

Abstract
Patients with Charcot–Marie–Tooth Type 2D (CMT2D), caused by dominant mutations in Glycl tRNA synthetase (GARS), present with progressive weakness, consistently in the hands, but often in the feet also. Electromyography shows denervation, and patients often report that early symptoms include cramps brought on by cold or exertion. Based on reported clinical observations, and studies of mouse models of CMT2D, we sought to determine whether weakened synaptic transmission at the neuromuscular junction (NMJ) is an aspect of CMT2D. Quantal analysis of NMJs in two different mouse models of CMT2D (GarsP278KY, GarsC201R), found synaptic deficits that correlated with disease severity and progressed with age. Results of voltage-clamp studies revealed presynaptic defects characterized by: (1) decreased frequency of spontaneous release without any change in quantal amplitude (miniature endplate current), (2) reduced amplitude of evoked release (endplate current) and quantal content, (3) age-dependent changes in the extent of depression in response to repetitive stimulation, and (4) release failures at some NMJs with high-frequency, long-duration stimulation. Drugs that modify synaptic efficacy were tested to see whether neuromuscular performance improved. The presynaptic action of 3,4 diaminopyridine was not beneficial, whereas postsynaptic-acting physostigmine did improve performance. Smaller mutant NMJs with correspondingly fewer vesicles and partial denervation that eliminates some release sites also contribute to the reduction of release at a proportion of mutant NMJs. Together, these voltage-clamp data suggest that a number of release processes, while essentially intact, likely operate suboptimally at most NMJs of CMT2D mice.
SIGNIFICANCE STATEMENT We have uncovered a previously unrecognized aspect of axonal Charcot–Marie–Tooth disease in mouse models of CMT2D. Synaptic dysfunction contributes to impaired neuromuscular performance and disease progression. This suggests that drugs which improve synaptic efficacy at the NMJ could be considered in treating the pathophysiology of CMT2D patients.
Keywords: 3,4 diaminopyridine; axonal neuropathy; neuromuscular transmission; physostigmine; quantal content; voltage clamp
Introduction
Charcot–Marie–Tooth (CMT) disease was described more than a century ago (Charcot and Marie, 1886; Tooth, 1886) and encompasses a large group of genetically and phenotypically heterogeneous diseases that affect the peripheral nerve at a worldwide frequency of at least 1/2500 (Skre, 1974). In this study, we focus on CMT2D, which is a type 2 axonal CMT (Dyck, 1975) caused by autosomal dominant mutations in Glycyl tRNA synthetase (GARS; Antonellis et al., 2003).
CMT2D patients present with a range of “classic” CMT2 symptoms that include slowly progressive, distal weakness with or without sensory abnormalities. The absence of sensory involvement may result in a diagnosis of distal spinal muscular atrophy type V, which is allelic to CMT2D (Antonellis et al., 2003; Sivakumar et al., 2005; Rohkamm et al., 2007). Clinical reports on CMT2D and other type 2 CMT patients describe electrophysiological abnormalities including aberrant spontaneous muscle activity, reduced compound muscle action potentials, and less commonly, decreased nerve conduction velocities (Sivakumar et al., 2005; Shen et al., 2011; Saporta and Shy, 2013). Some CMT2D patients report early symptoms that included cramping in hands and legs either in response to cold or upon exertion, and transient episodes of weakness and fatigue that slowly worsen with age (Sivakumar et al., 2005). These electrophysiological signatures and other symptoms of type 2 CMTs are typically attributed to axonal degeneration (Sivakumar et al., 2005), but are also consistent with possible neuromuscular junction (NMJ) dysfunction and transmission failure. To our knowledge, this possibility has not been systematically investigated for type 2 axonal CMTs. If synaptic defects are indeed an unrecognized factor in type 2 CMTs, even as a secondary effect of axonal pathology, it would open a possible new treatment avenue for patients. For example, patients with NMJ defects due to congenital myasthenias, are effectively treated with drugs that modulate synaptic efficacy (Engel et al., 2015) and if myasthenia-like deficits were a component of type 2 CMT, drugs to improve NMJ transmission could be considered.
The dominantly inherited mutations in GARS (Antonellis et al., 2003) underlying CMT2D have been successfully modeled through mouse genetics, with the identification of dominant Gars alleles that recapitulate most aspects of CMT2D, including axon atrophy and loss, denervation, muscle weakness, and consistently more severe symptoms in certain distal muscles than proximal ones (Seburn et al., 2006; Achilli et al., 2009; Motley et al., 2011; Sleigh et al., 2014). The severity of CMT2D varies widely among patients due, in part, to the specific GARS mutation that an individual carries (Antonellis et al., 2003; Del Bo et al., 2006; Dubourg et al., 2006; James et al., 2006), and a similar correlation exists for different mutant alleles of Gars mice (Seburn et al., 2006; Achilli et al., 2009; Sleigh et al., 2014). Here we focus on two mutant mouse strains, one referred to as GarsP278KY, which develops a severe peripheral neuropathy (Seburn et al., 2006), and the second, GarsC201R, which has a milder disease (Achilli et al., 2009). Both mutant strains are weaker than age-matched wild-types and weakness correlates with disease severity. Differences in muscle strength between the mild and severely affected CMTD2 mice may be partly due to early axon loss (∼30%) in the GarsP278KY mice that is not seen in the GarsC201R allele. However, motor axon number in GarsP278KY largely stabilizes after ∼5 weeks of age, whereas there is no reduction in axon number in GarsC201R nerves (Seburn et al., 2006; Achilli et al., 2009). Despite essentially stable axon counts, both alleles have a persistent, overt tremor that worsens gradually with age,(Seburn et al., 2006; Achilli et al., 2009; Motley et al., 2011; Sleigh et al., 2014) which could be caused by unreliable neuromuscular transmission at innervated terminals. Consistent with this idea is the previous finding that GarsP278KY muscle showed a more marked decrement in an integrated electromyogram (EMG) during tetanic contraction than wild-type (Seburn et al., 2006). In addition, both Gars mutant strains show muscle atrophy and some degree of morphological abnormality at the NMJ (Seburn et al., 2006; Motley et al., 2011; Sleigh et al., 2014) that may be indicative of ongoing synaptic dysfunction, but it is currently unknown whether intact NMJs function normally in CMT2D, or any other type 2 axonal CMT.
Materials and Methods
Mice.
The mice used in these experiments were obtained from research colonies maintained at The Jackson Laboratory. The two strains of Gars mutant mice are established models of CMT2D and are described in several previous publications (Seburn et al., 2006; Achilli et al., 2009; Motley et al., 2011; Stum et al., 2011). Briefly, one strain of mice (CAST;B6-GarsNmf249/JRwb; Stock no. 17540; Seburn et al., 2006) carry a spontaneous dominant mutation (insertion) that results in a P to KY substitution at amino acid 278 of the GlyRS protein. These mice hereafter referred to as GarsP278KY; develop overt disease symptoms by 2–3 weeks of age. A second strain of mice (C3H/HeH-GarsC201R; Achilli et al., 2009) carry an N-ethyl-N-nitrosourea (ENU)-induced dominant point mutation that causes a cysteine to arginine substitution at residue 201 of the GlyRS protein. These mice (hereafter GarsC201R) have a qualitatively similar, but generally milder CMT2D phenotype. Both GarsP278KY and GarsC201R mutant strains are routinely maintained by mating heterozygous male mutant mice to female wild-type mice. For these experiments, additional matings were set up using female mice homozygous for a transgene expressing yellow fluorescent protein (B6.Cg-Tg(Thy1-YFP)16Jrs/J; Stock no. 3709, hereafter YFP16; Feng et al., 2000). Litters from these matings produced the necessary controls (YFP16;Gars+/+), as well as either YFP16;GarsP278KY or YFP16;GarsC201R. Colonies of Gars mice also carrying the YFP transgene have been maintained for several years and neither the onset nor lifespan of either strain has changed. In addition, results of analyses performed on these mice are in good agreement with our previous results on mutants that did not carry the YFP transgene. All mice were maintained in the same vivarium on a 12 h light/dark cycle and were provided food and water ad libitum. Care and procedures were reviewed for compliance and approved by the Animal Care and Use Committee of The Jackson Laboratory.
NMJ immunohistochemistry and analysis of innervation status.
CMT2D mice had been crossed to mice carrying the transgene for yellow fluorescent protein that allowed visualization of the presynaptic nerve (for details, see Mice). Thus, for analysis, the levator auris longus (LAL) muscle was removed and placed in 2% paraformaldehyde for 15 min and then rinsed three times in PBS. Finally, to visualize postsynaptic acetylcholine receptors (AChRs) on the muscle cell surface, muscles were incubated with α-bungarotoxin conjugated with AlexaFluor 594 (NMJ only) or 647 (NMJ costaining with Bassoon, see below; (1/2000; Invitrogen) in cold PBS containing 2% BSA, 2% normal goat serum, and 0.1% Triton for 20 min. Muscles were then examined by fluorescent microscopy (Nikon E6000) at a magnification of 63×. In each muscle 100 NMJs were randomly viewed and classified as described previously (Seburn et al., 2006). Junctions where the nerve completely overlapped the AChRs on the muscle were defined as fully occupied, those with a portion of receptors clearly vacated by the presynaptic nerve were defined as partially occupied, and those with AChR plaques that had no associated nerve were defined as denervated.
Bassoon staining and quantification of release sites.
