

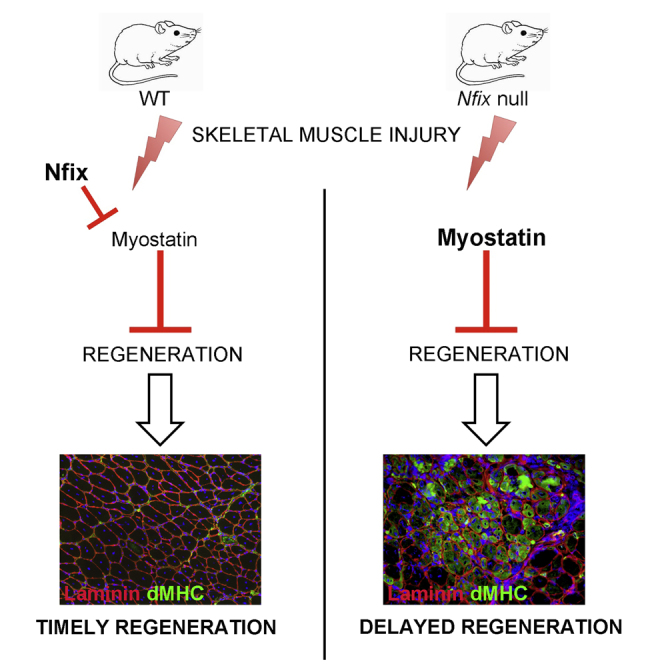
Summary
Nfix belongs to a family of four highly conserved proteins that act as transcriptional activators and/or repressors of cellular and viral genes. We previously showed a pivotal role for Nfix in regulating the transcriptional switch from embryonic to fetal myogenesis. Here, we show that Nfix directly represses the Myostatin promoter, thus controlling the proper timing of satellite cell differentiation and muscle regeneration. Nfix-null mice display delayed regeneration after injury, and this deficit is reversed upon in vivo Myostatin silencing. Conditional deletion of Nfix in satellite cells results in a similar delay in regeneration, confirming the functional requirement for Nfix in satellite cells. Moreover, mice lacking Nfix show reduced myofiber cross sectional area and a predominant slow twitching phenotype. These data define a role for Nfix in postnatal skeletal muscle and unveil a mechanism for Myostatin regulation, thus providing insights into the modulation of its complex signaling pathway.
Keywords: Myostatin, Nfix, regeneration, satellite cells, skeletal muscle
Graphical Abstract
Highlights
-
•
Muscle differentiation is delayed in Nfix-null satellite cells in vitro
-
•
Mice lacking Nfix in satellite cells show delayed regeneration upon injury
-
•
Nfix directly regulates Myostatin expression during regeneration
-
•
In vivo silencing of Myostatin rescues the regeneration deficit in Nfix-null mice
Rossi et al. highlight a function for Nfix during postnatal skeletal muscle regeneration and a link with Myostatin. During regeneration, Nfix regulates Myostatin expression, thus controlling the temporal progression of the regeneration process. When Nfix is deficient, regeneration is markedly delayed.
Introduction
Skeletal muscles play a critical role in voluntary movement, and proper regeneration of this tissue after injury can be severely compromised in a variety of contexts including neuromuscular disorders. In vertebrates, muscle progenitors originating from presomitic and cranial mesoderm give rise to “embryonic” or primary myogenesis, necessary to establish the basic muscle pattern, and “fetal” secondary myogenesis, characterized by growth and maturation of the muscle masses and by onset of innervation (Biressi et al., 2007a). These two waves of myogenesis are mediated by distinct embryonic and fetal myoblasts, respectively, each characterized by differentially expressed genes and properties of the myoblasts as well as the differentiated cells that they produce (Biressi et al., 2007b, Hutcheson et al., 2009). A key regulator of the embryonic and fetal classes is Nuclear Factor I X (Nfix), which is abundant in the fetus and almost absent in the embryo (Biressi et al., 2007b, Messina et al., 2010). Nfix drives the transcriptional changes from embryonic to fetal myogenesis by specifically repressing embryonic and activating fetal specific genes (Messina et al., 2010), and its role is partially conserved during evolution (Pistocchi et al., 2013).
Nfix is part of a family of highly conserved DNA-binding proteins that function as transcriptional activators and/or repressors: Nfia, Nfib, Nfic, and Nfix. NFI-binding sites are present in the promoter, enhancer and silencer regions of many cellular and viral genes (Kruse and Sippel, 1994), and NFI binding motifs were detected in promoters of genes expressed in different organs, such as brain (Bedford et al., 1998), lung (Bachurski et al., 1997), liver (Jackson et al., 1993), intestine (Xu et al., 2005), muscle (Spitz et al., 1997), connective tissue and skeletal elements (Szabó et al., 1995). Nfix-null mice are characterized by a reduced body size and inability to fully extend their limbs as well as CNS and skeletal deficits (Campbell et al., 2008, Driller et al., 2007). NFI binding sites are present in the promoters of the differentiation gene Myogenin and muscle-specific phosphofructo-kinase (Darville et al., 1992, Edmondson et al., 1992), and they can form a complex with Myogenin, thus increasing their affinity for muscle-specific genes (Funk and Wright, 1992, Johanson et al., 1999).
Another critical regulator of prenatal and postnatal myogenesis is Myostatin, a secreted factor of the TGFβ superfamily that was shown to be a negative regulator of muscle size, where loss-of-function mutations result in a dramatic increase in muscle mass in different species including human (McPherron et al., 1997, McPherron and Lee, 1997, Schuelke et al., 2004). Although interference of this pathway was reported to lead to functional improvement of dystrophic muscles in mice (Bogdanovich et al., 2002), the precise mode of action of this powerful signaling pathway as a potential therapeutic agent for myopathies remains unclear. Notably, the contextual role of Myostatin was highlighted, where in the embryo it regulates the balance between proliferation and differentiation of muscle progenitors (Manceau et al., 2008), whereas in the adult, opposing roles in regulating myogenic differentiation have been reported (Langley et al., 2002, Thomas et al., 2000, McCroskery et al., 2003, Taylor et al., 2001).
Postnatal myogenesis is assured by satellite cells (SCs) that are located between the basal lamina and the myofiber plasma membrane (Mauro, 1961). Although they account for less than 5% of total muscle nuclei in adult muscles, they play an indispensible role in the regeneration of adult skeletal muscle (Lepper et al., 2011, McCarthy et al., 2011, Murphy et al., 2011, Relaix and Zammit, 2012, Sambasivan et al., 2011).
Here, we report that absence of Nfix provokes an imbalance in skeletal muscle homeostasis and regeneration. Moreover, we identify Nfix as a regulator of Myostatin, where in vivo silencing of Myostatin in regenerating Nfix-null muscles rescues the regeneration defect. These findings have broad implications for the understanding of pathways regulating postnatal myogenesis and regeneration as well as providing insights into the role of Myostatin as a critical regulator of skeletal muscle differentiation.
