer.
Knowledge of the mechanism of cancer initiation by estrogens suggests that prevention of cancer can be achieved by blocking formation of the depurinating estrogen-DNA adducts. If the initiation of cancer is blocked, promotion, progression and development of the disease would be prevented. A variety of evidence suggests that cancer prevention could be achieved by use of the dietary supplements NAC and Res. Thus, use of these two dietary supplements could prove to be a widely applicable approach to cancer prevention.
Authors’ contributions
EC and ER jointly wrote the manuscript. They both reviewed articles and selected material to include in the review article. Both authors read and approved the final manuscript.
Acknowledgements
We thank Muhammad Zahid, Assistant Professor, for his continuing contributions to this research. Core support at the Eppley Institute was supported by grant P30 36727 from the National Cancer Institute.
Competing interests
The authors declare that they have no competing interests.
Abbreviations
- CAT
catechol
- COMT
catechol-O-methyltransferase
- CYP
cytochrome P450
- CYP19
aromatase
- DES
diethylstilbestrol
- E1
estrone
- E2
estradiol
- ER
estrogen receptor
- GSH
glutathione
- H
Harvey
- HES
hexestrol
- NAC
N-acetylcysteine
- NQO1
quinone reductase
- Res
resveratrol
Contributor Information
Ercole L. Cavalieri, Phone: 402-559-7237, Email: ecavalie@unmc.edu
Eleanor G. Rogan, Email: egrogan@unmc.edu
References
- 1.Nandi S. Role of hormones in mammary neoplasia. Cancer Res. 1978;38:4046–4049. [PubMed] [Google Scholar]
- 2.Lang R, Redmann U. Non-mutagenicity of some sex hormones in the Ames salmonella/microsome mutagenicity test. Mutat Res. 1979;67:361–365. doi: 10.1016/0165-1218(79)90033-8. [DOI] [PubMed] [Google Scholar]
- 3.Drevon C, Piccoli C, Montesano R. Mutagenicity assays of estrogenic hormones in mammalian cells. Mutat Res. 1981;89:83–90. doi: 10.1016/0165-1218(81)90134-8. [DOI] [PubMed] [Google Scholar]
- 4.Lang R, Reimann R. Studies for a genotoxic potential of some endogenous and exogenous sex steroids. I. Communication: examination for the induction of gene mutations using the Ames Salmonella/microsome test and the HGPRT test in V79 cells. Environ Mol Mutagen. 1993;21:272–304. doi: 10.1002/em.2850210311. [DOI] [PubMed] [Google Scholar]
- 5.Li JJ. Estrogen carcinogenesis in hamster tissues: update. Endocr Rev. 1993;14:94–95. doi: 10.1210/edrv-11-4-524. [DOI] [PubMed] [Google Scholar]
- 6.Furth J. Hormones as etiological agents in neoplasia. In: Becker FF, editor. Cancer a comprehensive treatise, 1 Etiology: chemical and physical carcinogenesis. New York: Cancer Plenum Press; 1982. pp. 89–134. [Google Scholar]
- 7.Li JJ, Li SA. Estrogen carcinogenesis in hamster tissues: a critical review. Endocr Rev. 1990;11:524–531. doi: 10.1210/edrv-11-4-524. [DOI] [PubMed] [Google Scholar]
- 8.Nandi S, Guzman RC, Yang J. Hormones and mammary carcinogenesis in mice, rats, and humans: a unifying hypothesis. Proc Natl Acad Sci. 1995;92:3650–3657. doi: 10.1073/pnas.92.9.3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Feigelson HS, Henderson BE. Estrogens and breast cancer. Carcinogenesis. 1996;17:2279–2284. doi: 10.1093/carcin/17.11.2279. [DOI] [PubMed] [Google Scholar]
- 10.Dickson RB, Stancel GM. Estrogen receptor-mediated processes in normal and cancer cells. J Natl Cancer Inst Monogr. 2000;27:135–145. doi: 10.1093/oxfordjournals.jncimonographs.a024237. [DOI] [PubMed] [Google Scholar]
