

Abstract
The endoplasmic reticulum (ER) is at the epicenter of astrocyte Ca2+ signaling. We sought to identify the molecular mechanism underlying store-operated calcium entry that replenishes ER stores in mouse Müller cells. Store depletion, induced through blockade of sequestration transporters in Ca2+-free saline, induced synergistic activation of canonical transient receptor potential 1 (TRPC1) and Orai channels. Store-operated TRPC1 channels were identified by their electrophysiological properties, pharmacological blockers, and ablation of the Trpc1 gene. Ca2+ release-activated currents (ICRAC) were identified by ion permeability, voltage dependence, and sensitivity to selective Orai antagonists Synta66 and GSK7975A. Depletion-evoked calcium influx was initiated at the Müller end-foot and apical process, triggering centrifugal propagation of Ca2+ waves into the cell body. EM analysis of the end-foot compartment showed high-density ER cisternae that shadow retinal ganglion cell (RGC) somata and axons, protoplasmic astrocytes, vascular endothelial cells, and ER–mitochondrial contacts at the vitreal surface of the end-foot. The mouse retina expresses transcripts encoding both Stim and all known Orai genes; Müller glia predominantly express stromal interacting molecule 1 (STIM1), whereas STIM2 is mainly confined to the outer plexiform and RGC layers. Elimination of TRPC1 facilitated Müller gliosis induced by the elevation of intraocular pressure, suggesting that TRPC channels might play a neuroprotective role during mechanical stress. By characterizing the properties of store-operated signaling pathways in Müller cells, these studies expand the current knowledge about the functional roles these cells play in retinal physiology and pathology while also providing further evidence for the complexity of calcium signaling mechanisms in CNS astroglia.
SIGNIFICANCE STATEMENT Store-operated Ca2+ signaling represents a major signaling pathway and source of cytosolic Ca2+ in astrocytes. Here, we show that the store-operated response in Müller cells, radial glia that perform key structural, signaling, osmoregulatory, and mechanosensory functions within the retina, is mediated through synergistic activation of transient receptor potential and Orai channels. The end-foot disproportionately expresses the depletion sensor stromal interacting molecule 1, which contains an extraordinarily high density of endoplasmic reticulum cisternae that shadow neuronal, astrocytic, vascular, and axonal structures; interface with mitochondria; but also originate store-operated Ca2+ entry-induced transcellular Ca2+ waves that propagate glial excitation into the proximal retina. These results identify a molecular mechanism that underlies complex interactions between the plasma membrane and calcium stores, and contributes to astroglial function, regulation, and response to mechanical stress.
Keywords: astrocyte end-feet, calcium, ER stores, reactive gliosis, store-operated channels, TRPC
Introduction
Ca2+ mobilization from astroglial endoplasmic reticulum (ER) stores plays a central and irreplaceable role in brain function. Regenerative openings of Ca2+ release channels, induced by mechanical, osmotic, lipid messenger, purinergic, and cholinergic stimuli, elevate the intracellular calcium concentration ([Ca2+]i) in astrocytes, which in turn modulates neuronal plasticity, signaling, pathology, and local blood flow (Khakh and McCarthy, 2015; Newman, 2015; Ryskamp et al., 2015). Although replenishment of the ER Ca2+ pool is essential for maintained astroglial excitability and function, the identity of ion channels that mediate store-operated Ca2+ entry (SOCE) and their roles in Ca2+ homeostasis, function, and pathology of native CNS astroglia remains largely unclear.
Previous investigations in amphibian and mammalian retinae indicated that SOCE might disproportionately contribute to tonic synaptic signaling in the presence of sustained illumination. For example, inhibition of SOC channels lowered photoreceptor [Ca2+]i and suppressed synaptic output to postsynaptic bipolar/horizontal neurons (Szikra et al., 2008, 2009). This suggests that SOCE, which operates under conditions when voltage-operated signals are closed yet potent clearance mechanisms continue to extrude Ca2+ (Duncan et al., 2006), might simultaneously alleviate ER stress caused by depleted Ca2+ levels and extend the dynamic range of the synapse in combination with Ca2+-induced Ca2+ release (Szikra et al., 2008; Križaj, 2012; Chen et al., 2014). It is not clear, however, how this mechanism might impact the function of Müller astroglia that ensheath every retinal neuron, and play crucial roles in neurotransmitter uptake and metabolic support (Reichenbach and Bringmann, 2010). ER Ca2+ release through retinal glial InsP3 and/or ryanodine channels amplifies the effects of light, mechanical stress, swelling, and metabotropic stimuli; it modulates glioneuronal and gliovascular feedback (Keirstead and Miller, 1995; Newman and Zahs, 1998; Newman, 2015), and is presumably followed by sequestration of Ca2+ entering cytosol through SOC channels into the ER. The identity of stromal interacting molecule (STIM) depletion sensors or SOC channels in native Müller glia or brain astrocytes has not been unequivocally ascertained. Although Orai and Stim transcripts were detected in cell lines (Barajas et al., 2008; Motiani et al., 2013), Orai-mediated Ca2+ release-activated (CRAC) currents (ICRAC) have never been recorded in mature astrocytes (Kettenman and Ransom, 2013). Instead, the main astroglial SOCE conduit is believed to be transient receptor potential 1 (TRPC1), a nonselective cation channel originally identified by its homology to the Drosophila photochannel (Verkhratsky et al., 2014). Whether TRPC1 is store operated under physiological conditions has been under debate as Trpc1 is prominently expressed in mouse rod photoreceptors and vascular smooth muscle cells, yet its ablation was shown to have no effect on SOCE in these cell types (Dietrich et al., 2007; Molnar et al., 2012).
We report that SOCE in adult native Müller cells is subserved by synergistic activation of TRPC1 and Orai channels. Loss of TRPC1 facilitated the gliotic response induced by increased intraocular pressure (IOP), suggesting that the channel might contribute to the glial mechanosusceptibility. SOCE triggered ICRAC and initiated transcellular Ca2+ waves that propagated from apical and end-foot processes toward the soma. These findings demonstrate that adult retinal astroglia simultaneously use two phylogenetically distinct signaling mechanisms to implement novel modes of retinal signal integration.
Materials and Methods
Animals.
Wild-type (WT) 129/SvImJ mice of either sex were purchased from The Jackson Laboratory. Knock-out (KO) mice deficient in TRPC1 or TRPC3 channels were generated in the laboratory of Lutz Birnbaumer (National Institute of Environmental Health Sciences; Liao et al., 2014) and double-KO (Trpc1/3−/−) mice were generated by breeding single-KO mice (Molnar et al., 2012). Trpc1−/− and Trpc1/3−/− mice are healthy, and show no overt signs of neurological or metabolic impairments or differences in spontaneous behavior. Homozygous and heterozygous knock-out mice were identified by PCR analysis of genomic DNA extracted from tail biopsy specimens using appropriate primer pairs. The animals were kept on a 12 h light/dark cycle, and were fed laboratory chow and water ad libitum. Mice [postnatal day 30 (P30) to P60] were killed before isolation and dissociation of retinae by inhalation with isoflurane and cervical dislocation. For Trpc1−/− experiments, ∼1.2-year-old mice were used. Animal handling, anesthetic procedures, and experiments were performed in accordance with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals and The Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research, and were approved by the Institutional Animal Care and Use Committees at the University of Utah. An abstract containing a portion of this work was published previously (Molnar et al., 2013).
Retinal cell dissociation and plating.
Müller cells were acutely dissociated as described previously (Szikra et al., 2009; Jo et al., 2015). In brief, retinae were dissected from enucleated mouse eyes in cold Leibovitz 15 medium (11 mg/ml; Life Technologies) supplemented with 20 mm d-glucose, 10 mm Na-HEPES, 2 mm Na-pyruvate, 0.3 mm Na-ascorbate, and 1 mm glutathione, pH 7.4, 300 mOsm (referred as L-15). The tissue was placed into L-15 containing papain (7 U/ml; Worthington) for 1 h at room temperature (RT). After digestion of the extracellular matrix, retinae were rinsed three times with cold L-15, placed on ice, and cut into 1–2 mm pieces. To mechanically dissociate retinal cells, two to three small retinal pieces were triturated in L-15 and plated on class coverslips coated with concanavalin A (0.2 mg/ml; Sigma-Aldrich). Coverslips were mounted on a stage of an upright microscope (Eclipse E600FN, Nikon) and visualized through a 40× (0.80 NA) water-immersion objective. Müller cells were identified by their typical elongated morphology (Fig. 1A). Cells were perfused with Ringer's solution containing the following (in mm): 125 NaCl, 2.5 KCl, 1.25 Na2HPO4, 2 CaCl2, 1.5 MgCl2, 25 Na-HEPES, 10 glucose, 0.5 l-glutamine, 1 pyruvic acid, 1 lactic acid, 0.3 ascorbic acid, and 0.5 glutathione, pH 7.4 (osmolarity, 300 mOsm). In Ca2+-free Ringer's solution, Ca2+ was replaced with 1 mm EDTA. All experiments were performed at room temperature.
Figure 1.

Depletion of ER Ca2+ stores triggers SOCE in retinal Müller glia. A, Bright-field image of representative, acutely dissociated mouse Müller glial cells. Subregions are indicated with capital letters: D, distal stalk; S, soma; P, proximal stalk; Ef, end-foot. Scale bar, 10 μm. B, Ca2+ imaging. Representative calibrated [Ca2+]i trace evoked during store depletion in Ca2+-free saline supplemented with CPA. SERCA inhibition elicited a small transient increase in cytosolic Ca2+ (white arrow) followed by store-dependent elevation in [Ca2+]i during the re-exposure to 2 mm Ca2+, consisting of a peak response (black arrow) followed by a sustained plateau. C, Comparison of depletion-evoked [Ca2+]i elevations in control conditions (black trace; n = 28, N = 4) and the presence of an inhibitory cocktail containing 2-APB (100 μm), SKF-96365 (10 μm), and Gd3+ (10 μm; red trace; n = 28, N = 4). D, Summary graph; average [Ca2+]i ± SEM. N.S., Nonsignificant. **p < 0.01.
Calcium imaging.
