

Summary
The Helicobacter pylori adhesin BabA binds mucosal ABO/Leb blood group (bg) carbohydrates. BabA facilitates bacterial attachment to gastric surfaces, increasing strain virulence and forming a recognized risk factor for peptic ulcers and gastric cancer. High sequence variation causes BabA functional diversity, but the underlying structural-molecular determinants are unknown. We generated X-ray structures of representative BabA isoforms that reveal a polymorphic, three-pronged Leb binding site. Two diversity loops, DL1 and DL2, provide adaptive control to binding affinity, notably ABO versus O bg preference. H. pylori strains can switch bg preference with single DL1 amino acid substitutions, and can coexpress functionally divergent BabA isoforms. The anchor point for receptor binding is the embrace of an ABO fucose residue by a disulfide-clasped loop, which is inactivated by reduction. Treatment with the redox-active pharmaceutic N-acetylcysteine lowers gastric mucosal neutrophil infiltration in H. pylori-infected Leb-expressing mice, providing perspectives on possible H. pylori eradication therapies.
Graphical abstract
Introduction
The gastric pathogen H. pylori chronically infects more than half of all people, leaving all infected with histological gastritis, and a subset of individuals with peptic ulcer disease, or gastric cancer, one of the most frequently lethal malignancies in many societies (Peek and Blaser, 2002; Polk and Peek, 2010). Chronic gastritis is associated with extensive gastric mucosal leucocyte infiltration (Rautelin et al., 1993). Yet neither strong immune response nor the stomach acidity clears established infections. This emphasizes H. pylori's extraordinary adaptation and adherence tropism for the gastric epithelium and overlying mucus layer, a niche hostile to most other microbes. H. pylori's ability to attach to the glycosylated gastric epithelial cell surfaces and overlying mucins facilitates host response management and acquisition of nutrients leached from the epithelium, and is directed by members of the “H. pylori outer membrane protein” family (“Hops”; pfam: PF01856; Alm et al., 2000). A prominent role is taken by the BabA adhesin, which binds to mono-(ABO) and difucosylated (Lewis b; Leb) derivatives of the type 1 chain lacto series carbohydrates (glycans) present at high density in glycolipids, glycoproteins, and mucins of the gastrointestinal (GI) tract (Ilver et al., 1998; Aspholm-Hurtig et al., 2004) (see Table S1 available online). In particular, tight BabA-mediated adherence is linked to disease-associated strains (Gerhard et al., 1999; Prinz et al., 2001), presumably because it potentiates delivery of secreted virulence effectors such as VacA and CagA, whose interference with host tissue signaling pathways results in tissue damage or worse, neoplastic transformation (reviewed by Hatakeyama and Higashi, 2005; Posselt et al., 2013). An important component of H. pylori's physiology is its increased genetic diversity (Kang and Blaser, 2006), with a particularly high rate of adaptive evolution in the BabA adhesin, where the high sequence diversity among clinical isolates functionally affects binding properties such as the ABO versus O binding preference (generalist versus specialist) and possibly also the 1,000-fold range of affinities of H. pylori strains for their Leb glycan receptors (Ka ∼108 to 1011 M−1) (Aspholm-Hurtig et al., 2004). This promotes emergence of highly individualized H. pylori strain variants that likely contribute to the extreme longevity of established infections and directs the infection and disease outcome (reviewed by Kang and Blaser, 2006; Suerbaum and Josenhans, 2007). BabA's adaptive traits are exemplified in the emergence of blood group specialist and generalist strains in accord with geographic differences in blood group prevalence (Aspholm-Hurtig et al., 2004), where the former are restricted to binding of Leb and are found in bg O dominant populations such as South American Amerindians, while the latter bind Leb as well as its GalNAc- and Gal-substituted blood group A and B derivatives (i.e., Aleb and Bleb). Though the multiple sequence analysis of BabA isoforms point to regions under positive, diversifying selection (Aspholm-Hurtig et al., 2004), the structural-molecular determinants that underlie BabA polymorphism are currently unknown.
Here we report X-ray structures of the adhesion domain of four representative BabA isoforms, including bg O specialist and bg A/B/O generalist type BabA proteins, alone and in complex with cognate ABO/Leb-type glycan receptors to establish a structural framework in which to understand the adhesion's extensive sequence diversity in functional and evolutionary contexts. We find a structurally plastic binding site that allows fast functional modification, illustrated by our finding that H. pylori can switch in ABO versus O bg binding preference by single amino acid substitutions in its carbohydrate binding domain (CBD). The anchor point for receptor binding is the embrace of the ABO fucose residue by a disulfide-clasped loop, which can be inactivated by the redox-active pharmaceutic N-acetylcysteine. This provides alternative perspectives on its possible pharmacological action and suggestions for improved treatment strategies.