Results
Altered Skeletal Muscle Phenotype in Nfix-Null Mice
We demonstrated previously that Nfix plays a critical role in the switch from embryonic to fetal myogenesis (Messina et al., 2010). Interestingly, Nfix-null mice are characterized by reduced body size and inability to fully extend their limbs, suggesting a possible muscular phenotype (Driller et al., 2007). To explore the role of Nfix in adult myogenesis, we examined its expression and observed that both myonuclei and Pax7+ SCs express Nfix (Figure 1A). Histological analysis of Nfix-null muscles showed a marked reduction in the cross sectional area of myofibers, although general muscle architecture remained normal compared to wild-type littermates (Figures 1B and 1C). In addition, Nfix-null SC-derived myoblasts were able to differentiate in vitro (Figure S1A), with no significant difference in proliferation and apoptosis rates with respect to the wild-type (Figures S1B and S1C). We reported previously that the embryonic marker slow MyHC is negatively regulated by Nfix, through repression of its activator Nfatc4 (Messina et al., 2010). Interestingly, adult muscles lacking Nfix were characterized by an increased expression of slow MyHC, which is evident in both typically slow-twitching muscles (such as the Soleus) and fast-twitching muscles (such as the EDL, Extensor Digitorum Longus) (Figure 1D). Notably, Nfix-null differentiated SC-derived myotubes in vitro also expressed higher levels of slow MyHC compared to wild-type (Figure S1D).
Figure 1.

Nfix Is Expressed by SCs and Its Absence Leads to a Reduced Myofiber Cross Sectional Area and to an Increased Slow MyHC Expression
(A) Immunofluorescence analysis of Nfix expression (green) in Pax7+ SCs (red) on a Tibialis anterior muscle section of a Mlc3f-lacz mouse, where myonuclei are in purple (β-gal). Hoechst was used to stain nuclei (n = 4 mice). The scale bar represents 25 μm.
(B) H&E staining on Tibialis anterior muscles from WT and Nfix-null mice (n = 5 mice). The scale bar represents 100 μm.
(C) Graphical representation of the myofiber cross sectional area (CSA) in WT and Nfix-null muscles. The plotted values represent the distribution of n = 65 measurements on five random microscope fields (∗∗∗p < 0.001 and two-tailed unpaired t test). The data are presented as mean ± whiskers from min to max.
(D) Western blot analysis of slow MyHC expression in WT and Nfix-null Soleus (SOL) and EDL muscles. The β-tubulin was used to normalize.
See also Figure S1.
SC Differentiation and Muscle Regeneration Are Delayed in the Absence of Nfix
To assess the role of Nfix during regeneration, we first examined single muscle fibers isolated from wild-type and Nfix-null mice. Freshly isolated wild-type and Nfix-null single fibers had comparable numbers of associated Pax7+ SCs (Figure S2A) and after 24 hr in culture the number of MyoD expressing cells was similar (Figure S2B). Analysis of SC proliferation and percentage of MyoD expression during the first 12 hr in culture revealed no difference between the two groups (Figures S2C and S2D). Although no significant alterations in numbers of Pax7+ and MyoD+ SCs were noted on Nfix-null myofibers, immunostaining for MyoD and Myogenin on wild-type and Nfix-null myofibers showed a marked reduction in the number of double Myod+/Myogenin+ cells in absence of Nfix (Figures 2A and 2B). Notably, this loss of differentiated cells in Nfix-null myofibers was more pronounced after 72 hr in culture, suggesting a possible involvement of Nfix during SC differentiation. Interestingly, the reduction in Myogenin+ cells was less evident at later stages (Figure 2B), thus suggesting a delay in muscle differentiation rather than a general block of the differentiation process.
Figure 2.

SCs Lacking Nfix Are Characterized by a Delayed Differentiation
(A) Immunofluorescence analysis of MyoD (green) and Myogenin (Myog, red) expression on single muscle fibers from WT and Nfix-null muscles after 24, 48, 72, and 96 hr in culture. Hoechst was used to stain nuclei (n = 3 WT and 3 Nfix-null mice). The scale bar represents 50 μm.
(B) Quantification of MyoD+/Myogenin+ and MyoD+/Myogenin− SCs associated with WT and Nfix-null myofibers at 24, 48, 72, and 96 hr in culture. The quantification is the result of three independent experiments on three WT and three Nfix-null mice. The data are presented as mean ± SD (not significant, ns; ∗∗∗p < 0.001 with n = 11 WT myofibers and n = 16 Nfix-null myofibers; and two-tailed unpaired t test).
See also Figure S2.
We reasoned that a delay in myogenic differentiation in absence of Nfix could result in an altered regeneration in vivo. We therefore injected the Tibialis anterior muscle of wild-type and Nfix-null mice with the snake venom cardiotoxin (CTX) to induce muscle regeneration. Following tissue damage, we observed a striking delay in muscle regeneration after injury of Nfix-null mice, as observed by persistent expression of developmental MyHC (Collins et al., 2005, Sartore et al., 1982, Schiaffino et al., 1986) (dMHC) expression (Figures 3A and 3D). In addition, the Nfix-null exhibited smaller regenerating myofibers compared to wild-type (Figure S2E). Accordingly, Myogenin expression was delayed and protracted in Nfix-null muscles (Figure 3B), in keeping with our in vitro observations (Figures 2A and 2B). Nevertheless, in spite of this altered timing, muscle regeneration appeared complete at later time points in the Nfix-null (28 days after injury, see Figure 3A). Interestingly, in Nfix-null mice, we observed a marked upregulation of Myostatin, a potent inhibitor of myogenesis, suggesting that this signaling pathway might be involved in the delayed muscle regeneration phenotype (Figure 3B). This was most evident from day 5 after CTX injection at a time when differentiation has already initiated, and it was accompanied by the upregulation of the Myostatin downstream effector phosphorylated Smad3 (pSmad3), particularly after 7 days (Figure 3C), suggesting a functional role for Myostatin signaling. These results clearly indicate a role for Nfix in regulating the proper timing of Myostatin expression and, in turn, of muscle regeneration upon injury. Analysis of Myostatin expression in uninjured muscles revealed no difference between wild-type and Nfix-null animals (data not shown), suggesting that the upregulation of Myostatin expression specifically occurs as a consequence of muscle injury.
Figure 3.

Absence of Nfix Leads to a Marked Impairment of Muscle Regeneration after Injury
(A) Immunofluorescence for dMHC (green) on regenerating WT and Nfix-null Tibialis anterior muscles 5, 7, 10, 14, and 28 days (d) following CTX injection. The laminin is marked in red and Hoechst was used to stain nuclei (n = 3 mice for each group). The scale bar represents 50 μm.
(B) Western blot analysis of Myogenin and Myostatin expression on WT and Nfix-null muscles 3, 5, 10, and 28 days (d) following CTX injection. The β-tubulin was used to normalize.
(C) Western blot analysis of pSmad3 and Smad3 expression on WT and Nfix-null muscles 5 and 7 days (d) following CTX injection. GAPDH was used to normalize.
(D) Quantification of the percentage of dMHC positive myofibers in WT and Nfix-null mice after 7 and 14 days following CTX injection (n = 6 mice for time point 7 and n = 5 mice for time point 14). The data are presented as mean ± SD (∗p < 0.05; ∗∗p < 0.01; and two-tailed unpaired t test).
See also Figure S2E.
