

Abstract
The ability of 17β-estradiol (E2) to enhance hippocampal object recognition and spatial memory depends on rapid activation of extracellular signal-regulated kinase (ERK) in the dorsal hippocampus (DH). Although this activation can be mediated by the intracellular estrogen receptors ERα and ERβ, little is known about the role that the membrane estrogen receptor GPER plays in regulating ERK or E2-mediated memory formation. In this study, post-training DH infusion of the GPER agonist G-1 enhanced object recognition and spatial memory in ovariectomized female mice, whereas the GPER antagonist G-15 impaired memory, suggesting that GPER activation, like E2, promotes hippocampal memory formation. However, unlike E2, G-1 did not increase ERK phosphorylation, but instead significantly increased phosphorylation of c-Jun N-terminal kinase (JNK) in the DH. Moreover, DH infusion of the JNK inhibitor SP600125 prevented G-1 from enhancing object recognition and spatial memory, but the ERK inhibitor U0126 did not. These data suggest that GPER enhances memory via different cell-signaling mechanisms than E2. This conclusion was supported by data showing that the ability of E2 to facilitate memory and activate ERK signaling was not blocked by G-15 or SP600125, which demonstrates that the memory-enhancing effects of E2 are not dependent on JNK or GPER activation in the DH. Together, these data indicate that GPER regulates memory independently from ERα and ERβ by activating JNK signaling, rather than ERK signaling. Thus, the findings suggest that GPER in the DH may not function as an estrogen receptor to regulate object recognition and spatial memory.
SIGNIFICANCE STATEMENT Although 17β-estradiol has long been known to regulate memory function, the molecular mechanisms underlying estrogenic memory modulation remain largely unknown. Here, we examined whether the putative membrane estrogen receptor GPER acts like the classical estrogen receptors, ERα and ERβ, to facilitate hippocampal memory in female mice. Although GPER activation did enhance object recognition and spatial memory, it did so by activating different cell-signaling mechanisms from ERα, ERβ, or 17β-estradiol. These data indicate that 17β-estradiol and GPER independently regulate hippocampal memory, and suggest that hippocampal GPER may not function as an estrogen receptor in the dorsal hippocampus. These findings are significant because they provide novel insights about the molecular mechanisms through which 17β-estradiol modulates hippocampal memory.
Keywords: ERK, G-1, hippocampus, JNK, object placement, object recognition
Introduction
Although sex steroid hormones, such as the potent estrogen 17β-estradiol (E2), influence the etiology and symptomatology of disorders, such as depression and dementia in women (Kessler et al., 2005; Yaffe et al., 2007), the neural mechanisms through which estrogens regulate cognitive function are not well understood. E2 can enhance hippocampal-dependent object recognition and spatial memory in female rodents by rapidly activating numerous cell-signaling cascades, including the extracellular signal-regulated kinase (ERK) pathway (Fernandez et al., 2008; Lewis et al., 2008; Boulware et al., 2013; Fortress et al., 2013; Pereira et al., 2014). Yet the estrogen receptors (ERs) that mediate these rapid effects remain unclear. The intracellular ERs, ERα, and ERβ, enhance hippocampal object recognition and spatial memory in female mice by activating ERK signaling in the dorsal hippocampus (DH) within 5 min (Boulware et al., 2013). However, little is known about the role of membrane ERs, such as GPER, in hippocampal memory formation.
GPER is a G-protein-coupled receptor (GPCR) previously known as GPR30 (Funakoshi et al., 2006). GPER is expressed at high levels in the brain, including within the plasma membrane of neurons in the hippocampus and prefrontal cortex (Brailoiu et al., 2007; Akama et al., 2013; Almey et al., 2014). Similar to other GPCRs, GPER can activate cell-signaling pathways, such as ERK/MAPK, Akt, and c-Jun N-terminal kinase (JNK; Chimento et al., 2012). Moreover, E2 reportedly binds GPER with high affinity in peripheral tissues (Thomas et al., 2005; Prossnitz et al., 2007), prompting a name change from GPR30 to GPER. However, some investigators maintain that GPER is not a true ER, but may instead collaborate in mediating the biological actions of estrogens (Levin, 2009; Langer et al., 2010).
GPER has been shown to affect hippocampal-dependent spatial working memory in studies using systemic injections of the GPER agonist G-1 or antagonist G-15 (Hammond et al., 2009; Hammond and Gibbs, 2011; Hawley et al., 2014). These studies found that G-1 enhances, whereas G-15 impairs, spatial memory in ovariectomized rats. However, their use of systemic injections does not permit definitive conclusions about the role of hippocampal GPER in memory formation. Furthermore, these studies did not examine the molecular mechanisms underlying the memory-enhancing effects of GPER. As such, the present study used DH infusions of G-1 and G-15 to pinpoint the role of DH GPER in regulating hippocampal memory and to determine whether similar cell-signaling mechanisms are necessary for GPER and E2 to enhance hippocampal memory.
Here, we report that activation of DH GPER enhances both object recognition and spatial memory in ovariectomized female mice, but that these effects depend on JNK, not ERK, signaling in the DH. This important role of JNK in GPER-induced regulation of memory is consistent with the involvement of JNK signaling in synaptic plasticity, neuronal regeneration, and brain development (Tararuk et al., 2006; Waetzig et al., 2006). Interestingly, the memory-enhancing effects of E2 were not dependent on either JNK or GPER activation in the DH. Collectively, these data suggest that GPER enhances hippocampal memory by activating different cell-signaling cascades than E2. Thus, GPER in the DH does not appear to mediate the beneficial effects of E2 on object recognition and spatial memory.
Materials and Methods
Subjects.
Subjects were female C57BL/6 mice (8–10 weeks of age) purchased from Taconic Biosciences. After surgery, mice were singly housed in a room with a 12 h light/dark cycle, and were allowed ad libitum access to food and water. All behavioral testing was performed between 9:00 A.M. and 6:00 P.M. in a quiet room with dim lights. All procedures were approved by the University of Wisconsin-Milwaukee Institutional Animal Care and Use Committee, and followed policies set forth by the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
General experimental design.
After recovery from cannula implantation and ovariectomy surgery, mice underwent behavioral testing in an object-recognition (OR) task to measure object recognition memory and an object placement (OP) task to measure spatial memory. These tasks were chosen because they are sensitive to E2, and the single training trial used for each is ideal for linking rapid biochemical alterations to memory formation. In ovariectomized mice, immediate post-training bilateral infusion of 5 μg E2 into the dorsal hippocampus enhances OR tested 48 h after infusion (Fernandez et al., 2008; Fan et al., 2010; Zhao et al., 2010, 2012; Boulware et al., 2013; Fortress et al., 2013, 2014; Pereira et al., 2014) and OP tested 24 h after infusion (Boulware et al., 2013; Fortress et al., 2014). Each task used unique sets of objects to maintain novelty and prevent interference from one task to the other. OR and OP testing were separated by 2 weeks to allow the hippocampus to fully recover from infusion. The order of testing varied for animals within each group. Two weeks after the final behavioral testing, mice were infused and the dorsal hippocampus was collected bilaterally 5, 10, 15, or 30 min later for Western blotting (Fig. 1).
Figure 1.
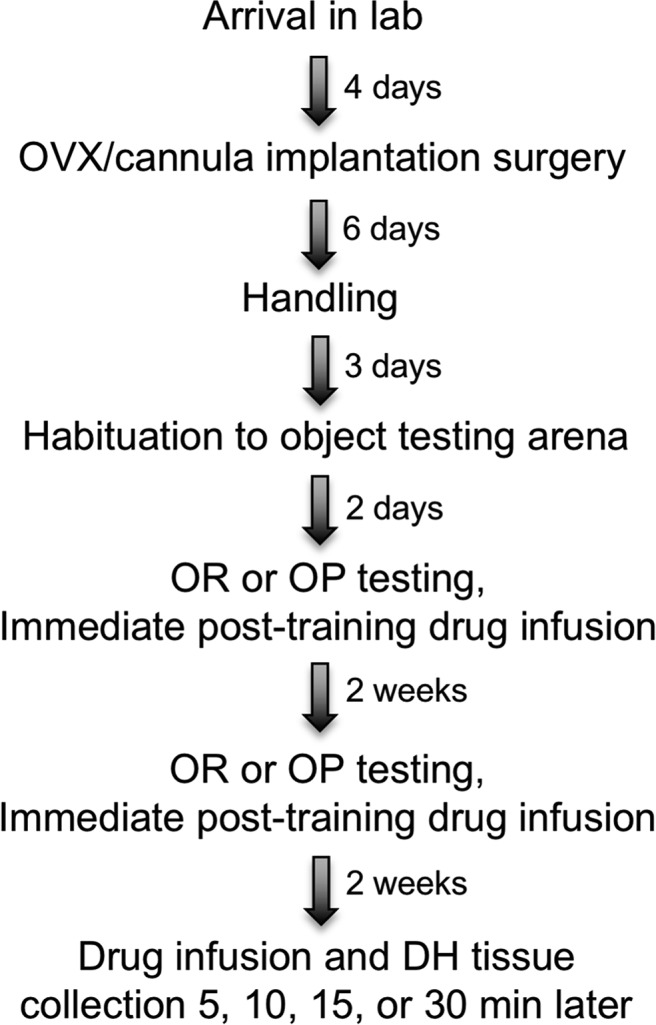
General experimental design for all studies. See text for details. OVX, Ovariectomy.
Surgery.
Four days after arrival in the laboratory, mice were bilaterally ovariectomized and implanted with chronic indwelling guide cannulae within the same surgical session as described previously (Boulware et al., 2013; Fortress et al., 2013, 2014). Mice were anesthetized with isoflurane gas (2% isoflurane in 100% oxygen) and secured in a stereotaxic apparatus (Kopf Instruments). Following ovariectomy, mice were implanted with guide cannulae (22 gauge; C232G, Plastics One) into the DH (−1.7 mm AP, ±1.5 mm ML, −2.3 mm DV) or DH and dorsal third ventricle [intracerebroventricular (i.c.v.); −0.9 mm AP, ±0.0 mm ML, −2.3 mm DV] as described previously (Boulware et al., 2013; Fortress et al., 2013, 2014). Dummy cannulae (C232DC, Plastics One) were inserted into all guide cannulae to preserve patency of the guide cannulae. Cannulae were fixed to the skull with dental cement (Darby Dental) that served to close the wound. Mice were allowed 6 d to recover from surgery before the start of behavioral testing.
Drugs and infusions.