Results
BabA Attains High-Affinity ABO bg Antigen Binding by Oligomerization
Secondary structure predictions indicate an autotransporter-like architecture for BabA, with a ∼50 kDa α-helical domain located upstream of a C-terminal β sheet region that is thought to form the transmembrane domain (residues 524–721) (Figure 1A). We previously found that residues 25–460 of the mature 721 residue BabA formed a stable, crystallizable fragment (hereafter dubbed BabAadhesin domain or BabAAD)(Subedi et al., 2014). Isothermal titration calorimetry (ITC) showed strain 17875 BabAAD bound Leb, with a monovalent binding interaction with a low micromolar dissociation constant (Kd) of 80.0 ± 7.1 μM (Figure 1B). This is in sharp contrast to the generally high Leb binding affinities reported for most H. pylori strains (Kd∼10−8 to 10−11 M−1; Aspholm-Hurtig et al., 2004) and the 0.39 nM Kd here measured for the full-length protein by means of surface plasmon resonance (SPR) (Figure 1C). Comparative binding studies and cross-linking data demonstrated BabA attains its high binding affinity by means of oligomerization in presence of the transmembrane domain (Figures 1C–1E). Whereas full-length BabA showed a single, exceedingly slow dissociation rate constant (2.32 E-4 s−1), as also reported by (Younson et al., 2009), the BabAAD shows biphasic dissociation kinetics composed of a similarly slow as well as a fast dissociating species (Figures 1C and 1D). Cross-linking in H. pylori outer membranes identified native BabA in 250 kDa oligomeric (presumably trimeric) complexes (Figure 1E). In contrast, BabAAD was found as a monomer with fast dissociation kinetics, and a minor fraction of a slow-dissociating oligomer (Figure S1A). Thus, full-length BabA's high apparent affinity for Leb is attributable to multivalent binding (avidity) and an associated slow dissociation rate constant, whereas BabAAD's biphasic binding profiles reflect a mixed oligomer and monomer population, likely due to destabilized oligomerization in the absence of the transmembrane domain.
Figure 1. BabAAD Interacts with Lewis b bg Antigens.
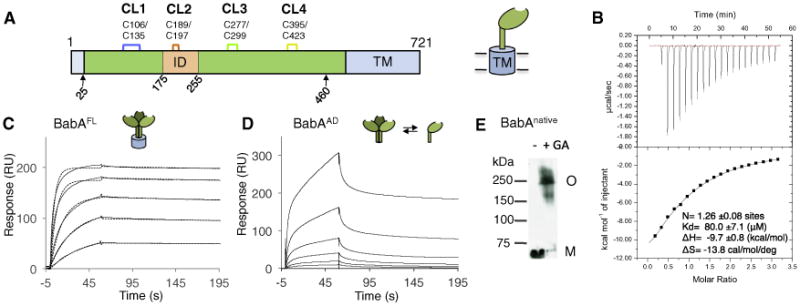
(A) Schematic of the BabA architecture. Arrows indicate the aa 25–460 BabA adhesin domain fragment (BabAAD). Abbreviations: CL, cysteine-clasped loops; TM, predicted transmembrane domain; ID, Bab insertion domain (Figures S2 and S3).
(B) ITC injection heats (upper) and normalized binding isotherm (lower) of the BabAAD titrated with Leb5.
(C) SPR sensorgram of full-length BabA (solid and dashed lines show raw and fitted binding curves, respectively, for 500, 250, 125, 62.5, 31.3, and 15.7 nM BabA, from the top down; with a dissociation constant Kd = 3.9E-10 ± 0.9E-10 (M), an association rate constant ka = 6.1E5 ± 1.4E5 (M−1 s−1), and slow dissociation rate constant, kd = 2.3E-4 ± 0.8E-4 (s−1).
(D) Similar SPR sensorgram of purified BabAAD binding to a Leb-coated chip; [BabA] as in (C).
(E) Immunoblot detection of BabA from glutaraldehyde(GA) crosslinked H.pylori 17875/Leb bacterial cells; M, monomer, O, BabA oligomer. See also Figure S1.