Specific Deletion of Nfix in SCs Recapitulates the Regeneration Defects
Since Nfix expression is not restricted to SCs, we investigated muscle regeneration after CTX injury in tamoxifen treated Tg:Pax7-CreERT2:Nfixfl/− mice (see Figure S3A) to examine the specific role of Nfix in Pax7+ SCs. Tamoxifen treated Tg:Pax7-CreERT2:Nfixfl/− mice were compared with both tamoxifen untreated mice and mice expressing only the Tg:Pax7-CreERT2 allele and treated with tamoxifen, as controls. To verify the efficacy of tamoxifen treatment, sections of regenerating mouse muscles from each group were stained for Nfix. In both Tg:Pax7-CreERT2:Nfix+/+ and tamoxifen untreated Tg:Pax7-CreERT2:Nfixfl/− mice, all the centrally nucleated myofibers were Nfix+, as expected (Figure S3C). In tamoxifen treated Tg:Pax7-CreERT2:Nfixfl/− mice, Nfix expression was completely abolished in regenerating myonuclei, while it was still present in non-muscle nuclei, thereby confirming the specificity of Tg:Pax7-CreERT2 deletion (Figure S3C). Efficiency of Nfix deletion was also measured by counting the percentage of Pax7+ cells that had excised Nfix and was calculated to be 96.2% ± 0.4 (Figure S3D). Notably, tamoxifen treated Tg:Pax7-CreERT2:Nfixfl/− mice showed a delayed regeneration when compared to both tamoxifen untreated floxed mice and tamoxifen treated Nfix+/+ mice (Figure 4A). This defective regeneration was evident in tamoxifen treated Tg:Pax7-CreERT2:Nfixfl/− mice at 7 and 14 days post-CTX injection where dMHC expression persisted, and H&E sections revealed the continued presence of necrotic and inflammatory areas (Figures 4A and S3E). Further, the analysis of Myogenin clearly showed protracted expression in the tamoxifen treated Tg:Pax7-CreERT2:Nfixfl/− mice, confirming the delay in the regenerative process (Figure 4B). Finally, the distribution of the cross sectional area of regenerating myofibers in the three groups supports the delayed regeneration phenotype of tamoxifen treated Tg:Pax7-CreERT2:Nfixfl/− mice, mostly evident after 14 days (Figure S3B). These data support the specific function of Nfix in SCs for proper timing of Myogenin expression as well as the regeneration process.
Figure 4.

Absence of Nfix in SCs Determines the Delay in Muscle Regeneration
(A) H&E staining and immunofluorescence for dMHC (green) on transverse sections of regenerating Tibialis anterior muscles from Tg:Pax7-CreERT2:Nfix+/+ and Tg:Pax7-CreERT2:Nfixfl/−mice with (TMX) or without (NO TMX) tamoxifen treatment. The muscles were collected and stained after 5, 7, and 14 days (d) from CTX injection. The laminin is marked in red and Hoechst was used to stain nuclei (n = 3 mice for each group). The scale bar represents 100 μm.
(B) Western blot analysis of Myogenin expression on regenerating muscles from Tg:Pax7-CreERT2:Nfix+/+ (Nfix+/+) and Tg:Pax7-CreERT2:Nfixfl/− mice (Nfixfl/−) with (TMX) or without (NO TMX) tamoxifen treatment after 3, 5, 7, and 14 days (d) from CTX injection. Myogenin and GAPDH in the right panel come from two separate gels but using the same samples and protein amount. GAPDH was used to normalize.
See also Figure S3.
Nfix Regulates Myostatin Expression in Differentiating Myoblasts
In keeping with the observation that Nfix-null mice exhibit delayed muscle regeneration, Myostatin expression was upregulated in the absence of Nfix (Figure 3B). Since Myostatin is a TGF-β family member with anti-myogenic properties (Elliott et al., 2012), its upregulation can explain the delayed regeneration observed in mice lacking Nfix. To address this point, we investigated the possibility that Nfix might directly regulate Myostatin expression in differentiating myoblasts. First, we analyzed Myostatin expression in differentiating SC-derived myoblasts. As shown in Figure 5A, Nfix acts as a suppressor of Myostatin in differentiating myoblasts, even in absence of the contribution of the myofiber.
Figure 5.

Nfix Regulates Myostatin Expression in Differentiating Myoblasts through a Direct Binding to its Promoter
(A) Real-time qPCR showing Myostatin upregulation in differentiating SC-derived myoblasts. The SCs were isolated by FACS and plated in differentiation medium for a time course analysis from 1 to 3 days (DM1, DM2, and DM3) (n = 4 DM1 WT, n = 2 DM1 Nfix-null, n = 5 DM2 WT, n = 3 DM2 Nfix-null, n = 4 DM3 WT, and n = 4 DM3 Nfix-null). The data are presented as mean ± SD (not significant, ns; ∗p < 0.05; ∗∗p < 0.01; and two-tailed unpaired t test).
(B) Western blot analysis of Nfix expression in C2C12 myoblasts transduced with a control vector (C2C12 scramble) or with a vector carrying a shRNA targeting Nfix (C2C12 shNfix). GAPDH was used to normalize.
(C) Real-time qPCR analysis of Myostatin expression in scramble and shNfix C2C12 in a time course from 3 to 7 days in differentiation medium. The values are plotted as relative expression and normalized to GAPDH (n = 3 independent samples for each time point). The data are presented as mean ± SD (not significant, ns; ∗p < 0.05; ∗∗p < 0.01; and two-tailed unpaired t test).
(D) ChIP on differentiated C2C12 transduced with a vector expressing a HA-tagged Nfix2 isoform to test binding to putative Nfix binding sites on Myostatin promoter located at −87/−212 bp and −274/−548 bp from transcription start site. Binding on Nfatc4 promoter and on an intergenic region were used as positive and negative controls, respectively. The data are means of two independent experiments and expressed as fold enrichment (mean ± SD) relative to the IgG signal (n = 3 independent ChIP).
(E) Real-time qPCR for Myostatin in WT and Nfix-null SC-derived myoblasts transduced with a control vector (scramble) or with a vector overexpressing the Nfix2 isoform (Nfix2-HA). The values are plotted as relative expression and normalized to GAPDH (n = 3 samples for each group). The data are presented as mean ± SD (∗p < 0.05; ∗∗p < 0.01; and two-tailed unpaired t test).
For biochemical analysis, we validated this result using the C2C12 myogenic cell line. C2C12 cells were transduced with a lentiviral vector carrying a small hairpin RNA targeting Nfix (shNfix) or a scrambled sequence as a control (scramble). The transduction effectively resulted in the downregulation of Nfix expression in shNfix-treated C2C12, as confirmed by western blot (Figure 5B). Importantly, Myostatin expression was upregulated in Nfix-silenced myotubes starting from day 5, confirming that Nfix was able to downregulate Myostatin expression in differentiating C2C12 myoblasts (Figure 5C).
Nfix can bind with high affinity to the palindromic consensus sequence TTGGC(N5)GCCAA (Kruse and Sippel, 1994), although NFI factors can also bind to hemi-binding sites (a single TTGGC or GCCAA site alone) (Gronostajski, 2000) with a lower affinity. In silico analysis using Genomatix Software allowed us to identify hypothetical Nfix hemi-binding sites in the Myostatin promoter that were used to design specific primers. Notably, the identified sites are flanked by putative binding sites for MEF-2, which is a known co-factor of Nfix (Messina et al., 2010).
To assess whether Nfix can bind directly and regulate the Myostatin promoter, we performed a chromatin immunoprecipitation (ChIP) assay on differentiating C2C12. Proliferating C2C12 myoblasts were transduced with a HA-tagged Nfix2 lentiviral vector and then ChIP analysis was performed after 4 days in differentiation medium on the identified Nfix binding domains of Myostatin promoter. As expected, Nfix bound to the Nfatc4 promoter as described previously (Messina et al., 2010), and it was also found to directly bind to Myostatin promoter (Figure 5D). To determine how Nfix binding affected Myostatin expression, we transduced wild-type and Nfix-null SC-derived myoblasts with a Nfix2-HA lentivirus. A significant decrease in Myostatin expression was noted following Nfix overexpression, both in wild-type and in Nfix-null myoblasts (Figure 5E), thereby confirming the repressive role of Nfix on Mstn expression.