Imaging experiments followed the protocols from the study by Ryskamp et al. (2011, 2014). Acutely dissociated retinal Müller cells were loaded with the ratiometric fluorescent dye fura-2 AM (Enzo Life Sciences) for 30 min. To allow complete de-esterification of intracellular AM esters, cells were incubated in dye-free L-15 medium for 30 min. Excitation for 340 and 380 nm filters (Semrock) was provided by a 150 W Xenon arc lamp (DG4, Sutter Instruments). Fluorescence emission was high-pass filtered at 510 nm and captured with cooled digital CCD cameras (Photometrics). Data acquisition and F340/F380 ratio calculation were performed by NIS Elements version 3.22 software (Nikon). Fluorescence imaging was performed on regions of interest (ROIs) that encompassed the cell body, the end-foot, and distal end regions, as well as end-foot and distal stalks, and were typically binned at 3 × 3 or 4 × 4. Background fluorescence was measured in similarly sized ROIs in neighboring areas devoid of cells. In a subset of experiments, the data are presented as 340/380 nm ratios. Calibration of free [Ca2+]i was performed using ionomycin (10 μm) and 10 mm Ca2+ to determine Rmin and Rmax, respectively. The apparent free [Ca2+]i was determined from the equation [Ca2+]i = ((R − Rmin)/(Rmax − R)) × (Fmax380/Fmin380) × Kd, where R is the ratio of emission intensity at 510 nm evoked by 340 nm excitation versus emission intensity at 510 nm evoked by 380 nm excitation; Rmin is the ratio at nominally free Ca2+; Rmax is the ratio at saturating Ca2+; and the Kd for Ca2+-fura 2 at room temperature was taken to be 224 nm (Neher, 1995). Ca2+ waves were studied with a 100× oil objective (1.2 NA) at 1 × 1 pixel binning. The average speed of wave propagation was calculated by plotting the time at wave initiation against the time it reached the cell body at the measured distance between the two subcellular compartments.
Electrophysiology.
Whole-cell voltage-clamp recordings were performed in acutely dissociated Müller glia (Jo et al., 2015). Borosilicate glass electrodes (1.2 mm outer diameter, 0.95 mm inner diameter, with internal filament; World Precision Instruments) were pulled with a vertical puller (PB-7, Narishige), and resistances were 7–9 MΩ. The pipette solution contained the following (in mm): 125 NaCl, 10 HEPES, 1 MgCl2, 10 EGTA, 4 ATP magnesium salt, 0.3 GTP sodium salt, pH 7.3, with or without 5.4 CaCl2 (∼295 Osm). The solution used in combined patch-clamp and Ca2+-imaging experiments contained the following (in mm): 120 Cs-gluconate, 1 CaCl2, 2 MgCl2, 20 HEPES, 11 EGTA, and 30 glucose together with 100 μm fura-2 pentapotassium salt, pH 7.4. Cells were continuously perfused with external solution; when divalent free, saline contained the following (in mm): 80 NaCl, 5 HEPES hemisodium salt, 0.63 Na2HPO4, 2 EDTA, 0.5 glutathione, 0.75 sodium pyruvate, 5 lactic acid, 0.3 ascorbic acid, 10 d-glucose, 0.5 l-glutamine, pH 7.4 (osmolarity of 300–303 mOsm was adjusted with mannitol). Recording pipettes were positioned with micromanipulators (MPC-200; Sutter Instruments). Liquid junction potentials were subtracted, pipette and membrane capacitances compensated, and currents were Bessel filtered at 2.9 kHz with EPC-10 (HEKA Elektronik) or Multiclamp 700B (Molecular Devices) amplifiers. Baseline currents were recorded at −80 mV with 1 Hz and stored on a computer using PatchMaster (HEKA Elektronik) or pCLAMP (Molecular Devices) acquisition software. Voltage ramps (−110 to +50 mV over 100 ms; 5 kHz sampling rate) were applied for the determination of the reversal potential. Subsequent analysis was performed with Igor Pro (WaveMetrics) and OriginPro 8.0 (OriginLab).
Ocular hypertension in vivo.
Mice were anesthetized with intraperitoneal injection of ketamine/xylazine and 1% tropicamide ophthalmic solution USP (Bausch & Lomb) was applied to the eyes to dilate the pupils. Anesthetic ophthalmic drops (0.5% proparacaine hydrochloride) were applied to the eyes to minimize ophthalmic pain during the procedure. IOP was elevated by injecting 2 μl polystyrene microbeads (10 μm diameter FluoSpheres, Invitrogen) into the anterior chamber of the left eyes, using a blunt-tip Hamilton syringe (Hamilton Company; Huang et al., 2011). The microbeads were resuspended in PBS at a final concentration of 7.2 × 106 beads/ml. The rate of injection was 2 μl over a 60 s period. The contralateral eye was injected with 2 μl of vehicle (PBS). Erythromycin 0.5% ophthalmic ointment (Bausch & Lomb) was applied to the cornea after surgery. IOPs were measured between 10:00 A.M. and noon with a tonometer (TonoLab) weekly until eyes were harvested at 1, 2, 3, or 8 weeks postinjection. IOPs were determined as the mean of 15–20 tonometer readings. Microbead treatment induced ocular hypertension in 12 of 12 wild-type and 14 of 14 TRPC1/3−/− eyes, while vehicle treatment had no effect on intraocular pressure.
Immunofluorescence was performed as described previously (Rentería et al., 2005; Huang et al., 2011). Mice were enucleated, and eyecups were prepared by dissecting corneas and lenses away. Retinal tissue from wild-type and TRPC1/3−/− retinae was harvested, mounted, and stained in parallel. First, eyecups were fixed by 4% PFA in 0.1 m phosphate buffer (PBS), pH 7.2, for 1 h, at room temperature then were washed 3 × 10 min in PBS. Fixed tissues were immersed for 1–3 h in 15% sucrose, followed by 12–16 h immersion in 30% sucrose at 4°C and optimum cutting temperature (Tissue-Tek, Sakura) embedding. Frozen blocks were sectioned at 12 μm and mounted on glass slides (Fisher Scientific). For immunocytochemistry, cryostat sections were thawed, washed in PBS, and placed for 30 min in blocking solution (10 ml of PBS, 30 μl of Triton X-100, 100 mg of bovine serum albumin, 100 μl of 10% w/v Na-azide). We used a monoclonal mouse anti-glutamine synthetase (GS; 1:1000; BD Biosciences) as a Müller cell marker and a polyclonal rabbit anti-GFAP primary antibody (1:1000; Dako) as a marker for gliosis. We used polyclonal STIM1 (1:500; Sigma-Aldrich) and STIM2 (1:500; Cell Signaling Technology) antibodies, which were validated in immunoblots. As reported previously (Molnar et al., 2012), our attempts to determine the subcellular localization of TRPC1 and TRPC3 using monoclonal and polyclonal antibodies from commercial suppliers (Alomone Labs, Sigma-Aldrich, Abcam, and Millipore), and custom-designed affinity-purified antibodies that targeted either the C terminus or the N terminus of mouse TRPC1 (Covance) were unsuccessful. Confirming doubts about TRPC1 antibody specificity/affinity (Rychkov and Barritt, 2007; Gilliam and Wensel, 2011), we found that TRPC1 antibodies from different manufacturers elicit different retinal staining patterns; moreover, signals in wild-type and TRPC1/3−/− retinae were identical (data not shown).
Primary antibodies were diluted in blocking solution and applied overnight at 4°C. After washing the primary antibodies 3 × 5 min in PBS fluorophore-conjugated secondary antibodies (goat anti-rabbit Alexa Fluor 488 and goat anti-mouse Alexa Fluor 594; 1:1000 respectively; Life Technologies) were applied for 1 h at room temperature. After a final set of washes in PBS, the sections were covered in Fluoromount G (Southern Biotechnology). Negative controls without a primary antibody showed no staining. Images (three to five per retina) were acquired on a confocal microscope (LSM 510, Zeiss; or FX1000, Olympus) using 488 nm Ar (10%) and 594 nm He/Ne (100%) lines for fluorophore excitation, suitable bandpass or long-pass filters for emission detection, and 40×/1.2 NA oil objectives.
GFAP expression levels in Müller cells were analyzed with ImageJ (NIH). ROIs encompassing the inner plexiform layer (IPL) and inner nuclear layer (INL) were determined for each immunofluorescence image. Threshold values were established to highlight the pixels showing GFAP immunolabeling within the ROI. The area fractions (percentage of highlighted pixels within the ROI) were measured and averaged per retina. After measuring the area fraction values for each retina, data were averaged for each group according to treatment and mouse strain, and are shown as the mean ± SEM.
Transmission electron microscopy.
Tissues were processed in buffered 2.5% glutaraldehyde and 1% paraformaldehyde, and postfixed in 1% buffered osmium tetroxide followed by embedding in epoxy resins. Müller cell end-feet were imaged at 2.18 nm resolution using automated transmission electron microscope (ATEM) imaging as a part of high-resolution mouse retinal connectome RC2 (Marc et al., 2013). The details of tissue processing, ATEM imaging, and computational connectome assembly are as described previously (Anderson et al., 2009, 2011a,b; Marc et al., 2013). The tissue for mouse retinal connectome RC2 was derived from MarcLab Bloc 8473-1, a portion of central retina from the right eye of a 6-month-old C57BL/6J female mouse fixed with an intravitreal injection of mixed aldehydes within 30 s after being killed. The injection fixative was room temperature 2.5% glutaraldehyde, 1% formaldehyde, 1 mm MgSO4, and 3% sucrose in 0.1 m cacodylate buffer at pH 7.4. After eyecup dissection, the eye was immersed in ice-cold fixative for 24 h, followed by osmication and en bloc uranyl acetate staining, dehydration, and embedding in epoxy resin, as described by Anderson et al. (2011b). Volume RC2 is composed of >1400 serial sections, each imaged with ∼1000–1100 overlapping 4K × 4K 16-bit image tiles/section with a GATAN UltraScan 4000 phosphorimaging camera. The volume is a virtual cylinder of 0.25–0.28 mm diameter spanning the outer nuclear layer (ONL) and the vitreal border of the retina. The image dataset was computationally built into an aligned volume using custom applications and software tools (Tasdizen et al., 2010), forming a volume of ∼48 terabytes, hosted on our Viking server and available via the Viking client (connectomes.utah.edu).
Solutions and reagents.
Most reagents were purchased from Sigma-Aldrich and VWR. SKF 96365 and 2-APB were from Tocris Bioscience; Synta66 and GSK7975A were from AOBIOUS, Inc.
Statistical analysis.
Data are expressed as the mean ± SEM, unless noted otherwise. Comparisons between two treatments in a cell/animal were determined with the paired t test; comparisons between different groups were evaluated by one-way ANOVA with post hoc Tukey's multiple comparison of means (Origin 8.0, OriginLab). p values <0.05 (*) were considered to be statistically significant; n indicates the number of cells, and N indicates the number of animals.