X-Ray Structure of the BabA Adhesin Domain
To gain molecular insight into BabA-mediated Leb binding, we determined the X-ray structure of BabAAD. Crystal growth was facilitated with one of two nanobodies (Nb-ER14 or Nb-ER19) generated against native BabA purified from H. pylori found to stabilize the recombinant BabAAD fragment (Figure S1B) (Subedi et al., 2014). Both Nbs specifically bound to BabA-expressing H. pylori cells, demonstrating that the conformation of Nb-stabilized BabAAD is equivalent to that of native BabA in the bacterial outer membrane (Figure S1C). The X-ray structures of the BabAAD in complex with Nb-ER14 or Nb-ER19 showed that BabA's ectodomain comprises a core domain consisting of seven α helices organized in a joined 4-helix (H2, H6, H7, H9) plus 3-helix (H3, H4, H5) bundle (hereafter referred to as 4+3-helix bundle) (Figures 2A and S1D; Table S2). Multiple sequence alignment (MSA) of Hop proteins and superimposition of BabAAD on the SabA ectodomain structure (SabA binds sialyl-lactose and sialylated Lewis antigens as sLex and sLea found in inflamed gatric tissues; Mahdavi et al., 2002; PDB: 4O5J; Pang et al., 2014; Figure S2) suggest that the 4+3-helix bundle topology is conserved among most Hop members. In SabA and a recently reported structure of the extracellular domain of strain J99 BabA (Hage et al., 2015), an additional α-helical coiled-coil domain is seen at a right angle to the 4+3-helix bundle domain (Figure S2). Compared to SabA, the BabAAD comprises an 80 residue insertion domain (Bab ID; residues 175–255) that forms a four-stranded β-plate (S3–S6) located between H4 and H5 of the 4+3-helix bundle (Figures 2A and S2). Hop family members hold a striking, well-conserved paired cysteine pattern, composed of two to four equivalent Cys pairs in BabA, BabB, BabC, HopZ, HopD, HopQ, HopM, and HopN; one in HopA; whereas two Cys-pairs are located in other positions in SabA and SabB (Figures 2B and S2A). Our MS analysis of native full-length BabA (Table S3) and the BabAAD crystal structure show that the four cysteine pairs form sequential disulfide bonds, three of which (except Cys189/Cys197) form the bases of extended Cys-clasped loops (CL1, CL3, and CL4) protruding from a common surface of BabA's ectodomain covering a surface area of ∼2,200 Å2 (Figures 2A and 2B). This area coincides with four of five BabA regions with increased sequence diversity, suggesting a role in adaptive radiation (Figure 3A) (Aspholm-Hurtig et al., 2004). A fifth diversity region lies at the opposing end of the BabA ectodomain and corresponds to the loop connecting H6 and H7 (residues 327–348) (Figure 3A).
Figure 2. Crystal Structure of BabAAD.
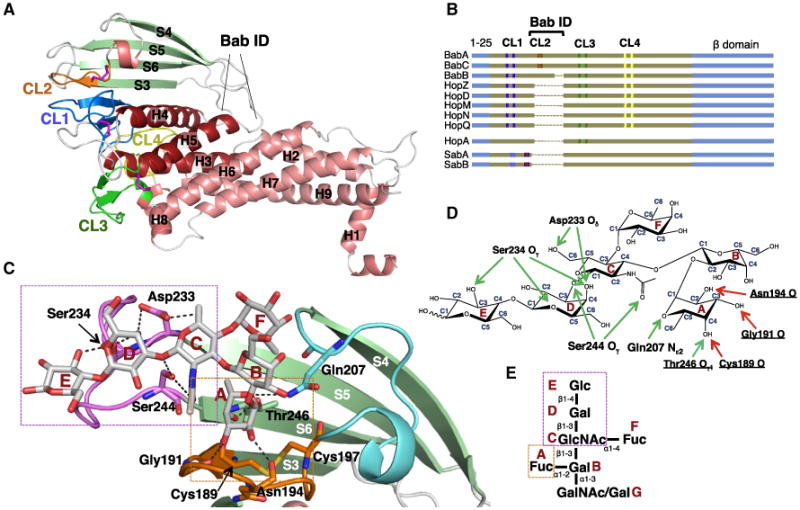
(A) X-ray structure of strain 17875 BabAAD (for clarity, Nb-ER19 is not shown, see Figure S1D). Helices and strands are colored red and green, respectively; Cys-bound loops CL1 (Cys106-Cys135), CL2 (Cys189-Cys197), CL3 (Cys277-Cys299), and CL4 (Cys395-Cys423) are colored blue, orange, green, and yellow, respectively. Bab ID: residues 175–255 (Figures S2 and S3).
(B) Schematic alignment of Cys-loop topology (vertical marks, colored as in Figures 1A and 1B) in the known or suspected Hop family adhesins. The α-helical ectodomain and β strand domains are colored brown and blue, respectively (sequence lengths not to scale, see Figure S2 for full MSA).
(C) Structure of 17875 BabAAD (colored as in Figure 2) bound to Leb6 (Table S1). Leb6 and interacting amino acids (labeled) are shown in stick representation (O, N, and S atoms are colored red, blue, and yellow, respectively). Two binding subsites can be identified: the α1-2 fucose binding pocket (boxed orange) formed by CL2 and T246 in strand S6, and the type 1 chain binding region (boxed magenta) formed by the loop connecting strands S5 and S6 (i.e., DL2, see Figure 3).
(D) H-bond network steering the 17875 BabAAD-Leb interaction. Side-chain- and main-chain-mediated H-bonds are depicted as green and red arrows, respectively.
(E) Schematic of ABO Lewis b bg antigens (see Table S1), with monosaccharides labeled A–G.
Figure 3. Structure of BabAAD Bound to Lewis b bg H Hexasaccharide.