Muscles were prepared, and presynaptic and postsynaptic components of the NMJ were visualized, as described above. Immunohistochemistry followed a published protocol (Nishimune, 2012). In addition, muscles were incubated overnight with a mouse primary antibody against the active zone protein Bassoon (1/1000; SAP7F407, Enzo Life Sciences) and fluorescently conjugated secondary (Zenon AlexaFluor 568 Mouse IgG2a, Life Technologies, catalog #Z2506). Images were collected as Z-series using a Leica SP5 confocal microscope. Bassoon-stained active zones were quantified using 3D reconstructions (Imaris v7.4.2, Bitplane) of NMJs. Using this software, each NMJ was reconstructed and then evaluated empirically by rotating and examining the 3D image from presynaptic and postsynaptic perspectives to determine that the reconstruction matched the actual staining. Briefly, for each image, the postsynaptic receptor area was rendered by smoothing and thresholding using background subtraction based on local contrast. The result was then filtered based on a minimal voxel size to eliminate any artifactual staining not associated with the terminal area. The volume and surface area of the rendered volume were determined by the software. Puncta stained with bassoon were identified and counted in a three-step process. First, it was determined on wild-type NMJs that a background subtraction that eliminated “spots” with a diameter >0.250 μm removed the majority of extra-junctional “puncta.” For each image, this result was then filtered to include only spots above a fixed intensity at the center of the spot. Finally, the spots needed to associate with the presynaptic side, within the perimeter defined by postsynaptic staining. The intensities used for filtering were determined empirically for each image by thresholding the appropriate channel and noting the value giving the best representation of visible staining. Using this approach gave wild-type results similar to those reported previously (Nishimune, 2012).
Muscle preparation and voltage-clamp.
Mice were anesthetized with isoflurane (2%, 300 ml/min) to remove the intact LAL muscle. This flat muscle controls the movement of the ear and is a well characterized mixed fiber-type muscle (Angaut-Petit et al., 1987). Muscles were placed immediately in a specially designed recording chamber and pinned onto the silastic such that solution could flow freely across the tissue on both sides. The recording chamber was perfused continuously with Ringer's solution comprised of (in mmol/L): 118 NaCl, 3.5 KCl, 2 CaCl2, 0.7 MgSO4 26.2, NaHCO3, 1.7 NaH2PO4, and 5.5 glucose, and equilibrated with 95%O2/5%CO2 to maintain a pH of 7.3–7.4. The solution was at room temperature (20°–22°C) for all experiments. Voltage-clamp experiments followed previously described methods (Rich et al., 2002; Wang et al., 2004, 2005). Once the muscle was pinned the tissue was stained by the addition of 4-(4-diethylaminostyryl)-N-methylpyridinium iodide (4-Di-2ASP; Magrassi et al., 1987) at a concentration of ∼2 μm for 2.5 min. This method provides staining sufficient for visualization of the superficial nerve terminals and the surface of the muscle fibers. A concentric bipolar electrode (FHC) was placed in contact with the nerve and connected to the stimulator (WPI A360). Stimulator output was capacitively coupled to the electrode to avoid potential damage from DC polarizing currents (Guyton and Hambrecht, 1974). The preparation was then placed under an upright epifluorescence microscope (Leica DMLFSA). Electrodes (5–10 MΩ) were filled with 3 m KCL and 10 ng/ml of sulforhodamine so that the electrode tip could be seen under the microscope. Synapses were located and fibers were impaled within 100 μm of the terminal and voltage-clamped to −50 mV. In initial experiments muscle fibers were crushed away from the endplate band to avoid movement induced by nerve stimulation (Glavinović, 1979; Wang et al., 2004, 2005), but in most experiments the muscle-specific sodium-channel blocker (μ-Conotoxin GIIIB, Alomone Labs; Cruz et al., 1985; Robitaille and Charlton, 1992) was introduced (∼1 μm) to eliminate muscle action potentials and contraction.
Wire-hang test.
A variation on the wire-hang test (Gomez et al., 1997; Rafael et al., 2000) was used to assess the response to drugs that enhance synaptic function via either presynaptic or postsynaptic mechanisms. Briefly, mice were placed on a 6” × 9” piece of wire mesh and then the mesh was inverted and held ∼6” above the countertop. The latency (seconds) to a fall was timed and recorded, up to a maximum of 1 min. Tests were performed at approximately the same time each day. Each mouse performed three trials in a given session and a rest period of at least 30 s was given between individual trials. To allow the mice time to adjust to the wire-hang test and learn how to perform, animals were given at least three consecutive daily practice sessions before drug administration and testing trials.
In drug/vehicle trials, drugs were prepared fresh from frozen stock on each day (in sterile PBS, 0.1 mg/kg- physostigmine; 2.5 mg/kg, 3,4 diaminopyridine (3,4-DAP; Sigma-Aldrich). Each mouse performed a pre-injection trial, and then was injected intraperitoneally and retested 60 min later. Mice were dosed three independent times in 5 d with intervening practice days. The best wire-hang score in three trials is reported for that day (3 trials/d on 3 test days). Performance was calculated as pre-injection/postinjection latency-to-fall, expressed as a percentage. The average percentage difference is calculated from the 3 independent days of injections.
Data collection and analysis.
Custom software was used for collection and analysis of synaptic currents. At each NMJ, spontaneous miniature endplate currents (MEPCs) were collected for 1 min. The nerve was then stimulated (0.5 Hz) and 15–20 individual evoked endplate currents (EPCs) were recorded. Quantal content was directly calculated by dividing the EPC amplitude by the average MEPC amplitude recorded for each synapse. We used the extent of EPC facilitation/depression during evoked trains (Zucker and Regehr, 2002) to indirectly evaluate probability of release following a protocol previously used at the mouse NMJ (Kong et al., 2009; Wang et al., 2010). We recorded 15–20 responses to a 10 pulse, 50 Hz train delivered to the nerve and 20 responses were recorded. The extent of depression/potentiation was calculated by dividing the averaged amplitude of the 10th pulse by the first pulse.
Electron microscopy.
For electron microscopy, muscles were processed as previously described (Burgess et al., 2010). In brief, muscles were fixed in 2% paraformaldehyde, 2% glutaraldehyde in 0.1 m cacodylate buffer. The endplate-containing region was isolated and embedded, and then sectioned for transmission electron microscopy. Seventy-five nanometer plastic sections were mounted on grids and viewed using a Jeol 1230 electron microscope equipped with a Hamamatsu digital camera system for image collection.
Statistics.
Unless otherwise noted averaged numbers are reported as mean ± SE. We used a nested ANOVA (using animal as a random factor) for genotype by age comparisons of electrophysiology, active zones, and vesicle data. This statistic controls for animal-to-animal variation and the effect of taking small samples to represent a larger population. We also evaluated interactions for 2 and 4 month electrophysiology data using a standard two-way ANOVA (genotype by age), but the results did not modify any conclusions so results derived from the more appropriate nested design are reported. A Student's t test was used for comparison of wire-hang and quantal content comparison of 70 Hz voltage-clamp data. In all cases, the statistical cutoff for declaring a significant difference was p < 0.05.
Results
Proximal LAL muscles in CMT2D mice show prevalent NMJ dysmorphology, but most terminals retain innervation
The proximal LAL muscle has not previously been examined in CMT2D mice, so before conducting voltage-clamp experiments we evaluated NMJ morphology and assessed innervation status. Compared with wild-type terminals (Fig. 1A,B) nearly all LAL terminals, in both GarsP278KY (Fig. 1C–I) and GarsC201R (Fig. 1J–L) muscles, have evident dysmorphology that includes more diffuse postsynaptic staining with less distinct guttering as well as thinner axons and presynaptic nerves. As with other previously studied muscles NMJ dysmorphology varies with disease severity (severe GarsP278KY > mildGarsC201R) and can range from subtle, near normal looking NMJs to clearly fragmented junctions (Fig. 1 compare J, K). At low-magnification an innervating presynaptic axon is evident at most NMJs in LAL muscles of even the more severely affected GarsP278KY mice (Fig. 1C–E), but at higher-magnification some of these junctions are found to be only partially innervated even in the mildly affected GarsC201R mice (Fig. 1G–I,L). Indeed, the most striking difference in LAL NMJs between CMT2D alleles was the extent of partial denervation (Fig. 1F). In GarsP278KY muscles from 2-month-old mice, more than one-half of the junctions were partially innervated, whereas in muscles of 4-month-old GarsC201R mice only ∼5% of NMJs had this status. There were also more than three times as many denervated junctions in the GarsP278KY than in the GarsC201R muscles (6.5 vs 1.9%) even though the latter were 2 months older. Synaptic function is likely to be compromised at junctions that are not fully occupied by a presynaptic terminal if other processes cannot compensate. Our analysis of the LAL predicts that innervation status could contribute to observed changes in synaptic function at GarsP278KY NMJs of 2-month-old mice where ∼50% of NMJs are incompletely innervated. In contrast, because ∼95% of NMJs in the GarsC201R LAL are fully innervated, even at 4 months, functional changes would be independent of innervation status at most NMJs.
Figure 1.

Morphological analysis of LAL muscle in CMT2D mice. Images shown are of LAL terminals in 2 month-old (A, B) wild-type, (C–I) GarsP278KY and (J–L) 4-month-old GarsC201R mice. Red shows α-bungarotoxin-stained postsynaptic ACh receptors, green is presynaptic nerve expressing YFP (see Materials and Methods). Innervation analysis (F) included examination of ≥100 terminals from each of three LAL muscles/genotype. Control wild-type (A, B) terminals show typical complex pretzel-like morphology and postsynaptic receptors are entirely apposed by the presynaptic nerve. At low-magnification (20×) it can be seen that (C–E) GarsP278KY terminals retain some innervation, but when viewed at higher-magnification (63×; G–I) nearly all terminals show some dysmorphology (G–I, upper and lower terminal) and many are only partially innervated (G–I, lower terminal). Occupancy was analyzed, and ∼10% of terminals were denervated but >50% showed some degree of partial denervation muscles. The LAL muscle in GarsC201R mice showed a similar phenotype to GarsP278KY muscles, with widespread dysmorphology at most terminals with severity ranging from (J) mild, near-normal to (K) fragmented, or (L) partially denervated. Overall, consistent with previous findings, GarsC201R LAL muscles had many (F) fewer denervated or partially denervated terminals than seen in GarsP278KY, but across both alleles at least 80–90% of NMJs retain at least partial innervation.