Results
ER store depletion evokes store-operated Ca2+ entry in mouse Müller glia
We used Ca2+ imaging to investigate SOCE in acutely dissociated adult Müller glia, which are easily identified by the cylindrical perikaryon emanating from an apical and basal process that could be distinguished by an expanded tuft and a bulbous end-foot, respectively (Fig. 1A). In previous studies (Ryskamp et al., 2014; Jo et al., 2015), we showed that dissociated and intact Müller cells can be distinguished by their immunoreactivity for glutamine synthetase, Kir4.1 subunits, and vanilloid transient receptor potential type V4 (TRPV4). Store-operated signals were measured by using the standard protocol that combines depletion of ER stores with the restoration of Ca2+ to the extracellular solution (Takemura and Putney, 1989; Molnar et al., 2012). The [Ca2+]i in cells superfused with Ca2+-free saline dropped from the average baseline of 193 ± 8 nm (n = 87, N = 10) to 61 ± 4 nm (n = 96, N = 10), suggesting a loss of steady-state Ca2+ influx. In the continued absence of extracellular Ca2+, cyclopiazonic acid (CPA; 5 μm), a reversible antagonist of sarcoplasmic-ER Ca2+ ATPases (SERCAs), typically elicited a small transient increase in [Ca2+]i (Fig. 1B, white arrow). As previously observed in photoreceptors (Szikra et al., 2008, 2009), the magnitude of CPA-mediated [Ca2+]i response varied in individual cells, reflecting cell-to-cell variability in the efficacy of Ca2+ sequestration and plasma membrane extrusion mechanisms. Subsequent add-back of control saline containing 2.6 mm [Ca2+]o resulted in [Ca2+]i “overshoots” that represent the defining hallmark of the SOCE response (Fig. 1B, black arrow). Although overshoot responses were observed in every glial compartment (i.e., the apical process, soma, and the end-foot), their amplitude was the largest in the end-foot, which also represented an origin site from which Ca2+ signals spread toward the cell body in the form of a transcellular Ca2+ wave (Fig. 2, Movie 1). SOCE-induced Ca2+ waves were initiated at the end-foot in 58% of cells (11 of 19 cells), showing mean propagation speeds of 4.3 ± 0.77 μm/s. Eight of 19 cells showed the initial response in the apical process, with propagation speeds of 4.3 ± 0.95 μm/s, whereas Ca2+ waves were never initiated from the cell body. We conjecture that SOCE-induced Ca2+ waves reflect rapid, SERCA-mediated refilling of internal ER stores associated with regenerative activation of Ca2+-sensitive InsP3 receptors (Newman and Zahs, 1998; Lipp et al., 2009).
Figure 2.

SOCE is associated with the induction of transcellular Ca2+ waves. A, Representative images of a depletion-induced Ca2+ wave that is initiated in both processes with propagation into the cell body. B, [Ca2+]i increases during the [Ca2+]i wave for each compartment (n = 19). C, D, The propagation velocity and rise time (time required for SOCE-mediated [Ca2+]i increase from 10% to 90% of maximum) is faster in the end-foot compared with the apical process and the cell body (p < 0.05; n = 20; N = 4).
Store depletion induces Ca2+ waves in the apical and end-foot processes that propagate centrifugally into the cell body.
To confirm that depletion-induced Ca2+ overshoots were mediated by SOC channels, we used a cocktail consisting of nonselective inhibitors of Orai and TRPC channels 2-APB (100 μm), SKF 96365 (10 μm), and Gd3+ (10 μm), which block SOCE in mouse photoreceptors (Molnar et al., 2012). The cocktail reduced the average depletion-evoked [Ca2+]i peak response from 460 ± 51 nm (n = 80 cells, N = 10 animals) to 269 ± 43 nm (∼40% reduction; n = 32, N = 5, p < 0.05; Fig. 1; see 4A) and reduced the rate of [Ca2+]i change Δ[Ca2+]i/dt during Ca2+ add-back from 3.22 ± 0.62 to 1.05 ± 0.07 nm/s (n = 28, N = 3, p < 0.05). Thus, dissociated mouse Müller cells express a prominent depletion-activated [Ca2+]i response that is susceptible to SOCE inhibition.
Given that the hyperpolarized membrane potential in mouse Müller glia (approximately −90 mV; Jo et al., 2015) constitutes a large driving force for Ca2+ influx and that absolute [Ca2+]i levels in “resting” mouse Müller glia are ∼2- to 4-fold higher compared with retinal neurons (Ryskamp et al., 2014), we hypothesized that baseline [Ca2+]MC is maintained by steady-state activation of SOC channels. However, the inhibitor cocktail did not affect baseline [Ca2+]i, which remained at 203 ± 19 nm (n = 25, N = 4; p = 0.89). Consistent with this, ablation of TRPC1/3 channels and selective blockers of Orai channels had no effect on resting [Ca2+]i (see below). Thus, the plasma membrane influx that maintains baseline [Ca2+]i is not likely to involve activation of Orai, TRP, or voltage-operated Ca2+ channels. The absence of steady-state SOC channel activation, observed as the insensitivity to pharmacological/genetic manipulation of putative SOC channels, additionally suggests that ER stores in dissociated Müller glia are not depleted of Ca2+.
Store depletion activates nonselective cation currents in Müller glia membranes
Given that commercially available TRP antibodies from major suppliers are unreliable (Gilliam and Wensel, 2011; Molnar et al., 2012), we aimed to uncover the identity of the SOC channel using electrophysiological criteria—Orai channels are almost exclusively permeable to Ca2+, whereas TRPCs are nonselective cation channels (Vig et al., 2006; Liao et al., 2014). Whole-cell currents were recorded from voltage-clamped cells either alone or in combination with Ca2+ imaging. In addition to the Ca2+ indicator (pentapotassium salt of fura-2), the pipette solution contained high Cs+, whereas the extracellular saline contained TEA/Cs+ to suppress the powerful K+ conductances that dominate the permeability of the Müller glial plasma membrane (Reichenbach and Bringmann, 2010). As shown in Figure 3A, the [Ca2+]i drop in 0 Ca2+/CPA was accompanied by a slowly developing inward current that was probably carried by Na+. The average maximal amplitude of the depletion-induced transmembrane current was −27.6 ± 5.8 pA (N = 12; Fig. 3B). The steady-state I–V relationship of this current was characterized by reversal at 0 mV, quasi-linearity, and slight inward rectification at positive potentials (Fig. 3C). The “rectification index” in the presence of CPA (I+60 mV/I−60 mV) was calculated to be 0.58 ± 0.23 (n = 4) and 0.67 ± 0.14 (n = 2; p > 0.05) for 0 Ca/CPA and 2 mm Ca/CPA currents, respectively. Na+ permeability, linearity, absence of robust inward rectification, and reversal properties of Müller SOC currents are indicative of TRPC channel activation. In cells held at −80 mV, the readdition of 2.6 mm Ca2+ caused a Ca2+ overshoot, and enhanced both inward and outward components of the transmembrane current, with a maximal amplitude of −49.5 ± 13.8 pA (n = 10 cells, N = 9 animals; Fig. 3A,B).
Figure 3.

Store depletion activates nonselective cation currents in Müller glia membranes. Whole-cell currents. A, Store depletion in 0 Ca2+/CPA induces a slow, gradually developing inward current that is followed by large transient increase during the re-exposure to [Ca2+]o. The holding potential was −80 mV. B, Representative examples of the depletion-induced inward current during sustained exposure to 0 Ca2+/CPA in the presence (1) and absence (2) of Mg2+ (DVF saline). C, Summary graph for the amplitude of the peak current induced by store depletion and re-exposure to 2 mm [Ca2+]o. Baseline currents recorded before Ca2+/CPA application were subtracted. D, Representative I–V curves taken under 0 mm Ca2+/CPA and 2 mm Ca2+/CPA conditions show reversal at 0 mV and quasi-linearity with slight inward rectification. Data are reported as the mean ± SEM. *p < 0.05.
Canonical TRP channels contribute to store-operated signals in mouse Müller glia
The main TRP channel candidate implicated in depletion-evoked Ca2+ signals in astrocytes is TRPC1, which, however, may also mediate agonist-induced Ca2+ entry and was suggested to contribute to mechanically induced glutamate release (Malarkey et al., 2008). Because Trpc1−/− retinae show a compensatory upregulation of Trpc3 transcription (Molnar et al., 2012), the store-operated responses were primarily studied in double KO Müller cells. Baseline [Ca2+]i levels in Trpc1/3−/− Müller glia were statistically indistinguishable from wild-type cells (217.3 ± 8.9 nm; n = 67 cells, N = 9 animals; p = 0.998). Store depletion in Trpc1/3−/− cells continued to evoke Ca2+ overshoots; however, averaging of peak response amplitudes showed a reduction to 297.9 ± 21 nm, an ∼45% decrease from wild-type cells (n = 64, N = 9, p < 0.05; Fig. 4A,B). Reduction in Δ[Ca2+]SOCE was associated with slower-onset kinetics in KO cells (1.68 ± 0.11 nm/s; p < 0.05). There was a tendency toward decreased Trpc1/3−/− responses in the high-amplitude range of SOC responses (Fig. 4E), suggesting that TRPC channels might be preferentially activated at stronger depletion levels. Similar to imaging data, electrophysiology revealed a reduction in the amplitude of the depletion-induced inward current in wild-type cells (27.6 ± 5.8 pA; n = 15 cells from N = 9 animals) compared with Trpc1/3−/− cells (15.5 ± 5.5 pA; n = 7; N = 3; p = 0.054), a ∼46% decrease. The residual store-operated current was inwardly rectifying, with a reversal potential (Erev) that was displaced in the positive direction (Erev = 7.75 mV; n = 4 cells, N = 2 animals; Fig. 4B), as expected if the current was mediated by Orai channels (see below). We conclude that Müller cells use TRPC1/3 channels for a substantial portion of their SOCE response.
Figure 4.

Canonical TRP channels contribute to store-operated signals in mouse Müller glia. A, The peak amplitude of the depletion-evoked Ca2+ response in Trpc1/3−/− cells (n = 14, N = 4) is reduced compared with that of WT controls (n = 16, N = 3). The SOC channel-inhibitor cocktail evokes a significant (p < 0.05) additional reduction in peak SOCE (n = 14, N = 4). B, Representative I–V curves taken during peak SOCE in 2 mm [Ca2+]i. C, The selective TRPV3 antagonist Pyr3 (5 μm) has no effect on the depletion-induced increase in fura-2 ratio. D, Summary graph for the Trpc1/3−/− cohort in the presence/absence of the inhibitor cocktail. E, TRPC channels disproportionately contribute to high-amplitude SOCE. Distribution of peak overshoot amplitudes in wild-type, Trpc1/3−/−, and cocktail-treated cells.
To determine the nature of the residual SOCE in Trpc1/3−/−, we repeated the experiment in the presence of inhibitory cocktail that blocks Orai and TRP channels. As shown in Figure 4, A and B, 2-APB/39365/Gd3+ reduced the peak store-operated response in Trpc1/3−/− Müller cells to 182.8 ± 13.0 nm (n = 33 cells, N = 8 animals; p < 0.05), values close to the baseline of 174 ± 13 nm (n = 33, N = 8; Fig. 3B). Indicating that another type of store-operated Ca2+ channel operates in tandem with TRPCs, the Δ[Ca2+]i rate for SOCE onset was further reduced by the antagonists to 0.58 ± 0.10 nm [Ca2+]i/s (p < 0.05).