- 11.Cavalieri EL, Rogan EG. Depurinating estrogen-DNA adducts in the etiology and prevention of breast and other human cancers. Future Oncol. 2010;6:75–91. doi: 10.2217/fon.09.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cavalieri E, Rogan E. Unbalanced metabolism of endogenous estrogens in the etiology and prevention of human cancer. J Steroid Biochem Mol Biol. 2011;125:169–180. doi: 10.1016/j.jsbmb.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cavalieri EL, Rogan EG. The etiology and prevention of breast cancer. Drug Discov Today Dis Mech. 2012;9:e55–e69. doi: 10.1016/j.ddmec.2013.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cavalieri E, Rogan E. The molecular etiology and prevention of estrogen-initiated cancers. Mol Aspects Med. 2014;36:1–55. doi: 10.1016/j.mam.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saeed M, Rogan E, Cavalieri E. Mechanism of metabolic activation and DNA adduct formation by the human carcinogen diethylstilbestrol: the defining link to natural estrogens. Int J Cancer. 2009;124:1276–1284. doi: 10.1002/ijc.24113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cavalieri EL, Li K-M, Balu N, Saeed M, Devanesan P, Higginbotham S, Zhao J, Gross ML, Rogan E. Catechol ortho-quinones: the electrophilic compounds that form depurinating DNA adducts and could initiate cancer and other diseases. Carcinogenesis. 2002;23:1071–1077. doi: 10.1093/carcin/23.6.1071. [DOI] [PubMed] [Google Scholar]
- 17.Zahid M, Saeed M, Rogan EG, Cavalieri EL. Benzene and dopamine catechol quinones could initiate cancer or neurogenic disease. Free Radic Biol Med. 2010;48:318–324. doi: 10.1016/j.freeradbiomed.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saeed M, Higginbotham S, Rogan E, Cavalieri E. Formation of depurinating N3adenine and N7guanine adducts after reaction of 1,2-naphthoquinone or enzyme-activated 1,2-dihydroxynaphthalene with DNA. Implications for the mechanism of tumor initiation by naphthalene. Chem Biol Interact. 2007;165:175–188. doi: 10.1016/j.cbi.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 19.Saeed M, Higginbotham S, Gaikwad N, Chakravarti D, Rogan E, Cavalieri E. Depurinating naphthalene-DNA adducts in mouse skin related to cancer initiation. Free Radic Biol Med. 2009;47:1075–1081. doi: 10.1016/j.freeradbiomed.2009.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cavalieri EL, Stack DE, Devanesan PD, Todorovic R, Dwivedy I, Higginbotham S, Johansson SL, Patil KD, Gross ML, Gooden JK, Ramanathan R, Cerny RL, Rogan EG. Molecular origin of cancer: catechol estrogen-3,4-quinones as endogenous tumor initiators. Proc Natl Acad Sci. 1997;94:10937–10942. doi: 10.1073/pnas.94.20.10937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dwivedy I, Devanesan P, Cremonesi P, Rogan E, Cavalieri E. Synthesis and characterization of estrogen 2,3- and 3,4-quinones. Comparison of DNA adducts formed by the quinones versus horseradish peroxidase-activated catechol estrogens. Chem Res Toxicol. 1992;5:828–833. doi: 10.1021/tx00030a016. [DOI] [PubMed] [Google Scholar]
- 22.Li K-M, Todorovic R, Devanesan P, Higginbotham S, Köfeler H, Ramanathan R, Gross ML, Rogan EG, Cavalieri EL. Metabolism and DNA binding studies of 4-hydroxyestradiol and estradiol-3,4-quinone in vitro and in female ACI rat mammary gland in vivo. Carcinogenesis. 2004;25:289–297. doi: 10.1093/carcin/bgg191. [DOI] [PubMed] [Google Scholar]
- 23.Saeed M, Zahid M, Gunselman SJ, Rogan E, Cavalieri E. Slow loss of deoxyribose from the N7deoxyguanosine adducts of estradiol-3,4-quinone and hexestrol-3′,4′-quinone. implications for mutagenic activity. Steroids. 2005;70:29–35. doi: 10.1016/j.steroids.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 24.Saeed M, Rogan E, Fernandez SV, Sheriff F, Russo J, Cavalieri E. Formation of depurinating N3Adenine and N7Guanine adducts by MCF-10F cells cultured in the presence of 4-hydroxyestradiol. Int J Cancer. 2007;120:1821–1824. doi: 10.1002/ijc.22399. [DOI] [PubMed] [Google Scholar]