Quantal analysis reveals abnormal synaptic transmission in CMT2D mice at 2 months of age
Motor endplate current recordings were made under voltage-clamp conditions to control for potential changes in passive properties of muscle fibers that could occur with disease-related changes in muscle activity (Lomo and Rosenthal, 1972). Our first set of experiments used muscles from 2-month-old GarsP278KY and GarsC201R mutants and respective wild-type littermate controls. At this age, we found qualitatively similar changes in synaptic function for both mutants, but as with other documented phenotypes, the dysfunction was more pronounced at NMJs of the GarsP278KY mice than at synapses of GarsC201R mutants with the milder disease.
At 2 months of age, both alleles show no difference in MEPC amplitude (quantal amplitude; Fig. 2A,G), but the frequency of spontaneous release was consistently and significantly lower than wild-type (Fig. 2B,H). The finding that quantal amplitude at mutant synapses is not different from wild-type indicates that both postsynaptic receptor density and the amount of acetylcholine loaded into individual vesicles is unaffected at CMT2D terminals.
Figure 2.

Quantal analysis. Voltage-clamp experiments were performed using LAL muscles from 2-month-old GarsP278KY, (top) and both 2- and 4-month-old GarsC201R mice (bottom). All recordings were made at a holding potential of −50 mV. At 2 months of age, mutant NMJs in both GarsP278KY and GarsC201R mice showed: no change in MEPC amplitude, (A, G) lower-frequency of spontaneous release, (B, H) reduced EPC amplitude, and quantal content (C, I, D, J), and significantly greater depression (10th/1st) in response to 50 Hz stimulation (E, K). At 4 months of age GarsC201R NMJs show no additional changes in spontaneous release (G′, H′) but a further reduction in EPC amplitude and quantal content (I′, J′) while repetitive stimulation produced less depression compared with 2 month mutant NMJs (K′). F, Increased extracellular calcium successfully increased spontaneous release frequency at 4-month-old mutant GarsC201R NMJ. LAL muscles of six different mice for each genotype/age were used. In each muscle recordings were made on 3–12 synapses (mean and mode = 8) for a total of between 47 and 58 synapses for each genotype/age. Comparisons made using nested ANOVA. *p < 0.05, mutant versus wild-type; **p < 0.05, 2 versus 4 months GarsC201R.
Evoked release was also affected at 2-month-old mutant NMJs, as both alleles showed significant decreases of ∼25% in EPC amplitude (Fig. 2C,I) and quantal content (Fig. 2D,J). Clinical diagnoses of NMJ dysfunction uses response to repetitive stimulation (Rich, 2006), so we also examined changes in EPC amplitude in response to a 10 pulse, 50 Hz stimulus train. In addition, changes in facilitation or depression with this protocol can be used to infer changes in release probability (Zucker and Regehr, 2002; Kong et al., 2009). Using the ratio of the amplitude of the 10th/1st EPC in the train we found that, on average, NMJs of GarsP278KY and GarsC201R mice showed significantly greater depression (Fig. 2E,K). For healthy synapses greater depression correlates with higher initial probability of release (Zucker and Regehr, 2002).
Finally, we examined time course measures of both MEPCs and EPCs and found no significant changes between mutant and control NMJs at 2 months of age for either allele. (Time constants in ms: MEPC control vs mutant- GarsP278KY: 0.79 ± 0.24 vs 0.74±.0.20, GarsC201R: 0.75 ± 0.12 vs 0.73 ± 0.12; EPC control vs mutant: GarsP278KY: 1.1 ± 0.20 vs 1.2±.0.24, GarsC201R: 1.2 ± 0.14 vs 1.2 ± 0.17). Additional time course measures (time-to-peak, half-width, 10–90 rise time) also showed no changes at synapses in either Gars mutant at 2 months of age.
Together, analysis of synaptic measures reveals changes in synaptic transmission in 2-month-old CMT2D mice that are characterized by decreased spontaneous release frequency and evoked release amplitude and greater EPC depression in response to 50 Hz stimulation. The absence of any change in MEPC amplitude or time course measures points to a presynaptic defect. As with other CMT2D phenotypes examined previously, synaptic changes in the CMT2D mice correlate with disease severity associated with different mutant alleles.
Quantal content is further reduced in GarsC201R LAL terminals between 2 and 4 months
Given the relationship between synaptic dysfunction and disease severity across mutant alleles, we were also interested in determining whether synaptic measures changed with age for a given allele. For these experiments, we aged an additional cohort of GarsC201R mice to 4 months. We chose the milder GarsC201R allele for this study because innervation status is near normal even at the older age (Fig. 1F).
The additional 2 months produced no significant changes in spontaneous release compared with younger mutants. MEPC amplitude recorded at GarsC201R NMJs was still not different from control (Fig. 2G,G′), and MEPC frequency remained reduced to approximately the same extent as measured at 2 months (Fig. 2H,H′). Because the reduction in frequency of spontaneous release was such a robust, consistent finding, we were interested in determining whether a mutant synapse could increase spontaneous release in response to higher extracellular calcium. Raw traces show increased MEPC frequency when recorded from the same LAL synapse (4-month-old, GarsC201R) at two different calcium concentrations: typical 2 mm (Fig. 2F, top), and 4 mm (Fig. 2F, bottom). Initial release frequency was 0.4 Hz, at the lower end of the range observed previously (∼0.8 Hz; Fig. 2H,H′), but doubling extracellular calcium increased release frequency to 1.0 Hz, within normal range for a wild-type synapse, a comparable relative increase to that reported previously for wild-type mice (Plomp et al., 2000). Average MEPC amplitude also increased from 1.9 to 2.3 mA.
Evoked release at GarsC201R synapses showed a further decline, as mean EPC amplitude and quantal content were significantly lower at 4 months compared with 2 month and age-matched wild-type values (Fig. 2I,I′,J,J′). Interestingly, the extent of depression in response to 50 Hz trains at the older mutant NMJs was significantly less (i.e., >10th/1st ratio) compared with 2 month mutant values, and >4 month wild-type control values (Fig. 2K,K′), indicating a decrease in the probability of release between 2 and 4 months.
Examination of time course measures (MEPC and EPC, data not shown) revealed neither an age-related effect of the GarsC201R mutation nor any difference from age-matched controls.
Dynamic frequency-related changes in evoked release show atypical response at mutant NMJs
Observations during the delivery of 50 Hz stimulus trains prompted a more detailed analysis of dynamics during the 10 pulse train. EPC amplitude was measured for each pulse of the train and expressed relative to the amplitude of the first EPC. The typical pattern of the 50 Hz frequency response averaged across wild-type littermate NMJs was similar regardless of age or strain (Fig. 3) and is characterized by potentiation of initial EPCs (2nd–4th response, ∼105% initial) followed by moderate depression (85–90% initial). However, this pattern differed between mutant alleles and changed between 2 and 4 months in GarsC201R muscles.
Figure 3.

Evoked response to repetitive stimulation. A–C, show plots of averaged normalized EPC amplitude (±SE) for each stimulus of a 10 pulse, 50 Hz stimulus train (average of 20 trains at 0.5 Hz for each NMJ). In contrast to wild-type synapses, mutant NMJs in 2-month-old animals showed reduced or absent potentiation of initial EPCs and somewhat steeper depression. At 4 months of age, initial EPCs showed potentiation similar to wild-type. EPC amplitude was measured for each stimulus and plotted relative to the amplitude of the first pulse. Numbers of animals/synapses are the same as for data shown in Figure 2. Plots in D–G show the first, second, and 10th raw traces from a series of 10 EPC trains recorded in response to a 70 pulse,70 Hz stimulus (1 s duration), delivered every 2 s. Starting quantal content (m) is shown for each of the four NMJs. Mutant NMJs showed marked steadily progressive decrements from the first to 10th train, whereas the wild-type decreased and stabilized. The mutant NMJs are easily identified by comparing response to the 10th stimulus train (lower traces) to wild-type. In addition, clear failures of release were evident (arrows) for NMJs from severe and 2-month-old mild GarsC201R muscles. Failures were present in at least one train for 5 of 11 mutant NMJs, but were never observed in wild-type. The 1 s duration, 70 Hz trains were recorded in nine different muscles/experiments (n = 5, 6, 3, and 2 NMJs from 4-month-old wild-type, 4M-GarsC201R, 2M-GarsC201R, and 2M-GarsP278KY, respectively).
At NMJs of 2-month-old CMT2D mice, both GarsP278KY (Fig. 3A) and GarsC201R (Fig. 3B) showed near complete absence of potentiation, barely evident only for the second response at NMJs of GarsC201R mice. In addition, EPCs at NMJs for both mutant alleles showed a slightly steeper depression relative to respective wild-type (Fig. 3A,B), and the effect is again more marked at GarsP278KY NMJs than at GarsC201R NMJs (Fig. 3A,B).
Given the additional reduction in quantal content at GarsC201R NMJs between 2 and 4 months, we expected the response pattern of 4 month mutant NMJs to appear more like that observed at GarsP278KY terminals. To our surprise, the typical early potentiation was now evident and the overall pattern was more similar to wild-type, but with even slightly greater initial potentiation and somewhat less marked depression (decreased probability of release) in later responses (Fig. 3C).