Native TRPC3 channels may function as SOC channels and members of the STIM1 interactome (Lintschinger et al., 2000; Vazquez et al., 2003; Miyano et al., 2010; Liao et al., 2014), but can also form heteromeric TRPC complexes and/or function independently of internal stores (Grimaldi et al., 2003; DeHaven et al., 2009; Antigny et al., 2011; Horinouchi et al., 2012). To assess the relative contribution of TRPC3 to the overall SOCE in Müller cells, we triggered the overshoot response in the presence of Pyr3 (ethyl-1-(4-(2,3,3-trichloroacrylamide)phenyl)-5-(trifluoromethyl)-1H-pyrazole-4-carboxylate), a blocker of TRPC3- and 1-oleoyl-2-acetyl-sn-glycerol-mediated currents (Kiyonaka et al., 2009). At 5 μm, the compound had no effect on SOCE amplitude or kinetics; the average fluorescence ratio in Pyr3-treated cells was 0.4107 ± 0.0531 (n = 11), which was not significantly different (p = 0.309) from 0.5058 ± 0.0758 (n = 7) in control conditions (Fig. 4C). These data suggest that the depletion-evoked current is mediated by TRPC1 acting in combination with another channel that shows sensitivity to pan-SOC blockers.
Orai channels contribute to SOCE in Müller glia
CRAC channels are activated in a STIM-dependent fashion following the reduction of free [Ca2+]ER (Prakriya and Lewis, 2015). To assess the contribution of these Ca2+-selective channels to Müller glial SOCE, we depleted ER stores in the presence of putative selective inhibitors Synta66 and GSK7975A. Synta66 (10 μm) attenuated peak SOCE from 511.0 ± 78.5 to 349.9 ± 40.1 nm (N = 2; p < 0.01), whereas the antagonist had no significant effect on basal [Ca2+]i (221.5 ± 29.2 nm in untreated control and 251.7 ± 31.3 nm in Synta66-treated cells, respectively; Fig. 5). Likewise, the SOCE response in wild-type cells was partially antagonized by GSK-7975A (10 μm; Fig. 5C–E). The residual SOCE in Orai-inhibited cells was abolished by 2-APB/SKF 96365/Gd3+ (Fig. 5A,B).
Figure 5.

Orai channels contribute to Müller glial SOCE. A, Comparison of store depletion-evoked [Ca2+]i responses in the presence of Orai and SOC inhibitors. The Orai inhibitor Synta66 (10 μm; blue trace) partially inhibits the peak and plateau overshoot response. Additional reduction is observed in the presence of the SOC inhibitor cocktail (red trace). B, Summary graph for the calibrated [Ca2+]i response during Orai and SOC channel blockade. Data are reported as the mean ± SEM. N.S., nonsignificant. *p < 0.05. C, Increase in temperature from RT to 32°C slightly increases the amplitude of SOCE responses (white bars) without changing the relative contribution of Orai channels (dappled bars denote responses in the presence of Synta66). D, Representative inward currents, induced by CPA in DVF saline. Synta66 and GSK-7975A partially inhibit the depletion-induced inward current. E, Averaged I–V curves of depletion-evoked transmembrane currents in untreated controls (n = 10, N = 3), Synta66-treated cells (n = 10, N = 3; blue trace), and GSK7975A-treated cells (n = 10, N = 2; orange trace). F, Summary graphs for depletion evoked current responses in the presence of Orai antagonists. Shown are mean ± SEM values of the amplitude of current at 100 mV. *p < 0.0.
ICRAC activation was optimized by measuring depletion-induced currents in divalent-free (DVF) extracellular solutions, which facilitate monovalent cation influx through Orai channels (Hoth and Penner, 1993; Lepple-Wienhues and Cahalan, 1996; Prakriya and Lewis, 2015). Under these conditions, CPA induced slowly developing transmembrane currents with a quasi-linear I–V relationship (Fig. 5C,D). Consistent with ICRAC, the amplitude of these currents was attenuated in the presence of Synta66 from −26.9 ± 7.4 to −6.95 ± 1.85 pA (72.5 ± 6.9%; Fig. 4C,D). To confirm this result, we conducted the experiments with GSK-7975A, which reduced the amplitude of the depletion-induced current by 34.76 ± 7.47% (Fig. 5C–E). These results show that Orai channels contribute to SOCE in Müller glia.
Core body temperatures activate STIM1, decouple STIM1 from Orai1 (Xiao et al., 2011), and may stimulate the TRPV4 thermochannel expressed in Müller cells (Ryskamp et al., 2014). Thus, ICRAC is suboptimally activated at RT (Somasundaram et al., 1996). To determine whether glial SOCE is regulated by temperature, we compared the amplitudes of overshoot responses in control and Synta66-treated cells. The increase in temperature from RT to 32°C resulted in a modest increase in SOCE that was not statistically significant (Fig. 5C). The SOCE amplitude at 32°C was 0.723 ± 0.165 and was reduced to 0.371 ± 0.058 in the presence of Synta66 (n = 6/9 cells). Despite a ∼49% reduction, the result did not reach significance due to the large variability in the response. These data suggest that the relative fraction of Orai versus TRPC [Ca2+]SOCE response is likely to persist at the core body temperature.
If Müller glial SOCE is subserved by complementary and independent activation of TRPC1 and Orai channels, then cells lacking TRPC1 should mediate the depletion-induced calcium response exclusively through ICRAC. To test this prediction, we recorded depletion-induced currents and [Ca2+]i elevations in Trpc1−/− cells. As shown in Figure 6A, the CPA-induced inward current in DVF saline was antagonized by Synta66 (10 μm). The significant (p < 0.05) increases in the amplitude of the depletion-activated inward current (n = 5 KO cells) were almost entirely suppressed in the presence of Synta66 (Fig. 6). Analysis of depletion-induced [Ca2+]i overshoots showed a similar pronounced susceptibility of Trpc1−/− SOCE to Synta66, with the residual [Ca2+]i in 2 mm Ca2+ saline not significantly different from the predepletion baseline (Fig. 6D; n = 12).
Figure 6.

Orai channels mediate the residual SOC current in Trpc1−/− Müller cells. A, B, Whole-cell recording. ICRAC elicited in DVF/CPA saline is almost completely inhibited by Synta66. C, D, Ca imaging. The Orai blocker inhibits [Ca2+]i overshoots induced by store depletion. Bars in C show the SEM for four simultaneously recorded cells (black trace, control; gray trace, Synta66). D, cumulative data for 12 single-KO cells (N = 3 animals).
STIM1 is localized to mouse Müller glia
The activation of store-operated channels requires prior conformational changes of STIM proteins, which function as detectors for Ca2+ levels within the ER lumen (Roos et al., 2005). Analysis of retinal transcripts with the RT-PCR showed that the two STIM isoforms, which exhibit differences in Ca2+ affinity, kinetics, and localization (Wang et al., 2014; Shalygin et al., 2015; Sogkas et al., 2015), are transcribed within the mouse retina, together with transcripts coding for the three known Orai isoforms (Fig. 7A). Consistent with previous reports (Da Silva et al., 2008; Gilliam and Wensel, 2011; Molnar et al., 2012), RT-PCR results also showed the presence of Trpc1 and Trpc3. To investigate the localization of STIM1 and STIM2 isoforms, we performed immunocytochemistry in vertical sections of the mouse retina and in acutely dissociated Müller cells. Antibody labeling showed STIM1 expression to be ubiquitous across all retinal strata (Fig. 7B), with prominent immunoreactivity (IR) in rod inner segments, the ONL and the outer plexiform layer (OPL); punctate staining within the IPL; and in retinal ganglion cell (RGC) somata and proximal dendrites. Double labeling with the Müller cell marker GS showed massive STIM1-GS colocalization in glial end-feet that shadow endothelial vessels within the inner limiting membrane (ILM), indicating that Müller glia predominantly express STIM1 (Fig. 7B,C). This is exemplified in the inset in Figure 7C, where STIM1 signals are superposed onto GS-IR glial end-feet and radial processes that extend into the IPL. STIM2 signals were more prominent within the OPL and RGC layer but, while largely excluded from apical/basal processes and end-feet, also weakly labeled Müller cell somata (Fig. 7). Thus, SOCE in Müller cells is likely to mainly use the canonical STIM1 mechanism.
Figure 7.

STIM expression in the mouse retina. A, Representative RT-PCR data demonstrating that the mouse retina expresses transcripts encoding Stim1, Stim2, Orai1–3, Trpc1, and Trpc3 genes. B, Dissociated cells labeled with STIM1 and STIM2 antibodies, together with the GS and DAPI (perikaryal) markers. STIM1 is expressed in the cell body, apical/distal stalks, and the end-foot. Weak STIM2-IR is observed in the cell body. Putative rods (asterisks) and amacrine cell (#) are also labeled. S, Soma; Ef, end-foot. Scale bar, 10 μm. C, Representative confocal images of vertical sections double labeled for STIM1, STIM2, and GS. STIM1-IR is observed across all retinal layers but is particularly strong in RGC somata and Müller end-feet (marked by GS–STIM1 colocalization, arrowheads). STIM2-IR is localized to the proximal INL, potentially corresponding to bipolar somata and dendritic processes (arrows). Insets 1 and 2 are magnified regions selected by corresponding rectangles in the merged images showing the presence of STIM1 and the absence of STIM2 in the end-foot. D, Representative retinal STIM immunoblots.