In Vivo Silencing of Myostatin Rescues the Regeneration Delay in Nfix-Null Mice
From the results indicated above, we predicted that regeneration defects observed in Nfix-null mice would be rescued by reducing Myostatin expression. Due to lethality of perinatal mice and difficulties in obtaining viable Nfix:Mstn double-null mice, we electroporated wild-type and Nfix-null regenerating Tibialis anterior muscles with control plasmids (scramble) or plasmids carrying an shRNA targeting Mstn (shmstn). Initial studies were carried out to verify the correct downregulation of Mstn expression after electroporation of the shmstn plasmid both in entire muscles and in re-isolated myoblasts (Figures S4A and 6C). We then electroporated scramble and shmstn plasmids 4 days after muscle injury, to obtain the maximal downregulation of Myostatin around day 6, which corresponds to the temporal window in which Myostatin was upregulated in Nfix-null mice (Figure 3B). Muscles were then isolated and processed at different time points after injury. Remarkably, we obtained a rescue of the regeneration defects of Nfix-null mice in muscles treated with the shmstn plasmid, but not with the scramble control vector (Figure 6A). This was particularly evident at 14 days after muscle injury, as shown by analysis of muscle sections stained for dMHC. The differences observed were even more striking in total muscle section reconstructions (Figure S4C). The analysis of the cross sectional area of regenerating myofibers revealed, as expected, a reduced cross sectional area for Nfix-null muscles with respect to wild-type. However, the regenerative delay phenotype was rescued upon Myostatin silencing; shmstn treated Nfix-null myofibers were comparable in size to scramble-treated wild-type (WT) fibers at the same time points (Figure S4B). This result highlights Myostatin involvement in the regeneration defects of Nfix-null mice, although from this experiment we cannot rule out the contribution of Myostatin to Nfix-null myofiber size in resting conditions. Strikingly, the regeneration rescue is supported by quantification of dMHC, whose expression was significantly reduced as a consequence of Myostatin silencing in Nfix-null mice (Figure 6B).
Figure 6.

In Vivo Silencing of Myostatin in Regenerating Nfix Null Muscles Rescues their Regeneration Defects
(A) Immunofluorescence analysis of dMHC expression (green) on regenerating WT and Nfix-null Tibialis anterior muscles after muscle electroporation with a control plasmid (scramble) or with a plasmid carrying an shRNA targeting Myostatin (shmstn). The muscles were electroporated after 4 days following CTX injection and were collected and stained after 7, 10, and 14 days (d) following CTX injection. The laminin is marked in red and Hoechst was used to stain nuclei (n = 3 mice for each group). The scale bar represents 100 μm.
(B) Quantification of the percentage of dMHC positive myofibers in Nfix-null mice electroporated with scramble or shmstn plasmids after 7 and 14 days following CTX injection (n = 5 mice for each group). The data are presented as mean ± SD (∗p < 0.05 and two-tailed unpaired t test).
(C) Real-time qPCR showing Myostatin expression in myoblasts re-isolated from Tibialis anterior electroporated with control (scramble) or shMyostatin (shMstn) plasmids. The myoblasts were isolated the day after in vivo transfection, and cultures were stopped 2 days later to perform qRT-PCR. The data are presented as mean ± SD.
See also Figure S4.
Discussion
Developmental and regenerative myogenesis share common and distinct features. Previous work from our group demonstrated that Nfix is responsible for the transcriptional switch from embryonic to fetal myogenesis during prenatal development (Messina et al., 2010). In the present study, we show that Nfix is required for maintaining muscle physiology postnatally and for the proper timing of muscle regeneration, where we report a link between Nfix and Mstn expression. Notably, the absence of Nfix leads to a dramatic delay in muscle regeneration in vivo, and this phenotype can be rescued by suppressing Mstn expression.
One of the main features that we observed in Nfix-null muscles was an altered muscular physiology characterized by a strikingly reduced muscle fiber size and an overexpression of the slow MyHC isoform. As already observed during prenatal development, the ability of Nfix to modulate slow MyHC expression is maintained after birth. Since Nfix plays important roles in brain and CNS development (Campbell et al., 2008), those phenotypes might directly or indirectly impact on the muscle phenotypes reported here. However, we show that upregulation of slow MyHC in the absence of Nfix is reproducible in SC-derived myotubes in culture, thus suggesting that the phenotype observed is cell-autonomously dependent on Nfix function in muscle. Moreover, ChIP experiments on C2C12 confirmed Nfix binding to Nfatc4 promoter, further suggesting that the mechanism of action is conserved between prenatal and postnatal life.
We also noted delayed differentiation of single fiber-associated myogenic cells that is accompanied by the modulation of Myogenin expression. This finding is consistent with the marked delay in regeneration observed in Nfix-null mice. Importantly, in addition to the persistent expression of dMyHC and to a delayed expression of Myogenin we observed a sustained expression of Mstn in injured muscles of Nfix-null mice. Myostatin (GDF8) is a potent inhibitor of myogenesis, where a dramatic increase in muscle mass was reported in Mstn-null mice (McPherron et al., 1997). However, the mode of action of Myostatin remains under debate for both prenatal and postnatal myogenesis (Manceau et al., 2008). Here, we show that the increased expression of Mstn in both Nfix silenced differentiating C2C12 cells and SC-derived myoblasts, as also in Nfix-null regenerating muscles, correlates with the delayed differentiation in vitro and in injured muscles. Myostatin is known to inhibit myoblast differentiation through inhibition of SC activation and proliferation (McCroskery et al., 2003, Taylor et al., 2001) and through repression of MyoD activity (Langley et al., 2002). Moreover, inhibition of Myostatin is known to accelerate muscle regeneration (McCroskery et al., 2005), thus supporting a causative relationship between the increase in Myostatin levels that we observed in regenerating Nfix-null mice and the delayed regeneration that characterizes them. We note that differences between WT and Nfix-null mice are apparent in muscles from day 5 after CTX injection, coinciding with the first difference in Mstn expression. Moreover, in vivo silencing of Mstn in adult Nfix-null muscles resulted in recovery of the correct timing of muscle regeneration after injury, confirming the causative relationship between Mstn sustained expression and the delayed regeneration observed in mice lacking Nfix. Importantly, the delayed regeneration was observed also in tamoxifen treated Tg:Pax7-CreERT2:Nfixfl/− mice, supporting the central role played by Nfix in SCs. Accordingly, isolated SC-derived Nfix-null myotubes express higher levels of Myostatin during differentiation and this is reversed upon Nfix re-expression. We therefore identified a mechanism of action for Nfix by showing that it can act as a direct regulator of Mstn. In light of contradictory reports on the mode of action of Myostatin, it is important to underscore the potentially distinct contextual roles of Myostatin (Manceau et al., 2008). Similar to the contextual roles for Notch signaling in adult myogenesis (Mourikis et al., 2012a, Mourikis et al., 2012b, Rios et al., 2011), recent studies suggest that Myostatin is necessary to regulate to proper balance between proliferation and differentiation of progenitor cells (George et al., 2013, Manceau et al., 2008, McCroskery et al., 2003).
Here, we suggest that Mstn regulation is temporally controlled for appropriate muscle differentiation and regeneration. Forced Mstn expression or silencing would inevitably led to a complete block in regeneration or to an accelerated differentiation at the expense of the proper muscle physiology.
In summary, this work demonstrates that Nfix acts through an inhibitory mechanism at the Mstn promoter in differentiating myoblasts, thereby influencing the commitment of SCs during differentiation and the proper timing of muscle regeneration. The mechanism identified not only broadens our knowledge on the function of Nfix in postnatal skeletal myogenesis, but also contributes to our understanding of molecular mechanisms involved in the regulation of Mstn.