- 25.Stack D, Byun J, Gross ML, Rogan EG, Cavalieri E. Molecular characteristics of catechol estrogen quinones in reactions with deoxyribonucleosides. Chem Res Toxicol. 1996;9:851–859. doi: 10.1021/tx960002q. [DOI] [PubMed] [Google Scholar]
- 26.Zahid M, Kohli E, Saeed M, Rogan E, Cavalieri E. The greater reactivity of estradiol-3,4-quinone versus estradiol-2,3-quinone with DNA in the formation of depurinating adducts. implications for tumor-initiating activity. Chem Res Toxicol. 2006;9:164–172. doi: 10.1021/tx050229y. [DOI] [PubMed] [Google Scholar]
- 27.Hinrichs B, Zahid M, Saeed M, Ali MF, Cavalieri EL, Rogan EG. Formation of diethylstilbestrol-DNA adducts in human breast epithelial cells and inhibition by resveratrol. J Steroid Biochem Mol Biol. 2011;127:276–281. doi: 10.1016/j.jsbmb.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jan S-T, Devanesan P, Stack D, Ramanathan R, Byun J, Gross ML, Rogan E, Cavalieri E. Metabolic activation and formation of DNA adducts of hexestrol, a synthetic non-steroidal carcinogenic estrogen. Chem Res Toxicol. 1998;11:412–419. doi: 10.1021/tx970141n. [DOI] [PubMed] [Google Scholar]
- 29.Saeed M, Gunselman SJ, Higginbotham S, Rogan E, Cavalieri E. Formation of the depurinating N3adenine and N7guanine adducts by reaction of DNA with hexestrol-3′-4′-quinone or enzyme-activated 3′-hydroxyhexestrol. Implications for a unifying mechanism of tumor initiation by natural and synthetic estrogens. Steroids. 2005;70:37–45. doi: 10.1016/j.steroids.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 30.Paxton MB. Leukemia risk associated with benzene exposure in the Pliofilm cohort. Environ Health Perspect. 1996;104(Suppl 6):1431–1436. doi: 10.1289/ehp.961041431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rinsky RA, Smith AB, Hornung R, Filloon TG, Young RJ, Okun AH, Landrigan PJ. Benzene and leukemia. An epidemiologic risk assessment. N Engl J Med. 1987;316:1044–1050. doi: 10.1056/NEJM198704233161702. [DOI] [PubMed] [Google Scholar]
- 32.Sabourin PJ, Bechtold WE, Griffith WC, Birnbaum LS, Lucier G, Henderson RF. Effect of exposure concentration, exposure rate, and route of administration on metabolism of benzene by F344 rats and B6C3F1 mice. Toxicol Appl Pharmacol. 1989;99:421–444. doi: 10.1016/0041-008X(89)90151-8. [DOI] [PubMed] [Google Scholar]
- 33.Snyder R, Kalf GF. A perspective on benzene leukemogenesis. Crit Rev Toxicol. 1994;24:177–209. doi: 10.3109/10408449409021605. [DOI] [PubMed] [Google Scholar]
- 34.Greenlee WF, Gross EA, Irons RD. Relationship between benzene toxicity and the disposition of 14C-labelled benzene metabolites in the rat. Chem Biol Interact. 1981;33:285–299. doi: 10.1016/0009-2797(81)90047-8. [DOI] [PubMed] [Google Scholar]
- 35.Rickert DE, Baker TS, Bus JS, Barrow CS, Irons RD. Benzene disposition in the rat after exposure by inhalation. Toxicol Appl Pharmacol. 1979;49:417–423. doi: 10.1016/0041-008X(79)90441-1. [DOI] [PubMed] [Google Scholar]
- 36.Sadler A, Subrahmanyam VV, Ross D. Oxidation of catechol by horseradish peroxidase and human leukocyte peroxidase: reactions of o-benzoquinone and o-benzosemiquinone. Toxicol Appl Pharmacol. 1988;93:62–71. doi: 10.1016/0041-008X(88)90025-7. [DOI] [PubMed] [Google Scholar]
- 37.National Toxicology Program. Toxicology and Carcinogenesis Studies of Naphthalene (CAS No. 91-20-3). In: F344/N Rats (Inhalation Studies), NTP Technical Report No. 500. Research Triangle Park, NC: NIH Publ. No. 01-4434. 2000
- 38.Herbst AL, Ulfelder H, Poskanzer DC. Adenocarcinoma of the vagina. Association of maternal stilbestrol therapy with tumor appearance in young women. N Engl J Med. 1971;284:878–881. doi: 10.1056/NEJM197104222841604. [DOI] [PubMed] [Google Scholar]