Presence of ER cisternae within the Müller cell end-feet suggests a specialized signaling compartment
Because of the intimate association of Müller cells with every class of cell in the inner retina, they are ideally positioned to respond to signaling involving vascular, glial, and neural components. The privileged activation of depletion-induced Ca2+ signals in end-feet (Fig. 1) together with the disproportionate expression of STIM1 in this compartment (Fig. 7) suggest that SOCE might have important functions in the regulation of Müller–vascular, Müller–astrocyte, and Müller–axonal interactions at the vitreal interface, and that SOCE-dependent signals propagating centrifugally in the form of Ca2+ waves might regulate function in the proximal retina. We searched for an ultrastructural correlate for the preferential Ca2+ store signaling within the Müller end-foot within the existing literature database but, surprisingly, were unable to locate evidence about the localization and distribution of such putative ER structures. Given the central importance of end-foot ER stores for astroglial signaling in general (Verkhratsky et al., 2014), together with their known roles in retinal neuroglial, gliovascular, volume, and metabolic homeostasis (Newman and Zahs, 1998; Pivneva et al., 2008; Jo et al., 2015; Newman, 2015), we examined EM sections from the RC2 mouse connectome that is currently under construction. Figure 8A shows a vertical section of the proximal inner retina, consisting of a Müller cell end-foot directly adjacent to a ganglion cell (blue), a retinal astrocyte (green), an axon fiber bundle, and pericyte/endothelial constituents of a blood vessel. Müller cell end-feet contain dense arrays of SER tubules representing 100–1000 more SER membrane per unit volume than any other cell in the retina. Classic SER cisternae in Müller cell end-feet are transformed into a massive collection of organized SER tubules with dentate cross sections (Marc et al., 2013), some as fine as 10 nm in diameter. At the vitreal interface and throughout the proximal fiber, the SER completely and densely surrounds mitochondria, indicating a role for SER–mitochondrial interactions in the regulation of the ICRAC and Ca2+ sequestration into each compartment (Watson and Parekh, 2012). The SER tubules are densely packed throughout terminal end-feet and also reach up into the IPL. These network tubules are particularly evident at the end-foot:astrocyte interface (Fig. 8B), where the tubules largely run parallel to the astrocyte membrane. Similarly, all neurons encased by Müller cell end-feet are surrounded by a dense mesh of SER tubules (Fig. 8C,D), and the basement membrane of endothelia (Fig. 8E,F) is directly adjacent to large arrays of tubules. We propose that the abundance of SER tubules within the Müller end-foot (outstripping any other retinal cell by two to three orders of magnitude) provides a potentially significant resource for tonic and activity-dependent release of Ca2+ and consequent STIM1-dependent activation of TRPC1/Orai channels.
Figure 8.

SER-rich Müller cell end-feet contact astrocytes, microglia, endothelia, and neurons in the mouse retina. A, Vertical section of the proximal, electron-dense end-foot of a Müller cell (MC) adjacent to the ocular vitreous (*), with abundant end-foot mitochondria (m). MCs are immediately adjacent to neurons such as ganglion cells (GC; blue), large bundles of axon fascicles (F), astrocytes (AsC; green), and endothelial cells (Endo) of capillaries (C). B, A horizontal section of six Müller cell proximal end-feet (MC1–6) approaching the vitreal border and surrounding an astrocyte. MC4 exemplifies the high MC SER tubule density with many 15–20 nm tubules running parallel to the plasmalemma next to the astrocyte. Mitochondria are embedded in dense SER networks. C, A horizontal section of six Müller cell proximal end-feet surrounding the initial ascending dendrite of a ganglion cell, and contacting both endothelial and microglial cells. D, High-resolution view of Müller cell end-foot MC4 from C demonstrating the high density of SER tubules near the ganglion cell. Large SER tubules tend to be dentate in shape (vertical arrows) and branch into very tiny tubules ∼15 nm in diameter near the membrane (slanted arrows). E, A horizontal section of a dozen SER-rich Müller cell end-feet with two (MC3, MC4, single arrows) forming extensive apposition to the basement membrane (yellow) of capillary endothelia. An astrocyte forms a small basement membrane apposition. F, High-resolution view of Müller cell end-foot MC1 from E demonstrating the high density of fine SER tubules near the astrocyte (vertical arrows) and cytoskeletal elements in the two cells. Actin filaments (10 nm) are positioned in bundles along the Müller cell membrane. Finer intermediate filaments (5–6 nm) form large axial bundles throughout the astrocyte. Scale markers on all panels, 1000 nm.
Ablation of TRPC1 channels enhances reactive gliosis
TRPC channels and Ca2+ stores might be involved in neurological disease (Selvaraj et al., 2012) and in mechanotransduction (Maroto et al., 2005). Given the proposed mechanosensory role for astrocyte TRPC1 (Malarkey et al., 2008), we asked whether TRPC1 ablation might have an impact on the reactive gliosis observed in a mouse glaucoma model (Huang et al., 2011). Microinjection of polystyrene microbeads resulted in comparable elevations in the IOP of wild-type and Trpc1/3−/− animals 1 week postinjection (WT eyes, 21.0 ± 1.0 mmHg, N = 9; KO eyes, 20.6 ± 0.8 mmHg, N = 9; Fig. 9A), suggesting that TRPC1/3 channels do not contribute to IOP regulation. Suggestive of reactive gliosis induced by mechanical trauma, both genotypes showed increased immunoreactivity for the reactive gliotic marker GFAP (Fig. 9B). GFAP immunoreactivity in Trpc1/3−/− microbead-injected eyes appeared to be stronger, with faster onset (Fig. 9C) compared with WT controls (N = 10 for WT and N = 14 for Trpc1/3−/−; p < 0.05). Thus, in contrast to other TRP isoforms that contribute to reactive gliosis upon activation or deletion (Ryskamp et al., 2014), TRPC1 might serve as a suppressor of the force-induced reactive response.
Figure 9.

Ablation of TRPC1/3 channels enhances gliosis in an acute mouse glaucoma model. A, Injection of polystyrene microbeads (10 μm) elevates IOP in WT and Trpc1/3−/− eyes. Averaged time course of intraocular pressure in wild-type and TRPC1/3−/− mice. Time point of microbead injection is referred to as a day 0. B, Representative retinal sections prepared from wild-type or TRPC1/3−/− mice and labeled for GFAP 8 weeks after the injection of microbeads. C, Averaged time course of the intensity of GFAP immunosignals in retinal sections from wild-type and TRPC1/3−/− mice.
Discussion
This study provides five major new insights regarding the physiology of mammalian Müller glia. First, we report that store-operated signaling represents a major signaling pathway and source of cytosolic Ca2+ in mouse Müller cells. Second, SOCE-associated [Ca2+]i signals constitute a trigger mechanism for propagating excitation from the inner/outer retina into Müller somata in the form of Ca2+ waves that follow store release and depletion. Third, depletion of Müller glial Ca2+ stores synergistically activates TRPC and Orai channels, probably through STIM1. Fourth, the high density and complex location-specific arrangements of ER cisternae and ER–mitochondrial contacts in the end-foot indicate a potentially key function for Ca2+ stores/SOCE in local cell interactions. Finally, our data suggest that glial TRPC1 channels may be sensitive to mechanical stress and modulate force-induced gliosis in the mammalian retina. Together, these pharmacological, imaging, electrophysiological, immunostaining, genetic, and ultrastructural data expand our understanding of Müller glial and retinal Ca2+ homeostasis, contribute to the current debate about astroglial SOCE, and provide insight into how glial signaling at the ILM could influence the physiology of the outer retina.
Depletion-induced Müller glial [Ca2+]i signals exhibit an electrophysiological and pharmacological profile of combined TRPC1 and Orai activation (Fig. 10). Many studies in heterologously expressing and native cells, including astrocytes, have questioned whether TRPC1 can mediate SOCE, as indicated by the following observations: (1) recombinant TRPC1 may be insensitive to store depletion (DeHaven et al., 2009); (2) RNAi-mediated knockdown of Trpc1 does not compromise SOCE (Storch et al., 2012); (3) genetic ablation of TRPC1 does not affect SOC signals in some cell types (Dietrich et al., 2007; Varga-Szabo et al., 2008; Molnar et al., 2012); (4) TRPC1 expression may not increase whole-cell currents induced by store depletion (Sinkins et al., 1998; Lintschinger et al., 2000); (5) TRPC1 may not mediate SOCE in some cortical astrocytes (Grimaldi et al., 2003); and (6) the founding member of the family, dTRP, is not store operated (Prakriya and Lewis, 2015). Our conclusion that TRPC1 partially mediates SOCE in Müller glia is based on the following: (1) I–V linearity of the CPA-sensitive inward current; (2) reversal at 0 mV; (3) permeability to Na+ in 2 mm Ca2+-containing saline; (4) slow-onset kinetics typical of SOC currents; and (5) susceptibility to nonselective TRP antagonists. In addition, (6) Trpc1/3−/− cells show reduced SOCE compared with wild-type cells, and Orai blockers inhibit SOCE in Trpc1−/− cells, whereas the TRPC3 blocker Pyr3 has no effect the depletion response; and (7) Ca2+ overshoots induced by re-exposure to 2 mm [Ca2+]o are only partially antagonized by Orai antagonists but (8) are blocked by nonselective TRPC inhibitors. Finally, Trpc1 mRNA and/or protein were detected in immortalized Müller cell cultures (Da Silva et al., 2008) and were localized to the upper part of the INL (Gilliam and Wensel, 2011; Molnar et al., 2012). The properties of residual store-activated signals in Trpc1/3−/− cells, inward rectification, sensitivity of the depletion-induced current to [Ca2++ Mg2+]o, and inhibition by Synta66/GSK-7975A indicate that Orai channels and ICRAC contribute the remaining ∼50% (30–70%) to the overall depletion-induced current and [Ca2+]i. The identity of the Orai isoform that supports ICRAC in Müller glia remains to be determined.
Figure 10.
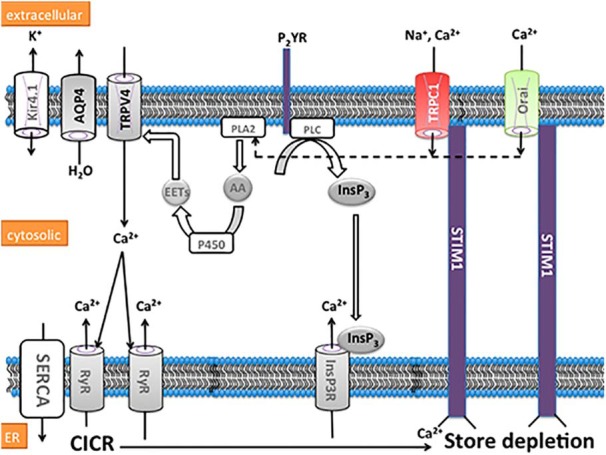
Schematic of proposed store-operated signaling mechanisms in end-feet of Müller glia. Partial or full depletion of the ER Ca2+ pool induced by activity-dependent lowering of [Ca2+]o, ryanodine receptor (RyR) opening and/or stimulation of the purinergic P2Y receptor-coupled pathway that includes phospholipase C (PLC) and InsP3R activation, induces conformational change in STIM1, triggering subsequent activation of plasma membrane TRPC1 and Orai channels. The resulting Ca2+ influx leads to the activation of phospholipase A2 (PLA2), cytochrome P450 (P450), and epoxyeicosatrienoic acid (EET)-sensitive TRPV4 channels, which form a functional complex with AQP4 water channels and Kir4.1 subunit-containing K+ channels. The stores are eventually calcium-replenished through SERCA pumps.