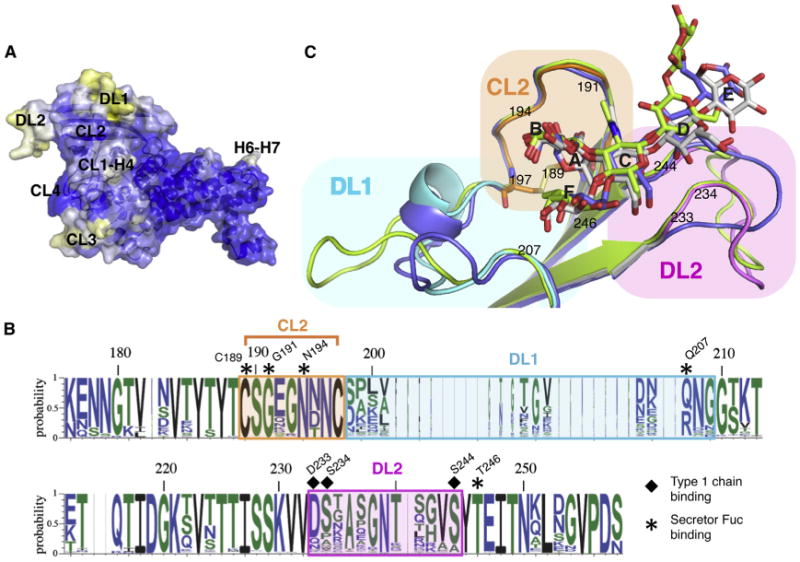
(A) Solvent-accessible surface of BabAAD, with blue, white, and yellow corresponding to high, medium, and low sequence conservation in multiple sequence alignment of 237 publicly available BabA sequences. Four out of five regions of increased sequence diversity map to the same side of the adhesin: (i) the loop connecting CL1 and H4 (CL1-H4; residues 136–146); (ii and iii) two loops in the Bab ID, e.g., DL1 (Diversification Loop 1; residues 200–210, connecting CL2 and S4) and DL2 (residues 234–242, connecting S5 and S6) (Figures 2D and S3); and (iv) CL3 (residues 279–299).
(B) Superimposition of strain 17875 BabAAD(colored as Figure 2C), with mutant BabAAD where the strain 17875 insertion domain is replaced by that of strain P437 (blue) or A730 (green).
(C) Sequence conservation plot of the BabA insertion domain. Residues that interact with the secretor fucose and core 1 moiety are highlighted by asterisks and squares, respectively. See also Figure S3 and Table S2.
Leb Binds a Structurally Heterogeneous Carbohydrate Binding Domain
BabA binds specifically to the lacto series glycans with a terminal fucose in α1-2 linkage (secretor Fuc) to the type 1 chain Galβ1-3GlcNAc core (Figure 2; Table S1) (Borén et al., 1993; Ilver et al., 1998). The Leb receptor is formed by addition of a second fucose, in α1-4 linkage to the type 1 chain GlcNAc residue (Figure 2E; Table S1). To gain structural insight into BabA's CBD, the 17875 BabAAD was crystallized together with the Leb hexasaccharide (“Leb6,” residues A–F; Figure 2; Tables S1 and S2). Leb6 is located at the tip of the β sheet insertion domain (Bab ID; Figure S4A) with its reducing end glucose (E) pointing away from the CBD (Figure 2C). The secretor fucose is bound in a defined pocket formed between Thr246 in strand S6 and the disulfide-clasped loop CL2. Up to five hydrogen bonds (H-bonds) anchor the secretor fucose in the CL2-enclosed pocket. Its C2 and C3 hydroxyl groups form H-bonds with the backbone carbonyl groups of Asn194 and Gly191, respectively, and its C4 hydroxyl forms H-bonds with both the Cys189 carbonyl and the Thr246 hydroxyl (Figures 2C and 2D). An additional H-bond can form between the fucose's endocyclic oxygen (O5) and the Gln207 side chain amide. Leb's subterminal α1-4 linked (Lewis) fucose does not make specific bonds with the adhesin (Figures 2C and 2D). Nevertheless, BabA shows about 2-fold stronger binding to Leb versus type 1 H neoglycoconjugates (Borén et al., 1993). In addition, BabA binds the GlcNAc-Gal-Glc moiety in the glycan core (C, D, and E, Figure 2E) via six H-bonds with the triad Asp233, Ser234, and Ser244 (Figures 2C and 2D). These “Asp-Ser-Ser-triad” interactions provide important specificity that distinguishes type 1 chain Leb-related glycans from type 2 chain glycans whose Gal-GlcNAc components are linked in β1-4 rather than β1-3 configuration (Table S1). Lack of BabA-type 2 chain glycan binding probably results from H-bond network disruption and steric interference with Asp233 and Ser234 (Figure S6E).