In Gars mutants observable weakness occurs during behaviors lasting seconds rather than milliseconds. Thus, we were also interested in the ability of mutant NMJs to sustain release for periods longer than the 200 ms duration of the 10 pulse 50 Hz trains. To accomplish this at NMJs where a sufficiently stable penetration was established, we also recorded EPCs in response to 10 consecutive stimulus trains of 1 s duration (70 pulses at 70 Hz) delivered once every 2 s. Successful recordings, of at least 10 stimulus trains per NMJ, were made in several muscles at 16 different NMJs including five wild-type and 11 mutants (2 GarsP278KY and 9 GarsC201R, 3 and 6 from 2- and 4-month-old muscles, respectively). Overall, qualitative examinations of 70 Hz raw traces reveal that mutant NMJs do not sustain release during the repeated 1 s stimulus as well as wild-type (Fig. 3D–G). If the response to the first train in a series is compared across genotype/age (D–G, top), a marked difference is only clearly evident for the severe GarsP278KY NMJ, but effects on each mutant NMJ becomes more marked with successive trains such that by the 10th train (D–G, bottom) release is clearly more severely reduced at mutant NMJs. In traces shown, release failures occur in the second train at the GarsP278KY NMJ (Fig. 3E, middle, arrows) and during the 10th train for both GarsP278KY and 2-month-old GarsC201R NMJs shown (Fig. 3E,F, bottom, arrows). We examined all individual 70 Hz traces for each NMJ to better evaluate the extent of the failures. Failures never occurred at wild-type NMJs, but at least one failure occurred during at least one stimulus train at 5 of 11 mutant NMJs. Of these five, two were from GarsP278KY muscles and three were recorded in 2-month-old old GarsC201R muscles. Failures for both GarsP278KY NMJs occurred as early as the second stimulus train and failures also occurred during the initial 10 stimuli of the train, whereas for GarsC201R NMJs (2 month), failures did not appear until the fourth train or later and failures were not seen within the initial 10 stimuli of any train. The 50 Hz analysis above suggested a compensatory response between 2 and 4 months for GarsC201R NMJs, and in keeping with this, examination of traces for 4-month-old GarsC201R NMJs revealed no clear failures, but traces included intermittent, very small EPCs (5–15 nA), not present in any wild-type traces, and these were more common toward the end of individual traces and in later (4–10th) trains.
We also calculated quantal content using the EPC averaged across trains (n = 10/NMJ) of the 70th EPC in the trains and compared mutant and wild-type values. Final quantal content at the end of the 70 Hz trains ranged between 21 and 49 for the five wild-type NMJs and between 5 and 22 for the 11 mutant synapses. Including all mutant data, the average mutant quantal content of the final 70 Hz EPC (mean 13.2 ± 5) was significantly lower than wild-type (mean 31.4 ± 12; t(14) = 4.4, p = 0.01) and this difference was also significant if the comparison was restricted to 4-month-old GarsC201R (mean 13.5 ± 3) and wild-type (t(9) = 3.5, p = 0.01).
Together the data on the response of Gars mutant NMJs to repetitive stimulation suggest impaired vesicle recycling compared with wild-type. The cumulative intra- and inter-train decrements shown in Figure 3 are consistent with such a deficiency and suggests multiple processes likely contribute to defective synaptic transmission.
As a final assessment of the severity of defects at Gars NMJs we reviewed all of the non-DAP experiments seeking fibers where we were able to record MEPCs (i.e., innervated NMJs), but were unable to evoke an EPC (data not included in Fig. 2). This scenario was found in 17 of 179 NMJs (7,6,4 GarsP278KY and GarsC201R 2 and 4 months, respectively) including one or more occurrence (max = 4) in 9 of the18 mutant muscles (6–12 NMJs recorded per muscle). In contrast, for wild-type this situation arose at only 4 of 184 NMJs in 4 of 18 muscles and never occurred more than once in any experiment (6–11 NMJs/experiment).
Is the observed reduction in quantal content associated with fewer active release sites?
Voltage-clamp data reveal apparent presynaptic changes that cause a reduced quantal content as well as a reduced capacity for sustained release for mice carrying either Gars mutant allele. To investigate whether a reduced number of release sites could account for the lower quantal content and MEPC frequency at mutant NMJs, we stained LAL terminals for Bassoon, a presynaptic protein present at NMJ release sites (tom Dieck et al., 1998; see Materials and Methods for details, based on Nishimune et al., 2004). We used only LAL muscles from 2-month-old GarsP278KY mice with the rationale that, if release sites were contributing, it would be clearly evident at synapses of the most severely affected muscles. Wild-type terminals showed complete apposition of YFP-positive presynaptic terminals with postsynaptic receptors and bassoon-stained puncta in association with the presynaptic nerve (Fig. 4A–D). Consistent with the smaller size of the mutant mice/muscles (Seburn et al., 2006; Achilli et al., 2009), and reports for NMJs in other muscles (Sleigh et al., 2014), the mean area of LAL end plates in GarsP278KY mice was significantly smaller (Fig. 4I, compare solid and dashed vertical lines). However, the range in counts of the bassoon-stained puncta was similar for wild-type and GarsP278KY terminals (Fig. 4I, solid vs open) and there was no significant change (p = 0.4) in either the average absolute number (Fig. 4I, solid and dashed horizontal lines) or density of release sites (p ≥ 0.17; Fig. 4J) compared with wild-type terminals. As expected from the initial morphological analysis of mutant NMJs (Fig. 1) 10 of the 34 terminals that were sampled from GarsP278KY muscles for this analysis had a portion of postsynaptic receptors not apposed by the presynaptic nerve and without bassoon-puncta. Although these terminals clearly lacked bassoon-stained puncta in the partially denervated regions, the relative proportion of the junction affected was not large (≤15%; Fig. 4H,K) so the effect on puncta counts was small. Counts for the 10 partially denervated junctions had a similar mean, median and range as those without any evident loss of presynaptic nerve (mean = 689 vs 720, median = 616 vs 632, range = 233–2049 vs 134–1840). Together, these data show that release sites in GarsP278KY muscles are retained so long as the presynaptic terminal persists. Therefore, at partially denervated LAL junctions release sites could be reduced in proportion to the extent of partial denervation. However, other factors must also contribute because half of the NMJs in 2-month-old GarsP278KY, and ∼85% in GarsC201R LAL muscles, retain complete innervation (Fig. 1).
Figure 4.

Quantification of bassoon-stained release sites in 2-month-old GarsP278KY LAL muscles. Process of visualization and quantification of bassoon-stained release sites is shown for a wild-type LAL terminal (A–D). Analysis included assessment of: (A) the postsynaptic receptor area, (B) presence of presynaptic nerve, and (C) bassoon-stained puncta. Puncta were identified using a three-step process to render a 3D image (D) for automated counting of release sites (Imaris; see Materials and Methods for details). Typical GarsP278KY NMJ (E–H) have more diffuse postsynaptic staining (E), apposed by a thinned presynaptic nerve (F), that nonetheless retains bassoon-stained puncta (G) at innervated locations (NB, the areas where YFP appears to not overlie bassoon labeling are due to the faint YFP signal in portions of the axon and the thresholding of the image; at higher gain YFP was detectable in the vicinity of all bassoon-stained puncta). However, as expected, when a portion of the presynaptic nerve vacates, bassoon-stained release sites are no longer evident (merged image H, arrow; expanded in K). GarsP278KY sample included a total of 34 synapses that included 10 terminals with small areas of evident partial denervation. I, Scatterplot shows NMJ area and counts of bassoon-positive puncta at individual synapses of GarsP278KY (solid) and wild-type (open) NMJs. Vertical and horizontal lines indicate sample means (GarsP278KY and wild-type, solid and dashed, respectively). Mean area of LAL NMJs was significantly smaller (p < 0.002) compared with wild-type (solid vs dashed vertical line). Individual puncta counts covered a similar range and neither mean counts (dashed and solid horizontal lines) nor density (J) were significantly different between genotypes (p = 0.4 and 0.17, respectively). Analysis included 5–10 (mean = 7) terminals from each of five LAL muscles of each genotype (n = 34 and 35, GarsP278KY and wild-type, respectively). Statistical comparisons used a nested ANOVA.
Vesicle number and localization
Given that a change in the number of release sites was insufficient to account for changes observed in evoked release, we next used electron microscopy to compare vesicle parameters at wild-type (Fig. 5A) and NMJs from the severely affected GarsP278KY (Fig. 5B). We analyzed electron micrographs of portions of 5–10 NMJs (median 8) from each of five animals per genotype. Consistent with the lack of difference in MEPC amplitude, vesicle size was similar for mutant and wild-type terminals (50 ± 0.71 vs 54 ± 1.2, respectively). Average vesicles counts tended to be lower at GarsP278KY NMJs compared with wild-type, but due to large inter-animal variation, for both genotypes, the difference did not quite reach our statistical cutoff for significance (p = 0.08; Fig. 5C, inset). However, consistent with light microscopy results above (Fig. 4I) and other work (Sleigh et al., 2014; their Figs. 3D, 6C; GarsC201R and GarsP278KY, respectively), GarsP278KY mutant synapses are smaller than wild-type and therefore, the areas of captured portions of mutant NMJs in our electron micrographs also tended to be smaller (mean = 14 vs 28 μm2, respectively, p = 0.04). When vesicle number is plotted against area (Fig. 5C) it is evident that mutant muscle had fewer terminals with areas >20 μm2 and vesicle counts >250 (Fig. 5C, ellipse), and a preponderance of small (<20 μm2) terminals with vesicle counts of <150 (Fig. 5C, inset). In general, mutant vesicle number still scaled with terminal area as the density of the vesicles (number/μm2) was not different from wild-type (Fig. 5D). However, there are several examples of mutant terminals with vesicle counts below any found at wild-type terminals (Fig. 5C, inset, ellipse). Thus, despite the fact that the comparison of average wild-type and mutant vesicle counts did not reach our statistical cutoff, we contend that quantal content is likely limited by vesicle number for at least some terminals among the generally smaller GarsP278KY mutant terminals. Note, however, that the micrograph in Figure 5B is an example from among those terminal portions that were clearly “depleted” in the GarsP278KY muscles. In a preliminary study we also examined two mutant muscles (5 NMJs each, data not shown) from the distal plantaris muscle, which has more severe NMJ dysmorphology than the proximal LAL, and terminals such as shown in Figure 5B were not found. Thus, such terminals should be considered worst-case, and if present at all, are not widespread even in the more severely affected distal plantaris of the mild GarsC201R allele.