During infusions, mice were gently restrained and dummy cannulae were replaced with an infusion cannula (C313I; DH: 28 gauge, extending 0.8 mm beyond the 1.5 mm guide; i.c.v., 28 gauge, extending 1.0 mm beyond the 1.8 mm guide) attached to PE50 polyethylene tubing that was mounted on a 10 μl Hamilton syringe. Infusions were controlled by a microinfusion pump (KDS Legato 180, KD Scientific) and conducted immediately post-training at a rate of 0.5 μl/min in the DH or 1 μl/2 min into the dorsal third ventricle as described previously (Boulware et al., 2013; Fortress et al., 2013, 2014). Infusion cannulae remained in place for 1 min after each infusion to prevent diffusion back up the cannula track. For studies in which E2 or G-1 was administered in combination with G-15 or a cell-signaling inhibitor, the antagonist or cell-signaling inhibitor was first infused bilaterally into the DH and then E2 or G-1 was infused intracerebroventricularly immediately afterward. We routinely use this triple infusion protocol to prevent possible damage to the DH from two DH infusions in rapid succession (Fernandez et al., 2008; Fan et al., 2010; Zhao et al., 2010, 2012; Boulware et al., 2013; Fortress et al., 2013). This protocol allows us to infuse compounds adjacent to the DH while inhibiting receptor or cell-signaling activation directly within the DH.
G-1 (1-[4-(6-bromobenzo[1,3]dioxol-5yl)-3a,4,5,9b-tetrahydro-3H-cyclopenta [c]quinolin-8-yl]-ethanone; Azano Biotech) was dissolved in 16% dimethylsulfoxide (DMSO) and infused at doses of 2 or 4 ng/hemisphere into the DH or 8 ng intracerebroventricularly. G-1 is a selective agonist for GPER that does not bind ERα and ERβ at concentrations up to 10 μm in vitro (Bologa et al., 2006; Blasko et al., 2009). The vehicle control for G-1 was 16% DMSO in 0.9% saline. G-15 ((3aS*,4R*,9bR*)-4-(6-bromo-1,3-benzodioxol-5-yl)-3a,4,5,9b-3H-cyclopenta[c]quinolone; Azano Biotech) was dissolved in 2% DMSO and infused at doses of 1.85, 3.7, and 7.4 ng/hemisphere into the DH. G-15 is a selective antagonist for GPER that also does not bind to ERα and ERβ at concentrations up to 10 μm in vitro (Dennis et al., 2009). The vehicle control for G-15 was 2% DMSO in 0.9% saline.
Cyclodextrin-encapsulated E2 (Sigma-Aldrich) was dissolved in 0.9% saline and infused at doses of 5 μg/hemisphere into the DH or 10 μg intracerebroventricular (Zhao et al., 2012; Boulware et al., 2013). The vehicle control for E2 was 2-hydroxypropyl-β-cyclodextrin (HBC, Sigma-Aldrich), dissolved in 0.9% saline using the same amount of cyclodextrin as E2 for infusions. The JNK inhibitor SP600125 (Anthra[1,9-cd]pyrazol-6(2H)-one; Sigma-Aldrich) was dissolved in 2% DMSO and infused at doses of 0.55 and 2.75 ng/hemisphere into the DH. SP600125 is a selective inhibitor for JNK that does not affect other MAPK family members such as ERK or p38 at concentrations <10 μm (Bennett et al., 2001). The vehicle control for SP600125 was 2% DMSO in 0.9% saline. The MEK inhibitor U0126 (1,4-diamino-2,3-dicyano-1,4-bis (o-aminophenylmercapto) butadiene; Promega) was dissolved in 25% DMSO and infused at a dose of 0.5 μg/hemisphere into the DH. This dose does not impair OR and OP memory by itself (Fernandez et al., 2008; Boulware et al., 2013), and therefore, any effects of U0126 in combination with E2 or G-1 cannot be attributed to a general memory-impairing effect of this compound. The vehicle control for U0126 was 25% DMSO in 0.9% saline.
Object recognition and object placement.
OR and OP were conducted to examine hippocampus-dependent object recognition and spatial memory. Both tasks have been shown to involve DH function (Baker and Kim, 2002; Luine et al., 2003; Frye et al., 2007; Cohen et al., 2013) and are sensitive to E2 treatment (Gresack and Frick, 2006; Zhao et al., 2010). Before the start of behavioral training, mice were handled (1 min/d) for 3 d to acclimate them to the experimenters. They were also familiarized with objects by placing a small Lego not used during testing in their home cage for 4 d. At the start of training, mice were habituated to the empty white arena (width, 60 cm; length, 60 cm; height, 47 cm) by allowing them to explore for 5 min/d for 2 consecutive days. On the third day, mice were habituated for 2 min in the arena, and then placed in a holding cage while two identical objects were placed near the northwest and northeast corners of the arena. Mice were then returned to the arena and allowed to freely explore the objects until they accumulated 30 s of investigation (or until a total of 20 min had elapsed). Immediately after this training, mice were infused and then returned to their home cage. After 24 or 48 h, memory was tested by allowing mice to accumulate 30 s exploring a novel object and an object identical to one of the familiar objects from training. Time spent with the objects and elapsed time to accumulate 30 s of exploration were recorded using ANYmaze tracking software (Stoelting). Because mice inherently prefer novelty, mice that remember the familiar training object spend more time than chance (15 s) investigating the novel object. Chance was set at 15 s because this is the value at which mice spend exactly the same amount of time with each object. As such, chance levels of performance represent no memory of the training objects. Because vehicle-infused female mice do not remember the familiar object 48 h after training (Gresack et al., 2007; Boulware et al., 2013; Fortress et al., 2014; Pereira et al., 2014), a 48 h delay was used to test the memory-enhancing effects of E2 and G-1. However, vehicle-infused female mice do remember the familiar object 24 h after training (Gresack et al., 2007; Fan et al., 2010; Zhao et al., 2012; Boulware et al., 2013), so the shorter 24 h delay was used to test the potential memory impairing effects of G-15 and cell-signaling inhibitors.
The OP task used the same apparatus and general procedure as OR, but instead of substituting a novel object for a training object during testing, one familiar object was moved to the southeast or southwest corner of the testing arena. Different objects were used in OP and OR. Because vehicle-infused females remember the original object placement after 4 h, but not 24 h (Boulware et al., 2013), we used the 24 h delay to test memory-enhancing effects of E2 and G-1, and the 4 h delay to test memory-impairing effects of G-15 and cell-signaling inhibitors. Two weeks separated OR and OP testing to allow acute effects of drug infusions to dissipate before the next infusion.
Western blotting.
Western blotting was performed as described previously (Fernandez et al., 2008; Boulware et al., 2013). To determine the effects of G-1 on DH cell signaling, mice were cervically dislocated and decapitated, and the DH was dissected bilaterally on an ice-cold plate 5, 15, or 30 min after infusion. To expose the DH, the overlying parietal, occipital, and temporal cortices were removed using a scalpel and forceps. Horizontal cuts were made at a 45° angle through each side of the DH at the level of the base of the superior colliculus. The fornix was then transected with the scalpel blade and the entire DH, including the dentate gyrus and cornu ammonis fields, was bilaterally removed with forceps and placed in a 1.5 ml microcentrifuge tube. Tissue samples were immediately weighed and frozen on dry ice, and then stored at −80°C until homogenization. To determine the effects of E2, on DH cell signaling, the DH was dissected bilaterally 5 or 10 min after infusion. In all other experiments, the DH was dissected bilaterally 5 min after infusion. DH tissues were resuspended to 50 μl/mg in lysis buffer and homogenized using a sonicator (Branson Sonifier 250) as described previously (Fortress et al., 2015). Proteins were then electrophoresed on 10% Tris-HCl precast gels (Bio-Rad) and transferred to PVDF membranes (Bio-Rad). Western blots were blocked with 5% skim milk and incubated with primary antibodies (phospho-ERK, phospho-Akt, phospho-PI3K, phospho-JNK, and phospho-ATF2, 1:1000; Cell Signaling Technology) overnight at 4°C. Blots were then incubated for 1 h at room temperature with a rabbit HRP-conjugated secondary antibody (1:5000; Cell Signaling Technology), and developed using West Dura chemiluminescent substrate (Pierce). A ChemiDoc MP gel imager (Bio-Rad) was used to detect signal correlating with protein expression. Densitometry was performed using Carestream Molecular Imaging Software (Carestream Healthcare). Blots then were stripped with 0.2 m NaOH and incubated with antibodies (total-ERK, total-Akt, total-PI3K, and total-JNK, 1:1000; β-actin, 1:5000; Cell Signaling Technology) for protein normalization. Data were represented as percentage immunoreactivity relative to vehicle controls. Treatment effects were measured within single gels (n = 5–8/group).
Statistics.
For OR and OP data, one-sample t tests were conducted using SPSS (IBM) to determine whether each group spent more time than chance (15 s) exploring the novel or moved object (Gresack and Frick, 2003; Gervais et al., 2013; Pereira et al., 2014). This analysis is essential to determine whether learning occurred within each group. For between-group comparisons within each behavioral experiment, one-way ANOVAs were conducted followed by Fisher's LSD post hoc tests using GraphPad Prism 6. Western blot data were also analyzed using one-way ANOVAs followed by Fisher's LSD post hoc tests and selected t tests. Significance was determined at p < 0.05.
Results
GPER regulates hippocampal memory
We first infused the GPER agonist G-1 into the DH to determine whether activation of GPER in the DH enhances OR and OP memory. Mice received bilateral DH infusion of vehicle (16% DMSO) or one of two doses of G-1 (2 or 4 ng/hemisphere) immediately after OR training. Forty-eight hours later, mice infused with vehicle or 2 ng G-1 spent no more time with the novel object than chance (15 s). In contrast, mice infused with 4 ng/hemisphere G-1 spent more time exploring the novel object than chance (t(10) = 3.4, p = 0.007; Fig. 2A), suggesting that 4 ng G-1 enhanced OR memory. One-way ANOVA indicated a significant main effect of treatment (F(2,30) = 3.4, p = 0.047) and post hoc tests revealed that mice infused with 4 ng, but not 2 ng, G-1 spent significantly more time with the novel object than mice infused with vehicle (p = 0.014). Elapsed time to accumulate 30 s of exploration did not differ among the groups (F(2,30) = 1.26, p > 0.05; vehicle = 721.4 ± 95.24; 2 ng G-1 = 807.5 ± 87.59; 4 ng G-1 = 613.1 ± 75.98). Mice also received bilateral DH infusion of vehicle, 2 ng G-1, or 4 ng G-1 immediately after OP training. Twenty-four hours later, mice infused with vehicle or 2 ng G-1 did not exhibit a preference for the moved object. However, as in OR, mice receiving 4 ng/hemisphere of G-1 spent significantly more time than chance with the moved object (t(9) = 3.81, p = 0.004; Fig. 2B), demonstrating enhanced spatial memory. One-way ANOVA revealed a significant main effect of treatment (F(2,27) = 4.08, p = 0.028), driven by the fact that the 4 ng G-1 group spent significantly more time with the moved object than the vehicle group (p = 0.009). Elapsed time to accumulate 30 s of exploration did not differ among the groups (F(2,27) = 1.62, p > 0.05; vehicle = 419.6 ± 47.8; 2 ng G-1 = 506.0 ± 72.76; 4 ng G-1 = 371.1 ± 33.61).