Experimental Procedures
Animal Models
Nfix-null mice were generated crossing heterozygous mice obtained from Prof. Richard M. Gronostajski (University of Buffalo) and were always compared to WT littermates or age-matched WT mice. To increase Nfix-null mice survival, their food was supplemented with a soft dough chow (Transgenic Dough Diet, Bioserv) starting from postnatal day (P)21, as described (Campbell et al., 2008). WT mice used as controls were fed with the same diet. Tg:Pax7-CreERT2 mice were described previously (Mourikis et al., 2012b). Nfixfl mice were obtained from Prof. Richard M. Gronostajski (Campbell et al., 2008). Mouse genotyping was performed by PCR (GoTaq DNA Polymerase, Promega) using the primers listed in Table S1. For muscle regeneration, 20 μl of 100 μM CTX from Naja mossambica (Sigma-Aldrich) were injected in the Tibialis anterior of 2-month-old mice. Induction of Nfix deletion in Tg:Pax7-CreERT2:Nfixfl/− was performed by multiple tamoxifen injections. Starting from P18, mice received three subcutaneous injections (one every 24 hr for 3 days) of 50 μg tamoxifen (20 mg/ml) per gram of mouse. At 1 week before CTX, mice were subjected to an intraperitoneal injection of 25 mg/ml tamoxifen (250 μγ per gram of mouse). Starting from the day after, mice were fed with a specific tamoxifen enriched or control diet (Harlan). A detailed scheme of tamoxifen treatment is described in Figure S3A.
Mice were kept in pathogen-free conditions and all procedures were conformed to Italian law (D. Lgs n° 2014/26, implementation of the 2010/63/UE) and approved by the University of Milan Animal Welfare Body and by the Italian Minister of Health.
Measurement of Myofiber Cross Sectional Area
Cross sectional area of the myofibers was calculated on section images obtained from Tibialis anterior muscles using ImageJ.
Cell Culture
C2C12 cells (ATCC) were cultured at 37°C, 5% CO2 in DMEM (Lonza) plus 20% fetal bovine serum (FBS) (Lonza), 2 mM L-Glutamine (Sigma-Aldrich), and 1% Penicillin/Streptomycin (Euroclone). Differentiation was induced plating 1.5 × 105 cells in 35 mm petri dishes with DMEM, 2% horse serum (Lonza), 2 mM L-Glutamine and 1% Penicillin/Streptomycin.
SC Isolation and Culture
SCs (myoblasts) were isolated from WT and Nfix-null mice at P7–P10. Forelimb, hindlimb, and diaphragm muscles were dissected, mechanically cut, and enzymatically digested at 37°C under constant shaking with a solution containing Collagenase I (100 μg/ml, Sigma-Aldrich), Dispase (500 μg/ml, Gibco), and DNaseI (100 μg/ml, Roche) in PBS (Sigma-Aldrich). Undigested tissue was precipitated for 5 min, and the supernatant was centrifuged for 5 min at 1,200 g. The cell pellet was resuspended in DMEM plus 10% FBS, 3% chick embryo extract, 2 mM L-Glutamine, 1% Penicillin/Streptomycin and preplated in 150 mm petri dishes for 1 hr. After pre-plating, the non-adherent, myoblast enriched population was collected and plated in collagen coated (Collagen from calf skin, Sigma-Aldrich) 90 mm petri dishes at a density of 15,000 cells per petri. After a few days in proliferation, the myoblasts were eventually plated at high density (250,000 cells in 35 mm dishes) in differentiation medium (DMEM, 5% horse serum, 2 mM L-Glutamine, and 1% Penicillin/Streptomycin).
For cell sorting experiments, tissues were processed similarly, and then cells were blocked with 10% donkey serum (Sigma-Aldrich) in PBS for 15 min at room temperature (RT). The cells were incubated 30 min on ice with primary antibody detecting SM/C-2.6 antigen, biotinylated, 1:200 (Fukada et al., 2004). After two washes, cells were incubated with Streptavidin-APC (BD Pharmingen, 1:500) and CD45-FITC (Rat Anti-Mouse, 30-F11, BD Pharmingen, 1:100), 20 min on ice. The cells were washed again and sorted with a MoFlo XDP cell sorter (Beckman Coulter) based on positivity for SM/C-2.6 antigen and negativity for CD45.
BrdU Incorporation
For BrdU incorporation, 3 × 104 SC-derived myoblasts were plated in 35 mm petri dishes. The cells were exposed for 1 hr to 50 μm BrdU (Amersham). The cells were then fixed with 95% EtOH-5% acetic acid (VWR) for 20 min at RT, incubated with PBS-1.5 M HCl (VWR) for 10 min at RT, washed twice with PBS, and permeabilized with 0.2% Triton in PBS. Primary antibody (1:100, anti-Bromo-deoxyuridine clone BU-1, Amersham) was incubated for 1 hr at 4°C. After four washes, secondary antibody (1:500) was incubated with Hoechst for 40 min at RT. The cells were washed twice with PBS before mounting.
ELISA Assay for Caspase 9 Concentration
Measurement of the apoptosis rate was performed with an ELISA kit for the detection of mouse Caspase-9 concentration (Cusabio Biotech), according to manufacturer’s instructions. Data were acquired with a spectrophotometer (wavelength 450 nm, Tecan). Calculation of results was performed using Curve Expert software.
Single Fiber Isolation and Culture
Single muscle fibers were isolated from gastrocnemius and quadriceps muscles as previously described (Rosenblatt et al., 1995). In brief, muscles were enzymatically dissociated with 0.2% Collagenase I (Sigma-Aldrich) in DMEM for 30 min at 37°C with constant rolling. Single fibers were isolated under a stereomicroscope by gently passing through fire-polished Pasteur pipettes with different sized apertures. Purified single fibers were transferred in new plates with SC growth medium, cultured at 37°C, 5% CO2, and fixed for staining with 4% paraformaldehyde every 24 hr from 0 to 96 hr. EdU incorporation and staining were performed using the Click-iT EdU Alexa Fluor 594 Imaging Kit (Life Technologies), according to manufacturer’s instruction.
Lentiviral Transduction
C2C12 myoblasts were transduced with a lentivirus carrying a scrambled sequence or a shNfix. Transduction was performed in suspension (in DMEM 20% FBS), at a MOI of 100 and in the presence of Polybrene (8 μg/ml, Sigma-Aldrich). After overnight (O/N) incubation, the medium was changed, and cells were treated with puromycin (2 μg/ml, Sigma-Aldrich). SC-derived myoblasts and C2C12 used for ChIP experiments were transduced with a lentiviral vector carrying a control sequence or a Nfix2-HA-tagged isoform, described in Messina et al. (2010).
Immunofluorescence and Histology
Muscles were isolated from tendon to tendon, rapidly passed in liquid nitrogen-cooled isopentane (VWR) for 1 min, then in liquid nitrogen for 2 min, and left at −80°C until processed. There were 7 μm sections that were cut with a cryostat (Leica) on positively charged glass slides (Superfrost Plus, Thermo Scientific). Muscle sections were stained with H&E (Sigma-Aldrich) according to standard protocols. For immunofluorescence analysis, cells or sections were fixed for 10 min with 4% paraformaldehyde in PBS at 4°C (apart from staining for dMHC that do not require fixation). Samples were then permeabilized with 1% BSA (Sigma-Aldrich)-0.2% Triton X-100 (Sigma-Aldrich) in PBS for 30 min and blocked with 10% goat serum (Sigma-Aldrich) in PBS for 30 min at RT. Primary antibodies were incubated O/N at 4°C in PBS. After two washes with PBS-1% BSA-0.2%Triton, samples were incubated with secondary antibodies (1:500, Jackson Laboratory. Fluorochromes used: 488, 649, 594, and 546) and Hoechst (1:500, Sigma-Aldrich) in PBS for 45 min at RT, washed twice with PBS-0.2% Triton and mounted with Fluorescence Mounting Medium (Dako).