Two loops in the BabA binding pocket, DL1 and DL2, comprise regions with high sequence diversity due to positive selection (Figures 3A and S3) (Aspholm-Hurtig et al., 2004). Analysis of 237 BabA alleles shows strict conservation of the fucose-binding residues in the CL2 pocket (Cys189, Gly191, Asn194, and Thr246) (Figure 3B). In contrast, however, the type 1 chain binding site is located in DL2 and reveals considerable allelic variation, including conservative substitutions of an H-bond donor/acceptor in position 233 (D/Q/N), but with a predicted loss of hydrogen bonding capacity in ∼35% and ∼18% of cases at positions 234 (S/P/A) and 244 (S/A), respectively. The X-ray structures of the BabA CBD of Alaskan and Peruvian strains A730 and P436 (Figures 3C and S3), respectively, show the sequence variation in DL1 and DL2 results in a high structural heterogeneity in BabA's Leb binding pocket. In both BabA isoforms, structural reorganization of the DL1 loop and/or mutation in the 233-234-244 triad results in detachment and reorientation of the glycan's reducing end (Figure 3C), and is associated with an ∼15- and ∼20-fold loss in monovalent binding affinity for P436 and A730, respectively (Figures S4B and S4C). The presented BabA isoforms indicate the Galα1-2Fuc conformation in the CL2-enclosed pocket forms the invariant component in a structurally highly polymorphic BabA-Leb interaction (Figure 3C).
CL2 Loop Conformation and High Affinity Lewis b Binding Are Redox Sensitive
To examine the functional importance of CL2 and other BabA Cys-Cys loops, we tested if Leb binding was affected by cystine reduction. First, RIA analyses with 125I-labeled Leb and strain 17875 bacterial cells pre-exposed to dithiothreitol (DTT) showed loss of Leb binding, with a half maximum effective concentration (EC50) of 10 mM DTT (Figure 4A). The EC50 increased to >40 mM when DTT and Leb were coincubated with the bacterial cells, which suggests that Leb binding shields the CL2 disulfide from the reducing agent. This agrees with the structural data showing that Leb blocks solvent accessibility of Cys189-Cys197 (Figure 2C). The redox-induced loss of Leb binding was fully reversible after reconditioning of inactivated (to exclude de novo synthesis) bacterial cells in nonreducing buffer (Figure 4A), which indicates that disulfide reduction does not irreversibly damage the structural prerequisites for Leb binding. In addition, far UV circular dichroism showed that the 17875 BabAAD retains its secondary structure content upon disulfide reduction (Figure S4D). Double Cys189Ala/Cys197Ala mutants (“BabACL2”) were made to test if loss of the CL2 disulfide alone might underlie the redox sensitivity of BabA-Leb binding. RIA tests showed that a chromosomal babACL2 mutant of H. pylori strain J166 had lost all Leb-binding activity (Figure 4B), even though the BabACL2 protein was expressed in similar levels to that of its WT parent and reached the cell surface as a folded protein (Figures 4 and S4E–S4G). However, residual Leb binding was seen when BabACL2 from strain 17875 was expressed from a plasmid in the babA knockout strain P1ΔbabA, showing that some BabA proteins can retain a low level of Leb binding in absence of the CL2 disulfide. Also, when tested for the DTT-susceptibility of Leb-binding, H. pylori clinical isolates were found to fall into two phenotypic groups, with EC50 values between 2 and 10 mM (Figure 4C). To characterize the residual Leb binding by CL2 mutant bacteria, cells expressing WT and CL2 mutant 17875 BabA were flowed over a SPR chip surface containing immobilized Leb. The kinetic profiles of the BabACL2 mutant showed a lowered association rate compared to WT (Figure 4D), whereas its slow dissociation rate was intact. This outcome indicates the Cys189Cys197 disulfide bond serves to constrain the CL2 loop into a high-affinity encounter conformation for binding of the Leb secretor fucose (Figures 3B and S6A).
Figure 4. The CL2 Disulfide Bond Is Crucial for High-Affinity Leb Binding.
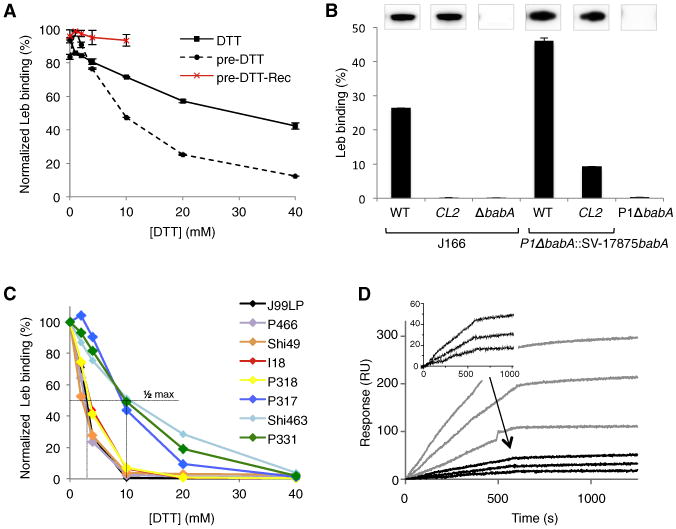
(A) Normalized radioimmunoassay (RIA) of H. pylori strain 17875/Leb binding to 125I-labeled Leb in presence of increasing concentrations of DTT, added prior to (dotted lines) or coincubated with (solid lines) Leb. DTT-exposed bacteria resuspended in DTT-free buffer for 2 hr, show recovery of Leb binding (red line). Data points show mean ± SD, n = 2.