Figure 2.

GPER activation enhances OR and OP memory. A, Mice receiving DH infusion of 4 ng/hemisphere G-1 (but not vehicle or 2 ng/hemisphere G-1) spent more time than chance (dashed line at 15 s) with the novel object 48 h after training. This group also spent more time with the novel object than vehicle, indicating enhanced memory for the familiar object (vehicle, n = 11; 2 ng G-1, n = 11; 4 ng G-1, n = 11). B, Similarly, mice infused with 4 ng G-1, but not vehicle or 2 ng G-1, spent significantly more time with the moved object than the vehicle group or than chance 24 h after OP training, indicating enhanced spatial memory (vehicle, n = 11; 2 ng G-1, n = 9; 4 ng G-1, n = 10). C, Mice receiving 7.4 ng G-15 exhibited impaired OR memory relative to vehicle and chance 24 h after DH infusion, whereas mice receiving vehicle, 1.85 ng G-15, or 3.7 ng G-15 did not (vehicle, n = 13; 1.85 ng G-15, n = 10; 3.7 ng G-15, n = 11; 7.4 ng G-15, n = 10). D, In OP, 7.4 ng G-15 impaired spatial memory relative to vehicle and chance 4 h after DH infusion, but no other dose of G-15 affected memory (vehicle, n = 16; 1.85 ng G-15, n = 13; 3.7 ng G-15, n = 13; 7.4 ng G-15, n = 14). E, Intracerebroventricular infusion of 8 ng G-1 significantly enhanced 48 h OR relative to vehicle (Veh) and chance, and DH infusion of 1.85 ng G-15 abolished this effect (Veh+Veh, n = 12; G-1+Veh, n = 11; G-1+G-15, n = 10). F, Similarly, G-15 prevented G-1 from enhancing OP relative to vehicle and chance (Veh+Veh, n = 15; G-1+Veh, n = 13; G-1+G-15, n = 12). Each bar represents the mean ± SEM time spent with the novel or moved object (*p < 0.05, **p < 0.01,***p < 0.001 relative to chance; #p < 0.05, ##p < 0.01 relative to vehicle). n.s., Non-significant.
Because these data suggested that activation of GPER facilitates hippocampal memory, we next examined effects of GPER antagonism on memory. Immediately after OR or OP training, mice received bilateral DH infusion of vehicle (2% DMSO) or one of three doses of G-15 (1.85, 3.7, or 7.4 ng/hemisphere). Memory was tested 24 h later for OR and 4 h later for OP because vehicle-infused ovariectomized female mice remember the familiar and moved objects at these delays (Boulware et al., 2013). Mice receiving vehicle (t(12) = 5.28, p = 0.0002), 1.85 ng G-15 (t(9) = 4.46, p = 0.002), or 3.7 ng G-15 (t(10) = 2.44, p = 0.035) spent significantly more time than chance (15 s) with the novel object 24 h after OR training (Fig. 2C), suggesting intact OR memory after treatment with a low dose of G-15. In contrast, mice receiving 7.4 ng G-15 did not prefer the novel object (t(9) = 0.006, p = 0.996; Fig. 1C), suggesting that this dose impaired OR memory. This conclusion was supported by a significant main effect of treatment (F(3,40) = 3.13, p = 0.036) and post hoc tests showing that mice infused with 7.4 ng G-15 spent significantly less time with the novel object than mice infused with vehicle (p = 0.006) or 1.85 ng G-15 (p = 0.04). No other groups differed from each other. The main effect of treatment was not significant for elapsed time to accumulate 30 s of exploration (F(3,40) = 2.35, p > 0.05; vehicle = 456.4 ± 43.56; 1.85 ng G-15 = 645.1 ± 62.11; 3.7 ng G-15 = 568.5 ± 57.09; 7.4 ng G-15 = 520.7 ± 45.84). In OP, mice receiving DH infusion of vehicle (t(15) = 3.72, p = 0.002), 1.85 ng G-15 (t(12) = 7.08, p < 0.0001), or 3.7 ng G-15 (t(12) = 2.84, p = 0.015) spent significantly more time than chance with the moved object, whereas mice infused with 7.4 ng G-15 did not (t(9) = 0.2, p = 0.84; Fig. 2D). The main effect of treatment was significant (F(3,52) = 7.61, p = 0.0003), due in part to the fact that the 7.4 ng G-15 group spent significantly less time with the moved object than the vehicle (p = 0.009) and 1.85 ng G-1 (p < 0.0001) groups. These data suggest that 7.4 ng G-15 impaired OP, as was seen with OR. The 1.85 ng G-15 group spent significantly more time with the moved object than the vehicle (p = 0.039) or 3.7 ng G-15 (p = 0.003) groups. Elapsed time to accumulate 30 s of exploration did not differ among the groups (F(3,52) = 1.1, p > 0.05; vehicle = 539.4 ± 41.76; 1.85 ng G-15 = 605.7 ± 57.83; 3.7 ng G-15 = 663.2 ± 56.18; 7.4 ng G-15 = 566.9 ± 54.04).
Finally, to confirm that G-15 antagonizes the effects of G-1, we examined whether G-15 could block G-1-induced memory-enhancement. To this end, we infused G-1 into the dorsal third ventricle and G-15 bilaterally into the DH. A dose of 8 ng G-1 was infused into the dorsal third ventricle because bilateral DH infusion of 4 ng/hemisphere G-1 enhanced memory in both tasks (Fig. 2A,B). A dose of 1.85 ng/ hemisphere G-15 was used because this dose had no detrimental effects on memory in both tasks and the 3.7 ng G-15 group did not differ from the 7.4 ng G-15 group in either task (Fig. 2C,D). Immediately after training in each task, mice received a DH infusion of vehicle (2% DMSO) or G-15 (1.85 ng/hemisphere) followed immediately by an intracerebroventricular infusion of vehicle (16% DMSO) or G-1 (8 ng). OR and OP retention were tested 48 and 24 h later, respectively. In both tasks, G-15 blocked the memory enhancing effects of G-1 (Fig. 2E,F). Only mice receiving G-1 + vehicle showed a significant preference for the novel object (t(10) = 5.17, p = 0.0004; Fig. 2E) and moved object (t(12) = 3.38, p = 0.005; Fig. 2F). The memory-enhancing effects of G-1 were also reflected in significant main effects of treatment for OR (F(2,30) = 5.57, p = 0.009) and OP (F(2,37) = 3.54, p = 0.039). Post hoc tests revealed that the G-1+Veh group spent significantly more time with the novel object (p = 0.026) and moved object (p = 0.019) than the vehicle group. In contrast, DH infusion of G-15 abolished the memory-enhancing effects of G-1, as illustrated by the fact that the G-1+G-15 group significantly differed from the G-1+Veh group, but not the Veh+Veh group, in OR (p = 0.003) and OP (p = 0.04). Elapsed time to accumulate 30 s of exploration did not differ among the groups for either OR (F(2,30) = 0.9, p > 0.05; Veh+Veh = 456.4 ± 31.49; G-1+Veh = 539.3 ± 65.77; G-1+G-15 = 448.4 ± 59.14) or OP (F(2,37) = 1.55, p > 0.05; Veh+Veh = 657.9 ± 80.64; G-1+Veh = 541.1 ± 63.0; G-1+G-15 = 487.6 ± 61.42). Collectively, these results demonstrate that GPER activation is necessary for G-1 to enhance hippocampal memory in female mice, and suggest that GPER regulates both OR and spatial memory.
G-1 does not activate ERK or PI3K/Akt signaling in the DH
We have shown previously that the enhanced OR and spatial memory induced by DH infusion of E2 or intracellular ER agonists requires phosphorylation of p42 ERK and PI3K/Akt in the DH (Fernandez et al., 2008; Fan et al., 2010; Boulware et al., 2013; Fortress et al., 2013). To determine whether GPER also enhances memory by activating these cell-signaling pathways, we first measured the effects of GPER activation on ERK phosphorylation. Mice received bilateral DH infusion of 4 ng G-1 and the DH was dissected bilaterally 5, 15, or 30 min later. In contrast to E2 (Fernandez et al., 2008; Boulware et al., 2013), G-1 infusion did not significantly increase levels of phospho-p42 ERK (F(3,16) = 0.72, p > 0.05) at any time point examined (Fig. 3A). G-1 also did not affect levels of phospho-p44 ERK (F(3,16) = 3.07, p < 0.05; Fig. 3A). We next examined activation of the PI3K/Akt signaling pathway. G-1 did not affect levels of phospho-PI3K (F(3,16) = 0.68; p > 0.05; Fig. 3B), but did have a significant effect on phospho-Akt (F(3,16) = 3.94, p < 0.05; Fig. 3C) such that levels of phospho-Akt were significantly decreased relative to vehicle 30 min after infusion (p < 0.05; Fig. 3C). These data are not consistent with the increase in PI3K and Akt phosphorylation we have observed 5 min after DH infusion of E2 (Fan et al., 2010). Collectively, these data indicate that GPER activation does not activate ERK or PI3K/Akt signaling in the DH, and suggest that the effects of GPER activation on DH cell signaling differ from those of E2 or classical ER agonists.
Figure 3.

GPER does not activate the ERK or PI3K/Akt signaling pathways. A, G-1 (4 ng/hemisphere) infusion did not increase DH p42 and p44 ERK phosphorylation relative to vehicle 5, 15, or 30 min after DH infusion (n = 5/group). B, G-1 infusion significantly reduced Akt phosphorylation levels relative to vehicle in the DH 30 min after infusion (n = 5/group). C, G-1 infusion did not alter PI3K phosphorylation relative to vehicle 5, 15, or 30 min after DH infusion (n = 5/group). Each bar represents the mean ± SEM percentage change from vehicle controls (*p < 0.05). Insets, Representative Western blots.