Figure 5.

Electron microscopic analysis of NMJs. Electron micrographs of NMJs from LAL muscles of 2-month-old (A) wild-type and (B) GarsP278KY mice were captured (5 animals per genotype, 5–10 NMJs (median 8 per muscle). Mutant NMJ shown is representative of a “severely” affected NMJ with a low-vesicle count. Note junctional folds and other synaptic specializations are still evident at mutant synapses. Analysis showed vesicle size was similar for mutant and wild-type terminals (50 vs 54 nm, respectively; data not shown). C, Scatterplot shows that the area of terminal portions analyzed scale with vesicle counts for both genotypes. Note the relative absence of large, high vesicle count terminals (ellipse) and the preponderance of small, low vesicle count NMJs in the mutant (C, lower quadrant; expanded in inset). Counts at some mutant NMJs were lower than values recorded at any wild-type NMJ (inset, ellipse). However, due to large inter-animal variability for both genotypes differences in average vesicle counts for wild-type and GarsP278KY NMJs (p = 0.08) were not statistically different (see Results). D, The density of the vesicles present in GarsP278KY terminals was not different from wild-type. Detailed analysis of vesicle location revealed no differences in the number of docked vesicles (within 20 nm), or within 200 nm for either absolute or normalized counts (per micron of membrane). E, Average counts of mitochondria (A, B, arrows) were significantly lower at GarsP278KY terminals (median14 vs 6, wild-type and GarsP278KY, respectively).
Figure 6.
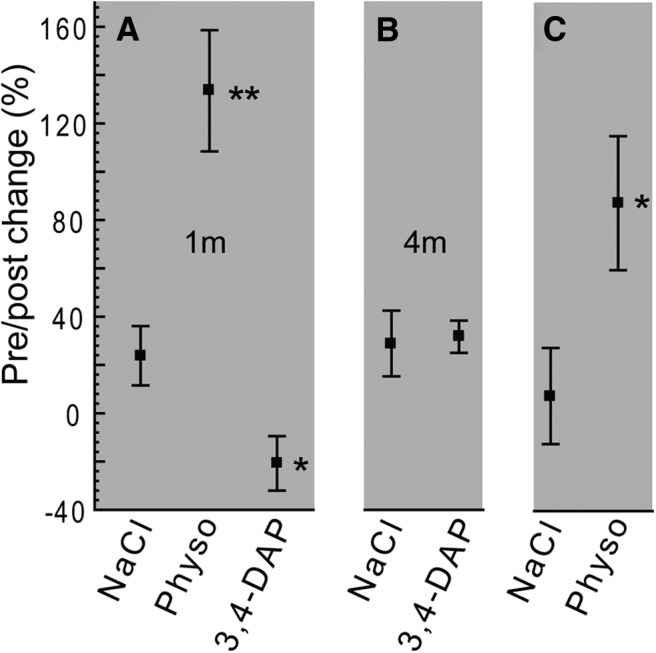
Wire-hang performance of CMT2D mice. Performance is expressed as the percentage change in wire-hang time after treatment ([(post-pre)/post]/100; see Materials and Methods for protocol details). A, GarsC201R mice (1-month-old) were able to perform better with physostigmine (postsynaptic), but showed a decrease in performance after administration of 3,4-DAP (presynaptic) or (B) no change to 3,4 DAP injection at 4 months of age. C, GarsP278KY mice showed a similar response to physostigmine, but were not tested with 3,4 DAP. Mice were 30- to 40-d-old for testing (1 and 4 month: GarsC201R, n = 6; +/+, n = 5; GarsP278KY, n = 5; +/+, n = 4). Wild-type (+/+) mice typically perform the task to completion (60 s max) and showed no drug-related change in performance at the dosages used (physostigmine, 0.1 mg/kg; 3,4 DAP, 2.5 mg/kg), so data are not shown. Pairwise comparisons drug-treated versus NaCl with Student's t test; **(t(9) = 4.2, p < 0.01; *t(8) ≥ 2.3, p ≤ 0.047).
To complete our electron microscopy analysis of vesicles we evaluated their location relative to the presynaptic membrane. We counted the number of vesicles within 200 nm of the presynaptic membrane and the number of docked vesicles (within 20 nm) and found no significant changes at mutant terminals for either absolute counts or counts normalized per micron of presynaptic membrane (Fig. 5D).
Finally, we also counted the number of identifiable mitochondria visible in electron micrographs. Interestingly, on average, there were significantly fewer mitochondria observed at GarsP278KY terminals compared with wild-type (Fig. 5E). However, there was wide variation in counts from terminal to terminal for both genotypes, but the median was also shifted (14 vs 6, GarsP278KY and wild-type, respectively) suggesting the reduction in the mean number of mitochondria observed at NMJs of CMT2D mice was not due to a small number of mutant terminals with very few or no mitochondria.
Together, electron microscopy analysis of NMJs suggests that smaller terminals with fewer vesicles likely contributes to lower quantal content for at least a portion of NMJs in muscles of the severe GarsP278KY mice. Vesicles at mutant terminals were of normal size and were localized to the presynaptic membrane similar to wild-type, suggesting vesicle processing/trafficking is operating normally at CMT2D NMJs.
Enhancing synaptic function improves wire-hang performance of CMT2D mice
Prior work has shown that CMT2D mice have significant muscle weakness (Seburn et al., 2006; Achilli et al., 2009; Motley et al., 2011). Our data reveal significant synaptic dysfunction that could contribute to weakness in the CMT2D mice, so we next tested whether enhancing synaptic function could improve in vivo performance of the Gars mutants in a task requiring strength. We first confirmed that mutant synapses were capable of responding to the two test drugs in vitro: 3,4-DAP (amifampridine), which acts presynaptically to increase quantal content (Thomsen and Wilson, 1983), and physostigmine (eserine), which acts postsynaptically to enhance current duration at postsynaptic receptors (Shaw et al., 1985). In vitro recordings confirmed that both drugs acted as predicted to increase EPC amplitude at 4 month GarsC201R NMJs (n ≥ 18 NMJs; see Fig. 7, DAP; physostigmine data not shown). We next evaluated whether the enhanced synaptic currents could translate to better whole animal performance on the wire-hang test. Mice first performed an initial wire-hang and then were injected with 3,4-DAP, physostigmine, or saline vehicle and retested 60 min later. We first tested the more mildly affected GarsC201R mice and found that administration of physostigmine significantly improved wire-hang times, whereas 3,4-DAP caused a significant decrease (Fig. 6A,1 month) or no change (Fig. 6B, 4 months) in performance. GarsP278KY mice also showed improved wire-hang times after physostigmine administration (Fig. 6C). However, because the more severe disease causes them to perform so poorly in general, (latency to fall ≤8 s) we did not test 3,4-DAP to see if it decreased performance in the GarsP278KY mice.
Figure 7.

Quantal analysis in the presence of 3.4 DAP. Voltage-clamp experiments were conducted on cohorts of 2- and 4-month-old GarsC201R and wild-type mice with 3,4 DAP added to the bath (20 μm). Dashed lines are wild-type, solid lines are GarsC201R, and 2 and 4 month data are shown with circles and triangles, respectively. Control values without DAP are re-plotted from Figure 2. A–E, The addition of 3,4 DAP to prolong presynaptic depolarization caused qualitatively similar changes at mutant and wild-type NMJs. Differences that existed between mutant and wild-type measures without 3,4 DAP (Fig. 2) were eliminated at 4-month-old mutant NMJs when 3,4 DAP was present, although quantal content was still somewhat lower. At 2-month-old mutant NMJs, quantal content remained significantly lower (p = 0.01) compared with wild-type. F, Quantal content calculated for the final (10th) 50 Hz pulse was reduced by 3,4 DAP in both wild-type and mutant.
The improved performance of CMT2D mice after administration of physostigmine, the lack of improved performance in the presence of 3,4-DAP, and the existence of evident failures in evoked release with 1 s long trains, are consistent with a presynaptic defect that can be counteracted by enhanced activation of postsynaptic receptors, but limited capacity for increasing or maintaining quantal content.