- 39.Li JJ, Li SA, Klicka JK, Parsons JA, Lam LK. Relative carcinogenic activity of various synthetic and natural estrogens in the Syrian hamster kidney. Cancer Res. 1983;43:5200–5204. [PubMed] [Google Scholar]
- 40.Liehr JG, Ballatore AM, Dague BB, Ulubelen AA. Carcinogenicity and metabolic activation of hexestrol. Chem Biol Interact. 1985;55:157–176. doi: 10.1016/S0009-2797(85)80125-3. [DOI] [PubMed] [Google Scholar]
- 41.Metzler M, McLachlan JA. Oxidative metabolism of the synthetic estrogens hexestrol and dienestrol indicates reactive intermediates. Adv Exp Med Biol. 1981;136:829–837. doi: 10.1007/978-1-4757-0674-1_65. [DOI] [PubMed] [Google Scholar]
- 42.Haaf H, Metzler M. In vitro metabolism of diethylstilbestrol by hepatic, renal and uterine microsomes of rats and hamsters.Effects of different inducers. Biochem Pharmacol. 1985;34:3107–3115. doi: 10.1016/0006-2952(85)90155-8. [DOI] [PubMed] [Google Scholar]
- 43.Blaich G, Göttlicher M, Cikryt P, Metzler M. Effects of various inducers on diethylstilbestrol metabolism, drug-metabolizing enzyme activities and the aromatic hydrocarbon (Ah) receptor in male Syrian golden hamster liver. J Steroid Biochem. 1990;35:201–204. doi: 10.1016/0022-4731(90)90275-W. [DOI] [PubMed] [Google Scholar]
- 44.Zhu BT, Conney AH. Functional role of estrogen metabolism in target cells: review and perspectives. Carcinogenesis. 1998;9:1–27. doi: 10.1093/carcin/19.1.1. [DOI] [PubMed] [Google Scholar]
- 45.Spink DC, Hayes CL, Young NR, Christou M, Sutter TR, Jefcoate CR, Gierthy JF. The effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin on estrogen metabolism in MCF-7 breast cancer cells: evidence for induction of a novel 17 beta-estradiol 4-hydroxylase. J Steroid Biochem Mol Biol. 1994;51:251–258. doi: 10.1016/0960-0760(94)90037-X. [DOI] [PubMed] [Google Scholar]
- 46.Hayes CL, Spink DC, Spink BC, Cao JQ, Walker NJ, Sutter TR. 17β-Estradiol hydroxylation catalyzed by human cytochrome P450 1B1. Proc Natl Acad Sci USA. 1996;93:9776–9781. doi: 10.1073/pnas.93.18.9776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Spink DC, Spink BC, Cao JQ, DePasquale JA, Pentecost BT, Fasco MJ, Li Y, Sutter TR. Differential expression of CYP1A1 and CYP1B1 in human breast epithelial cells and breast tumor cells. Carcinogenesis. 1998;19:291–298. doi: 10.1093/carcin/19.2.291. [DOI] [PubMed] [Google Scholar]
- 48.Männistö PT, Kaakkola S. Catechol-O-methyltransferase (COMT): biochemistry, molecular biology, pharmacology, and clinical efficacy of the new selective COMT inhibitors. Pharmacol Rev. 1999;51:593–628. [PubMed] [Google Scholar]
- 49.Yager JD. Catechol-O-methyltransferase: characteristics, polymorphisms and role in breast cancer. Drug Discov Today Dis Mech. 2012;9:e41–e46. doi: 10.1016/j.ddmec.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kappus H. Lipid peroxidation: Mechanisms, analysis, enzymology and biological relevance. In: Sies H, editor. Oxidative Stress. New York: Academic Press; 1985. pp. 273–310. [Google Scholar]
- 51.Gaikwad NW, Rogan EG, Cavalieri EL. Evidence from ESI-MS for NQO1-catalyzed reduction of estrogen ortho-quinones. Free Radic Biol Med. 2007;43:1289–1298. doi: 10.1016/j.freeradbiomed.2007.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gaikwad NW, Yang L, Rogan EG, Cavalieri EL. Evidence from ESI-MS for NQO2-catalyzed reduction of estrogen ortho-quinones. Free Radic Biol Med. 2009;46:253–262. doi: 10.1016/j.freeradbiomed.2008.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Talalay P, Dinkova-Kostova AT, Holtzclaw WD. Importance of phase 2 gene regulation in protection against electrophile and reactive oxygen toxicity and carcinogenesis. Adv Enzyme Regul. 2003;43:121–134. doi: 10.1016/S0065-2571(02)00038-9. [DOI] [PubMed] [Google Scholar]