Immunofluorescences on single fibers were performed in suspension. Single myofibers were collected in 1.5 ml tubes and fixed for 10 min with 4% paraformaldehyde in PBS at 4°C. Fixed myofibers were treated with PBS-1% Triton-10% goat serum for 30 min at RT. Primary antibodies were incubated O/N in PBS-1% goat serum. After two washes with a solution containing 1% BSA in PBS, single myofibers were incubated with secondary antibodies and Hoechst in PBS for 1 hr at RT, washed twice with PBS-1% BSA, and mounted on slides.
Images were acquired at RT using a Leica-DMI6000B fluorescence microscope equipped with 10× and 20× magnification objectives. Leica DFC365FX or DFC400 cameras were used for image capture. The Leica Application Suite software was used for acquisition. Single channel pictures were merged using Photoshop. The following primary antibodies and dilutions were used: rabbit anti-Nfix (1:200, Novus Biologicals); chicken anti-laminin (1:500, Abcam); rabbit anti-laminin (1:300, Sigma-Aldrich); mouse anti Pax7 (1:2, DSHB); mouse anti-total MyHC (MF20) (1:2, DSHB); chicken anti-β-gal (1:500, Abcam); mouse anti-dMHC, which detects the Myh3 isoform (1:40, Monosan); rabbit anti-MyoD (C-20) (1:60, Santa Cruz Biotechnology); and mouse anti-Myogenin (1:3, DSHB).
RNA Extraction, RT-, and Real-Time qPCR
Total RNA from cultured cells was extracted using the NucleoSpin kits RNA XS and II (Macherey-Nagel). Isolation of total RNA from muscles was performed with TRIzol Reagent (Invitrogen) according to the manufacturer’s instructions. RNA was quantified using a NanoPhotometer (Implen). There was 1 μg of total RNA for each sample that was retrotranscribed with the iScript Reverse Transcription Supermix for RT-quantitative (q)PCR (Bio-Rad) in a total volume of 20 μl. For qRT PCR, cDNA was diluted 1:10 and 5 μl of the diluted cDNA was loaded in a total volume of 20 μl (iTaq Universal, Bio-Rad). Primers used are listed in Table S2. The relative quantification of gene expression was determined by comparative CT method, and normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH).
ChIP Assay
For ChIP, 3.5 × 106 scramble C2C12 myoblasts per antibody were plated. The cells were maintained in culture for 1 day in DMEM, 20% FBS, and then induced to differentiate with DMEM, 2% horse serum, at a density of 1.5 × 106 cells in 90 mm petri dishes. Crosslink was performed at day 4 in differentiation with 1% formaldehyde (Sigma-Aldrich) in DMEM for 10 min. Crosslink was blocked with 0.125 M Glycine (Sigma-Aldrich) in PBS for 10 min while shaking. After two washes with PBS, cells were scraped with PBS containing protease inhibitors and phenylmethanesulfonyl fluoride (PMSF) and centrifuged for 15 min at 1,500 g at 4°C. The cell pellet was dounced 20 times in a 7 ml pestle in swelling buffer, then centrifuged for 10 min at 5,000 g at 4°C, and the nuclei pellet was sonicated with a Bioruptor Sonicator (Diagenode) for 15 min, with repeated cycles of 15 s of sonication and 15 s of resting. Samples were finally centrifuged for 10 min at 12,000 g at 4°C, and chromatin-enriched supernatant was used in the following passages. Chromatin was pre-cleared for 2 hr with Protein G Sepharose (PrG, 15 μl per sample) (Amersham) and rabbit serum (2.5 μl per sample) and for 2 hr with PrG previously blocked with BSA (10 μg/ml) and Salmon Sperm (1 μg/ml) (Sigma-Aldrich) on a rotating platform at 4°C. Chromatin was than incubated O/N with primary antibodies. The day after, immunocomplexes were precipitated by addition of blocked PrG for 3 hr on rotating platform at 4°C, then centrifuged for 2 min at 12,000 g at 4°C. Samples were then repeatedly washed and the antibody-protein-DNA complexes were eluted twice for 10 min at 65°C. Eluted samples were incubated O/N at 65°C with 10 μg RNase (Sigma-Aldrich) and 200 mM NaCl (Sigma-Aldrich) to reverse crosslinks. Finally, DNA was treated with Proteinase K (Sigma-Aldrich) (20 μg per sample) for 3 hr at 50°C and then extracted with phenol-chloroform. Obtained DNA samples were analyzed with qPCR and results were plotted as fold enrichment with respect to the IgG sample. Primers used are listed in Table S3.
There were 5 μg of the following primary antibodies that were used: rabbit anti-HA-probe (Y-11, Santa Cruz Biotechnology); normal rabbit IgG (Santa Cruz Biotechnology).
Western Blot
Total protein extracts were obtained from cells or homogenized tissues, lysed with RIPA or Tissue extraction buffer plus protease and phosphatase inhibitors for 30 min on ice. Samples were then centrifuged 10 min at 12,000 x g at 4°C, and the supernatant transferred into a new tube and quantified using the DC Protein Assay (Bio-Rad). There were 30 μg of total protein extracts that were loaded on a 7%–12% SDS (Sigma-Aldrich) acrylamide gel (Sigma-Aldrich) and blotted to a nitrocellulose membrane (Whatman, Protran Nitrocellulose Transfer Membrane). After 1 hr of blocking in milk, primary antibodies were incubated O/N. Blots were then washed and incubated with secondary antibodies (1:10,000, IgG-HRP, Bio-Rad) for 40 min at RT and washed again. Finally, bands were revealed with ECL detection reagent (Amersham) and exposed to an X-ray film (Hyperfilm ECL, Amersham). In some cases, blots were acquired using the Chemidoc software (Bio-Rad).
The following primary antibodies and dilutions were used: rabbit anti-Nfix (1:5,000, Geneka Biotechnology); mouse anti-β-tubulin (1:5,000, Covance); mouse anti-slow MyHC (Bad5, 1:2, DSHB); mouse anti-total MyHC (MF20, 1:5, DSHB); mouse anti-Myogenin (IF5D, 1:3, DSHB); rabbit anti-Myostatin (1:500, Millipore); mouse anti-GAPDH (1:5,000, Sigma-Aldrich); rabbit anti-Smad3 (1:1,000, Abcam); rabbit anti-pSmad3 (1:1,000, Abcam).
In Vivo Electroporation
In vivo electroporation was performed 4 days after CTX injection. WT and Nfix-null mice were anesthetized and 40 μg of control (scramble, Sigma-Aldrich), or shmstn (Sigma-Aldrich) plasmids were injected in a total volume of 20 μl in Tibialis anterior muscles. Muscles were then immediately electroporated using a pulse generator (ECM 830, BTX) equipped with 5 mm needle electrodes to generate 100 V pulses, with a fixed duration of 20 ms and an interval of 200 ms between the pulses.
Statistical Analysis
All data are expressed as mean ± SD. Data were graphed using GraphPad Prism and analyzed with the two-tailed unpaired Student’s t test. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; and confidence intervals 95%, alpha level 0.05.