(B) RIA Leb-binding of (left) the J166CL2 mutant (Cys189Ala and Cys197Ala), J166 WT, and J166ΔbabA; and (right) a babA deletion mutant of strain P1 (P1ΔbabA) and this strain conjugated with a shuttle vector expressing WT or CL2 mutant babA from strain 17875. Inlays show α-BabA immunoblots of the corresponding strains. Data points show mean ± SD, n = 2.
(C) RIA experiment showing relative Leb binding of H. pylori clinical isolates preincubated with DTT.
(D) SPR sensorgrams of the H. pylori strains with plasmid-based 17875 babA expression CL2 or WT BabA (black or gray response curves, respectively). Three dilutions of bacteria were flushed over immobilized Leb receptor conjugates, amplification of the CL2 curves are shown in inset. See also Figure S4.
The Redox-Active Pharmaceutic N-Acetylcysteine Attenuates Leb Binding and BabA-Mediated H. pylori Mucosal Adherence and Inflammation
Based on the critical role of CL2 in Leb binding, we tested if redox-active pharmaceutics could impair BabA mediated H. pylori mucosal adherence. We focused on N-acetylcysteine (N-acetyl-L-cysteine; NAC), a reducing agent proven to treat chronic obstructive pulmonary disease (COPD) and cystic fibrosis, and that in combination therapies with antibiotics has shown improved H. pylori clinical eradication rates, possibly due to NAC's mucolytic activity (by means of mucin disulfide reduction) (Cammarota et al., 2010; Makipour and Friedenberg, 2011). We tested if NAC treatment of H. pylori would interfere with Leb binding. First, fluorescently labeled H. pylori was exposed to NAC and applied to Leb expressing human gastric mucosa in vitro. Bacterial adherence was inhibited by 86% at 10 mg/mL NAC and almost eliminated by 20 mg/mL (Figure 5A). Next, NAC treatment was applied to test efficacy in detachment of adherent H. pylori, where 20 mg/mL NAC detached 50% of cells bound to gastric mucosa, and 10-fold more NAC was needed to remove remaining bacterial cells (Figure 5A). Pretreatment of gastric sections with upto 200 mg/mL NAC did not affect bacterial binding (Figure S5A), while RIA experiments showed that incubation of bacteria with 20 mg/mL NAC inactivated BabA-Leb binding (Figure 5B), arguing that NAC does not alter receptor availability but inhibits adhesive function of the bacteria. Quantitative MS analysis of tryptic digests of NAC-treated BabAAD protein showed an NAC dose-dependent reduction of the CL2 disulfide bond, with an EC50 of 9 mg/mL (Figures S5B and S5C). Accordingly, we attribute NAC's inhibition of BabA-dependent adherence to human gastric mucosa to disruption of BabA's critical CL2 disulfide bond. The increased NAC concentrations needed for bacterial detachment may reflect the competitive binding by Leb and its shielding protection of CL2 from NAC-mediated reduction (Figures 2C, 4A, 5A, and 5B).
Figure 5. Treatment of H. pylori cells with N-Acetylcysteine Blocks BabA Adherence.
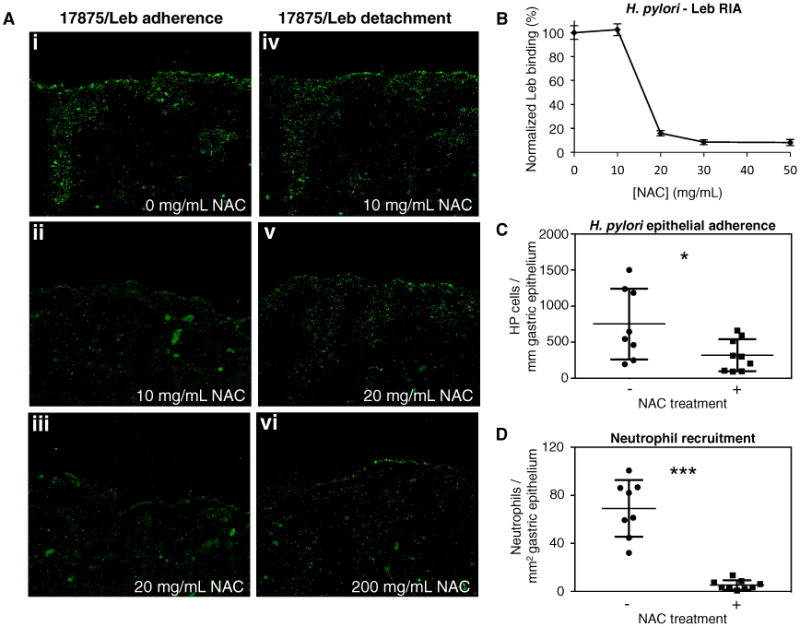
(A) FITC-labeled 17875/Leb bacteria binding to human gastric tissue sections. Prior to binding, bacteria were preincubated with 0, 10, or 20 mg/mL NAC (i, ii, and iii, respectively), resulting in 100%, 14%, and less than 5% adherent bacteria, respectively. In panels iv, v, and vi, tissue sections with bound bacteria are treated with 10, 20, or 200 mg/mL NAC, resulting in undetectable, 50%, and over 90% bacterial detachment, respectively.