Quantal analysis in the presence of 3,4 DAP
To assess the capacity of mutant NMJs to increase quantal release, we performed an additional set of voltage-clamp experiments using muscles from GarsC201R to assess NMJ function in the presence 20 μm 3,4 DAP. The data presented in Figure 7 show that mutant NMJs in 2- and 4-month-old LAL muscles are able to respond to the 3,4 DAP in a manner that is qualitatively similar to wild-type NMJs (note control values are replotted from Fig. 2). Lengthening the presynaptic depolarization increased EPC amplitude at mutant NMJs to values similar to control (Fig. 7C). However, mean quantal content at mutant 2-month-old NMJs remained significantly lower than the respective wild-type mean (Fig. 7D) and though it was still visibly reduced at 4-month-old mutant NMJs, it was not statistically different. Thus, in response to a single stimulus, mutant NMJs have the capacity to respond to 3,4 DAP and normalize EPC amplitude and increase quantal content to values at or near control. So why was wire-hang performance not improved by in vivo administration of 3,4 DAP? By the end of the 200 ms 50 Hz stimulation, the 20 μm 3,4 DAP reduced EPC amplitude to just 20% of initial values for both mutant and wild-type NMJs (Fig. 7E). To improve the wire-hang performance of the mutants would require an improvement in sustained release for up to a minute, so an equivalent in vivo dose would be expected to worsen performance. To examine this further we also calculated the quantal content of the last EPC (10th) in the 50 Hz train (Fig. 7F). Under non-drug control conditions quantal content of the 10th EPC is significantly lower at mutant NMJs than wild-type (Fig. 7F, compare control mutant vs wild-type). Importantly, introduction of 3,4 DAP reduces quantal content even further at mutant NMJs and even reduces wild-type quantal content to values similar to those seen at mutant NMJs. Thus, despite the capacity of mutant and wild-type NMJs to increase their quantal release in the presence of 3,4 DAP (Fig. 7C), release is not well sustained for even 200 ms of 50 Hz stimulation and, at least at the dose tested, would not be effective in the treatment of the presynaptic defect in the Gars mutant mice.
Discussion
The objective of this study was to determine whether NMJ dysfunction is a previously unrecognized aspect of CMT2D and could therefore present a novel treatment avenue using drugs available for treatment of neuromuscular disorders characterized by reduced presynaptic release. Overall, our data confirm the presence of a presynaptic defect at the mutant NMJs that likely contributes to muscle weakness and can be overcome by administration of the cholinesterase inhibitor physostigmine. To our knowledge, this is the first detailed examination of NMJ function in a type 2 axonal CMT.
We studied the relatively mildly affected proximal LAL muscle from animals carrying two different Gars mutations that produce disease phenotypes in mice that vary, as does CMT2D, from mild (GarsC201R) to severe (GarsP278KY). These new data describing synaptic defects are consistent with other previously described phenotypes (Seburn et al., 2006; Achilli et al., 2009; Motley et al., 2011; Stum et al., 2011; Sleigh et al., 2014) insofar as the different mutations did not produce distinct NMJ phenotypes, but rather produced changes that vary along a continuum both within and across genotype/age. As such, we propose these data represent at least a portion of a spectrum of NMJ dysfunction that, if present in patients, could present differently in each patient and to varying degrees at different times. The spectrum is evident within the LAL and would likely be expanded if examined across muscles because morphological data for the NMJ in CMT2D mice has consistently shown that distal muscles (lumbricals, tibialis anterior, plantaris) have more marked degeneration than NMJs in more proximal muscles (transversus abdominis, levator auris longus; Seburn et al., 2006; Achilli et al., 2009; Motley et al., 2011; Sleigh et al., 2014). Based on the more prevalent distal partial denervation alone, it is reasonable to assume that NMJ dysfunction in our mice is likely worse in distal muscles, a notion that is also consistent with disease presentation in humans (Saporta and Shy, 2013).
Together, these data indicate that the mutant GARS has a broad effect that leaves the fundamental release machinery intact but “weakens” NMJ function in the CMT2D mice. Overall, the results point clearly to a presynaptic problem that reduces quantal content, but examination of key determinants of quantal content did not reveal a primary mechanism. Instead, our data suggest that release processes overall are not functioning optimally and that dysfunction includes a spectrum of effects that for a given NMJ could include: (1) reduced terminal areas that may contain fewer total vesicles (inset Fig. 5C), and (2) morphological breakdown of the presynaptic nerve including some loss of associated release sites (Fig. 4K). For example, despite statistical differences in average vesicle number not reaching the 5% statistical cutoff, it seems clear that reduced vesicle number could contribute at some NMJs, at least in the severe GarsP278KY allele (Fig. 5C). However, if reduced vesicle number was the sole disease mechanism for mutant Gars we would also have expected administration of 3,4 DAP to cause greater relative depression at GarsC201R NMJs than at wild-type with 50 Hz stimulation. Instead the relative depression induced by the 3,4 DAP was near identical (Fig. 7E). Similarly, although NMJ area is significantly smaller in distal GarsP278KY muscles, in GarsC201R muscles NMJ areas are not different from wild-type and do not change between 1 and 3 months of age (Sleigh et al., 2014, their Fig. 3), yet in our experiments quantal content is reduced between 2 and 4 months. Thus, reduced NMJ area likely contributes, but is also insufficient as a primary mechanism. Finally, although we found partially denervated NMJs where a reduction in the number of release sites could contribute to reduced quantal content, this too seems insufficient to account entirely for the observed dysfunction in both alleles. Although ∼50% of NMJs in the LAL of the severe GarsP278KY allele are partially denervated, only ∼10% partial denervation is present in LAL of the mild GarsC201R allele at 4 months of age.
The paucity of mitochondria in mutant nerve terminals in our electron micrographs is also potentially interesting. Reduced mitochondria in terminals may influence synaptic transmission through a variety of mechanisms, including deficits in ATP production or changes in intracellular calcium dynamics (Stowers et al., 2002). The mitochondria that were present in GarsP278KY terminals were not vacuolated or otherwise obviously degenerating. Their reduced number may reflect a failure in axonal transport, biogenesis or turnover, but the association of MFN2 with CMT2A makes it clear that defects in neuronal mitochondria can directly lead to axonal neuropathy (Zuchner et al., 2004). The Gars gene in mammals encodes both the mitochondrial and cytosolic forms of the protein through alternative start codon usage (Shiba et al., 1994; Williams et al., 1995; Mudge et al., 1998). All mutations associated with CMT2D and neuropathy in humans and mice are in the common, downstream domains of the GARS protein that are shared by both isoforms. For most other tRNA synthetases, separate nuclear genes encode the mitochondrial and cytosolic enzymes and overall the genetics of tRNA synthetase-associated CMTs (Tolkunova et al., 2000; Scheper et al., 2007; Isohanni et al., 2010; Latour et al., 2010; McLaughlin et al., 2010; Vester et al., 2013) does not clearly indicate a mitochondrial basis for the neuropathies. Nonetheless, mitochondrial dysfunction, whether primary or secondary, also cannot be ruled out as contributing to the synaptic dysfunction we observed.
Our data also indicate ongoing, apparently compensatory changes in synaptic transmission at the NMJ. A variety of mechanisms could compensate for reduced synaptic currents to help maintain muscle function. The finding that the 4 month mutant NMJs again show some initial potentiation in response to 50 Hz activation may be an example of such compensation, but other changes may occur outside the synapse as well. We know that changes in muscle activity can modify passive properties of muscle (Lomo and Rosenthal, 1972) and such changes may occur in the CMT2D mice. Voltage-clamp measures are unaffected by changes in specific membrane resistance and capacitance or other changes (e.g., Na+ channels) that could alter muscle excitability. Thus, while our voltage-clamp data definitively establish the presence of synaptic defects in the CMT2D mice, it is possible that muscle fiber characteristics or other processes might increase or decrease the likelihood of transmission failures that could contribute to weakness.
Based on the combined results presented here, we propose that CMT2D, and perhaps other type 2 axonal CMTs, display synaptic dysfunction that could contribute both variably and intermittently to weakness or fatigue. In wild-type mice, NMJs operate in a “failsafe” manner, such that firing the nerve produces sufficient depolarization in the muscle to cause an action potential without fail. This requires a “safety factor” of excess synaptic current to insure reliable muscle contraction (Paton and Waud, 1967; Rich, 2006). In the CMT2D mice, we show that average quantal content of mutant NMJs is significantly lower than wild-type (Fig. 2), and based on examination of distributions of mutant quantal content values, we propose a model of CMT2D synaptic transmission (Fig. 8) where disease processes diminish presynaptic release to a variable extent at most, if not all, NMJs. Distributions of quantal content at mutant NMJs each show a distinct leftward shift relative to wild-type, including between 15 and 30% of NMJs (depending on age and genotype) with a quantal content lower than any recorded at wild-type NMJs (Fig. 8A, left of vertical dashed line). We suggest currents at these severely affected NMJs could regularly fail to initiate muscle action potentials. At the other end of the range, fully 40% of wild-type NMJs have quantal contents ≥50, whereas only ∼20% of values are in this range for NMJs in muscles of 2-month-old GarsC201R mutants, and for the NMJs in muscles of severe GarsP278KY and 4 month GarsC201R mice, exactly 2 NMJs fall in this range. The extent to which the population of mutant NMJs with quantal contents within normal range contributes to transmission failures and therefore weakness is unknown. However, based on the failures observed at some 2-month-old GarsC201R mutant NMJs during sustained 1 s stimulation at 70 Hz (Fig. 3) and previously reported EMG decrements during in situ tetanic muscle contractions with 700 ms 80 Hz stimulation (Seburn et al., 2006, their Fig. 3B) it seems likely such failure is substantial. The diagram in Figure 8B demonstrates how NMJs with severely reduced quantal content (e.g., GarsP278KY or 4 month GarsC201R) may not reach threshold for initiation of an action potential, even in the absence of failures. Actual raw traces from relatively mildly affected mutant NMJs in Figure 8C (also Fig. 3), demonstrate how depression during repetitive activation could reduce release below the safety factor and cause failure of muscle activation.
Figure 8.

