

Abstract
In continuation to our previous work, thiazolopyrimidines 2a–x were synthesized through intramolecular cyclization of 2-phenacylthio-dihydropyrimidine hydrobromides 1a–x using polyphosphoric acid. On the other hand, thiazolo[3,2-a]pyrimidine-3-one 3 was coupled with aryldiazonium salts or condensed with isatin to afford compounds 4a–c or 5, respectively. Chemical structure of the target compounds was substantiated by IR, FT-IR, 1H-, 13C and DEPT-13C NMR, MS as well as microanalyses. Moreover, the lipophilicity of the target compounds is expressed as Clog P. The antimicrobial screening of the test compounds 2a–x, 4a–c and 5 revealed moderate activity in comparison to reference drugs. Compounds 2a–c, 2e, 2o and 2v showed a gradual increase in their anti-inflammatory activity reaching its maximum at 5 h compared to indomethacin. Furthermore, the analgesic activity of compounds 2a–c, 2e, 2o and 2v revealed a maximum activity after 5 h of injection compared to aspirin and the LD50 of compounds 2e and 2v was determined.
Keywords: Thiazolopyrimidines, Synthesis, Antimicrobial, Anti-inflammatory, Analgesic
1. Introduction
Ever since the appearance of bacterial and fungal resistance toward the known chemotherapeutic agent and the amazing discovery of new chemotherapeutic molecule constitute a real challenge. Consequently, new potent chemotherapeutic agents are needed, and if possible, with new modes of actions (Sahu et al., 2008). In addition, microbial infections often produce pain and inflammation. Chemotherapeutic, analgesic and anti-inflammatory drugs are prescribed simultaneously in normal practice. Consequently, a compound possessing all three activities is recommended (Sahu et al., 2008). Hence, the treatment of pain continues to be the subject of considerable pharmaceutical and clinical research (Sahu et al., 2008).
In recent years, thiazolopyrimidines have been of interest due to their vital role in many biological activities (Viveka et al., 2012) such as antimicrobial (Ghorab et al., 2000, Sayed et al., 2006, Sherif et al., 1993, Tozkoparan et al., 1999, Youssef and Amin, 2012), anti-inflammatory (Dinakaran et al., 2012, Jeanneau-Nicolle et al., 1992, Sayed et al., 2006, Sherif et al., 1993, Tozkoparan et al., 1999), analgesic (Tozkoparan et al., 1999), antioxidant (Youssef and Amin, 2012), insecticides (Sherif et al., 1993), antidepressant (Tozkoparan et al., 1999), antiviral (Sayed et al., 2006), antitumor (Sayed et al., 2006, Tozkoparan et al., 1999), and calcium channel blocker activities (Balkan et al., 1992a, Balkan et al., 1996, Jeanneau-Nicolle et al., 1992, Tozkoparan et al., 1999). Additionally, it was reported that such a ring system has the ability to inhibit the enzyme 3′,5′-cyclic AMP phosphodiesterase (Sherif et al., 1993), to have a theophylline-like activity (Sherif et al., 1993) and to be active against virulent Lewis lung tumor in mice (Sherif et al., 1993).
In our previous work, a series of ethyl 6-methyl-4-(substituted)phenyl-2-(substituted)-phenacylthio-1,4-dihydropyrimidine-5-carboxylate hydrobromide 1a–x have been synthesized and evaluated for their antibacterial and antifungal activities (Hussein et al., 2011).
Enlightened by aforementioned data and in continuation to our previous work, we herein the synthesis of some new thiazolo[3,2-a]pyrimidine derivatives for the evaluation of their antimicrobial, anti-inflammatory and analgesic effects, if any. This work is concerned with the study of the effect of structural modification on the biological activities of the target compounds.
2. Results and discussion
2.1. Chemistry
Compounds 1a–x were cyclodehydrated into the corresponding thiazolo[3,2-a]pyrimidine compounds 2a–x by heating with freshly prepared polyphosphoric acid (PPA) for 2 h followed by neutralization with ammonia, Scheme 1.
Scheme 1.

Synthetic route for compounds 2a–x. (a) polyphosphoric acid, 110 °C, (b) NH4OH.
Basically, such products of 2a–x had been formulated as the 5H-thiazolo [3,2-a] pyrimidine structure A based on the comparison of 1H NMR spectral data for both compounds 1a–x and 2a–x (Balkan et al., 1992b, Sherif et al., 1993); Fig. 1.
Figure 1.
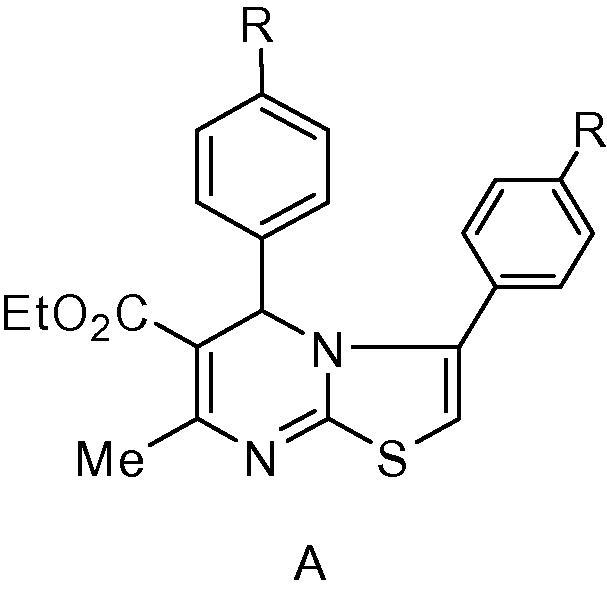
Structure of compound A.
1H NMR spectrum of compound 1b (Hussein et al., 2011), Fig. 2, show the C6-CH3 as singlet signal at δ 2.56 ppm and C4-H as singlet signal at δ 5.58 ppm. 1H NMR spectrum of compound 2b as an example, Fig. 2, showed a singlet signal (3H) at δ 2.50 ppm assigned for the C7-CH3 protons and singlet signal (1H) at δ 6.13 ppm assigned for the pyrimidine H-5. The appearance of the CH3 protons signal at the same position on both 1b and 2b and the down field shift for the pyrimidine H-5 in 2b, at δ 6.13 ppm compared to the pyrimidine H-4 in 1b, which appeared at δ 5.58 ppm, indicates that the environment around carbon-5 in 2b differs from that around carbon-4 in 1b. Consequently, we have assigned structure A, Fig. 1, for the reaction products 2a–x (Balkan et al., 1992b, Sherif et al., 1993).
Figure 2.

Structure of compounds 1b and 2b.
Structures of the synthesized compounds 2a–x were verified by IR, 1H-, 13C and DEPT-13C NMR, mass spectra as well as elemental microanalyses. IR spectra of compounds 2a–x showed strong absorption bands at 1680–1636 cm−1 for carbonyl groups of the ester and at 1589–1561 cm−1 indicating C N stretching vibration. 1H NMR spectra revealed a triplet (3H) of quartet (2H) at δ 1.12–1.20 and 4.00–4.13 ppm, respectively of the ethyl group and multiplet at δ 6.30–8.40 ppm indicating aromatic protons. On the other hand, 13C NMR spectrum of compound 2k showed a signal at δ 14.26 ppm corresponding to CH3 of the ethyl group, δ 57.16 ppm corresponding to CH2 of the ethyl group, δ 166.43 ppm indicating C-8a and δ 166.61 ppm attributed to the carbonyl group. Moreover, DEPT 13C NMR spectrum of compound 2k showed inversion of CH2 of the ethyl group and disappearance of all quaternary carbons while the CH3 and CH groups stay the same. Moreover, mass spectra (EI) of compounds 2b, 2d, 2e, 2i, 2j, 2n, 2o, 2q and 2u revealed the molecular ion peaks (M+) corresponding to their molecular weights and M++2 for compounds 2j, 2n, 2o, 2q and 2u. Physicochemical and spectral data of compounds 2a–x are shown in the experimental section.
The active methylene carbon atom in compound 3 (Sherif et al., 1993, Shiryaev et al., 2013) was coupled readily with equimolar amount of freshly prepared aryldiazonium salt to give the corresponding 2-arylhydrazono derivatives, compounds 4a–c. The spectroscopic properties of these products suggest their existence, to a significant extent, in the tautomeric azo forms; hydrazono and diazenyl isomers in ∼90:10%, respectively. The hydazono isomer is the major one due to stabilization through the formation of intramolecular hydrogen bonding, while the presence of diazenyl isomer was proved by the existence of highly deshielded (C2-H) in accordance with the reported data (Rida et al., 1986, Weisberger and Taylor, 1981), Scheme 2.
Scheme 2.

Synthetic route for compounds 4a–c and 5. (a) bromoacetic acid, (b) isatin, sod. acetate, (c) p-(un)substituted-aniline, sod. nitrite, conc. HCl, 0–5 °C.
Structures of the synthesized compounds 4a–c were verified by IR, 1H-, 13C and DEPT-13C NMR, mass spectra in addition to elemental microanalyses. IR spectra of compounds 4a–c showed NH at 3145–3220 cm−1, the ester carbonyl group at 1711–1717 cm−1 and the carbonyl at position 3 at 1693–1695 cm−1. 1H NMR spectra for compounds 4a–c were characterized by the appearance of singlet signal at δ 8.13–8.83 ppm corresponding to NH proton (exchangeable with D2O) for hydazono isomer. The most characteristic signals indicated the presence of the possible two isomeric forms were the signals for CH3 and CH2 of the ethyl group that appeared as two sets of triplet at δ 1.00–1.40 ppm and two sets of quartet at δ 3.50–4.40 ppm, respectively. On the other hand, 13C NMR spectrum, of compound 4c revealed the presence of two ethyl CH3 of the two isomers at δ 13.98 and 18.34 ppm, benzylic carbon at δ 20.69 ppm, 7-CH3 at δ 22.55 ppm, C-5 at δ 55.25 ppm, two ethyl CH2 of the two isomers at δ 58.44 and 60.63 ppm, C-6 at δ 109.69 ppm, C-2 of diazenyl isomer at δ 114.50 ppm, aromatic carbons at δ 120.69–139.54 ppm, C-7 at δ 139.71 ppm, and C-2 of hydrazono isomer at δ 151.77 ppm (C N). It also showed signals at δ 153.36, 161.03 and 165.26 ppm corresponding to C-8a, amidic C O and ester C O, respectively. Moreover, DEPT 13C NMR spectrum, of compound 4c was characterized by the presence of two signals at δ 14.00 ppm and δ 18.32 ppm corresponding to the two ethyl CH3 groups of the two isomers and inversion of two signals at δ 58.44 ppm and δ 60.63 ppm indicating the presence of two ethyl CH2 groups for the two isomers. In addition, all quaternary carbons disappeared while the signals of CH3 and CH stay the same. Furthermore, mass spectra (EI) of compounds 4b and 4c revealed the molecular ion peaks (M+) corresponding to their molecular weights and M++2 for compound 4c. Physicochemical and spectral data of compounds 4a–c are shown in the experimental section.