Although astrocyte SOCE is believed to predominantly involve TRPC1 (Kettenman and Ransom, 2013; Verkhratsky et al., 2014), cultured cortical astrocytes express Stim1 and Orai1 mRNA (Barajas et al., 2008; Moreno et al., 2012; Motiani et al., 2013), whereas hippocampal astrocytes express Trpc1, Orai2, and Orai3 but lack Orai1 (Ronco et al., 2014). It remains to be seen, however, how studies performed in neonatal astroglial cultures extrapolate to native astrocytes and Bergmann glia (Pivneva et al., 2008; Foo et al., 2011). Arachidonic acid, which activates Orai1-Orai3 heteromers (Thompson and Shuttleworth, 2013) and Ca2+ influx in Bergmann glia (Singaravelu et al., 2006), also increases [Ca2+]i in Müller cells; however, these signals are mainly mediated through the CYP450–TRPV4 pathway rather than the ARC pathway (Ryskamp et al., 2015).
Antibody labeling suggests that isoform 1 is the predominant retinal STIM, with punctate expression across every layer but particularly strong expression in RGC somata and Müller end-feet, whereas STIM2 appeared to be localized mainly to bipolar cells and RGCs, with weak staining of Müller perikarya. The precise sequence of molecular steps that underpin combined Orai and TRPC1 activation by STIM1 is unknown—it has been suggested that TRPC1 activation requires prior STIM1-dependent activation of Orai1 (Ong et al., 2007; Jardin et al., 2008; Kim et al., 2009), that Orai activation is necessary for TRPC1 insertion into the plasma membrane (Vaca, 2010; Cheng et al., 2011), and that Orai1 represents regulatory subunits of the TRPC–STIM complex (Liao et al., 2014); however, TRPC1 but not Orai channels can function independently of STIMs (DeHaven et al., 2009; Choi et al., 2015). Our observation that depletion-induced activation persists in the presence of selective Orai blockers suggests that TRPC channels can be directly activated by store depletion (possibly through charged residues within the N-terminal K-domain of the depletion sensor; Zeng et al., 2008). It is tempting to speculate that different channel activation mechanisms associated with SOCE might perform distinct computational roles in radial glia (i.e., transducing different ER depletion levels).
The TRPC1–Orai–STIM1 mechanism is likely to function in conjunction with other components of the Müller end-foot signaling “toolkit,” including Ca2+ release channels, SERCA2, TRPV4, aquaporin 4 (AQP4), Kir 4.1, plasma membrane Ca2+ ATPases, and mitochondria (Križaj et al., 2004; Jo et al., 2015). We found that end-foot domains that ensheath astrocytes, axons, and blood vessels are packed with ER cisternae, STIM1, and mitochondria. Unlike cultured cells, which express SER predominantly around the nucleus and primary astrocytes, where it is concentrated near the plasma membrane (Pivneva et al., 2008), the Müller end-foot combines massive intracellular ER with subsurface cisternae that mirror vasculature, astrocytes, and neurons. Accordingly, end-feet showed the largest calcium/current responses to store depletion and originated the majority of depletion-induced Ca2+ waves. Ca2+ waves in Müller end-feet are also triggered by light, mechanical stimuli, TRPV4 channels, ATP, caffeine, and by activity-dependent decreases in extracellular calcium levels (Keirstead and Miller, 1995; Newman and Zahs, 1998; Li et al., 2001; Ryskamp et al., 2014), indicating that Müller excitability is subserved by both calcium-induced calcium release and SOCE. Close apposition of end-foot ER and mitochondria points to possible regulation of local ATP supply through SOCE-dependent modulation of Ca2+-sensitive mitochondrial F0F1 ATPases, dehydrogenases, and motility, however, ICRAC can also be modulated by mitochondria (Watson and Parekh, 2012; Müller et al., 2014; Jackson and Robinson, 2015). The STIM1/Orai/TRPC1 association may also regulate the ER stress response (Prins et al., 2011) and contribute to the function of the end-foot “dystrophin-associated protein complex” (Vandebrouck et al., 2007; Sabourin et al., 2012).
The relatively high [Ca2+]i levels (∼200 nm) and noisy baseline in hyperpolarized (approximately −90 mV), “resting” cells were similar to previously reported values (Keirstead and Miller, 1995; Jo et al., 2015) and might reflect a unique intracellular environment that affects ratiometric calibration and/or an unknown Ca2+ influx mechanism that is independent of voltage and SOCE. Its resistance to Orai/TRPC blockers additionally suggests that ER stores in isolated cells are not Ca2+ depleted. While the precise physiological function for TRPC1 and Orai channels in radial glia remains to be elucidated, TRPC1 has recently been implicated in the regulation of astrocyte proliferation (Golovina, 2005) and malignant transformation (Bomben and Sontheimer, 2010). An intriguing aspect of TRPC1 expression in Müller cells was the increased glial resilience in response to mechanical stress imposed by elevated IOP. We found that IOP-induced reactive gliosis is exacerbated in Trpc1/3−/− retinae, possibly due to the mechanosensitivity of TRPC1 (Maroto et al., 2005; Malarkey et al., 2008). Of note, inflammatory signaling is compromised in patients with low ICRAC activity (Feske et al., 2006), suggesting that SOC channels might modulate both protective and proinflammatory responses.
Overall, this study provides pharmacological, genetic, and functional evidence that depletion-dependent Ca2+ homeostasis in freshly isolated adult radial astroglia involves synergistic yet independent activation of two phylogenetically distinct SOC channels. The replenishment of ER stores may be crucial for the long-term maintenance of cytosolic Ca2+ signaling and glial metabolism (Müller et al., 2014); however, we also found SOCE to be sufficient to initiate the propagation of transcellular Ca2+ waves that might transmit excitation from proximal and distal retinae into the Müller soma. Our results may provide insight into the early mechanisms through which radial glia support early neuronal migration in the retina, which depends critically on SOCE (Sugioka et al., 1999; Sugioka and Yamashita, 2003), and into mechanisms that modulate the retinal glial reactive response to mechanical stress and stretch (Wang et al., 2013; Križaj et al., 2014). Moreover, our data provide, for the first time, ultrastructural evidence of the intracellular compartment complexity within the Müller end-foot together with direct experimental evidence for ICRAC in adult astroglia. These findings expand the signaling repertoire within which ER influences Müller function and show that SOCE is well placed to mediate the interactions between extracellular stimuli that activate astroglial store release, Ca2+ homeostasis, and excitability.
Footnotes
This research was supported by National Institutes of Health (NIH) Grants EY-015128 and EY-002576 (to R.E.M.), EY-022076 and EY-013870 (to D.K.), and P30-EY-014800 (to R.E.M. and D.K.); Department of Defense Grant W81XWH-12-1-0244; the University of Utah Neuroscience Initiative; the Willard L. Eccles Foundation (to D.K.); and unrestricted support from Research to Prevent Blindness to the Moran Eye Institute. A.I. was supported by predoctoral NIH Vision Training Grant T32EY-024234. We thank Dr. Daniel Ryskamp (University of Texas Southwestern Medical Center, Dallas, TX) for comments on the manuscript.
The authors declare no competing financial interests.
References
- Anderson JR, Jones BW, Yang JH, Shaw MV, Watt CB, Koshevoy P, Spaltenstein J, Jurrus E, Kannan UV, Whitaker RT, Mastronarde D, Tasdizen T, Marc RE. A computational framework for ultrastructural mapping of neural circuitry. PLoS Biol. 2009;7:e1000074. doi: 10.1371/journal.pbio.1000074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson JR, Jones BW, Watt CB, Shaw MV, Yang JH, Demill D, Lauritzen JS, Lin Y, Rapp KD, Mastronarde D, Koshevoy P, Grimm B, Tasdizen T, Whitaker R, Marc RE. Exploring the retinal connectome. Mol Vis. 2011a;17:355–379. doi: 10.3390/molecules17010355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson JR, Mohammed S, Grimm B, Jones BW, Koshevoy P, Tasdizen T, Whitaker R, Marc RE. The Viking viewer for connectomics: scalable multi-user annotation and summarization of large volume data sets. J Microsc. 2011b;241:13–28. doi: 10.1111/j.1365-2818.2010.03402.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antigny F, Jousset H, König S, Frieden M. Thapsigargin activates Ca2+ entry both by store-dependent, STIM1/Orai1-mediated, and store-independent, TRPC3/PLC/PKC-mediated pathways in human endothelial cells. Cell Calcium. 2011;49:115–127. doi: 10.1016/j.ceca.2010.12.001. [DOI] [PubMed] [Google Scholar]
- Barajas M, Andrade A, Hernandez-Hernandez O, Felix R, Arias-Montaño JA. Histamine-induced Ca2+ entry in human astrocytoma U373 MG cells: evidence for involvement of store-operated channels. J Neurosci Res. 2008;86:3456–3468. doi: 10.1002/jnr.21784. [DOI] [PubMed] [Google Scholar]
- Bomben VC, Sontheimer H. Disruption of transient receptor potential canonical channel 1 causes incomplete cytokinesis and slows the growth of human malignant gliomas. Glia. 2010;58:1145–1156. doi: 10.1002/glia.20994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Križaj D, Thoreson WB. Intracellular calcium stores drive slow non-ribbon vesicle release from rod photoreceptors. Front Cell Neurosci. 2014;8:20. doi: 10.3389/fncel.2014.00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng KT, Liu X, Ong HL, Swaim W, Ambudkar IS. Local Ca2+ entry via Orai1 regulates plasma membrane recruitment of TRPC1 and controls cytosolic Ca2+ signals required for specific cell functions. PLoS Biol. 2011;9:e1001025. doi: 10.1371/journal.pbio.1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi HJ, Sun D, Jakobs TC. Isolation of intact astrocytes from the optic nerve head of adult mice. Exp Eye Res. 2015;137:103–110. doi: 10.1016/j.exer.2015.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da Silva N, Herron CE, Stevens K, Jollimore CA, Barnes S, Kelly ME. Metabotropic receptor-activated calcium increases and store-operated calcium influx in mouse Muller cells. Invest Ophthalmol Vis Sci. 2008;49:3065–3073. doi: 10.1167/iovs.07-1118. [DOI] [PubMed] [Google Scholar]