G-1 rapidly activates JNK signaling in the DH
We next investigated whether GPER activation could phosphorylate JNK in the DH. As a seven transmembrane domain receptor, GPER is comprised of heterotrimeric G-protein subunits Gαβγ (Filardo and Thomas, 2005), and the Gβγ-subunit plays a role in activating protein kinase cascades, such as ERK and JNK (Luttrell et al., 1999; Filardo and Thomas, 2005; Goldsmith and Dhanasekaran, 2007). Moreover, JNK is known to play an important role in synaptic plasticity, neuronal regeneration, and brain development (Tararuk et al., 2006; Waetzig et al., 2006). Therefore, we hypothesized that GPER might phosphorylate one or both of the two JNK isoforms (p46 and p54). Mice were bilaterally infused into the DH with vehicle or 4 ng G-1 and phosphorylation of the JNK isoforms was measured 5, 15, and 30 min later. G-1 significantly altered the phosphorylation of both the p46 (F(3,16) = 13.46, p < 0.0001; Fig. 4A) and p54 (F(3,16) = 6.34, p < 0.005; Fig. 4B) isoforms of JNK, such that phospho-protein levels were significantly higher than vehicle 5 min after infusion (p < 0.05). These effects were transient, as levels of both phosphorylated isoforms returned to baseline 15 min after infusion. We next examined phosphorylation of the downstream JNK transcription factor, activating transcription factor 2 (ATF2; Antoniou and Borsello, 2012). G-1 infusion also significantly altered levels of phospho-ATF2 (F(3,20) = 3.3, p < 0.05; Fig. 4C). Similar to both JNK isoforms, phospho-ATF2 levels were significantly increased relative to vehicle 5 min after DH infusion (p < 0.05), but not 15 or 30 min later.
Figure 4.

GPER activation increases JNK phosphorylation in the DH. A, B, DH infusion of G-1 (4 ng/hemisphere) significantly increased phosphorylation of the JNK p46 isoform (A) and p54 isoform (B) relative to vehicle within 5 min. Levels returned to baseline 15 min later (n = 5/group). C, G-1 infusion significantly increased phosphorylation of the downstream JNK transcription factor ATF2 relative to vehicle in the DH 5 min after infusion (n = 6/group). D, E, Intracerebroventricular infusion of 8 ng G-1 significantly increased levels of phosphorylated p46 JNK (D) and p54 JNK (E) relative to vehicle 5 min after infusion (n = 6/group). These effects were blocked by DH infusion of G-15, indicating that GPER activation is necessary for G-1 to activate JNK signaling (n = 6/group). F, Neither G-1 nor G-15 altered phosphorylation of either ERK isoform (n = 6/group). Each bar represents the mean ± SEM percentage change from vehicle (*p < 0.05, **p < 0.01, ***p < 0.001). Insets, Representative Western blots.
To confirm that the G-1-mediated JNK activation observed occurred via GPER activation, we next examined whether G-15 could block the effects of G-1 on JNK activation. Mice received DH infusion of vehicle or G-15 plus intracerebroventricular infusion of vehicle or G-1; DH tissue was collected 5 min later. Consistent with the effects of DH G-1 infusion (Fig. 4A,B), intracerebroventricular infusion of G-1 increased phosphorylation of both the p46 (F(2,15) = 4.96, p < 0.05; Fig. 4D) and p54 (F(2,15) = 7.89, p < 0.005; Fig. 4E) isoforms of JNK 5 min after infusion (p < 0.05 relative to vehicle). Infusion of G-15 into the DH completely blocked these effects (Fig. 4D,E), suggesting that GPER activation induces JNK phosphorylation in the DH. In contrast, neither G-1 alone nor G-1+G-15 significantly altered ERK phosphorylation (p42, F(2,15) = 0.58, p > 0.05; p44, F(2,15) = 0.65, p > 0.05; Fig. 4F), consistent with the lack of effect of DH G-1 infusion on ERK (Fig. 3A).
Activation of JNK is necessary for GPER to regulate hippocampal memory
Given the rapid activation of JNK by G-1, we next examined whether this activation is necessary for G-1 to enhance memory. To do so, we used the JNK activation inhibitor, SP600125. We first needed to determine a dose of SP600125 that did not block memory on its own to ensure that any effects of this drug resulted from an interaction with G-1 rather than a general impairing effect on memory. Therefore, we infused mice with vehicle (2% DMSO) or one of two doses of SP600125 (0.55 or 2.75 ng/hemisphere) immediately after OR or OP training. Mice receiving vehicle (t(6) = 3.27, p = 0.02) or either dose of SP600125 (0.55 ng, t(5) = 2.7, p = 0.043; 2.75 ng, t(7) = 3.46, p = 0.01) spent significantly more time than chance with the novel object 24 h after OR training (Fig. 5A), suggesting that neither dose of SP600125 impaired OR memory. Similarly, mice infused with vehicle (t(8) = 3.87, p = 0.005) or either dose of SP600125 (0.55 ng, t(9) = 3.45, p = 0.007; 2.75 ng, t(7) = 3.7, p = 0.008) spent significantly more time than chance with the moved object 4 h after OP training (Fig. 5B), indicating that neither dose impaired OP memory. The lack of difference among the groups in both tasks was confirmed by one-way ANOVAs in which the main effects of treatment were not significant for OR (F(2,18) = 0.998, p > 0.05) or OP (F(2,24) = 0.65, p > 0.05). Elapsed time to accumulate 30 s of object exploration did not differ among the groups for either OR (F(2,37) = 1.5, p > 0.05; vehicle = 916.6 ± 67.6; 0.55 ng SP600125 = 823.5 ± 147.7; 2.75 ng SP600125 = 701.7 ± 69.8) or OP (F(2,24) = 0.92, p > 0.05; vehicle = 557.3 ± 104.8; 0.55 ng SP600125 = 424.1 ± 51.7; 2.75 ng SP600125 = 424.1 ± 79.7). Because neither dose affected memory on its own, we selected the highest behaviorally ineffective dose of SP600125 (2.75 ng/hemisphere) for our remaining studies.
Figure 5.

JNK inhibition, but not ERK inhibition, blocks GPER-mediated memory enhancement. A, Twenty-four hours after training, mice receiving DH infusion of vehicle or either dose of SP600125 spent more time with the novel object than the vehicle group or than chance, suggesting that neither dose of SP600125 impaired OR memory (vehicle, n = 7; 0.55 ng SP600125, n = 6; 2.75 ng SP600125, n = 8). B, Similarly, neither dose of SP600125 impaired OP memory, as indicated by the fact that all groups spent more time with the moved object than chance and that neither SP600125 group differed from vehicle (vehicle, n = 9; 0.55 ng SP600125, n = 10; 2.75 ng SP600125, n = 8). C, Immediately after OR training, mice received DH infusion of vehicle, SP600125 (1.85 ng/hemisphere), or U0126 (0.5 μg/hemisphere) followed by intracerebroventricular infusion of vehicle or G-1 (8 ng). Intracerebroventricular infusion of G-1 significantly enhanced OR memory relative to vehicle and chance. SP600125 infusion blocked this effect, but U0126 did not (vehicle, n = 11; G-1+Veh, n = 8; G-1+SP, n = 9; G-1+U0126, n = 11). D, Immediately after OP training, mice received DH and intracerebroventricular infusions as described in C. As with OR, G-1 enhanced OP memory relative to vehicle and chance, an effect that was blocked by SP600125 but not U0126 (Veh+Veh, n = 14; G-1+Veh, n = 13; G-1+SP, n = 13; G-1+U0126, n = 16). Each bar represents the mean ± SEM time spent with the novel or moved object (*p < 0.05, **p < 0.01, ***p < 0.001 relative to chance; #p < 0.05, ##p < 0.01 relative to vehicle). n.s., Non-significant.
To test whether activation of JNK or ERK was necessary for G-1 to enhance memory, we next infused mice with G-1 plus 2.75 ng SP600125 or the ERK inhibitor U0126 at a dose (0.5 μg/hemisphere) that has no effect on OR or OP on its own (Fernandez et al., 2008; Boulware et al., 2013). Mice received DH infusion of vehicle (25% DMSO), 0.5 μg/hemisphere U0126, or 2.75 ng/hemisphere SP600125 plus intracerebroventricular infusion of vehicle (16% DMSO) or 8 ng G-1 immediately after OR and OP training. Memory in OR and OP was tested 48 and 24 h later, respectively. In both tasks, SP600125, but not U0126, blocked the memory-enhancing effects of G-1 (Fig. 5C,D). Mice receiving G-1+Veh showed a significant preference for the novel object (t(7) = 2.68, p = 0.032) or moved object (t(12) = 3.55, p = 0.004), whereas mice receiving Veh+Veh (novel object, t(8) = 0.6, p = 0.56; moved object, t(12) = 0.8, p = 0.44) or G-1+SP600125 did not (novel object, t(8) = 1.16, p = 0.28; moved object, t(12) = 0.3, p = 0.77), suggesting that JNK activation is necessary for G-1 to enhance memory. In contrast to the effects of SP600125, mice infused with G-1+U0126 spent significantly more time than chance with the novel object (t(10) = 3.44, p = 0.006) or moved object (t(15) = 3.81, p = 0.002), suggesting that ERK activation is not necessary for G-1 to enhance memory. These findings were supported by significant main effects of treatment for both tasks (OR, F(3,35) = 4.79, p = 0.007; OP, F(3,52) = 4.17, p = 0.01) and post hoc analyses showing that the G-1+Veh and G-1+U0126 groups spent significantly more time with the novel object (G-1+Veh, p = 0.011; G-1+U0126, p = 0.019) and moved object (G-1+Veh, p = 0.002; G-1+U0126, p = 0.015) than the Veh+Veh group, whereas the G-1+SP600125 group did not. Elapsed time to accumulate 30 s of exploration did not differ among the groups for OR (F(3,35) = 2.32, p > 0.05; Veh+Veh = 499.8 ± 62.86; G-1+Veh = 512.3 ± 89.87; G-1+SP600125 = 736.9 ± 71.12; G-1+U0126 = 533.8 ± 66.59) or OP (F(3,52) = 1.57, p > 0.05; Veh+Veh = 604.6 ± 78.71; G-1+Veh = 499.5 ± 56.65; G-1+SP600125 = 514.1 ± 51.54; G-1+U0126 = 661.8 ± 56.84).