Structure of compound 5 was verified by FT-IR, 1H NMR, mass spectra as well as elemental microanalyses. FT-IR spectrum of compound 5 showed characteristic bands at 3200 cm−1 indicating the presence of the NH group, 1715 cm−1 corresponding to ester C O and strong absorption band at 1697 cm−1 corresponding to both amidic C O and indole C O groups. 1H NMR spectrum showed disappearance of the active methylene protons of SCH2 signal at δ 3.85 ppm indicating condensation of isatin at C2 of compound 3. It also showed a multiplet signal at δ 6.83–8.96 ppm (9H) indicating aromatic protons (5 protons of phenyl ring + 4 protons of indole ring) as well as singlet signal at δ 11.16 ppm indicating the presence of the indole NH group (exchangeable with D2O). Moreover, mass spectrum (EI) of compound (5) revealed the molecular ion peak (M+) corresponding to its molecular weight. Physicochemical and spectral data of compound 5 are cited in the experimental section.
2.1.1. Lipophilicity
The lipophilicity of the target compounds 2a–x, 4a–c and 5 is expressed in the term of Clog P values (experimental section). Most of the target compounds exhibited high values for Clog P are the most active as antimicrobial agents.
2.2. Antimicrobial activities
2.2.1. Antibacterial activity
Results of the antibacterial activity of compounds 2a–x, Table 1, revealed that at a concentration of 100 μmol/mL most of the test compounds were active against most of the used bacterial strains. Compounds (2f, 2h, 2j–l and 2p–r; with Clog P from 6.30 to 8.28) were completely inactive against all used organisms while compound (2v; with Clog P 6.97) was active only against Bacillus cereus and compound (2x; with Clog P 6.80) was active only against Staphylococcus aureus.
Table 1.
Antibacterial activity of compounds (2a–x, 4a–c and 5) and chloramphenicol (inhibition zones in mm at 100 μmol/mL).
| Comp No. | S. aureus | B. cereus | E. coli | P. aeruginosa | S. marcescens |
|---|---|---|---|---|---|
| 2a | 8 | 18 | – | – | – |
| 2b | 8 | 18 | – | – | – |
| 2c | 11 | 17 | 10 | 13 | 12 |
| 2d | 8 | 18 | – | – | – |
| 2e | 13 | 22 | 13 | 13 | 16 |
| 2f | – | – | – | – | – |
| 2g | 16 | 21 | 13 | 18 | 18 |
| 2h | – | – | – | – | – |
| 2i | 10 | 12 | 13 | – | – |
| 2j | – | – | – | – | – |
| 2k | – | – | – | – | – |
| 2l | – | – | – | – | – |
| 2m | 12 | 12 | 12 | – | 16 pi |
| 2n | 8 | – | 12 | 13 | – |
| 2o | 11 | – | 11 | – | – |
| 2p | – | – | – | – | – |
| 2q | – | – | – | – | – |
| 2r | – | – | – | – | – |
| 2s | 11 | 17 | 10 | 14 | – |
| 2t | 11 | 12 | 10 | 10 | 12 |
| 2u | 10 | – | 10 | 10 | – |
| 2v | – | 12 | – | – | – |
| 2w | 11 | 15 | 10 | – | – |
| 2x | 11 | – | – | – | – |
| 4a | 8 | – | 8 | – | – |
| 4b | 8 | – | 8 | – | – |
| 4c | 10 | – | 8 | – | – |
| 5 | 10 | – | – | – | |
| CHL | 24 | 30 | 25 | 15 | 34 |
| Control (DMSO) | – | – | – | – | – |
Pi = partial inhibition. – = no inhibition. CHL = chloramphenicol as antibacterial standard. AUMC = Assiut University Mycological Center.
In addition, compounds 2a–x showed 33.3–66.7% antibacterial activity of that of chloramphenicol against S. aureus, 40.0–73.33% against B. cereus, 40.0–52.0% against Escherichia coli, 66.7–120.0% against Pseudomonas aeruginosa and 33.3–52.3% against Serratia marcescens. Moreover, the variation of the antibacterial activity with concentrations (MICs) is showed in Table 3. It was noted that, the most sensitive organisms to the test compounds were B. cereus and P. aeruginosa.
Table 3.
Antibacterial activity of compounds (2a–x) and choramphenicol (inhibition zone in mm and MICs given in brackets at 100 μmol/mL).
| Comp No. | S. aureus | B. cereus | E. coli | P. aeruginosa | S. marcescens |
|---|---|---|---|---|---|
| 2a | 8(50) | 10(3.125) | – | – | – |
| 2b | 8(25) | 12(100) | – | 13(100) | – |
| 2c | 8(12.5) | 9(6.25) | 8(6.25) | 8(6.25) | 10(50) |
| 2d | 8(100) | 10(0.78) | – | – | – |
| 2e | 8(3.125) | 8(12.5) | 10(50) | 10(50) | 11(50) |
| 2g | 10(25) | 11(25) | 8(25) | 10(50) | 10(50) |
| 2i | 10(50) | 12(100) | 8(50) | – | – |
| 2m | 8(50) | 10(50) | 8(50) | – | 16(100) p.i |
| 2n | 8(100) | – | 12(100) | 13(100) | – |
| 2o | 10(25) | – | 11(100) | – | – |
| 2s | 8(25) | 12(50) | 8(25) | 14(100) | – |
| 2t | 8(50) | 12(50) | 9(25) | 8(50) | 9(50) |
| 2u | 10(100) | – | 10(100) | 10(100) | – |
| 2v | 10(50) | – | – | – | – |
| 2w | 11(100) | 8(6.25) | 8(50) | – | – |
| 2x | 10(12.5) | – | – | – | – |
| CHL | 10(0.39) | 12(0.19) | 11(1.56) | 10(6.25) | 14(3.125) |
Pi = partial inhibition. – = no inhibition. CHL = chloramphenicol as antibacterial standard. AUMC = Assiut University Mycological Center.
Again, from Table 1, Table 3, compounds (2c, 2e, 2g and 2t; with Clog P 7.27, 7.05, 7.42 and 7.92, respectively) have a wide spectrum of the antibacterial activity as being able to inhibit all test bacterial organisms with MICs ranging from 50 to 3.125 μmol/mL. Compounds (2m and 2s; with Clog P 7.27 and 7.05, respectively) were effective against four out of five bacterial strains with MICs ranging from 100 to 25 μmol/mL.
A previous work that deals with design, synthesis and antimicrobial activity of the new S-phenacylated dihydrpyrimidine hydrobromide compounds (1a–x) was reported. Thiazolopyrimidines 2a–x were synthesized and tested for their antibacterial activity. The results indicated that, the cyclized compounds (2a–e, 2g, 2m and 2s–w; with Clog P from 6.47 to 7.92) showed an improvement in the antibacterial activity than the open analogs.
Moreover, the results of the antibacterial activity of compounds 4a–c and 5, Table 1 indicated that at a concentration of 100 μmol/mL the test compounds 4a–c were active against S. aureus and E. coli while compound 5 was active only against S. aureus. Compounds (4a–c and 5; with Clog P from 4.95 to 5.66) were completely inactive against B. cereus, P. aeruginosa and S. marcescens.
2.2.2. Antifungal activity
Results of the antifungal activity of compounds 2a–x, Table 2, indicated that at a concentration of 100 μmol/mL most of the test compounds were active against most of the used bacterial strains. Compounds (2b, 2j, 2l, 2p–r and 2v; with Clog P from 6.97 to 7.42) were completely inactive against all used organisms while compounds (2f, 2k and 2x; with Clog P 6.30, 7.92 and 6.80, respectively) were active only against Candida albicans. Compound (2c; with Clog P 7.27) possessed antifungal activity equal to that of the reference drug against Scopulariopsis brevicaulis while compound (2t; with Clog P 7.92) exceeded that of the reference drug against S. brevicaulis. In addition, the active compounds 2a–x showed 33.3–62.5% antifungal activity of that of Clotrimazole against C. albicans with MICs ranged from 12.5–100 μmol/mL, 33.3–79.2% against Geotrichum candidum with MICs ranged from 3.125 to 100 μmol/mL, 40.0% to 92.0% against Fusarium oxysporum with MICs ranged from 3.125 to 100 μmol/mL, 28.6% to 50.0% against Aspergillus flavus with MICs at 100 μmol/mL, 41.7–75.0% against Trichophyton rubrum with MICs ranged from 3.125 to 100 μmol/mL and 46.2 to 123.1 against S. brevicaulis with MICs ranged from 25 to 100 μmol/mL.
Table 2.