(B) Normalized RIA Leb-binding of H. pylori strain 17875/Leb when being subjected to increasing concentrations of N-acetylcysteine (0, 10, 20, 30, and 50 mg/mL) for 1 hr at 37°C. Data points show mean ± SD, n = 3.
(C and D) H. pylori epithelial adherence and neutrophil recruitment in gastric epithelium of mice treated for 2 weeks with 0 (−) or 40 (+) mg/day NAC in their drinking water. Data points show, per animal, mean bacterial counts (n = 3) per mm of immunostained gastric epithelium (C; Figure S5D), or mean neutrophil counts (n = 3) per mm2 of immunohistostained gastric sections (D; Figure S5E). Statistical comparison of the groups produced a Welch-corrected t(9) = 2.29, *p = 0.0475; and t(7) = 7.559, ***p = 0.0001, respectively. Horizontal lines show sample mean ± SD, n = 8 (−NAC) and 9 (+NAC). See also Figure S5.
We next tested for an effect of NAC during H. pylori infection of Leb-expressing transgenic mice (Falk et al., 1995). A 2 week treatment with 40 mg/day/animal of NAC in the drinking water caused a 2-fold reduction in H. pylori epithelial adherence (Figures 5C and S5D) and 13-fold lower neutrophil recruitment to the gastric mucosa of NAC-treated mice (Figures 5D and S5E). These results indicate that interference with BabA-mediated adherence reduces infection-induced mucosal inflammation, probably by diminishing the ability of adherence to facilitate delivery of proinflammatory effectors such as CagA, VacA, peptidoglycan, urease, etc. (Ishijima et al., 2011; Posselt et al., 2013). Previous work has indeed shown that Leb-dependent adherence promotes gastritis and suggested that it increases H. pylori virulence, rather than acting solely to promote colonization (Guruge et al., 1998). However, as disulfide bonds in proteins are widespread, we cannot exclude models in which NAC's observed in vivo inflammation dampening involves additional mechanisms that synergize with CL2 reduction to diminish virulence.
BabA Binding of bg A and B Antigens
The majority of H. pylori strains are generalists, which bind to Leb and also ALeb and BLeb gastric mucosal glycans, in contrast to specialists that bind bgO/Leb only (Aspholm-Hurtig et al., 2004). Cocrystals of 17875 BabAAD with ALeb5 or BLeb7 (Table S1) show their binding conformation closely resembles that seen for Leb6, particularly at the secretor fucose. The bg A GalNAc or bg B Gal residues are positioned in a shallow pocket above Gln207 and Cys197-Ser198 (Figures 6 and S6D), with additional hydrogen bonds between Gln207 and the GalNAc C2 N-acetylgroup or the Gal C2 hydroxyl. In the BLeb complex, the Glu192 carboxyl also binds the Gal C6 hydroxyl. ITC indicates a 2-fold increase in monovalent affinity for the 17875 BabAAD:BLeb7 interaction compared to 17875 BabAAD:Leb5 (Figure S6B). Cocrystallization of 17875 BabAAD with the A type 1 hexasaccharide (A6-1; Figure S6D; Table S1) showed that the GlcNAc-Gal-Glc core of the type 1 glycan forms an alternative H-bond network with the Asp233-Ser234-Ser244 triad, possibly caused by a slight reorientation of the ligand core in glycans lacking the α1-4-linked Lewis fucose (Figure S6D). ITC showed that absence of the Lewis fucose in the 17875 BabAAD:A6-1 complex results in an ∼3.5 and 2-fold lower monovalent affinity compared to complexes with BLeb7 and Leb5, respectively (Figure S6C). Overall, the crystal structures of BabAAD with Leb6, BLeb7, ALeb5, and A6-1 reiterate that CL2-mediated binding of the secretor fucose constitutes the strongest conformational constraint in the BabA-glycan receptor interactions, in accord with its dominant role in BabA-receptor binding in vivo.
Figure 6. BabA Binding to bg A and B Glycans.