We next examined the effects of JNK and ERK inhibition on G-1-mediated hippocampal cell signaling 5 min after infusion. Drug treatment altered phosphorylation of both p46 JNK (F(2,19) = 6.56, p < 0.01; Fig. 6A) and p54 JNK (F(2,19) = 6.47, p < 0.01; Fig. 6B). Consistent with the behavioral data, intracerebroventricular infusion of G-1 increased phosphorylation of both p46 JNK and p54 JNK relative to vehicle (p < 0.05; Fig. 6A,B). DH infusion of SP600125 abolished the effects of G-1 on p46 and p54 JNK (Fig. 6A,B). In contrast, G-1 and SP600125 did not significantly alter ERK phosphorylation (p42, F(2,18) = 0.02, p > 0.05; p44, F(2,18) = 0.46, p > 0.05; Fig. 6C). Unlike SP600125, U0126 did not block the GPER-mediated JNK activation 5 min after infusion (Fig. 6D,E). Whereas G-1 increased phosphorylation of both p46 JNK (F(2,15) = 4.44, p < 0.05; Fig. 6D) or p54 JNK (F(2,15) = 6.68, p < 0.01; Fig. 6E), U0126 did not block the effects of G-1 on p46 JNK (t(10) = 2.35, p < 0.05; Fig. 6D) and p54 JNK (t(10) = 2.34, p < 0.05; Fig. 6E). Moreover, neither G-1 nor U0126 infusion altered ERK activation (p42, F(2,15) = 0.67, p > 0.05; p44, F(2,15) = 0.81, p > 0.05; Fig. 6F). These data suggest that ERK activation does not influence G-1-induced hippocampal JNK activation. Together, these results support the conclusion that activation of JNK, but not ERK, signaling in the DH is essential for GPER to induce memory enhancement.
Figure 6.

JNK inhibition, but not ERK inhibition, blocks GPER-mediated cell signaling in the DH. A, B, Intracerebroventricular infusion of 8 ng G-1 increased phosphorylation of p46 JNK (A) and p54 JNK (B) relative to vehicle 5 min later. These effects were blocked by DH SP600125 infusion (n = 7/group). C, Neither G-1 nor SP600125 altered ERK phosphorylation (n = 7/group). D, E, The increase in p46 (D) and p54 (E) phosphorylation induced by intracerebroventricular infusion of 8 ng G-1 was not blocked by DH U0126 infusion (n = 6/group). F, Neither G-1 nor the behaviorally subeffective dose of U0126 altered ERK phosphorylation (n = 6/group). Each bar represents the mean ± SEM percentage change from vehicle (*p < 0.05, **p < 0.01). Insets, Representative Western blots.
GPER and JNK activation are not necessary for E2-mediated hippocampal memory
We have previously demonstrated that E2 enhances hippocampal memory via ERα- or ERβ-mediated ERK activation in the DH (Fernandez et al., 2008; Boulware et al., 2013). In contrast, the aforementioned data support the hypothesis that the G-1-induced enhancement of hippocampal memory is dependent on hippocampal JNK activation, rather than ERK activation. This conclusion begs the question of whether JNK or GPER activation is necessary for E2-induced memory enhancement. To address this issue, we first examined the effects of E2 on JNK signaling in the DH. Mice received bilateral DH infusion of vehicle or 5 μg/hemisphere E2, a dose that enhances OR and spatial memory in ovariectomized young and middle-aged mice (Fernandez et al., 2008; Fan et al., 2010; Boulware et al., 2013; Fortress et al., 2013, 2014). The DH was dissected bilaterally 5 or 10 min after infusion. DH E2 infusion did not alter DH p46 JNK (F(2,15) = 0.35, p > 0.05) or p54 JNK (F(2,15) = 1.44, p > 0.05) phosphorylation at either the 5 or 10 min time point (Fig. 7A,B), suggesting that E2 does not activate JNK in the DH. As in our previous studies (Fernandez et al., 2008; Zhao et al., 2010; Boulware et al., 2013; Fortress et al., 2013), DH E2 infusion increased phospho-p42 ERK (F(2,15) = 4.7, p < 0.05; Fig. 7C) levels 5 min after infusion (p < 0.05 relative to vehicle) but had no effect on p44 ERK (F(2,15) = 0.05, p > 0.05; Fig. 7C). These data suggest that E2 increases activation of p42 ERK, but not JNK, in the DH.
Figure 7.

GPER and JNK inhibition do not affect E2-mediated cell signaling in the DH. A, B, DH infusion of E2 (5 μg/hemisphere) did not alter levels of phospho-p46 JNK (A) or phospho-p54 JNK (B) 5 or 10 min later (n = 6/group). C, DH infusion of E2 (5 μg/hemisphere) significantly increased phosphorylation of p42 ERK, but not p44 ERK, relative to vehicle 5 min after infusion. Levels returned to baseline 10 min later (n = 6/group). D, Intracerebroventricular infusion of E2 (10 μg) increased phospho-p42 ERK levels relative to vehicle 5 min after infusion; this effect was not blocked by DH infusion of G-15 or SP600125. Intracerebroventricular infusion of E2 (10 μg) did not alter p44 ERK phosphorylation (n = 6/group). E, F, Intracerebroventricular infusion of E2 did not alter p46 JNK (E) or p54 JNK (F) phosphorylation 5 min after infusion whether infused with vehicle, G-15, or SP600125 (n = 6/group). Each bar represents the mean ± SEM percentage change from vehicle (*p < 0.05, **p < 0.01, ***p < 0.001). Insets, Representative Western blots.
Next, we investigated the effects of GPER and JNK inhibition on E2-mediated hippocampal cell signaling. Mice received intracerebroventricular and DH infusions, respectively, of vehicle + vehicle, E2+Veh, E2+SP600125, or E2+G-15, and DH tissue was collected 5 min later. As in our previous work (Fernandez et al., 2008; Zhao et al., 2010; Boulware et al., 2013; Fortress et al., 2013), intracerebroventricular infusion of E2 increased levels of phospho-p42 ERK (F(3,20) = 7.6, p < 0.01; Fig. 7D), but not phospho-p44 ERK (F(3,20) = 0.7, p > 0.05; Fig. 7D). Phospho-p42 ERK levels were increased relative to vehicle in all groups receiving E2 (p < 0.05), suggesting that DH infusion of G-15 or SP600125 did not prevent E2 from increasing p42 ERK activation (G-15, p < 0.001; SP600125, p < 0.05; Fig. 7D). As with DH infusion, intracerebroventricular infusion of E2 did not alter phosphorylation of p46 JNK (F(3,20) = 0.74, p > 0.05; Fig. 7E) or p54 JNK (F(3,20) = 0.96, p > 0.05; Fig. 7F), whether alone or in combination with DH infusion of G-15 and SP600125. Together, these data provide additional evidence that E2 does not rapidly phosphorylate JNK in the DH and demonstrate that activation of JNK or GPER is not necessary for E2 to phosphorylate p42 ERK in the DH.
Given these findings, the next logical step was to determine whether JNK and GPER activation play a role in E2-mediated hippocampal memory enhancement. To do so, we infused mice immediately after OR and OP training with vehicle, G-15 (1.85 ng/hemisphere), or SP600125 (2.75 ng/hemisphere) into the DH followed by infusion of vehicle or E2 (10 μg) into the dorsal third ventricle. OR and OP retention were tested 48 and 24 h later, respectively. In both tasks, mice receiving E2+Veh showed a significant preference for the novel object (t(10) = 4.12, p = 0.002; Fig. 8A) and moved object (t(16) = 3.87, p = 0.001; Fig. 8B), in agreement with our previous work (Boulware et al., 2013; Fortress et al., 2014). Consistent with the lack of JNK activation observed above (Fig. 7E,F), SP600125 did not prevent E2 from enhancing OR or OP memory (Fig. 8A,B), as mice receiving E2+SP600125 spent significantly more time with the novel object (t(14) = 3.31, p = 0.005) and moved object (t(13) = 3.89, p = 0.002) than chance. Interestingly, G-15 also did not block E2-induced memory enhancements in either task (Fig. 8A,B), as demonstrated by the fact that mice receiving E2+G-15 spent significantly more time with the novel object (t(11) = 2.62, p = 0.02) and moved object (t(11) = 5.6, p = 0.0002) than chance. Accordingly, the main effects of treatment were significant for both tasks (OR, F(3,50) = 3.51, p = 0.02; OP, F(3,54) = 4.88, p = 0.005). Post hoc tests revealed that all E2-treated groups differed significantly from the vehicle group for both tasks (all p values < 0.001). However, the E2-treated groups did not differ from each other. Elapsed time to accumulate 30 s of exploration did not differ among the groups for OR (F(3,49) = 1.17, p > 0.05; Veh+Veh = 540.3 ± 64.04; E2+Veh = 639.4 ± 73.66; E2+G-15 = 468.2 ± 55.25; E2+SP600125 = 577.9 ± 56.08) or OP (F(3,54) = 0.942, p > 0.05; Veh+Veh = 557.7 ± 44.25; E2+Veh = 661.6 ± 64.37; E2+G-15 = 672.4 ± 38.56; E2+SP600125 = 606.3 ± 57.78). Together, these results suggest that neither JNK nor GPER activation in the DH are necessary for E2 to enhance hippocampal memory.
Figure 8.