- 54.Cavalieri E, Saeed M, Zahid M, Cassada E, Snow D, Miljkovic M, Rogan E. Mechanism of DNA depurination by carcinogens in relation to cancer initiation. IUBMB Life. 2012;64:169–179. doi: 10.1002/iub.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pullman A, Pullman B. Molecular electrostatic potential of the nucleic acids. Q Rev Biophys. 1981;14:289–380. doi: 10.1017/S0033583500002341. [DOI] [PubMed] [Google Scholar]
- 56.Chakravarti D, Pelling JC, Cavalieri EL, Rogan EG. Relating aromatic hydrocarbon-induced DNA adducts and H-ras mutations in mouse skin papillomas: the role of apurinic sites. Proc Natl Acad Sci USA. 1995;92:10422–10426. doi: 10.1073/pnas.92.22.10422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cavalieri E, Rogan E. Mechanisms of tumor initiation by polycyclic aromatic hydrocarbons in mammals. In: Neilson AH, editor. The handbook of environmental chemistry, PAHs and related compounds. Heidelberg: Springer-Verlag; 1998. pp. 81–117. [Google Scholar]
- 58.Devanesan PD, RamaKrishna NVS, Padmavathi NS, Higginbotham S, Rogan EG, Cavalieri EL, Marsch GA, Jankowiak R, Small GJ. Identification and quantitation of 7,12-dimethylbenz[a]anthracene-DNA adducts formed in mouse skin. Chem Res Toxicol. 1993;6:364–371. doi: 10.1021/tx00033a018. [DOI] [PubMed] [Google Scholar]
- 59.Cavalieri EL, Rogan EG, Li KM, Todorovic R, Ariese F, Jankowiak R, Grubor N, Small GJ. Identification and quantification of the depurinating DNA adducts formed in mouse skin treated with dibenzo[a, l]pyrene (DB[a, l]P), or its metabolites and in rat mammary gland treated with DB[a, l]P. Chem Res Toxicol. 2005;18:976–983. doi: 10.1021/tx049682k. [DOI] [PubMed] [Google Scholar]
- 60.Todorovic R, Devanesan P, Rogan E, Cavalieri E. Identification and quantification of stable DNA adducts of dibenzo[a, l]pyrene or its metabolites in vitro, and in mouse skin and rat mammary gland. Chem Res Toxicol. 2005;18:984–990. doi: 10.1021/tx049681s. [DOI] [PubMed] [Google Scholar]
- 61.Rogan EG, Devanesan PD, RamaKrishna NV, Higginbotham S, Padmavathi NS, Chapman K, Cavalieri EL, Jeong H, Jankowiak R, Small GJ. Identification and quantitation of benzo[a]pyrene–DNA adducts formed in mouse skin. Chem Res Toxicol. 1993;6:356–363. doi: 10.1021/tx00033a017. [DOI] [PubMed] [Google Scholar]
- 62.Colapietro AM, Goodell AL, Smart RL. Characterization of benzo[a]pyrene-initiated mouse skin papillomas for Ha-ras mutations and protein kinase C levels. Carcinogenesis. 1993;14:2289–2295. doi: 10.1093/carcin/14.11.2289. [DOI] [PubMed] [Google Scholar]
- 63.Chakravarti D, Mailander P, Li KM, Higginbotham S, Zhang HL, Gross MS, Cavalieri E, Rogan E. Evidence that a burst of DNA depurination in SENCAR mouse skin induces error-prone repair and forms mutations in the H-ras gene. Oncogene. 2001;20:7945–7953. doi: 10.1038/sj.onc.1204969. [DOI] [PubMed] [Google Scholar]
- 64.Mailander PC, Meza JL, Higginbotham S, Chakravarti D. Induction of A.T to G.C mutations by erroneous repair of depurinated DNA following estrogen treatment of the mammary gland of ACI rats. J Steroid Biochem Mol Biol. 2006;101:204–215. doi: 10.1016/j.jsbmb.2006.06.019. [DOI] [PubMed] [Google Scholar]
- 65.Bolton JL, Shen L. p-Quinone methides are the major decomposition products of catechol estrogen o-quinones. Carcinogenesis. 1998;17:925–929. doi: 10.1093/carcin/17.5.925. [DOI] [PubMed] [Google Scholar]
- 66.Liehr JG, Fang WF, Sirbasku DA, Ari-Ulubelen A. Carcinogenicity of catechol estrogens in Syrian hamsters. J Steroid Biochem. 1986;24:353–356. doi: 10.1016/0022-4731(86)90080-4. [DOI] [PubMed] [Google Scholar]
- 67.Li JJ, Li SA. Estrogen carcinogenesis in Syrian hamster tissues: role of metabolism. Fed Proc. 1987;46:1858–1863. [PubMed] [Google Scholar]
- 68.Newbold RR, Liehr JG. Induction of uterine adenocarcinoma in CD-1 mice by catechol estrogens. Cancer Res. 2000;60:235–237. [PubMed] [Google Scholar]