Author Contributions
Conceptualization, G.R. and G.M.; Methodology, S.A.; Investigation, G.R., S.A., C.B., S.M., and C.V.; Writing – Original Draft, G.R.; Writing – Review & Editing, S.T., G.C., and G.M.; Supervision, G.M.; and Funding Acquisition, G.M.
Acknowledgments
We thank G. Maroli for helpful discussion. We are also grateful to M. Magistroni for technical assistance, C. Villa (FRACTAL) for the Cytometry service, P. Mourikis for help with the tamoxifen protocol, F. Relaix and H. Amthor for their help and exchange of animal models, and R. Gronostajski for the kind exchange of information and animal models. G.R. conducted this study as partial fulfillment of her PhD in Cellular and Molecular Biology, San Raffaele University, Milan. This work received funding from the European Community, ERC StG2011 (RegeneratioNfix 280611), and the Italian Ministry of University and Research (MIUR-Futuro in Ricerca 2010).
Published: February 25, 2016
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Supplemental Information includes four figures and three tables and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2016.02.014.
Supplemental Information
References
- Bachurski C.J., Kelly S.E., Glasser S.W., Currier T.A. Nuclear factor I family members regulate the transcription of surfactant protein-C. J. Biol. Chem. 1997;272:32759–32766. doi: 10.1074/jbc.272.52.32759. [DOI] [PubMed] [Google Scholar]
- Bedford F.K., Julius D., Ingraham H.A. Neuronal expression of the 5HT3 serotonin receptor gene requires nuclear factor 1 complexes. J. Neurosci. 1998;18:6186–6194. doi: 10.1523/JNEUROSCI.18-16-06186.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biressi S., Molinaro M., Cossu G. Cellular heterogeneity during vertebrate skeletal muscle development. Dev. Biol. 2007;308:281–293. doi: 10.1016/j.ydbio.2007.06.006. [DOI] [PubMed] [Google Scholar]
- Biressi S., Tagliafico E., Lamorte G., Monteverde S., Tenedini E., Roncaglia E., Ferrari S., Ferrari S., Cusella-De Angelis M.G., Tajbakhsh S., Cossu G. Intrinsic phenotypic diversity of embryonic and fetal myoblasts is revealed by genome-wide gene expression analysis on purified cells. Dev. Biol. 2007;304:633–651. doi: 10.1016/j.ydbio.2007.01.016. [DOI] [PubMed] [Google Scholar]
- Bogdanovich S., Krag T.O., Barton E.R., Morris L.D., Whittemore L.A., Ahima R.S., Khurana T.S. Functional improvement of dystrophic muscle by myostatin blockade. Nature. 2002;420:418–421. doi: 10.1038/nature01154. [DOI] [PubMed] [Google Scholar]
- Campbell C.E., Piper M., Plachez C., Yeh Y.T., Baizer J.S., Osinski J.M., Litwack E.D., Richards L.J., Gronostajski R.M. The transcription factor Nfix is essential for normal brain development. BMC Dev. Biol. 2008;8:52. doi: 10.1186/1471-213X-8-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins C.A., Olsen I., Zammit P.S., Heslop L., Petrie A., Partridge T.A., Morgan J.E. Stem cell function, self-renewal, and behavioral heterogeneity of cells from the adult muscle satellite cell niche. Cell. 2005;122:289–301. doi: 10.1016/j.cell.2005.05.010. [DOI] [PubMed] [Google Scholar]
- Darville M.I., Antoine I.V., Rousseau G.G. Characterization of an enhancer upstream from the muscle-type promoter of a gene encoding 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase. Nucleic Acids Res. 1992;20:3575–3583. doi: 10.1093/nar/20.14.3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driller K., Pagenstecher A., Uhl M., Omran H., Berlis A., Gründer A., Sippel A.E. Nuclear factor I X deficiency causes brain malformation and severe skeletal defects. Mol. Cell. Biol. 2007;27:3855–3867. doi: 10.1128/MCB.02293-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edmondson D.G., Cheng T.C., Cserjesi P., Chakraborty T., Olson E.N. Analysis of the myogenin promoter reveals an indirect pathway for positive autoregulation mediated by the muscle-specific enhancer factor MEF-2. Mol. Cell. Biol. 1992;12:3665–3677. doi: 10.1128/mcb.12.9.3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott B., Renshaw D., Getting S., Mackenzie R. The central role of myostatin in skeletal muscle and whole body homeostasis. Acta Physiol. (Oxf.) 2012;205:324–340. doi: 10.1111/j.1748-1716.2012.02423.x. [DOI] [PubMed] [Google Scholar]
- Fukada S., Higuchi S., Segawa M., Koda K., Yamamoto Y., Tsujikawa K., Kohama Y., Uezumi A., Imamura M., Miyagoe-Suzuki Y. Purification and cell-surface marker characterization of quiescent satellite cells from murine skeletal muscle by a novel monoclonal antibody. Exp. Cell Res. 2004;296:245–255. doi: 10.1016/j.yexcr.2004.02.018. [DOI] [PubMed] [Google Scholar]
- Funk W.D., Wright W.E. Cyclic amplification and selection of targets for multicomponent complexes: myogenin interacts with factors recognizing binding sites for basic helix-loop-helix, nuclear factor 1, myocyte-specific enhancer-binding factor 2, and COMP1 factor. Proc. Natl. Acad. Sci. USA. 1992;89:9484–9488. doi: 10.1073/pnas.89.20.9484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George R.M., Biressi S., Beres B.J., Rogers E., Mulia A.K., Allen R.E., Rawls A., Rando T.A., Wilson-Rawls J. Numb-deficient satellite cells have regeneration and proliferation defects. Proc. Natl. Acad. Sci. USA. 2013;110:18549–18554. doi: 10.1073/pnas.1311628110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gronostajski R.M. Roles of the NFI/CTF gene family in transcription and development. Gene. 2000;249:31–45. doi: 10.1016/s0378-1119(00)00140-2. [DOI] [PubMed] [Google Scholar]
- Hutcheson D.A., Zhao J., Merrell A., Haldar M., Kardon G. Embryonic and fetal limb myogenic cells are derived from developmentally distinct progenitors and have different requirements for beta-catenin. Genes Dev. 2009;23:997–1013. doi: 10.1101/gad.1769009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson D.A., Rowader K.E., Stevens K., Jiang C., Milos P., Zaret K.S. Modulation of liver-specific transcription by interactions between hepatocyte nuclear factor 3 and nuclear factor 1 binding DNA in close apposition. Mol. Cell. Biol. 1993;13:2401–2410. doi: 10.1128/mcb.13.4.2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johanson M., Meents H., Ragge K., Buchberger A., Arnold H.H., Sandmöller A. Transcriptional activation of the myogenin gene by MEF2-mediated recruitment of myf5 is inhibited by adenovirus E1A protein. Biochem. Biophys. Res. Commun. 1999;265:222–232. doi: 10.1006/bbrc.1999.1390. [DOI] [PubMed] [Google Scholar]
- Kruse U., Sippel A.E. The genes for transcription factor nuclear factor I give rise to corresponding splice variants between vertebrate species. J. Mol. Biol. 1994;238:860–865. doi: 10.1006/jmbi.1994.1343. [DOI] [PubMed] [Google Scholar]