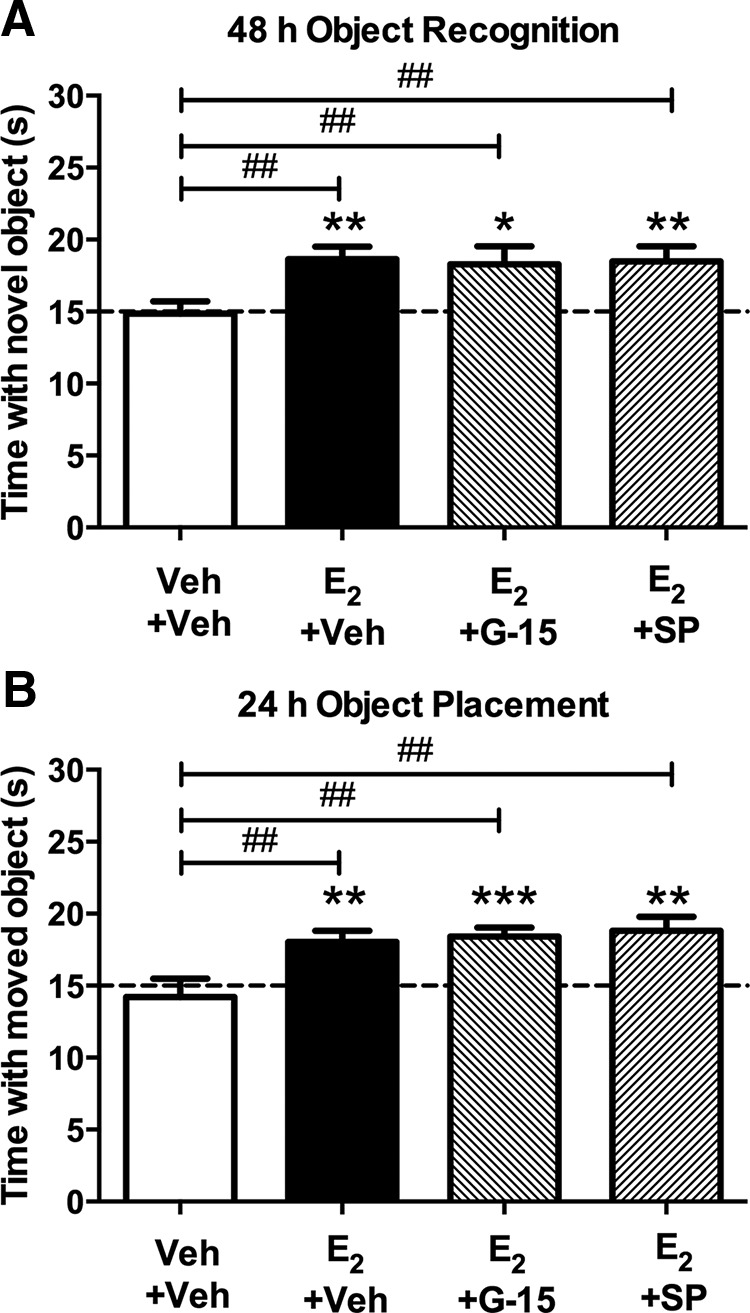
GPER and JNK activation in the DH are not necessary for E2 to enhance memory. A, Immediately after OR training, mice received DH infusion of vehicle, G-15 (1.85 ng/hemisphere), or SP600125 (2.75 ng/hemisphere) followed by intracerebroventricular infusion of vehicle or E2 (10 μg). Intracerebroventricular infusion of E2 significantly enhanced OR memory relative to vehicle and chance, and these effects were not blocked by G-15 or SP600125 (Veh+Veh, n = 16; E2+Veh, n = 11; E2+G-15, n = 12; E2+SP600125, n = 15). B, Immediately after OP training, mice received DH and intracerebroventricular infusions as described in A. Similar to OR, E2 enhanced OP memory relative to vehicle and chance, an effect that was not blocked by G-15 or SP600125 (Veh+Veh, n = 15; E2+Veh, n = 17; E2+G-15, n = 12; E2+SP600125, n = 14). Each bar represents the mean ± SEM time spent with the novel or moved object (*p < 0.05, **p < 0.01, ***p < 0.001 relative to chance; ##p < 0.01 relative to vehicle).
Discussion
The present study demonstrates that activation of GPER in the DH enhances both OR and spatial memory in ovariectomized mice, suggesting that hippocampal GPER can influence hippocampal memory formation. Moreover, the findings provide the first evidence that GPER, a putative membrane-associated ER, regulates hippocampal memory in an E2-independent manner. This conclusion is supported by several findings. First, unlike E2 and intracellular ER agonists (Boulware et al., 2013), DH GPER activation did not increase ERK phosphorylation in the DH, but rather increased JNK phosphorylation. Second, the memory-enhancing effects of GPER activation were blocked by inhibition of JNK, but not ERK, in the DH. Third, E2 infusion increased ERK, but not JNK, phosphorylation in the DH. Finally, the memory-enhancing effects of E2 were blocked by inhibition of ERK, but not of JNK or GPER. Together with our previous work (Fernandez et al., 2008; Boulware et al., 2013; Fortress et al., 2013), these data indicate that E2 enhances hippocampal memory in female mice by activating ERK, whereas GPER does so by activating JNK. As such, the data suggest that GPER activation in the DH is not involved in the memory-enhancing effects of E2.
Our findings showing that G-1 enhanced OR and OP memory are consistent with previous studies demonstrating that systemic injections of G-1 enhanced spatial memory in ovariectomized rats (Hammond et al., 2009; Hammond and Gibbs, 2011; Hawley et al., 2014). These studies did not permit conclusions about the role of hippocampal GPER in memory because systemic treatments do not specifically target the hippocampus. Therefore, we used DH infusions of G-1 to pinpoint the role of hippocampal GPER in regulating memory. To ensure that the effects of G-1 were specific to GPER, we tested whether G-15 could antagonize the effects of G-1, as some studies have indicated that G-1 can act in a GPER-independent manner (Kang et al., 2010; Wang et al., 2012). In contrast to those studies, we found that G-15 infusion into the DH prevented G-1 from enhancing OR and OP memory, and from increasing JNK phosphorylation. These data suggest that the effects of G-1 on memory and JNK activation are mediated by GPER in the DH. Interestingly, higher doses of G-15 impaired both OR and OP memory on their own. This finding is consistent with previous data showing that chronic systemic treatment with G-15 dose-dependently impaired spatial working memory in ovariectomized rats (Hammond et al., 2012). Together, these data suggest that GPER in the DH can mediate hippocampal memory formation.
Because the molecular mechanisms through which GPER influences hippocampal memory have not been investigated previously, a primary goal was to pinpoint possible downstream effectors of GPER activation in the DH. Based on our previous findings showing that p42 ERK activation is necessary for E2 and agonists of ERα and ERβ to enhance OR and OP memory (Fernandez et al., 2008; Boulware et al., 2013), we initially hypothesized that p42 ERK phosphorylation would be necessary for G-1 to enhance memory. This hypothesis was supported by data showing that systemic administration of G-1 increased p42 and p44 ERK phosphorylation in ovariectomized female mouse hippocampus (Hart et al., 2014) and bath-applied G-15 blocked an E2-induced increase in ERK phosphorylation and excitatory synaptic transmission (Kumar et al., 2015). Moreover, GPER can activate ERK signaling in pancreatic β cells and ERK cannot be activated in islets of GPER knock-out mice (Maggiolini and Picard, 2010; Sharma and Prossnitz, 2011). In contrast to these data, we found that G-1 did not affect p42 or p44 ERK phosphorylation in the DH at any time point. Although contradictory to the aforementioned studies, this result is consistent with data from vascular smooth muscle cells showing that E2, but not G-1, increases ERK phosphorylation (Ortmann et al., 2011). To further explore effects of G-1 on ERK signaling, we measured effects of G-1 on PI3K and Akt phosphorylation, based on our previous findings that PI3K/Akt signaling is necessary for E2 to activate DH ERK and enhance OR memory (Fan et al., 2010; Fortress et al., 2013). Additionally, GPER can regulate Akt signaling in numerous cell lines (Moriarty et al., 2006; Maggiolini and Picard, 2010) and in rat aorta tissue (Jang et al., 2013). However, as with ERK, we found that DH infusion of G-1 did not increase PI3K or Akt phosphorylation in the DH at any time point. Rather, Akt phosphorylation was decreased 30 min after infusion, the reason for which is unclear. Nevertheless, the fact that G-1 did not increase PI3K or Akt phosphorylation in the DH as was observed after E2 infusion (Fan et al., 2010; Fortress et al., 2013) indicates that intracranial administration of G-1 in vivo does not activate multiple ERK-related signaling kinases in the female mouse DH.
Consistent with these biochemical data, we found that ERK inhibition did not prevent G-1 from enhancing OR or OP memory. These results demonstrate that ERK activation is not necessary for GPER to enhance hippocampal memory in female mice. Although this finding is novel as it relates to memory, it is consistent with reports from peripheral tissues showing that the ERK inhibitors U0126 and PD98059 do not prevent G-1 from inducing endothelium-dependent vasorelaxation in the rat aorta (Jang et al., 2013) or DNA synthesis in human epithelial cells (Holm et al., 2011). Although these few examples do not permit general conclusions about the role of ERK in mediating the cellular effects of GPER activation, the present data support the conclusion that ERK is not involved GPER-mediated memory regulation.
Given the unexpected lack of a role for ERK in GPER-induced memory enhancement, we sought to identify other signaling pathways through which GPER may mediate memory. We focused on JNK because this MAPK is activated by various G-proteins (Goldsmith and Dhanasekaran, 2007) and is involved in regulating synaptic plasticity (Tararuk et al., 2006; Waetzig et al., 2006; Kim et al., 2007). We found that GPER activation led to rapid phosphorylation of both JNK isoforms in the DH, an effect that was blocked by DH infusion of the JNK inhibitor SP600125, but not U0126. G-1 also increased phosphorylation of the downstream JNK transcription factor ATF2, suggesting that the G-1-induced phosphorylation of JNK also activated nuclear transcription. Importantly, we found that activation of JNK, but not ERK, in the DH is necessary for GPER to faciliate memory in both the OR and OP tasks.
Although JNK has been studied in the context of cellular stress and apoptosis (Kyriakis and Avruch, 2001; Reinecke et al., 2013), it also plays an important role in synaptic plasticity, neuronal regeneration, and development in the CNS (Tararuk et al., 2006; Waetzig et al., 2006). However, its role in learning and memory remains unclear, as existing data provide conflicting results. For example, some studies suggest an important role of JNK activation in long-term inhibitory avoidance memory and in short-term synaptic plasticity and long-term depression (Bevilaqua et al., 2007; Li et al., 2007; Carboni et al., 2008). However, other data indicate that JNK negatively regulates short-term memory in the hippocampus (Bevilaqua et al., 2003). Although our findings cannot speak directly to the inconsitencies in the JNK literature, our results provide much needed additional information on the role of JNK in hippocampal memory. These data suggest that JNK is an essential mediator of GPER-induced memory modulation.
Evidence that GPER is an estrogen receptor comes from data collected in peripheral tissues showing that E2 binds GPER with high affinity (Revankar et al., 2005; Moriarty et al., 2006; Prossnitz et al., 2007). However, other evidence suggests that GPER acts independently of E2. For example, a study using endothelial cells from ERα/ERβ-deficient mice found that E2 could not activate cAMP or ERK pathways, despite the presence of GPER (Pedram et al., 2006). Moreover, COS-7 and Chinese hamster ovary cells transfected with GPER failed to signal in response to E2 (Otto et al., 2008). Furthermore, rapid extranuclear E2 signaling in breast cancer cells involved ERα and ERβ, but not GPER (Madak-Erdogan et al., 2008). Additionally, the neuroprotective effects of E2 in post-ischemic injury are not dependent on GPER (Lamprecht and Morrison, 2014). Data such as these have led some investigators to maintain that GPER is not a true ER, but rather collaborates with ERs to mediate the biological actions of estrogens (Levin, 2009). Such arguments have stimulated extensive debate about whether GPER functions as a true ER (Langer et al., 2010). The present study adds to the debate by showing that GPER and E2 in the DH do not enhance memory via the same cell-signaling mechanisms. Indeed, these data suggest that GPER activation in the DH is not necessary for E2 to enhance object recognition and spatial memory formation. However, this study cannot exclude other potential interactions between E2 and GPER in the DH and elsewhere in the brain.