- 69.Miller WR, O’Neill J. The importance of local synthesis of estrogen within the breast. Steroids. 1987;50:537–548. doi: 10.1016/0039-128X(87)90037-7. [DOI] [PubMed] [Google Scholar]
- 70.Simpson ER, Mahendroo MS, Means GD, Kilgore MW, Hinshelwood MM, Graham-Lorence S, Amarneh B, Ito Y, Fisher CR, Michael MD, et al. Aromatase cytochrome P450, the enzyme responsible for estrogen biosynthesis. Endocr Rev. 1994;15:342–355. doi: 10.1210/edrv-15-3-342. [DOI] [PubMed] [Google Scholar]
- 71.Jefcoate CR, Liehr JG, Santen RJ, Sutter TR, Yager JD, Yue W, Santner SJ, Tekmal R, Demers L, Pauley R, Naftolin F, Mor G, Berstein L. Tissue-specific synthesis and oxidative metabolism of estrogens. In: Cavalieri E, Rogan E, editors. JNCI monograph 27: Estrogens as endogenous carcinogens in the Breast and Prostate. Oxford: University Press; 2000. pp. 95–112. [DOI] [PubMed] [Google Scholar]
- 72.Santner SJ, Feil PD, Santen RJ. In situ estrogen production via the estrone sulfatase pathway in breast tumors: relative importance versus the aromatase pathway. J Clin Endocrinol Metab. 1984;59:29–33. doi: 10.1210/jcem-59-1-29. [DOI] [PubMed] [Google Scholar]
- 73.Pasqualini JR, Chetrite G, Blacker D, Feinstein MC, Delalonde L, Talbi M, Maloche C. Concentrations of estrone, estradiol, and estrone sulfate and evaluation of sulfatase and aromatase activities in pre- and post-menopausal breast cancer patients. J Clin Endocrinol Metab. 1996;81:1460–1464. doi: 10.1210/jcem.81.4.8636351. [DOI] [PubMed] [Google Scholar]
- 74.Lu F, Zahid M, Saeed M, Cavalieri EL, Rogan EG. Estrogen metabolism and formation of estrogen-DNA adducts in estradiol-treated MCF-10F cells. The effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin induction and catechol-O-methyltransferase inhibition. J Steroid Biochem Mol Biol. 2007;105:150–158. doi: 10.1016/j.jsbmb.2006.12.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lu F, Zahid M, Wang C, Saeed M, Cavalieri EL, Rogan EG. Resveratrol prevents estrogen-DNA adduct formation and neoplastic transformation in MCF-10F cells. Cancer Prev Res. 2008;1:135–145. doi: 10.1158/1940-6207.CAPR-08-0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mitrunen K, Hirvonen A. Molecular epidemiology of sporadic breast cancer. The role of polymorphic genes involved in oestrogen biosynthesis and metabolism. Mutat Res. 2003;544:9–41. doi: 10.1016/S1383-5742(03)00016-4. [DOI] [PubMed] [Google Scholar]
- 77.Singh S, Zahid M, Saeed M, Gaikwad NW, Meza JL, Cavalieri EL, Rogan EG, Chakravarti D. NAD(P)H:quinone oxidoreductase 1 Arg139Trp and Pro187Ser polymorphism imbalance estrogen metabolism towards DNA adduct formation in human mammary epithelial cells. J Steroid Biochem Mol Biol. 2009;117:56–66. doi: 10.1016/j.jsbmb.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cavalieri EL, Devanesan P, Bosland MC, Badawi AF, Rogan EG. Catechol estrogen metabolites and conjugates in different regions of the prostate of noble rats treated with 4-hydroxyestradiol: implications for estrogen-induced initiation of prostate cancer. Carcinogenesis. 2002;23:329–333. doi: 10.1093/carcin/23.2.329. [DOI] [PubMed] [Google Scholar]
- 79.Cavalieri EL, Kumar S, Todorovic R, Higginbotham S, Badawi AF, Rogan EG. Imbalance of estrogen homeostasis in kidney and liver of hamsters treated with estradiol: implications for estrogen-induced initiation of renal tumors. Chem Res Toxicol. 2001;14:1041–1050. doi: 10.1021/tx010042g. [DOI] [PubMed] [Google Scholar]
- 80.Devanesan P, Santen RJ, Bocchinfuso WP, Korach KS, Rogan EG, Cavalieri EL. Catechol estrogen metabolites and conjugates in mammary tumors and hyperplastic tissue from estrogen receptor alpha knock out (ERKO)/Wnt 1 mice; implications for initiation of mammary tumors. Carcinogenesis. 2001;22:1573–1576. doi: 10.1093/carcin/22.9.1573. [DOI] [PubMed] [Google Scholar]