Antifungal activity of compounds (2a–x, 4a–c and 5) and clotrimazole (inhibition zones in mm at 100 μmol/mL).
| Comp No. | C. albicans | G. candidum | F. oxysporum | A. flavus | T. rubrum | S. brevicaulis |
|---|---|---|---|---|---|---|
| 2a | 13 | 14 | 13 | 8 | 18 | 12 |
| 2b | – | – | – | – | – | – |
| 2c | 15 | 15 | 22 | 12 | 22 | 26 |
| 2d | 13 | 16 | 13 | – | 22 | 15 |
| 2e | 14 | 19 | 22 | 13 | 27 | 16 |
| 2f | 11 | – | – | – | – | – |
| 2g | 14 | 15 | 18 | 14 | 25 | 22 |
| 2h | 12 | 8 | 15 | – | 16 | 18 |
| 2i | – | 10 | 10 | – | 16 p.i | 22 |
| 2j | – | – | – | – | – | – |
| 2k | 8 | – | – | – | – | – |
| 2l | – | – | – | – | – | – |
| 2m | 14 | 14 | 16 | 12 | 16 | 20 |
| 2n | - | 13 | 12 | – | – | 14 |
| 2o | 13 | 14 pi | 10 | – | – | 15 |
| 2p | – | – | – | – | – | – |
| 2q | – | – | – | – | – | – |
| 2r | – | – | – | – | – | – |
| 2s | 10 | 16 | 13 | – | 18 | 16 |
| 2t | 14 | 17 | 23 | 13 | 22 | 32 |
| 2u | 8 | 14 | 18 | – | 16 pi | 16 |
| 2v | – | – | – | – | – | – |
| 2w | 13 | 14 | 14 | – | 15 | 16 |
| 2x | 8 | – | – | – | – | – |
| 4a | – | – | – | – | – | – |
| 4b | – | – | – | – | – | – |
| 4c | – | – | – | – | – | – |
| 5 | – | – | – | – | – | – |
| CLO | 24 | 24 | 25 | 28 | 36 | 26 |
| Control (DMSO) | – | – | – | – | – | – |
Pi = partial inhibition. - = no inhibition. CLO = clotrimazole antifungal standard. AUMC = Assiut University Mycological Center.
Moreover, the results indicated that compounds (2a, 2c, 2e, 2g, 2m and 2t; with Clog P 6.55, 7.27, 7.05, 7.42, 7.27 and 7.92, respectively) have a wide spectrum of antifungal activity being able to inhibit all test fungal organisms with MICs ranging from 100 to 3.125 μmol/mL. Compounds (2d, 2h, 2s, 2u and 2w; with Clog P 6.47, 8.28, 7.05, 7.77 and 7.55, respectively) were effective against five out of six fungal strains with MICs ranging from 100 to 3.125 μmol/mL, Table 4.
Table 4.
Antifungal activity of compounds (2a–x) and clotrimazole (inhibition zone in mm and MICs given in brackets at 100 μmol/mL).
| Comp No. | C. albicans | G. candidum | F. oxysporum | A. flavus | T. rubrum | S. brevicaulis |
|---|---|---|---|---|---|---|
| 2a | 8(50) | 8(25) | 8(12.5) | 8(100) | 8(12.5) | 12(100) |
| 2c | 8(25) | 8(3.125) | 10(6.25) | 12(100) | 8(3.125) | 8(25) |
| 2d | 8(12.5) | 8(3.125) | 10(6.25) | – | 8(3.125) | 16(50) p.i |
| 2e | 10(12.5) | 10(6.25) | 10(3.125) | 13(100) | 10(3.125) | 8(25) |
| 2f | 10(25) | – | – | – | – | – |
| 2g | 8(12.5) | 8(12.5) | 10(25) | 14(100) | 10(25) | 10(25) |
| 2h | 12(100) | 8(50) | 8(25) | – | 10(50) | 8(25) |
| 2i | – | 10(100) | 10(100) | – | 16(100) p.i | 8(50) |
| 2k | 8(100) | – | – | – | – | – |
| 2m | 14(100) | 10(50) | 10(50) | 12(100) | 10(50) | 20(100) |
| 2n | – | 10(12.5) | 8(50) | – | – | 14(50) |
| 2o | 13(100) | 8(25) | 10(100) | – | – | 13(50) |
| 2s | 10(100) | 12(12.5) | 13(100) | – | 13(50) | 16(100) |
| 2t | 10(50) | 12(25) | 12(25) | 13(100) | 8(25) | 11(25) |
| 2u | 8(100) | 10(12.5) | 10(50) | – | 8(50) | 23(50) p.i |
| 2w | 8(50) | 8(6.25) | 8(25) | – | 12(50) | 8(50) |
| 2x | 8(25) | – | – | – | – | – |
| CLO | 12(1.56) | 12(0.39) | 15(0.39) | 18(0.05) | 10(0.05) | 24(1.56) p.i |
Pi = partial inhibition. – = no inhibition. CLO = clotrimazole antifungal standard. AUMC = Assiut University Mycological Center.
Also, in continuation to our previous work (Hussein et al., 2011), thiazolopyrimidines 2a–x were synthesized and tested for their antifungal activity. The results revealed that, the cyclized compounds (2a, 2c–e, 2g, 2h, 2m, 2o, 2s–u and 2w; with Clog P from 6.47 to 8.28) showed an improvement in the antifungal activity than the open analogs.
On the other hand, the results of the antifungal activity of compounds (4a–c and 5; with Clog P from 4.95 to 5.66), indicated that at a concentration of 100 μmol/mL all the compounds were completely inactive against all the test microorganisms.
It is noteworthy to mention that, within each series, an increase in the Clog P values is accompanied by an increase in the antimicrobial activity. The most active compounds comprise in their structures an electron withdrawing group (R1 = halogen; R2 = halogen) while the least active compounds comprise in their structures an electron donating group (R1 = H, CH3; R2 = OCH3, CH3).
2.3. Pharmacological screening
2.3.1. Anti-inflammatory activity
The most active compounds 2a–e, 2o and 2t–x as antimicrobial agents were selected for the evaluation of their in vivo anti-inflammatory effects.
The results of anti-inflammatory activity (Table 5), revealed that all the test compounds showed a gradual increase of anti-inflammatory activity up to its maximum at 5 h ranging from 73.72:97.58% with compounds 2a–c and 2v being the most active giving 92.86:97.58% of that of indomethacin.
Table 5.
Percentage of edema inhibition of compounds (2a–e, 2o, 2t–x) and indomethacin on carrageenan induced paw edema in rats.
| Comp. No. | Edema inhibition (%)±SE |
|||||
|---|---|---|---|---|---|---|
| 0.5 h | 1 h | 2 h | 3 h | 4 h | 5 h | |
| Negative control | – | – | – | – | – | – |
| Indomethacin | 16.07 ± 0.29⁎ | 35.76 ± 0.00⁎⁎ | 58.03 ± 0.17⁎⁎ | 75.69 ± 0.29⁎⁎ | 88.26 ± 0.17⁎⁎ | 89.39 ± 0.17⁎⁎ |
| 2a | 8.15 ± 0.17⁎⁎⁎ | 28.69 ± 0.17⁎⁎ | 41.97 ± 0.29⁎⁎ | 75.69 ± 0.29⁎⁎ | 74.04 ± 0.17⁎⁎ | 83.01 ± 0.17⁎⁎ |
| 2b | 8.15 ± 0.17∗∗∗ | 28.69 ± 0.17⁎⁎ | 41.97 ± 0.17⁎⁎ | 67.59 ± 0.00⁎⁎ | 81.13 ± 0.17⁎⁎ | 83.01 ± 0.17⁎⁎ |
| 2c | 11.99 ± 0.33⁎ | 32.12 ± 0.44⁎⁎ | 51.64 ± 0.29⁎⁎ | 64.83 ± 0.17⁎⁎ | 83.40 ± 0.17⁎⁎ | 85.06 ± 0.17⁎⁎ |
| 2d | 0.00 ± 0.17⁎⁎⁎⁎ | 14.35 ± 0.00⁎ | 25.91 ± 0.17⁎⁎ | 46.03 ± 0.17⁎⁎ | 57.44 ± 0.29⁎⁎ | 65.90 ± 0.33⁎⁎ |
| 2e | 4.08 ± 0.00⁎⁎⁎⁎ | 17.99 ± 0.17⁎⁎ | 32.30 ± 0.29⁎⁎ | 51.38 ± 0.00⁎⁎ | 64.54 ± 0.29⁎⁎ | 72.28 ± 0.17⁎⁎ |
| 2o | 8.15 ± 0.02∗∗ | 21.41 ± 0.02⁎⁎ | 45.26 ± 0.02⁎⁎ | 59.48 ± 0.03⁎⁎ | 71.63 ± 0.03⁎⁎ | 78.67 ± 0.02⁎⁎ |
| 2t | 4.08 ± 0.00⁎⁎⁎⁎ | 14.35 ± 0.00⁎ | 25.91 ± 0.17⁎⁎ | 54.13 ± 0.17⁎⁎ | 64.53 ± 0.29⁎⁎ | 68.07 ± 0.29⁎⁎ |
| 2u | 4.08 ± 0.00⁎⁎⁎⁎ | 17.99 ± 0.17⁎⁎ | 32.30 ± 0.29⁎⁎ | 48.62 ± 0.20⁎⁎ | 59.86 ± 0.17⁎⁎ | 68.07 ± 0.29⁎⁎ |
| 2v | 16.07 ± 0.00⁎ | 35.76 ± 0.00⁎⁎ | 58.03 ± 0.50⁎⁎ | 67.59 ± 0.29⁎⁎ | 78.72 ± 0.29⁎⁎ | 87.23 ± 0.29⁎⁎ |
| 2w | 4.08 ± 0.00⁎⁎⁎⁎ | 17.99 ± 0.17⁎⁎ | 32.30 ± 0.29⁎⁎ | 48.62 ± 0.17⁎⁎ | 64.53 ± 0.29⁎⁎ | 65.90 ± 0.33⁎⁎ |
| 2x | 4.08 ± 0.00⁎⁎⁎⁎ | 17.99 ± 0.17⁎⁎ | 32.30 ± 0.29⁎⁎ | 46.03 ± 0.17⁎⁎ | 59.86 ± 0.17⁎⁎ | 68.07 ± 0.29⁎⁎ |
Significant difference at p < 0.01 versus control value.
Significant difference at p < 0.001 versus control value.
Significant difference at p < 0.05 versus control value.
non significantly different at p > 0.05 versus control value (student’s test).
It is noteworthy to mention that, the result of anti-inflammatory activity indicates that most of the compounds that showed the highest anti-inflammatory activity having in their structure R = p-Cl–C4H4 or p-OCH3C6H4 moieties indicate the crucial role of halogen or the OCH3 in the anti-inflammatory activity. These results comply with the previously reported findings of Kalgutkar et al. (2000).