Little is known about how specific ERs mediate the effects of E2 on memory. Estrogen loss at menopause has been associated with increased risk of age-related memory decline and dementia (Zandi et al., 2002; Yaffe et al., 2007), yet estrogen therapies carry health risks that preclude their use for alleviating memory dysfunction. Identifying the molecular mechanisms through which estrogens affect memory may reveal new targets for the development of drugs that mimic the memory-enhancing effects of estrogens without harmful side effects. The present study provides the first evidence that GPER activation can enhance hippocampal memory in a JNK-dependent manner, and that E2-mediated object recognition and spatial memory enhancement is independent of GPER and JNK activation in the DH. Although these findings do not support a role for DH GPER in the memory-enhancing effects of E2, the fact that GPER activation enhances hippocampal memory in a manner similar to E2 may suggest promising new avenues for the development of novel therapies that reduce the risk of memory decline and dementia in menopausal women.
Footnotes
This work was supported by the University of Wisconsin-Milwaukee, R01DA038042, a University of Wisconsin-Milwaukee Research Growth Initiative Award to K.M.F., and a UWM Distinguished Graduate Student Fellowship and Department of Psychology Summer Research Fellowship to J.K. We thank Dr Ashley Fortress and Jennifer Tuscher for critical comments on this paper, as well as Maciej Miaskowski for assistance with behavioral testing.
The authors declare no competing financial interests.
References
- Akama KT, Thompson LI, Milner TA, McEwen BS. Post-synaptic density-95 (PSD-95) binding capacity of G-protein-coupled receptor 30 (GPR30), an estrogen receptor that can be identified in hippocampal dendritic spines. J Biol Chem. 2013;288:6438–6450. doi: 10.1074/jbc.M112.412478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almey A, Cannell E, Bertram K, Filardo E, Milner TA, Brake WG. Medial prefrontal cortical estradiol rapidly alters memory system bias in female rats: ultrastructural analysis reveals membrane-associated estrogen receptors as potential mediators. Endocrinology. 2014;155:4422–4432. doi: 10.1210/en.2014-1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoniou X, Borsello T. The JNK signalling transduction pathway in the brain. Front Biosci. 2012;4:2110–2120. doi: 10.2741/528. [DOI] [PubMed] [Google Scholar]
- Baker KB, Kim JJ. Effects of stress and hippocampal NMDA receptor antagonism on recognition memory in rats. Learn Mem. 2002;9:58–65. doi: 10.1101/lm.46102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett BL, Sasaki DT, Murray BW, O'Leary EC, Sakata ST, Xu W, Leisten JC, Motiwala A, Pierce S, Satoh Y, Bhagwat SS, Manning AM, Anderson DW. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci U S A. 2001;98:13681–13686. doi: 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevilaqua LR, Kerr DS, Medina JH, Izquierdo I, Cammarota M. Inhibition of hippocampal Jun N-terminal kinase enhances short-term memory but blocks long-term memory formation and retrieval of an inhibitory avoidance task. Eur J Neurosci. 2003;17:897–902. doi: 10.1046/j.1460-9568.2003.02524.x. [DOI] [PubMed] [Google Scholar]
- Bevilaqua LR, Rossato JI, Clarke JH, Medina JH, Izquierdo I, Cammarota M. Inhibition of c-Jun N-terminal kinase in the CA1 region of the dorsal hippocampus blocks extinction of inhibitory avoidance memory. Behav Pharmacol. 2007;18:483–489. doi: 10.1097/FBP.0b013e3282ee7436. [DOI] [PubMed] [Google Scholar]
- Blasko E, Haskell CA, Leung S, Gualtieri G, Halks-Miller M, Mahmoudi M, Dennis MK, Prossnitz ER, Karpus WJ, Horuk R. Beneficial role of the GPR30 agonist G-1 in an animal model of multiple sclerosis. J Neuroimmunol. 2009;214:67–77. doi: 10.1016/j.jneuroim.2009.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bologa CG, Revankar CM, Young SM, Edwards BS, Arterburn JB, Kiselyov AS, Parker MA, Tkachenko SE, Savchuck NP, Sklar LA, Oprea TI, Prossnitz ER. Virtual and biomolecular screening converge on a selective agonist for GPR30. Nat Chem Biol. 2006;2:207–212. doi: 10.1038/nchembio775. [DOI] [PubMed] [Google Scholar]
- Boulware MI, Heisler JD, Frick KM. The memory-enhancing effects of hippocampal estrogen receptor activation involve metabotropic glutamate receptor signaling. J Neurosci. 2013;33:15184–15194. doi: 10.1523/JNEUROSCI.1716-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brailoiu E, Dun SL, Brailoiu GC, Mizuo K, Sklar LA, Oprea TI, Prossnitz ER, Dun NJ. Distribution and characterization of estrogen receptor G protein-coupled receptor 30 in the rat central nervous system. J Endocrinol. 2007;193:311–321. doi: 10.1677/JOE-07-0017. [DOI] [PubMed] [Google Scholar]
- Carboni S, Boschert U, Gaillard P, Gotteland JP, Gillon JY, Vitte PA. AS601245, a c-Jun NH2-terminal kinase (JNK) inhibitor, reduces axon/dendrite damage and cognitive deficits after global cerebral ischaemia in gerbils. Br J Pharmacol. 2008;153:157–163. doi: 10.1038/sj.bjp.0707574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chimento A, Sirianni R, Casaburi I, Ruggiero C, Maggiolini M, Andò S, Pezzi V. 17β-Estradiol activates GPER- and ESR1-dependent pathways inducing apoptosis in GC-2 cells, a mouse spermatocyte-derived cell line. Mol Cell Endocrinol. 2012;355:49–59. doi: 10.1016/j.mce.2012.01.017. [DOI] [PubMed] [Google Scholar]
- Cohen SJ, Munchow AH, Rios LM, Zhang G, Asgeirsdóttir HN, Stackman RW., Jr The rodent hippocampus is essential for nonspatial object memory. Curr Biol. 2013;23:1685–1690. doi: 10.1016/j.cub.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis MK, Burai R, Ramesh C, Petrie WK, Alcon SN, Nayak TK, Bologa CG, Leitao A, Brailoiu E, Deliu E, Dun NJ, Sklar LA, Hathaway HJ, Arterburn JB, Oprea TI, Prossnitz ER. In vivo effects of a GPR30 antagonist. Nat Chem Biol. 2009;5:421–427. doi: 10.1038/nchembio.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan L, Zhao Z, Orr PT, Chambers CH, Lewis MC, Frick KM. Estradiol-induced object memory consolidation in middle-aged female mice requires dorsal hippocampal extracellular signal-regulated kinase and phosphatidylinositol 3-kinase activation. J Neurosci. 2010;30:4390–4400. doi: 10.1523/JNEUROSCI.4333-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez SM, Lewis MC, Pechenino AS, Harburger LL, Orr PT, Gresack JE, Schafe GE, Frick KM. Estradiol-induced enhancement of object memory consolidation involves hippocampal extracellular signal-regulated kinase activation and membrane-bound estrogen receptors. J Neurosci. 2008;28:8660–8667. doi: 10.1523/JNEUROSCI.1968-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filardo EJ, Thomas P. GPR30: a seven-transmembrane-spanning estrogen receptor that triggers EGF release. Trends Endocrinol Metab. 2005;16:362–367. doi: 10.1016/j.tem.2005.08.005. [DOI] [PubMed] [Google Scholar]
- Fortress AM, Fan L, Orr PT, Zhao Z, Frick KM. Estradiol-induced object recognition memory consolidation is dependent on activation of mTOR signaling in the dorsal hippocampus. Learn Mem. 2013;20:147–155. doi: 10.1101/lm.026732.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortress AM, Kim J, Poole RL, Gould TJ, Frick KM. 17β-Estradiol regulates histone alterations associated with memory consolidation and increases Bdnf promoter acetylation in middle-aged female mice. Learn Mem. 2014;21:457–467. doi: 10.1101/lm.034033.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortress AM, Heisler JD, Frick KM. The mTOR and canonical Wnt signaling pathways mediate the mnemonic effects of progesterone in the dorsal hippocampus. Hippocampus. 2015;25:616–629. doi: 10.1002/hipo.22398. [DOI] [PubMed] [Google Scholar]
- Frye CA, Duffy CK, Walf AA. Estrogens and progestins enhance spatial learning of intact and ovariectomized rats in the object placement task. Neurobiol Learn Mem. 2007;88:208–216. doi: 10.1016/j.nlm.2007.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funakoshi T, Yanai A, Shinoda K, Kawano MM, Mizukami Y. G protein-coupled receptor 30 is an estrogen receptor in the plasma membrane. Biochem Biophys Res Commun. 2006;346:904–910. doi: 10.1016/j.bbrc.2006.05.191. [DOI] [PubMed] [Google Scholar]
- Gervais NJ, Jacob S, Brake WG, Mumby DG. Systemic and intra-rhinal-cortical 17-β estradiol administration modulate object-recognition memory in ovariectomized female rats. Horm Behav. 2013;64:642–652. doi: 10.1016/j.yhbeh.2013.08.010. [DOI] [PubMed] [Google Scholar]
- Goldsmith ZG, Dhanasekaran DN. G protein regulation of MAPK networks. Oncogene. 2007;26:3122–3142. doi: 10.1038/sj.onc.1210407. [DOI] [PubMed] [Google Scholar]
- Gresack JE, Frick KM. Male mice exhibit better spatial working and reference memory than females in a water-escape radial arm maze task. Brain Res. 2003;982:98–107. doi: 10.1016/S0006-8993(03)03000-2. [DOI] [PubMed] [Google Scholar]
- Gresack JE, Frick KM. Post-training estrogen enhances spatial and object memory consolidation in female mice. Pharmacol Biochem Behav. 2006;84:112–119. doi: 10.1016/j.pbb.2006.04.013. [DOI] [PubMed] [Google Scholar]