NMJ dysfunction in CMT2D mice. A, Plot of individual values for quantal content calculated for NMJs of each of the four experimental groups reveal CMT2D distributions are shifted to lower values compared with combined wild-type sample (white). Release at a proportion of terminals is severely reduced (≤20, left of dashed vertical line), whereas the majority is mildly or moderately affected with quantal content values shifted to the left of the wild-type mean (∼50), whereas the highest wild-type values (60–90) are largely absent for mutant NMJs. Note also, the 2 to 4 month progression at GarsC201R NMJs (yellow vs red) with the latter overlapping the distribution for severe GarsP278KY. In B, we show a hypothetical model of how the widespread NMJ dysfunction could produce variable and intermittent muscle weakness in CMT2D. Normal wild-type NMJs have a significant safety factor such that EPCs reliably depolarize the muscle above the threshold for an action potential (AP). In the CMT2D mice, terminals with a mild/moderate reduction in release (mild) would have a reduced safety factor and EPCs may intermittently fail to initiate an AP, whereas EPCs at severely affected terminals (severe) consistently do not reach the AP threshold. Note also, that because EPCs at affected terminals would initiate APs closer to their peak, APs would be slightly delayed (dotted AP) and EMG recordings of an affected muscle would be expected to display significant “jitter,” a clinical measure used to identify transmission failure. C, Repetitive activation increases the extent and variability of transmission failure (Fig. 3). This scenario is depicted by example raw EPC traces of the 50 Hz response of moderately affected mutant terminals and typical wild-type. The horizontal dashed line through these traces indicates the EPC amplitude equivalent to a quantal content of 20 for the wild-type NMJ (equivalent to vertical dashed line in A). This cutoff was selected because all values measured at wild-type NMJs exceeded it, whereas ∼20% of the values for each mutant population were below it. For the 50 Hz trains shown wild-type the amplitude of final EPCs persists well above this level, but depression at mutant NMJs is sufficient to approach this value for the four to five final EPCs (Fig. 3D–G).
Footnotes
This work was supported by R03-NS081334 (K.L.S.), RO1-NS054154 (R.W.B.). We thank the Scientific Services at The Jackson laboratory, particularly Pete Finger, for muscle sample preparation and technical assistance with electron microscopy, and Mark Lessard for technical assistance with 3D imaging and quantification of bassoon-stained junctions. The Scientific Services at Jackson are supported in part by NCI Cancer Center Support (CA034196); Bangor Christian High School, honor biology students Dylan Merchant, Zach Palmeter, and Cody Collins for their assistance with analysis of electron micrographs; and summer student Lauren Budd for her contribution to bassoon immunohistochemistry and Sam Murray for assistance with collection of wire-hang data.
The authors declare no competing financial interests.
References
- Achilli F, Bros-Facer V, Williams HP, Banks GT, AlQatari M, Chia R, Tucci V, Groves M, Nickols CD, Seburn KL, Kendall R, Cader MZ, Talbot K, van Minnen J, Burgess RW, Brandner S, Martin JE, Koltzenburg M, Greensmith L, Nolan PM, et al. An ENU-induced mutation in mouse glycyl-tRNA synthetase (GARS) causes peripheral sensory and motor phenotypes creating a model of Charcot-Marie-Tooth type 2D peripheral neuropathy. Dis Model Mech. 2009;2:359–373. doi: 10.1242/dmm.002527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angaut-Petit D, Molgo J, Connold AL, Faille L. The levator auris longus muscle of the mouse: a convenient preparation for studies of short- and long-term presynaptic effects of drugs or toxins. Neurosci Lett. 1987;82:83–88. doi: 10.1016/0304-3940(87)90175-3. [DOI] [PubMed] [Google Scholar]
- Antonellis A, Ellsworth RE, Sambuughin N, Puls I, Abel A, Lee-Lin SQ, Jordanova A, Kremensky I, Christodoulou K, Middleton LT, Sivakumar K, Ionasescu V, Funalot B, Vance JM, Goldfarb LG, Fischbeck KH, Green ED. Glycyl tRNA synthetase mutations in Charcot-Marie-Tooth disease type 2D and distal spinal muscular atrophy type V. Am J Hum Genet. 2003;72:1293–1299. doi: 10.1086/375039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess RW, Cox GA, Seburn KL. Neuromuscular disease models and analysis. Methods Mol Biol. 2010;602:347–393. doi: 10.1007/978-1-60761-058-8_20. [DOI] [PubMed] [Google Scholar]
- Charcot JM, Marie P. Sur une forme particulière d'atrophie musculaire progressive, souvent familiale, debutant par les pieds et les jambes et atteignant plus tard les mains. Rev Med. 1886;6:97–138. [Google Scholar]
- Cruz LJ, Gray WR, Olivera BM, Zeikus RD, Kerr L, Yoshikami D, Moczydlowski E. Conus geographus toxins that discriminate between neuronal and muscle sodium channels. J Biol Chem. 1985;260:9280–9288. [PubMed] [Google Scholar]
- Del Bo R, Locatelli F, Corti S, Scarlato M, Ghezzi S, Prelle A, Fagiolari G, Moggio M, Carpo M, Bresolin N, Comi GP. Coexistence of CMT-2D and distal SMA-V phenotypes in an Italian family with a GARS gene mutation. Neurology. 2006;66:752–754. doi: 10.1212/01.wnl.0000201275.18875.ac. [DOI] [PubMed] [Google Scholar]
- Dubourg O, Azzedine H, Yaou RB, Pouget J, Barois A, Meininger V, Bouteiller D, Ruberg M, Brice A, LeGuern E. The G526R glycyl-tRNA synthetase gene mutation in distal hereditary motor neuropathy type V. Neurology. 2006;66:1721–1726. doi: 10.1212/01.wnl.0000218304.02715.04. [DOI] [PubMed] [Google Scholar]
- Dyck PJ. Definition and basis of classification of hereditary neuropathy with neuronal atrophy and degeneration. In: Dyck PJ, Thomas PK, Lambert EH, editors. Peripheral Neuropathy. Philadelphia, PA: WB Saunders; 1975. pp. 825–867. [Google Scholar]
- Engel AG, Shen XM, Selcen D, Sine SM. Congenital myasthenic syndromes: pathogenesis, diagnosis, and treatment. Lancet Neurol. 2015;14:420–434. doi: 10.1016/S1474-4422(14)70201-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/S0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Glavinović MI. Voltage clamping of unparalysed cut rat diaphragm for study of transmitter release. J Physiol. 1979;290:467–480. doi: 10.1113/jphysiol.1979.sp012784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez CM, Maselli R, Gundeck JE, Chao M, Day JW, Tamamizu S, Lasalde JA, McNamee M, Wollmann RL. Slow-channel transgenic mice: a model of postsynaptic organellar degeneration at the neuromuscular junction. J Neurosci. 1997;17:4170–4179. doi: 10.1523/JNEUROSCI.17-11-04170.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyton DL, Hambrecht FT. Theory and design of capacitor electrodes for chronic stimulation. Med Biol Eng. 1974;12:613–620. doi: 10.1007/BF02477223. [DOI] [PubMed] [Google Scholar]
- Isohanni P, Linnankivi T, Buzkova J, Lönnqvist T, Pihko H, Valanne L, Tienari PJ, Elovaara I, Pirttilä T, Reunanen M, Koivisto K, Marjavaara S, Suomalainen A. DARS2 mutations in mitochondrial leucoencephalopathy and multiple sclerosis. J Med Genet. 2010;47:66–70. doi: 10.1136/jmg.2009.068221. [DOI] [PubMed] [Google Scholar]
- James PA, Cader MZ, Muntoni F, Childs AM, Crow YJ, Talbot K. Severe childhood SMA and axonal CMT due to anticodon binding domain mutations in the GARS gene. Neurology. 2006;67:1710–1712. doi: 10.1212/01.wnl.0000242619.52335.bc. [DOI] [PubMed] [Google Scholar]
- Kong L, Wang X, Choe DW, Polley M, Burnett BG, Bosch-Marcé M, Griffin JW, Rich MM, Sumner CJ. Impaired synaptic vesicle release and immaturity of neuromuscular junctions in spinal muscular atrophy mice. J Neurosci. 2009;29:842–851. doi: 10.1523/JNEUROSCI.4434-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latour P, Thauvin-Robinet C, Baudelet-Méry C, Soichot P, Cusin V, Faivre L, Locatelli MC, Mayençon M, Sarcey A, Broussolle E, Camu W, David A, Rousson R. A major determinant for binding and aminoacylation of tRNA(Ala) in cytoplasmic Alanyl-tRNA synthetase is mutated in dominant axonal Charcot-Marie-Tooth disease. Am J Hum Genet. 2010;86:77–82. doi: 10.1016/j.ajhg.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomo T, Rosenthal J. Control of ACh sensitivity by muscle activity in the rat. J Physiol. 1972;221:493–513. doi: 10.1113/jphysiol.1972.sp009764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magrassi L, Purves D, Lichtman JW. Fluorescent probes that stain living nerve terminals. J Neurosci. 1987;7:1207–1214. doi: 10.1523/JNEUROSCI.07-04-01207.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin HM, Sakaguchi R, Liu C, Igarashi T, Pehlivan D, Chu K, Iyer R, Cruz P, Cherukuri PF, Hansen NF, Mullikin JC, Biesecker LG, Wilson TE, Ionasescu V, Nicholson G, Searby C, Talbot K, Vance JM, Züchner S, NISC Comparative Sequencing Program et al. Compound heterozygosity for loss-of-function lysyl-tRNA synthetase mutations in a patient with peripheral neuropathy. Am J Hum Genet. 2010;87:560–566. doi: 10.1016/j.ajhg.2010.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motley WW, Seburn KL, Nawaz MH, Miers KE, Cheng J, Antonellis A, Green ED, Talbot K, Yang XL, Fischbeck KH, Burgess RW. Charcot-Marie-Tooth-linked mutant GARS is toxic to peripheral neurons independent of wild-type GARS levels. PLoS Genet. 2011;7:e1002399. doi: 10.1371/journal.pgen.1002399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudge SJ, Williams JH, Eyre HJ, Sutherland GR, Cowan PJ, Power DA. Complex organisation of the 5′-end of the human glycine tRNA synthetase gene. Gene. 1998;209:45–50. doi: 10.1016/S0378-1119(98)00007-9. [DOI] [PubMed] [Google Scholar]