2.3.2. Analgesic activity
The most active compounds 2a–c, 2e, 2o and 2v with regards to their anti-inflammatory activity were selected for testing their analgesic activity in mice using the hot plate method (Janssen and Jageneau, 1957) in comparison to aspirin as a reference drug.
Results of the analgesic activity (Table 6), revealed that compounds 2e and 2v showed a gradual increase of analgesic activity up to its maximum after 5 h of injection with 84.04% and 82.26% of that of aspirin respectively, whereas the maximum activity of compound 2o appears after 4 h of injection with 62.64% of the aspirin activity. On the other hand, compounds 2a and 2b revealed the maximum activity at 3 h interval with 35.36% and 37.80% of that of aspirin, respectively while compound 2c was the least active giving 34.39% of aspirin at 5 h interval.
Table 6.
Analgesic activity of compounds (2a–c, 2e, 2o and 2v) and aspirin using the hot-plate test.
| Comp No. | The average reaction time (s) at different time intervals after compound administration ± SE |
|||||
|---|---|---|---|---|---|---|
| 0.5 h | 1 h | 2 h | 3 h | 4 h | 5 h | |
| Control | 24.00 ± 2.08 | 25.00 ± 1.73 | 22.67 ± 1.20 | 22.67 ± 1.20 | 23.33 ± 1.20 | 23.00 ± 1.15 |
| Aspirin | 26.33 ± 3.18⁎ | 40.00 ± 2.89⁎⁎ | 68.00 ± 4.73⁎⁎ | 82.67 ± 2.73⁎⁎ | 91.67 ± 4.06⁎⁎ | 94.00 ± 3.22⁎⁎ |
| 2a | 24.33 ± 2.40⁎ | 25.33 ± 2.40⁎ | 28.00 ± 2.08⁎ | 29.00 ± 1.53⁎ | 26.33 ± 1.76⁎ | 27.33 ± 1.45⁎ |
| 2b | 24.00 ± 1.53⁎ | 28.00 ± 1.53⁎ | 29.33 ± 1.45⁎ | 31.00 ± 2.30⁎ | 31.33 ± 1.86⁎ | 31.33 ± 1.86⁎ |
| 2c | 25.00 ± 2.08⁎ | 26.33 ± 1.86⁎ | 30.33 ± 2.91⁎ | 25.33 ± 2.96⁎ | 29.00 ± 2.08⁎ | 32.33 ± 1.76⁎ |
| 2e | 38.00 ± 3.79⁎⁎⁎ | 45.33 ± 3.18⁎⁎ | 60.67 ± 5.61⁎⁎ | 57.00 ± 4.73⁎⁎ | 69.00 ± 3.51⁎⁎ | 79.00 ± 0.58⁎⁎ |
| 2o | 17.00 ± 1.16⁎ | 21.00 ± 0.58⁎ | 25.33 ± 2.60⁎ | 38.33 ± 1.67⁎⁎ | 57.00 ± 2.00⁎⁎ | 55.33 ± 3.38⁎⁎ |
| 2v | 25.33 ± 2.60⁎ | 40.00 ± 2.89⁎⁎ | 68.00 ± 2.08⁎⁎ | 75.67 ± 2.40⁎⁎ | 71.67 ± 4.80⁎⁎ | 77.33 ± 3.28⁎⁎ |
Non significantly different at p > 0.05 versus control value (student’s test).
Significantly different at p < 0.001 versus control value.
p < 0.01 versus control value.
The previous finding of analgesic activity indicates compounds with the highest analgesic activity having R = p-Cl–C4H4, p-OCH3C6H4 or p-CH3C6H4 moieties.
2.3.3. Acute toxicity (LD50)
The most active compounds 2e and 2v with regards to their anti-inflammatory and analgesic activity were selected for testing their acute toxicity using aspirin as a reference drug.
The median lethal dose (LD50) of compounds 2e and 2v was determined in mice according to a reported method (Sztaricskai et al., 1999) and was found to be nontoxic up to >1 g/kg whereas the LD50 of aspirin equals to 533 mg/kg (i.p.) (Abdel-Alem et al., 1980).
3. Conclusion
This work was devoted for the syntheses of some new thiazolo[3,2-a]pyrimidine derivatives 2a–x, 4a–c and 5. The structures of the target compounds were confirmed. The target compounds were tested for their antimicrobial activity and the most active compounds were screened for their MICs. They showed moderate activity against most of the used bacterial and fungal strains. Moreover, the lipophilicity of the target compounds is expressed as Clog P and the most active antimicrobial compounds comprise in their structures an electron withdrawing group (R1 = halogen; R2 = halogen) while the least active compounds comprise in their structures an electron donating group (R1 = H, CH3; R2 = OCH3, CH3).
In addition, the most active compounds 2a–c, 2e, 2o and 2v as antimicrobial were tested for their anti-inflammatory effects and they showed a gradual increase in their anti-inflammatory activity reaching its maximum at 5 h in the carrageenan induced paw edema test. The p-Cl–C4H4 or p-OCH3C6H4 derivatives showed the highest anti-inflammatory activity, which indicate the crucial role of halogen or OCH3 groups.
Furthermore, the most active compounds 2a–c, 2e, 2o and 2v with regards to their anti-inflammatory activity were selected for testing their analgesic activity. The tested compounds (2e and 2v) showed a gradual increase in their analgesic activity up to its maximum after 5 h of injection. In addition, the LD50 of them was determined in mice and was found nontoxic up to >1 g/kg whereas the LD50 of aspirin is 533 mg/kg (i.p.).
4. Experimental
Melting points were determined on an electrothermal melting point apparatus (Stuart Scientific, Model SMP1, Staffordshire, UK) and were uncorrected. Monitoring of the chemical reactions was carried out by TLC using precoated silica gel plates (kieselgel 0.25 mm, 60 G F254, Merk, Darmstadt, Germany) and CHCl3/CH3COCH3 (9:1 v/v) as the mobile phase. Visualization of the spots was effected by ultraviolet lamp (Spectroline Model CM-10, Seattle, USA), at wavelengths 254 and 365 nm and/or iodine stain.
IR and FT-IR spectra were carried out as KBr disks on a Shimadzu IR-470 spectrometer (Shimadzu, Kyoto, Japan) and Thermo scientific 912A0683 spectrometer (Thermo scientific, Japan) respectively, at the Faculty of Pharmacy, Assiut University, Assiut, Egypt. 1H NMR spectra were scanned on a Varian EM-360L NMR spectrophotometer (60 MHz, Varian, CA, USA) at the Faculty of Pharmacy, Assiut University. 1H, 13C and DEPT 13C NMR spectra were scanned on JEOLJNM-LA series FT-NMR system (400 MHz, JEOL, Tokyo, Japan) at the Assiut University Unit of Trace Analysis, Assiut University, Assiut, Egypt. Chemical shifts are expressed in δ-values (ppm) relative to tetramethylsilane (TMS) as an internal standard, using CDCl3 or DMSOd6 as solvents, and deuterium oxide was used for the detection of the exchangeable protons.
Mass spectra were recorded on a JMS-600H mssroute mass spectrometer (JEOL, Tokyo, Japan) at the Assiut University Unit of Trace Analysis, Assiut University, Assiut, Egypt and on Gas chromatography Mass, Quadrable-2010 Plus (Shimadzo, Kyoto, Japan) at the Micro Analytical Center, Faculty of Science, Cairo University, Cairo, Egypt.
Elemental microanalyses were performed on a Vario EL-CHNS Elemental Analyzer (Elementary Analysis GmbH, Germany), at Assiut University Unit of Trace Analysis, Assiut University, Assiut, Egypt and on a Vario elemental analyzer III (Vario, Hanau, Germany) at the Micro Analytical Center, Faculty of Science, Cairo University, Cairo, Egypt.
4.1. Calculations of logarithm partition coefficient values (Clog P)
The log P values of the target compounds 2a–x, 4a–c and 5, were computed with a routine method called the calculated log P (Clog P) contained in a PC-software package (McLogP 2.0, BioByte Corp., CA, USA). A representation of the molecular structure where the hydrogens were omitted or “suppressed” (SMILES notation) entered into the program, which computes the log P bases on Crippen’s fragmentation (Leo, 1993).
Most of chemicals used were of commercial grade: Aniline, anhydrous sodium acetate (El Nasr Pharm. Co. Egypt), p-chloroaniline, isatin (Aldrich, Germany), bromoacetic acid and p-methyl aniline (Fluka, Germany). All other chemicals and solvents are of the reagent grade.
4.2. General method for synthesis of ethyl 3,5-di(substituted)phenyl-7-methyl-5H-thiazolo[3,2-a]pyrimidine-6-carboxylate, 2a–x
The appropriate compound, 1a–x, was heated at 110–120 °C for 2 h with 10–15 times their weight of freshly prepared polyphosphoric acid (Dennler and Frasca, 1966). The reaction mixture was cooled, poured with stirring into ice-water, neutralized with concentrated ammonia and stirred overnight until crystallization was complete. The separated solid product was filtered, washed with water, dried and crystallized from hexane except compounds 2h, 2i, 2n, 2q, 2w that were crystallized from ethyl acetate–hexane mixture (2:1), Scheme 1.
4.2.1. Ethyl 3-(4-bromophenyl)-7-methyl-5-phenyl-5H-thiazolo[3,2-a]pyrimidine-6-carboxylate, 2b
m.p. 154–155 °C; yield 83%;Rƒ. 0.28 (CHCl3:CH3COCH3 9:1); Clog P 7.42; IR (KBr) (cm−1): 1676 (C O), 1560 (C N); 1H NMR (CDCl3): (ppm) 1.15 (3H, t, CH2CH3), 2.50 (3H, s, CH3), 4.06 (2H, q, CH2CH3), 6.13 (2H, s, pyr. H-5, thia H-2), 6.53–7.56 (9H, m, Ar–H); EI-MS m/z (%) 455 (M+, 26). Anal. Calcd (%) for C22H19BrN2O2S: C, 58.03; H, 4.21; N, 6.15. Found, C, 58.00; H, 4.19; N, 6.06.