- Langley B., Thomas M., Bishop A., Sharma M., Gilmour S., Kambadur R. Myostatin inhibits myoblast differentiation by down-regulating MyoD expression. J. Biol. Chem. 2002;277:49831–49840. doi: 10.1074/jbc.M204291200. [DOI] [PubMed] [Google Scholar]
- Lepper C., Partridge T.A., Fan C.M. An absolute requirement for Pax7-positive satellite cells in acute injury-induced skeletal muscle regeneration. Development. 2011;138:3639–3646. doi: 10.1242/dev.067595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manceau M., Gros J., Savage K., Thomé V., McPherron A., Paterson B., Marcelle C. Myostatin promotes the terminal differentiation of embryonic muscle progenitors. Genes Dev. 2008;22:668–681. doi: 10.1101/gad.454408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauro A. Satellite cell of skeletal muscle fibers. J. Biophys. Biochem. Cytol. 1961;9:493–495. doi: 10.1083/jcb.9.2.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy J.J., Mula J., Miyazaki M., Erfani R., Garrison K., Farooqui A.B., Srikuea R., Lawson B.A., Grimes B., Keller C. Effective fiber hypertrophy in satellite cell-depleted skeletal muscle. Development. 2011;138:3657–3666. doi: 10.1242/dev.068858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCroskery S., Thomas M., Maxwell L., Sharma M., Kambadur R. Myostatin negatively regulates satellite cell activation and self-renewal. J. Cell Biol. 2003;162:1135–1147. doi: 10.1083/jcb.200207056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCroskery S., Thomas M., Platt L., Hennebry A., Nishimura T., McLeay L., Sharma M., Kambadur R. Improved muscle healing through enhanced regeneration and reduced fibrosis in myostatin-null mice. J. Cell Sci. 2005;118:3531–3541. doi: 10.1242/jcs.02482. [DOI] [PubMed] [Google Scholar]
- McPherron A.C., Lee S.J. Double muscling in cattle due to mutations in the myostatin gene. Proc. Natl. Acad. Sci. USA. 1997;94:12457–12461. doi: 10.1073/pnas.94.23.12457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPherron A.C., Lawler A.M., Lee S.J. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature. 1997;387:83–90. doi: 10.1038/387083a0. [DOI] [PubMed] [Google Scholar]
- Messina G., Biressi S., Monteverde S., Magli A., Cassano M., Perani L., Roncaglia E., Tagliafico E., Starnes L., Campbell C.E. Nfix regulates fetal-specific transcription in developing skeletal muscle. Cell. 2010;140:554–566. doi: 10.1016/j.cell.2010.01.027. [DOI] [PubMed] [Google Scholar]
- Mourikis P., Gopalakrishnan S., Sambasivan R., Tajbakhsh S. Cell-autonomous Notch activity maintains the temporal specification potential of skeletal muscle stem cells. Development. 2012;139:4536–4548. doi: 10.1242/dev.084756. [DOI] [PubMed] [Google Scholar]
- Mourikis P., Sambasivan R., Castel D., Rocheteau P., Bizzarro V., Tajbakhsh S. A critical requirement for notch signaling in maintenance of the quiescent skeletal muscle stem cell state. Stem Cells. 2012;30:243–252. doi: 10.1002/stem.775. [DOI] [PubMed] [Google Scholar]
- Murphy M.M., Lawson J.A., Mathew S.J., Hutcheson D.A., Kardon G. Satellite cells, connective tissue fibroblasts and their interactions are crucial for muscle regeneration. Development. 2011;138:3625–3637. doi: 10.1242/dev.064162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pistocchi A., Gaudenzi G., Foglia E., Monteverde S., Moreno-Fortuny A., Pianca A., Cossu G., Cotelli F., Messina G. Conserved and divergent functions of Nfix in skeletal muscle development during vertebrate evolution. Development. 2013;140:1528–1536. doi: 10.1242/dev.076315. [DOI] [PubMed] [Google Scholar]
- Relaix F., Zammit P.S. Satellite cells are essential for skeletal muscle regeneration: the cell on the edge returns centre stage. Development. 2012;139:2845–2856. doi: 10.1242/dev.069088. [DOI] [PubMed] [Google Scholar]
- Rios A.C., Serralbo O., Salgado D., Marcelle C. Neural crest regulates myogenesis through the transient activation of NOTCH. Nature. 2011;473:532–535. doi: 10.1038/nature09970. [DOI] [PubMed] [Google Scholar]
- Rosenblatt J.D., Lunt A.I., Parry D.J., Partridge T.A. Culturing satellite cells from living single muscle fiber explants. In Vitro Cell. Dev. Biol. Anim. 1995;31:773–779. doi: 10.1007/BF02634119. [DOI] [PubMed] [Google Scholar]
- Sambasivan R., Yao R., Kissenpfennig A., Van Wittenberghe L., Paldi A., Gayraud-Morel B., Guenou H., Malissen B., Tajbakhsh S., Galy A. Pax7-expressing satellite cells are indispensable for adult skeletal muscle regeneration. Development. 2011;138:3647–3656. doi: 10.1242/dev.067587. [DOI] [PubMed] [Google Scholar]
- Sartore S., Gorza L., Schiaffino S. Fetal myosin heavy chains in regenerating muscle. Nature. 1982;298:294–296. doi: 10.1038/298294a0. [DOI] [PubMed] [Google Scholar]
- Schiaffino S., Gorza L., Dones I., Cornelio F., Sartore S. Fetal myosin immunoreactivity in human dystrophic muscle. Muscle Nerve. 1986;9:51–58. doi: 10.1002/mus.880090108. [DOI] [PubMed] [Google Scholar]
- Schuelke M., Wagner K.R., Stolz L.E., Hübner C., Riebel T., Kömen W., Braun T., Tobin J.F., Lee S.J. Myostatin mutation associated with gross muscle hypertrophy in a child. N. Engl. J. Med. 2004;350:2682–2688. doi: 10.1056/NEJMoa040933. [DOI] [PubMed] [Google Scholar]
- Spitz F., Salminen M., Demignon J., Kahn A., Daegelen D., Maire P. A combination of MEF3 and NFI proteins activates transcription in a subset of fast-twitch muscles. Mol. Cell. Biol. 1997;17:656–666. doi: 10.1128/mcb.17.2.656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabó P., Moitra J., Rencendorj A., Rákhely G., Rauch T., Kiss I. Identification of a nuclear factor-I family protein-binding site in the silencer region of the cartilage matrix protein gene. J. Biol. Chem. 1995;270:10212–10221. doi: 10.1074/jbc.270.17.10212. [DOI] [PubMed] [Google Scholar]
- Taylor W.E., Bhasin S., Artaza J., Byhower F., Azam M., Willard D.H., Jr., Kull F.C., Jr., Gonzalez-Cadavid N. Myostatin inhibits cell proliferation and protein synthesis in C2C12 muscle cells. Am. J. Physiol. Endocrinol. Metab. 2001;280:E221–E228. doi: 10.1152/ajpendo.2001.280.2.E221. [DOI] [PubMed] [Google Scholar]
- Thomas M., Langley B., Berry C., Sharma M., Kirk S., Bass J., Kambadur R. Myostatin, a negative regulator of muscle growth, functions by inhibiting myoblast proliferation. J. Biol. Chem. 2000;275:40235–40243. doi: 10.1074/jbc.M004356200. [DOI] [PubMed] [Google Scholar]
- Xu H., Uno J.K., Inouye M., Collins J.F., Ghishan F.K. NF1 transcriptional factor(s) is required for basal promoter activation of the human intestinal NaPi-IIb cotransporter gene. Am. J. Physiol. Gastrointest. Liver Physiol. 2005;288:G175–G181. doi: 10.1152/ajpgi.00396.2004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.