- 81.Rogan EG, Badawi AF, Devanesan PD, Meza JL, Edney JA, West WW, Higginbotham SM, Cavalieri EL. Relative imbalances in estrogen metabolism and conjugation in breast tissue of women with carcinoma: potential biomarkers of susceptibility to cancer. Carcinogenesis. 2003;24:697–702. doi: 10.1093/carcin/bgg004. [DOI] [PubMed] [Google Scholar]
- 82.Singh S, Chakravarti D, Edney JA, Hollins RR, Johnson PJ, West WW, Higginbotham SM, Cavalieri EL, Rogan EG. Relative imbalances in the expression of estrogen-metabolizing enzymes in the breast tissue of women with breast carcinoma. Oncol Rep. 2005;14:1091–1096. [PubMed] [Google Scholar]
- 83.Gaikwad NW, Yang L, Muti P, Meza JL, Pruthi S, Ingle JN, Rogan EG, Cavalieri EL. The molecular etiology of breast cancer: evidence from biomarkers of risk. Int J Cancer. 2008;122:1949–1957. doi: 10.1002/ijc.23329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gaikwad NW, Yang L, Pruthi S, Ingle JN, Sandhu N, Rogan E, Cavalieri E. Urine biomarkers of risk in the molecular etiology of breast cancer. Breast Cancer Basic Clin Res. 2009;3:1–8. doi: 10.4137/bcbcr.s2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pruthi S, Yang L, Sandhu NP, Ingle JN, Beseler CL, Suman VJ, Cavalieri EL, Rogan EG. Evaluation of serum estrogen-DNA adducts as potential biomarkers of breast cancer risk. J Steroid Biochem Mol Biol. 2012;132:73–79. doi: 10.1016/j.jsbmb.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gail MH, Brinton LA, Byar DP, Corle DK, Green SB, Schairer C, Mulvihill JJ. Protective individualized probabilities of developing breast cancer for white females who are being examined annually. J Natl Cancer Inst. 1989;81:1879–1886. doi: 10.1093/jnci/81.24.1879. [DOI] [PubMed] [Google Scholar]
- 87.Zahid M, Beseler CL, Hall JB, LeVan T, Cavalieri EL, Rogan EG. Unbalanced estrogen metabolism in ovarian cancer. Int J Cancer. 2014;134:2414–2423. doi: 10.1002/ijc.28565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zahid M, Goldner W, Beseler CL, Rogan EG, Cavalieri EL. Unbalanced estrogen metabolism in thyroid cancer. Int J Cancer. 2013;133:2642–2649. doi: 10.1002/ijc.28275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yang L, Gaikwad N, Meza J, Cavalieri E, Muti P, Trock B, Rogan E. Novel biomarkers for risk of prostate cancer-results from a case-control study. Prostate. 2009;69:41–48. doi: 10.1002/pros.20850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gaikwad NW, Yang L, Weisenburger DD, Vose J, Beseler C, Rogan EG, Cavalieri E. Urinary biomarkers suggest that estrogen-DNA adducts may play a role in the aetiology of non-Hodgkin lymphoma. Biomarkers. 2009;14:502–512. doi: 10.3109/13547500903121715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zahid M, Gaikwad N, Rogan EG, Cavalieri EL. Inhibition of depurinating estrogen-DNA adducts formation by natural compounds. Chem Res Toxicol. 2007;20:1947–1953. doi: 10.1021/tx700269s. [DOI] [PubMed] [Google Scholar]
- 92.De Flora S, et al. Mechanisms of anticarcinogenesis: The example of N-acetylcysteine. In: Ioannides C, Lewis DFV, et al., editors. Drugs, diet and disease, mechanistic approaches to cancer. Herts: Prentice Hall; 1994. pp. 151–202. [Google Scholar]
- 93.De Flora S, Cesarone CF, Balansky RM, Albini A, D’Agostini F, Bennicelli C, Bagnasco M, Camoirano A, Scatolini L, Rovida A, Izzotti A. Chemopreventive properties and mechanisms of N-acetylcysteine. The experimental background. J Cell Biochem Suppl. 1995;22:33–41. doi: 10.1002/jcb.240590806. [DOI] [PubMed] [Google Scholar]