4.2.2. Ethyl 3-(4-methoxyphenyl)-7-methyl-5-phenyl-5H-thiazolo[3,2-a]pyrimidine-6-carboxylate, 2d
m.p. 135–137 °C; yield 88%; Rƒ. 0.58 (CHCl3:CH3COCH3 9:1); Clog P 6.47; IR (KBr) (cm−1): 1661 (C O), 1574 (C N); 1H NMR (CDCl3): (ppm) 1.15 (3H, t, CH2CH3), 2.50 (3H, s, CH3), 4.06 (2H, q, CH2CH3), 6.13 (2H, s, pyr. H-5, thia H-2), 6.53–7.56 (9H, m, Ar–H); EI-MS m/z (%) 406 (M+, 51.82), 407 (M++1, 50), 408 (M++2, 49). Anal. Calcd (%) for C23H22N2O3S: C, 67.96; H, 5.46; N, 6.89. Found, C, 67.83; H, 5.47; N, 6.83.
4.2.3. Ethyl 7-methyl-3-(4-methylphenyl)-5-phenyl-5H-thiazolo[3,2-a]pyrimidine-6-carboxylate, 2e
m.p. 110–112 °C; yield 89%; Rƒ. 0.28 (CHCl3:CH3COCH3 9:1); Clog P 7.05; IR (KBr) (cm−1): 1679 (C O), 1564 (C N); 1H NMR (CDCl3): (ppm) 1.21 (3H, t, CH2CH3), 2.47 (3H, s, –C6H4–CH3), 2.58 (3H, s, CH3), 4.12 (2H, q, CH2CH3), 6.18 (1H, s, pyr. H-5), 6.24 (1H, s, thia H-2), 6.69–7.66 (9H, m, Ar–H); EI-MS: m/z (%) 390 (M+, 23), 391 (M++1, 6), 392 (M++2, 2). Anal. Calcd (%) for C23H22N2O2S: C, 70.74; H, 5.68; N, 7.17. Found, C, 70.63; H, 5.59; N, 7.26.
4.2.4. Ethyl 7-methyl-3-(4-nitrophenyl)-5-phenyl-5H-thiazolo[3,2-a]pyrimidine-6-carboxylate, 2f
m.p. 142 °C; yield 72%; Rƒ. 0.24 (CHCl3:CH3COCH3 9:1); Clog P 6.30; IR (KBr) (cm−1): 1678 (C O), 1589 (C N); 1H NMR (CDCl3): (ppm) 1.15 (3H, t, CH2CH3), 2.40 (3H, s, CH3), 4.06 (2H, q, CH2CH3), 6.09 (1H, s, pyr. H-5), 6.24 (1H, s, thia H-2), 6.66–8.33 (9H, m, Ar–H). Anal. Calcd (%) for C22H19N3O4S: C, 62.69; H, 4.54; N, 9.97. Found, C, 62.66; H, 4.75; N, 9.92.
4.2.5. Ethyl 5-(4-bromophenyl)-7-methyl-3-phenyl-5H-thiazolo[3,2-a]pyrimidine-6-carboxylate, 2g
m.p. 111–113 °C; yield 70%; Rƒ. 0.36 (CHCl3:CH3COCH3 9:1); Clog P 7.42; IR (KBr) (cm−1): 1679 (C O), 1570 (C N); 1H NMR (CDCl3): δ 1.15 (3H, t, CH2CH3), 2.46 (3H, s, CH3), 4.06 (2H, q, CH2CH3), 6.12 (2H, s, pyr. H-5, thia H-2), 6.40–7.53 (9H, m, Ar–H). Anal. Calcd (%) for C22H19BrN2O2S: C, 58.03; H, 4.21; N, 6.15. Found, C, 57.83; H, 4.30; N, 6.06.
4.2.6. Ethyl 3,5-di(4-bromophenyl)-7-methyl-5H-thiazolo[3,2-a]pyrimidine-6-carboxylate, 2h
m.p. 192–193 °C; yield 90%; Rƒ. 0.36 (CHCl3:CH3COCH3 9:1); Clog P 8.28; IR (KBr) (cm−1): 1678 (C O), 1562 (C N); 1H NMR (CDCl3): (ppm) 1.12 (3H, t, CH2CH3), 2.46 (3H, s, CH3), 4.00 (2H, q, CH2CH3), 6.00 (1H, s, pyr. H-5), 6.03 (1H, s, thia H-2), 6.39–7.50 (9H, m, Ar–H). Anal. Calcd (%) for C22H18Br2N2O2S: C, 49.46; H, 3.40; N, 5.24. Found, C, 49.63; H, 3.69; N, 5.07.
4.2.7. Ethyl 5-(4-bromophenyl)-3-(4-chlorophenyl)-7-methyl-5H-thiazolo[3,2-a]pyrimidine-6-carboxylate, 2i
m.p. 185–187 °C; yield 85%; Rƒ. 0.36 (CHCl3:CH3COCH3 9:1); Clog P 8.13; IR (KBr) (cm−1): 1678 (C O), 1564 (C N); 1H NMR (CDCl3): (ppm) 1.15 (3H, t, CH2CH3), 2.43 (3H, s, CH3), 4.03 (2H, q, CH2CH3), 6.00 (1H, s, pyr. H-5), 6.15 (1H, s, thia H-2), 6.42–7.50 (8H, m, Ar–H); EI-MS m/z (%) 489 (M+, 10), 490 (M++1, 0.28). Anal. Calcd (%) for C22H18BrClN2O2S: C, 53.95; H, 3.70; N, 5.72. Found, C, 53.70; H, 3.63; N, 6.02.
4.2.8. Ethyl 5-(4-bromophenyl)-3-(4-methoxyphenyl)-7-methyl-5H-thiazolo[3,2-a]pyrimidine-6-carboxylate, 2j
m.p. 185–187 °C; yield 85%; Rƒ. 0.32 (CHCl3:CH3COCH3 9:1); Clog P 7.34; IR (KBr) (cm−1): 1679 (C O), 1565 (C N); 1H NMR (CDCl3): (ppm) 1.15 (3H, t, CH2CH3), 2.40 (3H, s, CH3), 3.81 (3H, s, OCH3), 4.03 (2H, q, CH2CH3), 6.06 (2H, s, pyr. H-5, thia H-2), 6.50–7.30 (8H, m, Ar–H); EI-MS m/z (%) 485 (M+, 4), 486 (M++1, 18), 487 (M++2, 7). Anal. Calcd (%) for C23H21BrN2O3S: C, 56.91; H, 4.36; N, 5.77. Found, C, 56.87; H, 4.40; N, 6.10.
4.2.9. Ethyl 5-(4-bromophenyl)-7-methyl-3-(4-methylphenyl)-5H-thiazolo[3,2-a]pyrimidine-6-carboxylate, 2k
m.p. 183–185 °C; yield 88%; Rƒ. 0.36 (CHCl3:CH3COCH3 9:1); Clog P 7.92; IR (KBr) (cm−1): 1678 (C O), 1564 (C N); 1H NMR (CDCl3): (ppm) 1.22 (3H, t, CH2CH3), 2.46 (3H, s, –C6H4–CH3), 2.48 (3H, s, CH3), 4.11 (2H, q, CH2CH3), 6.18 (2H, s, pyr. H-5, thia H-2), 6.68–7.31 (8H, m, Ar–H); 13C NMR (CDCl3): (ppm) 14.26 (CH2CH3); 21.37 (–C6H4–CH3), 23.59 (7-CH3), 57.16 (CH2CH3), 59.77 (C-5), 100.66 (C-6), 102.50 (C-2), 122.02–139.89 (Ar–C + Ar–CH), 141.17 (C-3), 156.05 (C-7), 166.43 (C-8a), 166.61 (CO); DEPT 13C NMR (CDCl3): (ppm) 14.24(CH2CH3), 21.38 (7-CH3), 23.62 (–C6H4–CH3), 57.16 (inverted CH2CH3), 59.77 (C-5), 102.55 (C-2), 128.25–131.25 (Ar–CH). Anal. Calcd (%) for C23H21BrN2O2S: C, 58.85; H, 4.51; N, 5.97. Found, C, 58.82; H, 4.57; N, 6.33.
4.2.10. Ethyl 5-(4-bromophenyl)-7-methyl-3-(4-nitrophenyl)-5H-thiazolo[3,2-a]pyrimidine-6-carboxylate, 2l
m.p. 194–195 °C; yield 87%; Rƒ. 0.22 (CHCl3:CH3COCH3 9:1); Clog P 7.16; IR (KBr) (cm−1): 1679 (C O), 1564 (C N); 1H NMR (CDCl3): (ppm) 1.15 (3H, t, CH2CH3), 2.50 (3H, s, CH3), 4.10 (2H, q, CH2CH3), 6.06 (1H, s, pyr. H-5), 6.27 (1H, s, thia H-2), 6.50–8.40 (8H, m, Ar–H). Anal. Calcd (%) for C22H18BrN3O4S: C, 52.81; H, 3.63; N, 8.40. Found, C, 52.99; H, 3.87; N, 8.02.
4.2.11. Ethyl 5-(4-chlorophenyl)-7-methyl-3-phenyl-5H-thiazolo[3,2-a]pyrimidine-6-carboxylate, 2m
m.p. 155–156 °C; yield 85%; Rƒ. 0.38 (CHCl3:CH3COCH3 9:1); Clog P 7.27; IR (KBr) (cm−1): 1680 (C O), 1563 (C N); 1H NMR (CDCl3): (ppm) 1.16 (3H, t, CH2CH3), 2.50 (3H, s, CH3), 4.13 (2H, q, CH2CH3), 6.15 (2H, s, pyr. H-5, thia H-2), 6.63–7.63 (9H, m, Ar–H). Anal. Calcd (%) for C22H19ClN2O2S: C, 64.30; H, 4.66; N, 6.82. Found, C, 64.47; H, 4.71; N, 6.60.