- DeHaven WI, Jones BF, Petranka JG, Smyth JT, Tomita T, Bird GS, Putney JW., Jr TRPC channels function independently of STIM1 and Orai1. J Physiol. 2009;587:2275–2298. doi: 10.1113/jphysiol.2009.170431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich A, Kalwa H, Storch U, Mederos y Schnitzler M, Salanova B, Pinkenburg O, Dubrovska G, Essin K, Gollasch M, Birnbaumer L, Gudermann T. Pressure-induced and store-operated cation influx in vascular smooth muscle cells is independent of TRPC1. Pflugers Arch. 2007;455:465–477. doi: 10.1007/s00424-007-0314-3. [DOI] [PubMed] [Google Scholar]
- Duncan JL, Yang H, Doan T, Silverstein RS, Murphy GJ, Nune G, Liu X, Copenhagen D, Tempel BL, Rieke F, Krizaj D. Scotopic visual signaling in the mouse retina is modulated by high-affinity plasma membrane calcium extrusion. J Neurosci. 2006;26:7201–7211. doi: 10.1523/JNEUROSCI.5230-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, Hogan PG, Lewis RS, Daly M, Rao A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. 2006;441:179–185. doi: 10.1038/nature04702. [DOI] [PubMed] [Google Scholar]
- Foo LC, Allen NJ, Bushong EA, Ventura PB, Chung WS, Zhou L, Cahoy JD, Daneman R, Zong H, Ellisman MH, Barres BA. Development of a method for the purification and culture of rodent astrocytes. Neuron. 2011;71:799–811. doi: 10.1016/j.neuron.2011.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilliam JC, Wensel TG. TRP channel gene expression in the mouse retina. Vision Res. 2011;51:2440–2452. doi: 10.1016/j.visres.2011.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golovina VA. Visualization of localized store-operated calcium entry in mouse astrocytes. Close proximity to the endoplasmic reticulum. J Physiol. 2005;564:737–749. doi: 10.1113/jphysiol.2005.085035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimaldi M, Maratos M, Verma A. Transient receptor potential channel activation causes a novel form of [Ca2+]i oscillations and is not involved in capacitative Ca2+ entry in glial cells. J Neurosci. 2003;23:4737–4745. doi: 10.1523/JNEUROSCI.23-11-04737.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horinouchi T, Higashi T, Higa T, Terada K, Mai Y, Aoyagi H, Hatate C, Nepal P, Horiguchi M, Harada T, Miwa S. Different binding property of STIM1 and its novel splice variant STIM1L to Orai1, TRPC3, and TRPC6 channels. Biochem Biophys Res Commun. 2012;428:252–258. doi: 10.1016/j.bbrc.2012.10.034. [DOI] [PubMed] [Google Scholar]
- Hoth M, Penner R. Calcium release-activated calcium current in rat mast cells. J Physiol. 1993;465:359–386. doi: 10.1113/jphysiol.1993.sp019681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Xing W, Ryskamp DA, Punzo C, Križaj D. Localization and phenotype-specific expression of ryanodine calcium release channel in C57BL6 and DBA/2J mouse strains. Exp Eye Res. 2011;93:700–709. doi: 10.1016/j.exer.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson JG, Robinson MB. Reciprocal regulation of mitochondrial dynamics and calcium signaling in astrocyte processes. J Neurosci. 2015;35:15199–15213. doi: 10.1523/JNEUROSCI.2049-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jardin I, Lopez JJ, Salido GM, Rosado JA. Orai1 mediates the interaction between STIM1 and hTRPC1 and regulates the mode of activation of hTRPC1-forming Ca2+ channels. J Biol Chem. 2008;283:25296–25304. doi: 10.1074/jbc.M802904200. [DOI] [PubMed] [Google Scholar]
- Jo AO, Ryskamp DA, Phuong TT, Verkman AS, Yarishkin O, MacAulay N, Križaj D. TRPV4 and AQP4 channels synergistically regulate cells volume and calcium homeostasis in retinal Müller glia. J Neurosci. 2015;35:13525–13537. doi: 10.1523/JNEUROSCI.1987-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keirstead SA, Miller RF. Calcium waves in dissociated retinal glial (Müller) cells are evoked by release of calcium from intracellular stores. Glia. 1995;14:14–22. doi: 10.1002/glia.440140104. [DOI] [PubMed] [Google Scholar]
- Kettenman H, Ransom B. Neuroglia. Oxford, UK: Oxford UP; 2013. [Google Scholar]
- Khakh BS, McCarthy KD. Astrocyte calcium signaling: from observations to functions and the challenges therein. Cold Spring Harb Perspect Biol. 2015;7:a020404. doi: 10.1101/cshperspect.a020404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MS, Hong JH, Li Q, Shin DM, Abramowitz J, Birnbaumer L, Muallem S. Deletion of TRPC3 in mice reduces store-operated Ca2+ influx and the severity of acute pancreatitis. Gastroenterology. 2009;137:1509–1517. doi: 10.1053/j.gastro.2009.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyonaka S, Kato K, Nishida M, Mio K, Numaga T, Sawaguchi Y, Yoshida T, Wakamori M, Mori E, Numata T, Ishii M, Takemoto H, Ojida A, Watanabe K, Uemura A, Kurose H, Morii T, Kobayashi T, Sato Y, Sato C, Hamachi I, Mori Y. Selective and direct inhibition of TRPC3 channels underlies biological activities of a pyrazole compound. Proc Natl Acad Sci U S A. 2009;106:5400–5405. doi: 10.1073/pnas.0808793106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Križaj D. Calcium stores in vertebrate photoreceptors. Adv Exp Med Biol. 2012;740:873–889. doi: 10.1007/978-94-007-2888-2_39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Križaj D, Liu X, Copenhagen DR. Expression of calcium transporters in the retina of the tiger salamander (Ambystoma tigrinum) J Comp Neurol. 2004;475:463–480. doi: 10.1002/cne.20170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Križaj D, Ryskamp DA, Tian N, Tezel G, Mitchell CH, Slepak VZ, Shestopalov VI. From mechanosensitivity to inflammatory responses: new players in the pathology of glaucoma. Curr Eye Res. 2014;39:105–119. doi: 10.3109/02713683.2013.836541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepple-Wienhues A, Cahalan MD. Conductance and permeation of monovalent cations through depletion-activated Ca2+ channels (ICRAC) in Jurkat T cells. Biophys J. 1996;71:787–794. doi: 10.1016/S0006-3495(96)79278-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Holtzclaw LA, Russell JT. Müller cell Ca2+ waves evoked by purinergic receptor agonists in slices of rat retina. J Neurophysiol. 2001;85:986–994. doi: 10.1152/jn.2001.85.2.986. [DOI] [PubMed] [Google Scholar]
- Liao Y, Abramowitz J, Birnbaumer L. The TRPC family of TRP channels: roles inferred (mostly) from knockout mice and relationship to ORAI proteins. Handb Exp Pharmacol. 2014;223:1055–1075. doi: 10.1007/978-3-319-05161-1_14. [DOI] [PubMed] [Google Scholar]
- Lintschinger B, Balzer-Geldsetzer M, Baskaran T, Graier WF, Romanin C, Zhu MX, Groschner K. Coassembly of Trp1 and Trp3 proteins generates diacylglycerol- and Ca2+-sensitive cation channels. J Biol Chem. 2000;275:27799–27805. doi: 10.1074/jbc.M002705200. [DOI] [PubMed] [Google Scholar]
- Lipp S, Wurm A, Pannicke T, Wiedemann P, Reichenbach A, Chen J, Bringmann A. Calcium responses mediated by type 2 IP3-receptors are required for osmotic volume regulation of retinal glial cells in mice. Neurosci Lett. 2009;457:85–88. doi: 10.1016/j.neulet.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malarkey EB, Ni Y, Parpura V. Ca2+ entry through TRPC1 channels contributes to intracellular Ca2+ dynamics and consequent glutamate release from rat astrocytes. Glia. 2008;56:821–835. doi: 10.1002/glia.20656. [DOI] [PubMed] [Google Scholar]
- Marc RE, Jones BW, Watt CB, Anderson JR, Sigulinsky C, Lauritzen S. Retinal connectomics: towards complete, accurate networks. Prog Retin Eye Res. 2013;37:141–162. doi: 10.1016/j.preteyeres.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroto R, Raso A, Wood TG, Kurosky A, Martinac B, Hamill OP. TRPC1 forms the stretch-activated cation channel in vertebrate cells. Nat Cell Biol. 2005;7:179–185. doi: 10.1038/ncb1218. [DOI] [PubMed] [Google Scholar]
- Miyano K, Morioka N, Sugimoto T, Shiraishi S, Uezono Y, Nakata Y. Activation of the neurokinin-1 receptor in rat spinal astrocytes induces Ca2+ release from IP3-sensitive Ca2+ stores and extracellular Ca2+ influx through TRPC3. Neurochem Int. 2010;57:923–934. doi: 10.1016/j.neuint.2010.09.012. [DOI] [PubMed] [Google Scholar]
- Molnar T, Barabas P, Birnbaumer L, Punzo C, Kefalov V, Križaj D. Store-operated channels regulate intracellular calcium in mammalian rods. J Physiol. 2012;590:3465–3481. doi: 10.1113/jphysiol.2012.234641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molnar T, Frye AM, Barabas P, Ryskamp DA, Križaj D. The molecular mechanisms of store-operated calcium entry in Müller glia. Invest Ophthalmol Vis Sci. 2013;54:6344. [Google Scholar]
- Moreno C, Sampieri A, Vivas O, Peña-Segura C, Vaca L. STIM1 and Orai1 mediate thrombin-induced Ca(2+) influx in rat cortical astrocytes. Cell Calcium. 2012;52:457–467. doi: 10.1016/j.ceca.2012.08.004. [DOI] [PubMed] [Google Scholar]
- Motiani RK, Hyzinski-García MC, Zhang X, Henkel MM, Abdullaev IF, Kuo YH, Matrougui K, Mongin AA, Trebak M. STIM1 and Orai1 mediate CRAC channel activity and are essential for human glioblastoma invasion. Pflugers Arch. 2013;465:1249–1260. doi: 10.1007/s00424-013-1254-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller MS, Fox R, Schousboe A, Waagepetersen HS, Bak LK. Astrocyte glycogenolysis is triggered by store-operated entry and provides metabolic energy for cellular calcium homeostasis. Glia. 2014;62:526–534. doi: 10.1002/glia.22623. [DOI] [PubMed] [Google Scholar]
- Neher E. The use of fura-2 for estimating Ca buffers and Ca fluxes. Neuropharmacology. 1995;34:1423–1442. doi: 10.1016/0028-3908(95)00144-U. [DOI] [PubMed] [Google Scholar]