- Nishimune H. Active zones of mammalian neuromuscular junctions: formation, density, and aging. Ann N Y Acad Sci. 2012;1274:24–32. doi: 10.1111/j.1749-6632.2012.06836.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimune H, Sanes JR, Carlson SS. A synaptic laminin-calcium channel interaction organizes active zones in motor nerve terminals. Nature. 2004;432:580–587. doi: 10.1038/nature03112. [DOI] [PubMed] [Google Scholar]
- Paton WD, Waud DR. The margin of safety of neuromuscular transmission. J Physiol. 1967;191:59–90. doi: 10.1113/jphysiol.1967.sp008237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plomp JJ, Vergouwe MN, Van den Maagdenberg AM, Ferrari MD, Frants RR, Molenaar PC. Abnormal transmitter release at neuromuscular junctions of mice carrying the tottering alpha(1A) Ca(2+) channel mutation. Brain. 2000;123:463–471. doi: 10.1093/brain/123.3.463. [DOI] [PubMed] [Google Scholar]
- Rafael JA, Nitta Y, Peters J, Davies KE. Testing of SHIRPA, a mouse phenotypic assessment protocol, on Dmd(mdx) and Dmd(mdx3cv) dystrophin-deficient mice. Mamm Genome. 2000;11:725–728. doi: 10.1007/s003350010149. [DOI] [PubMed] [Google Scholar]
- Rich MM. The control of neuromuscular transmission in health and disease. Neuroscientist. 2006;12:134–142. doi: 10.1177/1073858405281898. [DOI] [PubMed] [Google Scholar]
- Rich MM, Waldeck RF, Cork LC, Balice-Gordon RJ, Fyffe RE, Wang X, Cope TC, Pinter MJ. Reduced endplate currents underlie motor unit dysfunction in canine motor neuron disease. J Neurophysiol. 2002;88:3293–3304. doi: 10.1152/jn.00270.2002. [DOI] [PubMed] [Google Scholar]
- Robitaille R, Charlton MP. Presynaptic calcium signals and transmitter release are modulated by calcium-activated potassium channels. J Neurosci. 1992;12:297–305. doi: 10.1523/JNEUROSCI.12-01-00297.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohkamm B, Reilly MM, Lochmüller H, Schlotter-Weigel B, Barisic N, Schöls L, Nicholson G, Pareyson D, Laurà M, Janecke AR, Miltenberger-Miltenyi G, John E, Fischer C, Grill F, Wakeling W, Davis M, Pieber TR, Auer-Grumbach M. Further evidence for genetic heterogeneity of distal HMN type V, CMT2 with predominant hand involvement and Silver syndrome. J Neurol Sci. 2007;263:100–106. doi: 10.1016/j.jns.2007.06.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saporta MA, Shy ME. Inherited peripheral neuropathies. Neurol Clin. 2013;31:597–619. doi: 10.1016/j.ncl.2013.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheper GC, van der Klok T, van Andel RJ, van Berkel CG, Sissler M, Smet J, Muravina TI, Serkov SV, Uziel G, Bugiani M, Schiffmann R, Krägeloh-Mann I, Smeitink JA, Florentz C, Van Coster R, Pronk JC, van der Knaap MS. Mitochondrial aspartyl-tRNA synthetase deficiency causes leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation. Nat Genet. 2007;39:534–539. doi: 10.1038/ng2013. [DOI] [PubMed] [Google Scholar]
- Seburn KL, Nangle LA, Cox GA, Schimmel P, Burgess RW. An active dominant mutation of glycyl-tRNA synthetase causes neuropathy in a Charcot-Marie-Tooth 2D mouse model. Neuron. 2006;51:715–726. doi: 10.1016/j.neuron.2006.08.027. [DOI] [PubMed] [Google Scholar]
- Shaw KP, Aracava Y, Akaike A, Daly JW, Rickett DL, Albuquerque EX. The reversible cholinesterase inhibitor physostigmine has channel-blocking and agonist effects on the acetylcholine receptor-ion channel complex. Mol Pharmacol. 1985;28:527–538. [PubMed] [Google Scholar]
- Shen H, Barry DM, Dale JM, Garcia VB, Calcutt NA, Garcia ML. Muscle pathology without severe nerve pathology in a new mouse model of Charcot-Marie-Tooth disease type 2E. Hum Mol Genet. 2011;20:2535–2548. doi: 10.1093/hmg/ddr152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiba K, Schimmel P, Motegi H, Noda T. Human glycyl-tRNA syntheta: wide divergence of primary structure from bacterial counterpart and species-specific aminoacylation. J Biol Chem. 1994;269:30049–30055. [PubMed] [Google Scholar]
- Sivakumar K, Kyriakides T, Puls I, Nicholson GA, Funalot B, Antonellis A, Sambuughin N, Christodoulou K, Beggs JL, Zamba-Papanicolaou E, Ionasescu V, Dalakas MC, Green ED, Fischbeck KH, Goldfarb LG. Phenotypic spectrum of disorders associated with glycyl-tRNA synthetase mutations. Brain. 2005;128:2304–2314. doi: 10.1093/brain/awh590. [DOI] [PubMed] [Google Scholar]
- Skre H. Genetic and clinical aspects of Charcot-Marie-Tooth's disease. Clin Genet. 1974;6:98–118. doi: 10.1111/j.1399-0004.1974.tb00638.x. [DOI] [PubMed] [Google Scholar]
- Sleigh JN, Grice SJ, Burgess RW, Talbot K, Cader MZ. Neuromuscular junction maturation defects precede impaired lower motor neuron connectivity in Charcot-Marie-Tooth type 2D mice. Hum Mol Genet. 2014;23:2639–2650. doi: 10.1093/hmg/ddt659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stowers RS, Megeath LJ, Górska-Andrzejak J, Meinertzhagen IA, Schwarz TL. Axonal transport of mitochondria to synapses depends on Milton, a novel Drosophila protein. Neuron. 2002;36:1063–1077. doi: 10.1016/S0896-6273(02)01094-2. [DOI] [PubMed] [Google Scholar]
- Stum M, McLaughlin HM, Kleinbrink EL, Miers KE, Ackerman SL, Seburn KL, Antonellis A, Burgess RW. An assessment of mechanisms underlying peripheral axonal degeneration caused by aminoacyl-tRNA synthetase mutations. Mol Cell Neurosci. 2011;46:432–443. doi: 10.1016/j.mcn.2010.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen RH, Wilson DF. Effects of 4-aminopyridine and 3,4-diaminopyridine on transmitter release at the neuromuscular junction. J Pharmacol Exp Ther. 1983;227:260–265. [PubMed] [Google Scholar]
- Tolkunova E, Park H, Xia J, King MP, Davidson E. The human lysyl-tRNA synthetase gene encodes both the cytoplasmic and mitochondrial enzymes by means of an unusual alternative splicing of the primary transcript. J Biol Chem. 2000;275:35063–35069. doi: 10.1074/jbc.M006265200. [DOI] [PubMed] [Google Scholar]
- tom Dieck S, Sanmartí-Vila L, Langnaese K, Richter K, Kindler S, Soyke A, Wex H, Smalla KH, Kämpf U, Fränzer JT, Stumm M, Garner CC, Gundelfinger ED. Bassoon, a novel zinc-finger CAG/glutamine-repeat protein selectively localized at the active zone of presynaptic nerve terminals. J Cell Biol. 1998;142:499–509. doi: 10.1083/jcb.142.2.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tooth HH. The peroneal type of progressive muscular atrophy. London: HK Lewis; 1886. [Google Scholar]
- Vester A, Velez-Ruiz G, McLaughlin HM, Lupski JR, Talbot K, Vance JM, Züchner S, Roda RH, Fischbeck KH, Biesecker LG, Nicholson G, Beg AA, Antonellis A. A loss-of-function variant in the human histidyl-tRNA synthetase (HARS) gene is neurotoxic in vivo. Hum Mutat. 2013;34:191–199. doi: 10.1002/humu.22210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Engisch KL, Li Y, Pinter MJ, Cope TC, Rich MM. Decreased synaptic activity shifts the calcium dependence of release at the mammalian neuromuscular junction in vivo. J Neurosci. 2004;24:10687–10692. doi: 10.1523/JNEUROSCI.2755-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Li Y, Engisch KL, Nakanishi ST, Dodson SE, Miller GW, Cope TC, Pinter MJ, Rich MM. Activity-dependent presynaptic regulation of quantal size at the mammalian neuromuscular junction in vivo. J Neurosci. 2005;25:343–351. doi: 10.1523/JNEUROSCI.3252-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Wang Q, Engisch KL, Rich MM. Activity-dependent regulation of the binomial parameters p and n at the mouse neuromuscular junction in vivo. J Neurophysiol. 2010;104:2352–2358. doi: 10.1152/jn.00460.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams J, Osvath S, Khong TF, Pearse M, Power D. Cloning, sequencing and bacterial expression of human glycine tRNA synthetase. Nucleic Acid Res. 1995;23:1307–1310. doi: 10.1093/nar/23.8.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuchner S, Mersiyanova IV, Muglia M, Bissar-Tadmouri N, Rochelle J, Dadali EL, Zappia M, Nelis E, Patitucci A, Senderek J, Parman Y, Evgrafov O, Jonghe PD, Takahashi Y, Tsuji S, Pericak-Vance MA, Quattrone A, Battaloglu E, Polyakov AV, Timmerman V, et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet. 2004;36:449–451. doi: 10.1038/ng1341. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]