4.2.12. Ethyl 3-(4-bromophenyl)-5-(4-chlorophenyl)-7-methyl-5H-thiazolo[3,2-a]pyrimidine-6-carboxylate, 2n
m.p. 157–158 °C; yield 76%; Rƒ. 0.42 (CHCl3:CH3COCH3 9:1); Clog P 8.13; IR (KBr) (cm−1): 1636 (C O), 1562 (C N); 1H NMR (CDCl3): (ppm) 1.20 (3H, t, CH2CH3), 2.48 (3H, s, CH3), 4.12 (2H, q, CH2CH3), 6.12 (1H, s, pyr. H-5), 6.22 (1H, s, thia H-2), 6.66–7.72 (8H, m, Ar–H). EI-MS m/z (%) 489 (M+, 51), 490 (M++1, 25), 491 (M++2, 29). Anal. Calcd (%) for C22H18BrClN2O2S: C, 53.95; H, 3.70; N, 5.72. Found, C, 53.83; H, 3.89; N, 5.59.
4.2.13. Ethyl 3,5-di(4-chlorophenyl)-7-methyl-5H-thiazolo[3,2-a]pyrimidine-6-carboxylate, 2o
m.p. 154–155 °C; yield 91%; Rƒ. 0.39 (CHCl3:CH3COCH3 9:1); Clog P 7.98; IR (KBr) (cm−1): 1677 (C O), 1564 (C N); 1H NMR (CDCl3): (ppm) 1.15 (3H, t, CH2CH3), 2.46 (3H, s, CH3), 4.06 (2H, q, CH2CH3), 6.03 (1H, s, pyr. H-5), 6.12 (1H, s, thia H-2), 6.60–7.48 (8H, m, Ar–H). EI-MS: m/z (%) 445 (M+, 12), 446 (M++1, 20), 447 (M++2, 5). Anal. Calcd (%) for C22H18Cl2N2O2S: C, 59.33; H, 4.07; N, 6.29. Found, C, 59.48; H, 4.00; N, 6.30.
4.2.14. Ethyl 5-(4-chlorophenyl)-3-(4-methoxyphenyl)-7-methyl-5H-thiazolo[3,2-a]pyrimidine-6-carboxylate, 2p
m.p. 154–156 °C; yield 75%; Rƒ. 0.36 (CHCl3:CH3COCH3 9:1); Clog P 7.19; IR (KBr) (cm−1): 1680 (C O), 1561 (C N); 1H NMR (CDCl3): (ppm) 1.15 (3H, t, CH2CH3), 2.46 (3H, s, CH3), 3.78 (3H, s, OCH3), 4.06 (2H, q, CH2CH3), 6.06 (2H, s, pyr. H-5, thia H-2), 6.50–7.13 (8H, m, Ar–H). Anal. Calcd (%) for C23H21ClN2O3S: C, 62.65; H, 4.80; N, 6.35. Found, C, 62.42; H, 4.75; N, 6.10.
4.2.15. Ethyl 5-(4-chlorophenyl)-7-methyl-3-(4-methylphenyl)-5H-thiazolo[3,2-a]pyrimidine-6-carboxylate, 2q
m.p. 100–102 °C; yield 80%; Rƒ. 0.44 (CHCl3:CH3COCH3 9:1); Clog P 7.77; IR (KBr) (cm−1): 1679 (C O), 1567 (C N); 1H NMR (CDCl3): (ppm) 1.15 (3H, t, CH2CH3), 2.39 (6H, s, 2CH3), 4.06 (2H, q, CH2CH3), 6.15 (1H, s, pyr. H-5), 6.18 (1H, s, thia H-2), 6.48–7.39 (8H, m, Ar–H). Anal. Calcd (%) for C23H21ClN2O2S: C, 65.01; H, 4.98; N, 6.59. Found, C, 65.83; H, 4.91; N, 6.23.
4.2.16. Ethyl 5-(4-chlorophenyl)-7-methyl-3-(4-nitrophenyl)-5H-thiazolo[3,2-a]pyrimidine-6-carboxylate, 2r
m.p. 120–121 °C; yield 92%; Rƒ. 0.32 (CHCl3:CH3COCH3 9:1); Clog P 7.01; IR (KBr) (cm−1): 1679 (C O), 1565 (C N); 1H NMR (CDCl3): δ 1.20 (3H, t, CH2CH3), 2.46 (3H, s, CH3), 4.13 (2H, q, CH2CH3), 6.13 (1H, s, pyr. H-5), 6.50 (1H, s, thia H-2), 6.60–8.36 (8H, m, Ar–H). Anal. Calcd (%) for C22H18ClN3O4S: C, 57.96; H, 3.98; N, 9.22. Found, C, 57.79; H, 4.00; N, 8.79.
4.2.17. Ethyl 7-methyl-5-(4-methylphenyl)-3-phenyl-5H-thiazolo[3,2-a]pyrimidine-6-carboxylate, 2s
m.p. 150–151 °C; yield 88%; Rƒ. 0.40 (CHCl3:CH3COCH3 9:1); Clog P 7.05; IR (KBr) (cm−1): 1679 (C O), 1567 (C N); 1H NMR (CDCl3): (ppm) 1.15 (3H, t, CH2CH3), 2.16 (3H, s, C6H5–CH3), 2.46 (3H, s, CH3), 4.00 (2H, q, CH2CH3), 6.03 (2H, s, pyr. H-5, thia H-2), 6.39–7.47 (9H, m, Ar–H). Anal. Calcd (%) for C23H22N2O2S: C, 70.74; H, 5.68; N, 7.17. Found, C, 70.51; H, 6.38; N, 7.08.
4.2.18. Ethyl 3-(4-bromophenyl)-7-methyl-5-(4-methylphenyl)-5H-thiazolo[3,2-a]pyrimidine-6-carboxylate, 2t
m.p. 174–176 °C; yield 79%; Rƒ. 0.42 (CHCl3:CH3COCH3 9:1); Clog P 7.92; IR (KBr) (cm−1): 1678 (C O), 1563 (C N); 1H NMR (CDCl3): (ppm) 1.20 (3H, t, CH2CH3), 2.20 (3H, s, –C6H4–CH3), 2.50 (3H, s, CH3), 4.06 (2H, q, CH2CH3), 6.00 (1H, s, pyr. H-5), 6.11 (1H, s, thia H-2), 6.50–7.66 (8H, m, Ar–H). Anal. Calcd (%) for C23H21BrN2O2S: C, 58.85; H, 4.51; N, 5.97. Found, C, 58.84; H, 4.57; N, 6.15.
4.2.19. Ethyl 3-(4-chlorophenyl)-7-methyl-5-(4-methylphenyl)-5H-thiazolo[3,2-a]pyrimidine-6-carboxylate, 2u
m.p. 173–175 °C; yield 85%; Rƒ. 0.39 (CHCl3:CH3COCH3 9:1); Clog P 7.77; IR (KBr) (cm−1): 1677 (C O), 1566 (C N); 1H NMR (CDCl3): δ 1.16 (3H, t, CH2CH3), 2.20 (3H, s, –C6H4–CH3), 2.42 (3H, s, CH3), 4.06 (2H, q, CH2CH3), 6.03 (1H, s, pyr. H-5), 6.10 (1H, s, thia H-2), 6.50–7.50 (8H, m, Ar–H). EI-MS m/z (%) 424 (M+, 53), 425 (M++1, 20), 426 (M++2, 36). Anal. Calcd (%) for C23H21ClN2O2S: C, 65.01; H, 4.98; N, 6.59. Found, C, 65.06; H, 5.10; N, 6.61%.
4.2.20. Ethyl 3-(4-methoxyphenyl)-7-methyl-5-(4-methylphenyl)-5H-thiazolo[3,2-a]pyrimidine-6-carboxylate, 2v
m.p. 162–164 °C; yield 89%; Rƒ. 0.33 (CHCl3:CH3COCH3 9:1); Clog P 6.97; IR (KBr) (cm−1): 1668 (C O), 1567 (C N); 1H NMR (CDCl3): (ppm) 1.13 (3H, t, CH2CH3), 2.17 (3H, s, –C6H4–CH3), 2.46 (3H, s, CH3), 3.75 (3H, s, OCH3), 4.00 (2H, q, CH2CH3), 5.94 (1H, s, pyr. H-5), 6.00 (1H, s, thia H-2), 6.42–7.00 (8H, m, Ar–H). Anal. Calcd (%) for C24H24N2O3S: C, 68.55; H, 5.75; N, 6.66. Found, C, 67.93; H, 6.79; N, 6.49.
4.2.21. Ethyl 7-methyl-3,5-di(4-methylphenyl)-5H-thiazolo[3,2-a]pyrimidine-6-carboxylate, 2w
m.p. 195–196 °C; yield 88%; Rƒ. 0.39 (CHCl3:CH3COCH3 9:1); Clog P 7.55; IR (KBr) (cm−1): 1680 (C O), 1570 (C N); 1H NMR (CDCl3): (ppm) 1.16 (3H, t, CH2CH3), 2.20 (3H, s, –C6H4–CH3), 2.50 (6H, s, –C6H4–CH3, CH3), 4.06 (2H, q, CH2CH3), 6.03 (1H, s, pyr. H-5), 6.10 (1H, s, thia H-2), 6.50–8.00 (8H, m, Ar–H). Anal. Calcd (%) for C24H24N2O2S: C, 71.26; H, 5.98; N, 6.93. Found, C, 71.13; H, 5.95; N, 6.64.
4.2.22. Ethyl 7-methyl-5-(4-methylphenyl)-3-(4-nitrophenyl)-5H-thiazolo[3,2-a]pyrimidine-6-carboxylate, 2x
m.p. 134–135 °C; yield 91%; Rƒ. 0.33 (CHCl3:CH3COCH3 9:1); Clog P 6.80; IR (KBr) (cm−1): 1652 (C O), 1560 (C N); 1H NMR (CDCl3): δ 1.16 (3H, t, CH2CH3), 2.20 (3H, s, –C6H4–CH3), 2.42 (3H, s, CH3), 4.03 (2H, q, CH2CH3), 6.00 (1H, s, pyr. H-5), 6.23 (1H, s, thia H-2), 6.42–8.31 (8H, m, Ar–H). Anal. Calcd (%) for C23H21N3O4S: C, 63.43; H, 4.86; N, 9.65. Found, C, 62.88; H, 5.38; N, 9.51.