- Newman EA. Glial cell regulation of neuronal activity and blood flow in the retina by release of gliotransmitters. Philos Trans R Soc Lond B Biol Sci. 2015;370:20140195. doi: 10.1098/rstb.2014.0195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EA, Zahs KR. Modulation of neuronal activity by glial cells in the retina. J Neurosci. 1998;18:4022–4028. doi: 10.1523/JNEUROSCI.18-11-04022.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong HL, Cheng KT, Liu X, Bandyopadhyay BC, Paria BC, Soboloff J, Pani B, Gwack Y, Srikanth S, Singh BB, Gill D, Ambudkar IS. Dynamic assembly of TRPC1-STIM1-Orai1 ternary complex is involved in store-operated calcium influx. Evidence for similarities in store-operated and calcium release-activated calcium channel components. J Biol Chem. 2007;282:9105–9116. doi: 10.1074/jbc.M608942200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pivneva T, Haas B, Reyes-Haro D, Laube G, Veh RW, Nolte C, Skibo G, Kettenmann H. Store-operated Ca2+ entry in astrocytes: different spatial arrangement of endoplasmic reticulum explains functional diversity in vitro and in situ. Cell Calcium. 2008;43:591–601. doi: 10.1016/j.ceca.2007.10.004. [DOI] [PubMed] [Google Scholar]
- Prakriya M, Lewis RS. Store-operated calcium channels. Physiol Rev. 2015;95:1383–1436. doi: 10.1152/physrev.00020.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prins D, Groenendyk J, Touret N, Michalak M. Modulation of STIM1 and capacitative Ca2+ entry by the endoplasmic reticulum luminal oxidoreductase ERp57. EMBO Rep. 2011;12:1182–1188. doi: 10.1038/embor.2011.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichenbach A, Bringmann A. Müller cells in the healthy and diseased retina. New York: Springer; 2010. [DOI] [PubMed] [Google Scholar]
- Rentería RC, Strehler EE, Copenhagen DR, Križaj D. Ontogeny of plasma membrane Ca2+ ATPase isoforms in the neural retina of the postnatal rat. Vis Neurosci. 2005;22:263–274. doi: 10.1017/S0952523805223027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronco V, Grolla AA, Glasnov TN, Canonico PL, Verkhratsky A, Genazzani AA, Lim D. Differential deregulation of astrocytic calcium signalling by amyloid-β, TNFα, IL-1β and LPS. Cell Calcium. 2014;55:219–229. doi: 10.1016/j.ceca.2014.02.016. [DOI] [PubMed] [Google Scholar]
- Roos J, DiGregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, Zhang S, Safrina O, Kozak JA, Wagner SL, Cahalan MD, Veliçelebi G, Stauderman KA. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol. 2005;169:435–445. doi: 10.1083/jcb.200502019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rychkov G, Barritt GJ. TRPC1 Ca(2+)-permeable channels in animal cells. Handb Exp Pharmacol. 2007;179:23–52. doi: 10.1007/978-3-540-34891-7_2. [DOI] [PubMed] [Google Scholar]
- Ryskamp DA, Witkovsky P, Barabas P, Huang W, Koehler C, Akimov NP, Lee SH, Chauhan S, Xing W, Rentería RC, Liedtke W, Krizaj D. The polymodal ion channel transient receptor potential vanilloid 4 modulates calcium flux, spiking rate, and apoptosis of mouse retinal ganglion cells. J Neurosci. 2011;31:7089–7101. doi: 10.1523/JNEUROSCI.0359-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryskamp DA, Jo AO, Frye AM, Vazquez-Chona F, MacAulay N, Thoreson WB, Križaj D. Swelling and eicosanoid metabolites differentially gate TRPV4 channels in retinal neurons and glia. J Neurosci. 2014;34:15689–15700. doi: 10.1523/JNEUROSCI.2540-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryskamp DA, Iuso A, Križaj D. TRPV4 links inflammatory signaling and neuroglial swelling. Channels. 2015;9:70–72. doi: 10.1080/19336950.2015.1017998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabourin J, Harisseh R, Harnois T, Magaud C, Bourmeyster N, Déliot N, Constantin B. Dystrophin/α1-syntrophin scaffold regulated PLC/PKC-dependent store-operated calcium entry in myotubes. Cell Calcium. 2012;52:445–456. doi: 10.1016/j.ceca.2012.08.003. [DOI] [PubMed] [Google Scholar]
- Selvaraj S, Sun Y, Watt JA, Wang S, Lei S, Birnbaumer L, Singh BB. Neurotoxin-induced ER stress in mouse dopaminergic neurons involves downregulation of TRPC1 and inhibition of AKT/mTOR signaling. J Clin Invest. 2012;122:1354–1367. doi: 10.1172/JCI61332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalygin A, Skopin A, Kalinina V, Zimina O, Glushankova L, Mozhayeva GN, Kaznacheyeva E. STIM1 and STIM2 proteins differently regulate endogenous store-operated channels in HEK293 cells. J Biol Chem. 2015;290:4717–4727. doi: 10.1074/jbc.M114.601856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singaravelu K, Lohr C, Deitmer JW. Regulation of store-operated calcium entry by calcium-independent phospholipase A2 in rat cerebellar astrocytes. J Neurosci. 2006;26:9579–9592. doi: 10.1523/JNEUROSCI.2604-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinkins WG, Estacion M, Schilling WP. Functional expression of Trpc1: a human homologue of the Drosophila Trp channel. Biochem J. 1998;331:331–339. doi: 10.1042/bj3310331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sogkas G, David Stegner D, Shahzad N. Syed SN, Timo Vogtle T, Eduard Rau E, Britta Gewecke B, Reinhold E. Schmidt RE, Bernhard Nieswandt B, Gessner JE. Cooperative and alternate functions for STIM1 and STIM2 in macrophage activation and in the context of inflammation. Immun Inflamm Dis. 2015;3:154–170. doi: 10.1002/iid3.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somasundaram B, Mahaut-Smith MP, Floto RA. Temperature-dependent block of capacitative Ca2+ influx in the human leukemic cell line KU-812. J Biol Chem. 1996;271:26096–26104. doi: 10.1074/jbc.271.42.26096. [DOI] [PubMed] [Google Scholar]
- Storch U, Forst AL, Philipp M, Gudermann T, Mederos y Schnitzler M. Transient receptor potential channel 1 (TRPC1) reduces calcium permeability in heteromeric channel complexes. J Biol Chem. 2012;287:3530–3540. doi: 10.1074/jbc.M111.283218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugioka M, Yamashita M. Calcium signaling to nucleus via store-operated system during cell cycle in retinal neuroepithelium. Neurosci Res. 2003;45:447–458. doi: 10.1016/S0168-0102(03)00004-X. [DOI] [PubMed] [Google Scholar]
- Sugioka M, Zhou WL, Hofmann HD, Yamashita M. Ca2+ mobilization and capacitative Ca2+ entry regulate DNA synthesis in cultured chick retinal neuroepithelial cells. Int J Dev Neurosci. 1999;17:163–172. doi: 10.1016/S0736-5748(99)00027-1. [DOI] [PubMed] [Google Scholar]
- Szikra T, Cusato K, Thoreson WB, Barabas P, Bartoletti TM, Krizaj D. Depletion of calcium stores regulates calcium influx and signal transmission in rod photoreceptors. J Physiol. 2008;586:4859–4875. doi: 10.1113/jphysiol.2008.160051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szikra T, Barabas P, Bartoletti TM, Huang W, Akopian A, Thoreson WB, Krizaj D. Calcium homeostasis and cone signaling are regulated by interactions between calcium stores and plasma membrane ion channels. PLoS One. 2009;4:e6723. doi: 10.1371/journal.pone.0006723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemura H, Putney JW., Jr Capacitative calcium entry in parotid acinar cells. Biochem J. 1989;258:409–412. doi: 10.1042/bj2580409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasdizen T, Koshevoy P, Grimm BC, Anderson JR, Jones BW, Watt CB, Whitaker RT, Marc RE. Automatic mosaicking and volume assembly for high-throughput serial-section transmission electron microscopy. J Neurosci Methods. 2010;193:132–144. doi: 10.1016/j.jneumeth.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JL, Shuttleworth TJ. Molecular basis of activation of the arachidonate-regulated Ca2+ (ARC) channel, a store-independent Orai channel, by plasma membrane STIM1. J Physiol. 2013;591:3507–3523. doi: 10.1113/jphysiol.2013.256784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaca L. SOCIC: the store-operated calcium influx complex. Cell Calcium. 2010;47:199–209. doi: 10.1016/j.ceca.2010.01.002. [DOI] [PubMed] [Google Scholar]
- Vandebrouck A, Sabourin J, Rivet J, Balghi H, Sebille S, Kitzis A, Raymond G, Cognard C, Bourmeyster N, Constantin B. Regulation of capacitative calcium entries by alpha1-syntrophin: association of TRPC1 with dystrophin complex and the PDZ domain of alpha1-syntrophin. FASEB J. 2007;21:608–617. doi: 10.1096/fj.06-6683com. [DOI] [PubMed] [Google Scholar]
- Varga-Szabo D, Authi KS, Braun A, Bender M, Ambily A, Hassock SR, Gudermann T, Dietrich A, Nieswandt B. Store-operated Ca(2+) entry in platelets occurs independently of transient receptor potential (TRP) C1. Pflugers Arch. 2008;457:377–387. doi: 10.1007/s00424-008-0531-4. [DOI] [PubMed] [Google Scholar]
- Vazquez G, Wedel BJ, Trebak M, St John Bird G, Putney JW., Jr Expression level of the canonical transient receptor potential 3 (TRPC3) channel determines its mechanism of activation. J Biol Chem. 2003;278:21649–21654. doi: 10.1074/jbc.M302162200. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A, Reyes RC, Parpura V. TRP channels coordinate ion signalling in astroglia. Rev Physiol Biochem Pharmacol. 2014;166:1–22. doi: 10.1007/112_2013_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vig M, Beck A, Billingsley JM, Lis A, Parvez S, Peinelt C, Koomoa DL, Soboloff J, Gill DL, Fleig A, Kinet JP, Penner R. CRACM1 multimers form the ion-selective pore of the CRAC channel. Curr Biol. 2006;16:2073–2079. doi: 10.1016/j.cub.2006.08.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Fan J, Zhang M, Sun Z, Xu G. Gene expression changes under cyclic mechanical stretching in rat retinal glial (Müller) cells. PLoS One. 2013;8:e63467. doi: 10.1371/journal.pone.0063467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Wang Y, Zhou Y, Hendron E, Mankarella S, Andrake S, Rothberg BS, Soboloff J, Gill D. Distinct Orai-coupling domains in STIM1 and STIM2 define the Orai-activating site. Nat Commun. 2014;5:3183. doi: 10.1038/ncomms4183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson R, Parekh AB. Mitochondrial regulation of CRAC channel-driven cellular responses. Cell Calcium. 2012;52:52–56. doi: 10.1016/j.ceca.2012.02.003. [DOI] [PubMed] [Google Scholar]
- Xiao B, Coste B, Mathur J, Patapoutian A. Temperature-dependent STIM1 activation induces Ca2+ influx and modulates gene expression. Nat Chem Biol. 2011;7:351–358. doi: 10.1038/nchembio.558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng W, Yuan JP, Kim MS, Choi YJ, Huang GN, Worley PF, Muallem S. STIM1 gates TRPC channels, but not Orai1, by electrostatic interaction. Mol Cell. 2008;32:439–448. doi: 10.1016/j.molcel.2008.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Store depletion induces Ca2+ waves in the apical and end-foot processes that propagate centrifugally into the cell body.