- Gresack JE, Kerr KM, Frick KM. Life-long environmental enrichment differentially affects the mnemonic response to estrogen in young, middle-aged, and aged female mice. Neurobiol Learn Mem. 2007;88:393–408. doi: 10.1016/j.nlm.2007.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond R, Gibbs RB. GPR30 is positioned to mediate estrogen effects on basal forebrain cholinergic neurons and cognitive performance. Brain Res. 2011;1379:53–60. doi: 10.1016/j.brainres.2010.11.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond R, Mauk R, Ninaci D, Nelson D, Gibbs RB. Chronic treatment with estrogen receptor agonists restores acquisition of a spatial learning task in young ovariectomized rats. Horm Behav. 2009;56:309–314. doi: 10.1016/j.yhbeh.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond R, Nelson D, Kline E, Gibbs RB. Chronic treatment with a GPR30 antagonist impairs acquisition of a spatial learning task in young female rats. Horm Behav. 2012;62:367–374. doi: 10.1016/j.yhbeh.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart D, Nilges M, Pollard K, Lynn T, Patsos O, Shiel C, Clark SM, Vasudevan N. Activation of the G-protein coupled receptor 30 (GPR30) has different effects on anxiety in male and female mice. Steroids. 2014;81:49–56. doi: 10.1016/j.steroids.2013.11.004. [DOI] [PubMed] [Google Scholar]
- Hawley WR, Grissom EM, Moody NM, Dohanich GP, Vasudevan N. Activation of G-protein-coupled receptor 30 is sufficient to enhance spatial recognition memory in ovariectomized rats. Behav Brain Res. 2014;262:68–73. doi: 10.1016/j.bbr.2014.01.006. [DOI] [PubMed] [Google Scholar]
- Holm A, Baldetorp B, Olde B, Leeb-Lundberg LM, Nilsson BO. The GPER1 agonist G-1 attenuates endothelial cell proliferation by inhibiting DNA synthesis and accumulating cells in the S and G2 phases of the cell cycle. J Vasc Res. 2011;48:327–335. doi: 10.1159/000322578. [DOI] [PubMed] [Google Scholar]
- Jang EJ, Seok YM, Arterburn JB, Olatunji LA, Kim IK. GPER-1 agonist G1 induces vasorelaxation through activation of epidermal growth factor receptor-dependent signalling pathway. J Pharm Pharmacol. 2013;65:1488–1499. doi: 10.1111/jphp.12113. [DOI] [PubMed] [Google Scholar]
- Kang L, Zhang X, Xie Y, Tu Y, Wang D, Liu Z, Wang ZY. Involvement of estrogen receptor variant ER-alpha36, not GPR30, in nongenomic estrogen signaling. Mol Endocrinol. 2010;24:709–721. doi: 10.1210/me.2009-0317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler RC, Chiu WT, Demler O, Merikangas KR, Walters EE. Prevalence, severity, and comorbidity of 12-month DSM-IV disorders in the national comorbidity survey replication. Arch Gen Psychiatry. 2005;62:617–627. doi: 10.1001/archpsyc.62.6.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MJ, Futai K, Jo J, Hayashi Y, Cho K, Sheng M. Synaptic accumulation of PSD-95 and synaptic function regulated by phosphorylation of serine-295 of PSD-95. Neuron. 2007;56:488–502. doi: 10.1016/j.neuron.2007.09.007. [DOI] [PubMed] [Google Scholar]
- Kumar A, Bean LA, Rani A, Jackson T, Foster TC. Contribution of estrogen receptor subtypes, ERalpha, ERbeta, and GPER1 in rapid estradiol-mediated enhancement of hippocampal synaptic transmission in mice. Hippocampus. 2015;25:1556–1566. doi: 10.1002/hipo.22475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev. 2001;81:807–869. doi: 10.1152/physrev.2001.81.2.807. [DOI] [PubMed] [Google Scholar]
- Lamprecht MR, Morrison B., 3rd GPR30 activation is neither necessary nor sufficient for acute neuroprotection by 17β-estradiol after an ischemic injury in organotypic hippocampal slice cultures. Brain Res. 2014;1563:131–137. doi: 10.1016/j.brainres.2014.03.037. [DOI] [PubMed] [Google Scholar]
- Langer G, Bader B, Meoli L, Isensee J, Delbeck M, Noppinger PR, Otto C. A critical review of fundamental controversies in the field of GPR30 research. Steroids. 2010;75:603–610. doi: 10.1016/j.steroids.2009.12.006. [DOI] [PubMed] [Google Scholar]
- Levin ER. G protein-coupled receptor 30: estrogen receptor or collaborator? Endocrinology. 2009;150:1563–1565. doi: 10.1210/en.2008-1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis MC, Kerr KM, Orr PT, Frick KM. Estradiol-induced enhancement of object memory consolidation involves NMDA receptors and protein kinase A in the dorsal hippocampus of female C57BL/6 mice. Behav Neurosci. 2008;122:716–721. doi: 10.1037/0735-7044.122.3.716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XM, Li CC, Yu SS, Chen JT, Sabapathy K, Ruan DY. JNK1 contributes to metabotropic glutamate receptor-dependent long-term depression and short-term synaptic plasticity in the mice area hippocampal CA1. Eur J Neurosci. 2007;25:391–396. doi: 10.1111/j.1460-9568.2006.05300.x. [DOI] [PubMed] [Google Scholar]
- Luine VN, Jacome LF, Maclusky NJ. Rapid enhancement of visual and place memory by estrogens in rats. Endocrinology. 2003;144:2836–2844. doi: 10.1210/en.2003-0004. [DOI] [PubMed] [Google Scholar]
- Luttrell LM, Daaka Y, Lefkowitz RJ. Regulation of tyrosine kinase cascades by G-protein-coupled receptors. Curr Opin Cell Biol. 1999;11:177–183. doi: 10.1016/S0955-0674(99)80023-4. [DOI] [PubMed] [Google Scholar]
- Madak-Erdogan Z, Kieser KJ, Kim SH, Komm B, Katzenellenbogen JA, Katzenellenbogen BS. Nuclear and extranuclear pathway inputs in the regulation of global gene expression by estrogen receptors. Mol Endocrinol. 2008;22:2116–2127. doi: 10.1210/me.2008-0059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maggiolini M, Picard D. The unfolding stories of GPR30, a new membrane-bound estrogen receptor. J Endocrinol. 2010;204:105–114. doi: 10.1677/JOE-09-0242. [DOI] [PubMed] [Google Scholar]
- Moriarty K, Kim KH, Bender JR. Minireview: estrogen receptor-mediated rapid signaling. Endocrinology. 2006;147:5557–5563. doi: 10.1210/en.2006-0729. [DOI] [PubMed] [Google Scholar]
- Ortmann J, Veit M, Zingg S, Di Santo S, Traupe T, Yang Z, Völzmann J, Dubey RK, Christen S, Baumgartner I. Estrogen receptor-alpha but not -beta or GPER inhibits high glucose-induced human VSMC proliferation: potential role of ROS and ERK. J Clin Endocrinol Metab. 2011;96:220–228. doi: 10.1210/jc.2010-0943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto C, Rohde-Schulz B, Schwarz G, Fuchs I, Klewer M, Brittain D, Langer G, Bader B, Prelle K, Nubbemeyer R, Fritzemeier KH. G protein-coupled receptor 30 localizes to the endoplasmic reticulum and is not activated by estradiol. Endocrinology. 2008;149:4846–4856. doi: 10.1210/en.2008-0269. [DOI] [PubMed] [Google Scholar]
- Pedram A, Razandi M, Levin ER. Nature of functional estrogen receptors at the plasma membrane. Mol Endocrinol. 2006;20:1996–2009. doi: 10.1210/me.2005-0525. [DOI] [PubMed] [Google Scholar]
- Pereira LM, Bastos CP, de Souza JM, Ribeiro FM, Pereira GS. Estradiol enhances object recognition memory in Swiss female mice by activating hippocampal estrogen receptor alpha. Neurobiol Learn Mem. 2014;114:1–9. doi: 10.1016/j.nlm.2014.04.001. [DOI] [PubMed] [Google Scholar]
- Prossnitz ER, Arterburn JB, Sklar LA. GPR30: a G protein-coupled receptor for estrogen. Mol Cell Endocrinol. 2007;265–266:138–142. doi: 10.1016/j.mce.2006.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinecke K, Herdegen T, Eminel S, Aldenhoff JB, Schiffelholz T. Knockout of c-Jun N-terminal kinases 1, 2 or 3 isoforms induces behavioural changes. Behav Brain Res. 2013;245:88–95. doi: 10.1016/j.bbr.2013.02.013. [DOI] [PubMed] [Google Scholar]
- Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science. 2005;307:1625–1630. doi: 10.1126/science.1106943. [DOI] [PubMed] [Google Scholar]
- Sharma G, Prossnitz ER. Mechanisms of estradiol-induced insulin secretion by the G protein-coupled estrogen receptor GPR30/GPER in pancreatic β-cells. Endocrinology. 2011;152:3030–3039. doi: 10.1210/en.2011-0091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tararuk T, Ostman N, Li W, Björkblom B, Padzik A, Zdrojewska J, Hongisto V, Herdegen T, Konopka W, Courtney MJ, Coffey ET. JNK1 phosphorylation of SCG10 determines microtubule dynamics and axodendritic length. J Cell Biol. 2006;173:265–277. doi: 10.1083/jcb.200511055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas P, Pang Y, Filardo EJ, Dong J. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology. 2005;146:624–632. doi: 10.1210/en.2004-1064. [DOI] [PubMed] [Google Scholar]
- Waetzig V, Zhao Y, Herdegen T. The bright side of JNKs:multitalented mediators in neuronal sprouting, brain development and nerve fiber regeneration. Prog Neurobiol. 2006;80:84–97. doi: 10.1016/j.pneurobio.2006.08.002. [DOI] [PubMed] [Google Scholar]
- Wang C, Lv X, Jiang C, Davis JS. The putative G-protein coupled estrogen receptor agonist G-1 suppresses proliferation of ovarian and breast cancer cells in a GPER-independent manner. Am J Transl Res. 2012;4:390–402. [PMC free article] [PubMed] [Google Scholar]
- Yaffe K, Barnes D, Lindquist K, Cauley J, Simonsick EM, Penninx B, Satterfield S, Harris T, Cummings SR. Endogenous sex hormone levels and risk of cognitive decline in an older biracial cohort. Neurobiol Aging. 2007;28:171–178. doi: 10.1016/j.neurobiolaging.2006.10.004. [DOI] [PubMed] [Google Scholar]
- Zandi PP, Carlson MC, Plassman BL, Welsh-Bohmer KA, Mayer LS, Steffens DC, Breitner JC. Hormone replacement therapy and incidence of Alzheimer disease in older women: the Cache County study. JAMA. 2002;288:2123–2129. doi: 10.1001/jama.288.17.2123. [DOI] [PubMed] [Google Scholar]
- Zhao Z, Fan L, Frick KM. Epigenetic alterations regulate estradiol-induced enhancement of memory consolidation. Proc Natl Acad Sci U S A. 2010;107:5605–5610. doi: 10.1073/pnas.0910578107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Fan L, Fortress AM, Boulware MI, Frick KM. Hippocampal histone acetylation regulates object recognition and the estradiol-induced enhancement of object recognition. J Neurosci. 2012;32:2344–2351. doi: 10.1523/JNEUROSCI.5819-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]