4.3. General method for synthesis of ethyl 5-phenyl-2,3-dioxo-2-(un)substituted phenyl hydrazono-7-methyl-2,3-dihydro-5H-thiazolo[3,2-a]pyrimidine-6-carboxylate, 4a–c
The appropriate aryldiazonium chloride (10 mmol) was prepared by adding concentrated hydrochloric acid (3 mL) to the appropriate aromatic amine (10 mmol) at 0–5 °C and treating the resulting hydrochloride solution with a cold solution of sodium nitrite (0.69 g, 10 mmol; in 5 mL water). Then, it was added drop wise with constant stirring at 0–5 °C to a cold solution of compound 3 (3.16 gm, 10 mmol,) in an absolute ethanol (80 mL), containing sodium acetate (3.02 gm, 37 mmol). The reaction mixture was stirred at room temperature for 2 h then diluted with cold water (30 mL) whereby the resultant crude product which precipitated was filtered, washed with water, dried and crystallized from absolute ethanol, Scheme 2.
4.3.1. Ethyl 7-methyl-3-oxo-5-phenyl-2-(2-phenylhydrazono)-2,3-dihydro-5H-thiazolo[3,2-a]pyrimidine-6-carboxylate, 4a
m.p. 130–131 °C; yield 85%; Rƒ. 0.41 (CHCl3:CH3COCH3 9:1); Clog P 4.95; IR (KBr) (cm−1): 1717 (ester C O), 1603 (C N); 1H NMR (CDCl3): (ppm) 1.23 (two sets, 6H, t, CH2CH3), 2.60 (3H, s, CH3), 4.03 (two sets, 4H, q, CH2CH3), 6.33 (1H, s, pyr. H-5), 7.10–7.76 (11H, m, Ar–H, C2-H), 8.83 (1H, s, NH; exchangeable with D2O). Anal. Calcd (%) for C22H20N4O3S: C, 62.84; H, 4.79; N, 13.32. Found, C, 62.55; H, 4.70; N, 13.39.
4.3.2. Ethyl 2-[2-(4-chlorophenyl)hydrazono]-7-methyl-3-oxo-5-phenyl-2,3-dihydro-5H-thiazolo[3,2-a]pyrimidine-6-carboxylate, 4b
m.p. 125–126 °C; yield 62%; Rƒ. 0.39 (CHCl3:CH3COCH3 9:1); Clog P 5.66; IR (KBr) (cm−1): 1710 (ester C O), 1598 (C N); 1H NMR (CDCl3, 400 MHz): (ppm) 1.23 (two sets, 6H, t, CH2CH3), 2.60 (3H, s, CH3), 4.03 (two sets, 4H, q, CH2CH3), 6.30 (1H, s, pyr. H-5), 7.00–7.70 (11H, m, Ar–H, C2-H), 8.13 (1H, s, NH; exchangeable with D2O). EI-MS m/z (%) 454.95 (M+, 27), 455.95 (M++1, 38), 456.90 (M++2, 10). Anal. Calcd (%) for C22H19ClN4O3S: C, 58.08; H, 4.21; N, 12.32. Found, C, 57.19; H, 4.62; N, 12.00.
4.3.3. Ethyl 7-methyl-2-[2-(4-methylphenyl)hydrazono]-3-oxo-5-phenyl-2,3-dihydro-5H-thiazolo[3,2-a]pyrimidine-6-carboxylate, 4c
m.p. 136–137 °C; yield 88%; Rƒ. 0.45 (CHCl3:CH3COCH3 9:1); Clog P 5.45; IR (KBr) (cm−1): 1711 (ester C O), 1604 (C N); 1H NMR (CDCl3, 400 MHz): (ppm) 1.15 (two sets, 6H, t, CH2CH3), 2.19 (3H, s, –C6H4–CH3), 2.45 (3H, s, CH3), 3.66 (2H, q, CH2CH3), 4.04 (2H, q, CH2CH3), 6.12 (1H, s, pyr. H-5), 6.95–7.35 (11H, m, Ar–H, C2-H), 8.62 (1H, s, NH; exchangeable with D2O); 13C NMR (CDCl3): (ppm) 13.98 (CH2CH3), 18.34 (CH2CH3), 20.69 (–C6H4–CH3), 22.55 (7-CH3); 55.25 (C-5), 58.44 (CH2CH3), 60.63 (CH2CH3), 109.69 (C-6), 114.50 (C2-H), 120.69–139.71 (Ar–C + Ar–CH), 151.77 (C-7), 153.36 (C-8a), 161.03 (C-3), 165.26 (ester C O); DEPT 13C NMR (CDCl3): δ 14.00 (CH2CH3), 18.32 (CH2CH3), 20.73 (–C6H4–CH3), 22.59 (7-CH3), 55.25 (C-5), 58.44 (inverted CH2CH3), 60.63 (inverted CH2CH3), 114.52 (C-2), 128.02–132.76 (Ar–CH). Anal. Calcd (%) for C23H22N4O3S: C, 63.58; H, 5.10; N, 12.89. Found, C, 63.53; H, 5.09; N, 12.63.
4.4. Synthesis of ethyl 5-phenyl-3-oxo-2-isatinylidene-7-methyl-2,3-dihydro-5H-thiazolo[3,2-a]pyrimidine-6-carboxylate, compound, 5
To a solution of compound 3 (0.63 g, 2 mmol) in glacial acetic acid (20 ml); isatin (0.35 g, 2.4 mmol) and fused sodium acetate (0.57 g, 7 mmol) were added. The reaction mixture was heated under reflux for 4 h, during which the product was partly crystallized out. After cooling, the reddish compound was filtered, washed with ethanol, dried and crystallized from ethanol, Scheme 2 5; m.p. 298–299 °C; yield 62%; Rƒ. 0.32 (CHCl3:CH3COCH3 9:1); Clog P 4.95; IR (KBr) (cm−1): 1684 (ester C O), 1599 (C N), 3170 (NH); 1H NMR (CDCl3, 400 MHz): (ppm) 1.26 (3 H, t, CH2CH3); 2.50 (3H, s, CH3); 4.20 (2H, q, CH2CH3); 6.33 (1H, s, pyr. H-5); 6.83–8.96 (9 H, m, Ar–H); 11.16 (1 H, s, NH; exchangeable with D2O). EI-MS: m/z (%) 445 (M+, 44), 446 (M++1, 12), 447 (M++2, 5). Anal. Calcd (%) for C22H19ClN4O3S: C, 64.71; H, 4.30; N, 9.43. Found, C, 64.81; H, 4.45; N, 9.33.
5. Biological testing
5.1. Antimicrobial activity
5.1.1. Antibacterial activity
Compounds 2a–x, 4a–c and 5 were tested for their antibacterial activity in vitro, in comparison with chloramphenicol as a reference drug using the standard agar cup diffusion method (Janssen and Jageneau, 1957, Sherif et al., 1993) at the Assiut University Mycological Center (AUMC), Faculty of Science, Assiut University, Assiut, Egypt. Five bacterial species representing both Gram-positive and Gram-negative strains which are common contaminants of the environments in Egypt some of which are involved in human and animal diseases were selected. The used bacterial strains are S. aureus (AUMC No. B-54) and B. cereus (AUMC No. B-5) as representatives of Gram-positive strains, and E. coli (AUMC No. B-53), P. aeruginosa (AUMC No. B-73), as well as S. marcescens (AUMC No. B-55) as representatives of Gram-negative strains (Janssen and Jageneau, 1957).
To prepare inocula for bioassay, bacterial strains were individually cultured for 24 h in 100 ml conical flasks containing 30 ml nutrient broth medium. Bioassay was done in 10 cm sterile plastic Petri plates in which bacterial suspension (1 m/plate) and 15 ml Nutrient agar medium (15 ml/plate) were poured. After solidification of the media, 5 mm diameter cavities were cut in the solidified agar (4 cavities/plate) using sterile cork borer. Tested compounds dissolved in dimethyl sulfoxide (DMSO) at 100 μmol/ml were pipetted in the cavities (20 μl/cavity). Cultures were then incubated at 28 ± 2 °C for 48 h. Results were taken as the diameter (in mm) of inhibition zone around cavities and the results are cited in Table 1.
5.1.2. Antifungal activity
The synthesized compounds 2a–x, 4a–c and 5 were tested for their antifungal activity in vitro, in comparison with clotrimazole as a reference drug using the standard agar cup diffusion method (Janssen and Jageneau, 1957, Sherif et al., 1993) at the Assiut University Mycological Center (AUMC), Faculty of Science, Assiut University, Assiut, Egypt. Six pathogenic, phytogenic, or food-poisoning fungal species were used in the present study: C. albicans (AUMC No. 418), G. candidum (AUMC No. 226), A. flavus (AUMC No. 1276), T. rubrum (AUMC No. 1804), S. brevicaulis (AUMC No. 729), and F. oxysporum (AUMC No. 5119) (Janssen and Jageneau, 1957).
To prepare inocula for bioassay, Fungi were grown for 7 days in 100 ml conical flasks containing 30 ml sabouraud’s dextrose broth. Bioassay was done in 10 cm sterile plastic Petri plates in which fungal suspension (1 ml/plate) and 15 ml sabouraud’s dextrose agar medium (15 ml/plate) were poured. After solidification of the media, 5 mm diameter cavities were cut in the solidified agar (4 cavities/plate) using sterile cork borer. Tested compounds dissolved in dimethyl sulfoxide (DMSO) at 100 μmol/ml were pipetted in the cavities (20 μl/cavity). Cultures were then incubated at 28 ± 2 °C up to 7 days. Results were read as the diameter (in mm) of inhibition zone around cavities and the results are cited in Table 2.
5.1.3. The minimum inhibitory concentrations (MICs)
The test compounds giving positive results were diluted with DMSO to prepare a series of descending concentrations and were similarly assayed as mentioned before and the least concentration (below which no activity) was recorded as the MIC. The results are cited in Table 3, Table 4.