- 94.Townsend DM, Tew KD, Tapiero H. The importance of glutathione in human disease. Biomed Pharmacother. 2003;57:145–155. doi: 10.1016/S0753-3322(03)00043-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cao K, Stack DE, Ramanathan R, Gross ML, Rogan EG, Cavalieri EL. Synthesis and structure elucidation of estrogen quinones conjugated with cysteine, N-acetylcysteine, and glutathione. Chem Res Toxicol. 1998;11:909–916. doi: 10.1021/tx9702291. [DOI] [PubMed] [Google Scholar]
- 96.Cao K, Devanesan PD, Ramanathan R, Gross ML, Rogan EG, Cavalieri EL. Covalent binding of catechol estrogens to glutathione catalyzed by horseradish peroxidase, lactoperoxidase, or rat liver microsomes. Chem Res Toxicol. 1998;11:917–924. doi: 10.1021/tx9702300. [DOI] [PubMed] [Google Scholar]
- 97.Samuni AM, Chuang EY, Krishna MC, Stein W, DeGraff W, Russo A, Mitchell JB. Semiquinone radical intermediate in catecholic estrogen-mediated cytotoxicity and mutagenesis: chemoprevention strategies with antioxidants. Proc Natl Acad Sci USA. 2003;100:5390–5395. doi: 10.1073/pnas.0930078100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zahid M, Saeed M, Ali MF, Rogan EG, Cavalieri EL. N-acetylcysteine blocks formation of cancer-initiating estrogen-DNA adducts in cells. Free Radic Biol Med. 2010;49:392–400. doi: 10.1016/j.freeradbiomed.2010.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Venugopal D, Zahid M, Mailander PC, Meza JL, Rogan EG, Cavalieri EL, Chakravarti E. Reduction of estrogen-induced transformation of mouse mammary epithelial cells by N-acetylcysteine. J Steroid Biochem Mol Biol. 2008;109:22–30. doi: 10.1016/j.jsbmb.2007.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Jang M, Cai L, Udeani GO, Slowing KV, Thomas CF, Beecher CW, Fong HH, Farnsworth NR, Kinghorn AD, Mehta RG, Moon RC, Pezzuto JM. Cancer chemopreventive activity of resveratrol, a natural product derived from grapes. Science. 1997;275:218–220. doi: 10.1126/science.275.5297.218. [DOI] [PubMed] [Google Scholar]
- 101.Aziz MH, Kumar R, Ahmad N. Cancer chemoprevention by resveratrol: in vitro and in vivo studies and the underlying mechanisms (review) Int J Oncol. 2003;23:17–28. [PubMed] [Google Scholar]
- 102.Stivala LA, Savio M, Carafoli F, Perucca P, Bianchi L, Maga G, Forti L, Pagnoni UM, Albini A, Prosperi E, Vannini V. Specific structural determinants are responsible for the antioxidant activity and the cell cycle effects of resveratrol. J Biol Chem. 2001;276:22586–22594. doi: 10.1074/jbc.M101846200. [DOI] [PubMed] [Google Scholar]
- 103.Guengerich FP, Chunb YJ, Kima D, Gillam EMJ, Shimada T. Cytochrome P450 1B1: a target for inhibition in anticarcinogenesis strategies. Mutat Res. 2003;523–524:173–182. doi: 10.1016/S0027-5107(02)00333-0. [DOI] [PubMed] [Google Scholar]
- 104.Floreani M, Napoli E, Quintieri L, Palatini P. Oral administration of trans-resveratrol to guinea pigs increases cardiac DT-diaphorase and catalase activities, and protects isolated atria from menadione toxicity. Life Sci. 2003;7224:2741–2750. doi: 10.1016/S0024-3205(03)00179-6. [DOI] [PubMed] [Google Scholar]
- 105.Zahid M, Gaikwad NW, Ali MF, Saeed M, Yang L, Rogan EG, Cavalieri EL. Prevention of estrogen-DNA adducts in MCF-10F cells by resveratrol. Free Radic Biol Med. 2008;45:136–145. doi: 10.1016/j.freeradbiomed.2008.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zahid M, Saeed M, Beseler C, Rogan EG, Cavalieri EL. Resveratrol and N-acetylcysteine block the cancer-initiating step in MCF-10F cells. Free Radic Biol Med. 2011;50:78–85. doi: 10.1016/j.freeradbiomed.2010.10.662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Muran P, Muran S, Beseler CL, Cavalieri EL, Rogan EG, Zahid M. Breast health and reducing breast cancer risk, a functional medicine approach. J Altern Complement Med. 2015;21:321–326. doi: 10.1089/acm.2014.0365. [DOI] [PubMed] [Google Scholar]