5.2. Pharmacological screening
5.2.1. Anti-inflammatory activity
Male adult albino rats (130–150 g) were obtained from the animal house, Faculty of Medicine, Assiut University, Assiut, Egypt. Animals were housed in separate cages, 6 animals each, in temperature-controlled rooms at 25 °C. Animals were allowed free access to rodent chow and water and maintained at a 12 h light/dark cycle. Work was conducted in accordance with the internationally accepted principles for laboratory animals’ use, care as found in the European Community Guidelines (Nargund et al., 1994).
Indomethacin (Liometacin® vial, Nile Company), Carrageenan (Sigma, USA), sodium carboxymethylcellulose (NaCMC) and normal saline were obtained from the local market.
For determination of anti-inflammatory activity, all tested compounds and the reference drug were suspended in 1% sodium carboxymethylcellulose (NaCMC) solution in normal saline. Suspensions of the test compounds, reference drug and 1% (NaCMC)-saline solution (negative control) were injected i.p.(1 mL each).
Compounds 2a–e, 2o and 2t–x were evaluated for their anti-inflammatory activity according to the carrageenan induced paw edema method (Nargund et al., 1994) in comparison to indomethacin as a reference drug. The test is based on the pedal inflammation in rat paws induced by subplantar injection of carrageenan suspension (0.2 mL of 1% solution in normal saline) into the right hind of the rats. Male adult albino rats were divided into groups, each of three animals. The thickness of rat paw was measured by a Vernier caliper (SMIEC, China) before and 1 h after carrageenan injection to detect the carrageenan induced inflammation. Each tested compound, at a dose of 54 μmol/kg, was injected i.p. to thirteen different groups of rats 1 h after carrageenan injection. The control group received a vehicle (1% NaCMC solution in normal saline), while the reference group received indomethacin i.p. at 54 μmol/kg.
The difference between the thicknesses of the two paws was taken as a measure of edema. The measurement was carried out at 0.5, 1, 2, 3, 4, and 5 h after injection of the test compounds, reference drug, and control. The results are listed in Table 5.
5.2.2. Analgesic activity
The analgesic activity of the most active anti-inflammatory compounds 2a–c, 2e and 2v was evaluated in mice using the hot plate method (Janssen and Jageneau, 1957) in comparison to aspirin as a reference drug. In this method, the time taken by the mouse to lick its feet or to jump within a plexiglass cylinder placed on a hot plate surface (55 °C) was determined. The reaction time was taken as the end point and the increase in hot plate latency was taken as a measure of the analgesic activity.
All tested compounds and the reference drug were suspended in 1% sodium carboxymethylcellulose (NaCMC) saline solution. Suspensions of the tested compounds, reference drug and 1% sodium carboxymethylcellulose (NaCMC) saline solution (negative control) were injected i.p.(1 mL each).
Male adult albino mice (25–30 g) were divided into eight groups, each of three animals. Solutions or suspensions of the test compounds and the reference drug in 1% sodium carboxymethylcellulose (NaCMC) solution were injected i.p. in a dose level of 300 μmol/kg into mice. The control group of animals was similarly treated with 1% sodium carboxymethylcellulose (NaCMC) saline solution. The reaction time was evaluated directly 0.5, 1, 2, 3, 4 and 5 h of first injection. The results are cited in Table 6.
5.2.3. Acute toxicity (LD50)
The median lethal dose (LD50) of the most active anti-inflammatory and analgesic compounds 2e and 2v was determined in mice.
Groups of male adult albino mice, each of three animals (25–30 g), were injected i.p. with graded doses of the test compounds. The percentage mortality in each group of animals was determined 24 h later to the injection (Sztaricskai et al., 1999).
Footnotes
Peer review under responsibility of King Saud University.

References
- Abdel-Alem A.M., Abdel-Kader M.A., El-Koussi A.A. Synthesis and anti-inflammatory activity of certain new N, N′-oxamides. Pharmazie. 1980;35:394–398. [PubMed] [Google Scholar]
- Balkan A., Uma S., Ertan M., Wiegrebe W. Thiazolo[3,2-a]pyrimidine derivatives as calcium antagonists. Pharmazie. 1992;47:687–688. [PubMed] [Google Scholar]
- Balkan A., Ertan M., Burgemeister T. Synthesis and structural evaluations of thiazolo[3,2-a]pyrimidine derivatives. Arch. Pharm. 1992;325:499–501. [Google Scholar]
- Balkan A., Tozkoparan B., Ertan M., Sara Y., Ertekin N. New thiazolo[3,2-a] pyrimidine derivatives, synthesis and calcium antagonistic activities. Boll. Chim. Farm. 1996;135:648–652. [PubMed] [Google Scholar]
- Dennler E.B., Frasca A.R. Synthesis of indazoles using polyphosphoric acid—I. Tetrahedron. 1966;22:3131–3141. [Google Scholar]
- Dinakaran V.S., Bomma B., Srinivasan K.K. Fused pyrimidines: the heterocycle of diverse biological and pharmacological significance. Der Pharm. Chem. 2012;4:255–265. [Google Scholar]
- Ghorab M.M., Abdel-Gawad S.M., El-Gaby M.S.A. Synthesis and evaluation of some new fluorinated hydroquinazoline derivatives as antifungal agents. IL Farmaco. 2000;55:249–255. doi: 10.1016/s0014-827x(00)00029-x. [DOI] [PubMed] [Google Scholar]
- Hussein M.A., Abdel Moty S.G., Abdel Aziz S.A., Abou-Salim M.A. Synthesis and antimicrobial activity of new substituted dihydropyrimidine derivatives. Bull. Pharm. Sci., Assiut Univ. 2011;34:37–58. [Google Scholar]
- Janssen P.A., Jageneau A.H. A new series of potent analgesics: dextro 2:2-diphenyl-3-methyl-4-morpholino-butyrylpyrrolidine and related amides. I. Chemical structure and pharmacological activity. J. Pharm. Pharmacol. 1957;9:381–400. [PubMed] [Google Scholar]
- Jeanneau-Nicolle E., Benoit-Guyod M., Namil A., Leclerc G. New thiazolo[3,2-a]pyrimidine derivatives, synthesis and structure–activity relationships. Eur. J. Med. Chem. 1992;27:115–120. [Google Scholar]
- Kalgutkar A.S., Marnett A.B., Crews B.C., Remmel R.P., Marnett L.J. Ester and amide derivatives of the nonsteroidal antiinflammatory drug, indomethacin, as selective cyclooxygenase-2 inhibitors. J. Med. Chem. 2000;43:2860–2870. doi: 10.1021/jm000004e. [DOI] [PubMed] [Google Scholar]
- Leo A.J. Calculating log Poct from structures. Chem. Rev. 1993;93:1281–1306. [Google Scholar]
- Nargund L.V., Reddy G.R., Hariprasad V. Anti-inflammatory activity of substituted 1,3,4-oxodiazoles. J. Pharm. Sci. 1994;83:246–248. doi: 10.1002/jps.2600830226. [DOI] [PubMed] [Google Scholar]
- Rida S.M., Salama H.M., Labouta I.M., Ghany Y.S. Syntheses and in vitro antimicrobial activities of thiazolo-[3,2-a]benzimidazol-3(2H)-ones. Pharmazie. 1986;41:324–326. doi: 10.1002/chin.198640178. [DOI] [PubMed] [Google Scholar]
- Sahu S.K., Banerjee M., Samantray A., Behera C., Azam M.A. Synthesis, analgesic, anti-inflammatory and antimicrobial activities of some novel pyrazoline derivatives. Trop. J. Pharm. Res. 2008;7:961–968. [Google Scholar]
- Sayed H.H., Shamroukh A.M., Rashad A.E. Synthesis and biological evaluation of some pyrimidine, pyrimido[2,1-b][1,3]thiazine and thiazolo[3,2-a]pyrimidine derivatives. Acta Pharm. 2006;56:231–244. [PubMed] [Google Scholar]
- Sherif S.M., Youssef M.M., Mobarak K.M., Abdel-Fattah A.M. A convenient synthesis of thiazolopyrimidines, thiazolodipyrimidines and heterocyclothiazolopyrimidines. Tetrahedron. 1993;49:9561–9572. [Google Scholar]
- Shiryaev A.K., Baranovskaya N.S., Eremin M.S. Synthesis of 5H-thiazolo[3,2-a]pyrimidines. Chem. Heterocycl. Compd. 2013;48:1550–1554. [Google Scholar]
- Sztaricskai F., Takács I.E., Pusztai F., Szabó G., Csípõ I. Antiulcer effect of the N-and O-β-d-glucopyranosides of 5-aminosalicylic acid. Arch. Pharm. 1999;332:321–326. doi: 10.1002/(sici)1521-4184(19999)332:9<321::aid-ardp321>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- Tozkoparan B., Ertan M., Kelicen P., Demirdamar R. Synthesis and anti-inflammatory activities of some thiazolo[3,2-a]pyrimidine derivatives. IL Farmaco. 1999;54:588–593. doi: 10.1016/s0014-827x(99)00068-3. [DOI] [PubMed] [Google Scholar]
- Viveka S., Dinesha, Laxmeshwar S.S., Nagaraja G.K. Ethyl 7-methyl-5-(4-methylphenyl)-3-oxo-2-{[3-(3,4-dichlorophenyl)-1-phenyl-1H-pyrazol-4-yl]methylidene}-2,3-dihydro-5H-[1,3]thiazolo[3,2-a]pyrimidine-6-carboxylate. Molbank. 2012;3(1–5):M776. [Google Scholar]
- Weisberger A., Taylor E.C. In: Benzimidazoles and Congeneric Tricyclic Compounds. Preston P.N., editor. John Wiely and Sons; New York: 1981. pp. 204–208. [Google Scholar]
- Youssef M.M., Amin M.A. Microwave assisted synthesis of some new thiazolopyrimidine, thiazolodipyrimidine and thiazolopyrimidothiazolopyrimidine derivatives with potential antioxidant and antimicrobial activity. Molecules. 2012;17:9652–9667. doi: 10.3390/molecules17089652. [DOI] [PMC free article] [PubMed] [Google Scholar]