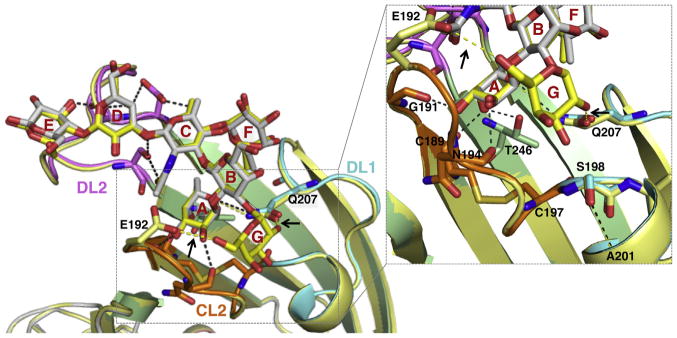
Overlay of the structures of BabAAD bound to Leb6 (colored as Figure 3B) or BLeb7 (Table S1; yellow). Leb6 and BLeb7 are shown in stick representation, as are glycan-binding amino acids (shown for BabAAD Leb6 complex, and Glu192 and Gln207 for the BLeb7 complex). Hydrogen bonds present in both the Leb6 and BLeb7 interaction or specific to BabAAD:BLeb7 (see arrows) are shown in black or yellow dashed lines, respectively. Inset shows detail (rotated up by ∼45°) of the subpocket binding the bg B Gal. Complexes with ALeb5 or A6-1 are shown in Figure S6D.
DL1 Polymorphism Affects Receptor Preference
We previously showed that H. pylori isolates exhibit high polymorphism in Leb binding strength and ABO preference, apparently reflecting adaptive selection (Aspholm-Hurtig et al., 2004). Although most adherent strains produce generalist BabA proteins, able to bind ALeb, BLeb, and Leb glycans, others (the majority South American Amerindian) are Leb-only specialists. The 17875 BabAAD crystal structures revealed that DL1, the 9–22 residue long diversity loop immediately downstream of the CL2, shapes a binding pocket for the extra GalNAc and Gal derivatives of bg A and B glycans (Figure 6),making it a prime candidate region for modulation of receptor preference. To test this hypothesis, the DL1 loop of generalist strain 17875 (residues aa 198–208) was replaced with that of specialist strain S831, which does not bind A- or BLeb (Figures 7A and S3) (Aspholm-Hurtig et al., 2004). This recombinant “BabAAD;DL1-S831” adopts the Leb-specialist binding characteristics of its S831 donor parent: (1) although the S831 DL1 graft is active for Leb binding (Figures 7B and S7A), titration with BLeb7 showed no binding heat signal in ITC (Figure S7B); and (2) competition with soluble Leb5, but not ALeb5, suppressed BabAAD;DL1-S831 binding to a Leb-coated SPR chip in a concentration dependent manner (Figures 7B and 7C). To visualize the molecular basis for receptor preferences, the recombinant specialist protein BabAAD;DL1-S831 was crystallized in complex with Leb6 and its structure compared to that of its generalist parent BabAAD in complex with Leb6 or BLeb7 (Figure 7A). The DL1 loops in 17875 and S831 adopt a highly similar backbone conformation, reflected in an almost identical binding conformation and H-bond pattern for Leb in the 17875 parent BabA and S831 DL1-grafted BabA (Figure 7A). The most noticeable differences in the recombinant S831 specialist and generalist 17875 parent result from the Leu-Pro (S831) versus Ser-Lys (17875) residues immediately downstream of CL2. Residues Lys199 to Thr202 in 17875 form an α-helical turn (Figures 3B and 3C) capped by an H-bond of Ser198 with the Ala201 backbone amide (Figure 7A). Leu-Pro in the S831 specialist causes loss of the capping hydrogen bond and rotation of residue 198 into the binding pocket that accommodates bg A and B GalNAc and Gal in generalist BabA (Figure 7A). Thus, inward rotation of a bulky 198 residue (Leu198 in S831) will interfere with the protruding bg A or B Gal derivatives. Analysis of 67 BabA proteins with known bg preference profiles (Aspholm-Hurtig et al., 2004) identified that Asp, Asn, or Leu at position 198 combined with Pro at 199 (11 Asp-Pro, 4 Asn-Pro, and 1 Leu-Pro) as a conserved property in 16 out of 18 specialist strains. In most Latin American strains, specialist adaption is also accompanied by a 7–11 residue insertion in DL1, as well as a replacement of Gln207 with Arg (Figure S3). The BabA ID phylogeny reveals the characteristic specialist bulky residue198-Pro199 pairs in several polyphyletic branches, e.g., in S831, P454, or P447 (Figure S3), which suggests mechanisms for functional selection and convergence to specialist phenotypes. In comparison, generalist strains show a higher diversity in positions 198 and 199, with 18 Ser-Pro, 9 Ser-Lys, 4 Ser-Glu, and 4 Ser-Ile pairs in the 35 cases examined. Four strains with specialist genotype in the 198–199 positions show an unexpected generalist phenotype (P436, P439, P442, and P452; Figure S3). The X-ray structure of BabAAD;ID-P436 in complex with BLeb7 shows the loss of interaction with the type 1 chain core (also seen in the BabAAD;ID-P436–Leb6 complex; Figure 3B) results in the ligand pivoting around the secretor fucose such that the bg B Gal is projected above rather than in clash with the D198 residue (Figure S7F). Strikingly, all four defiant specialists show loss of function mutations in the D-S-S triad in DL2 (Figure S3), suggesting the noncanonical generalist binding by a compensatory rotation of the ABO Leb ligands is a common mechanism in these strains.
Figure 7. Molecular Determinants of BabA bg Preference.
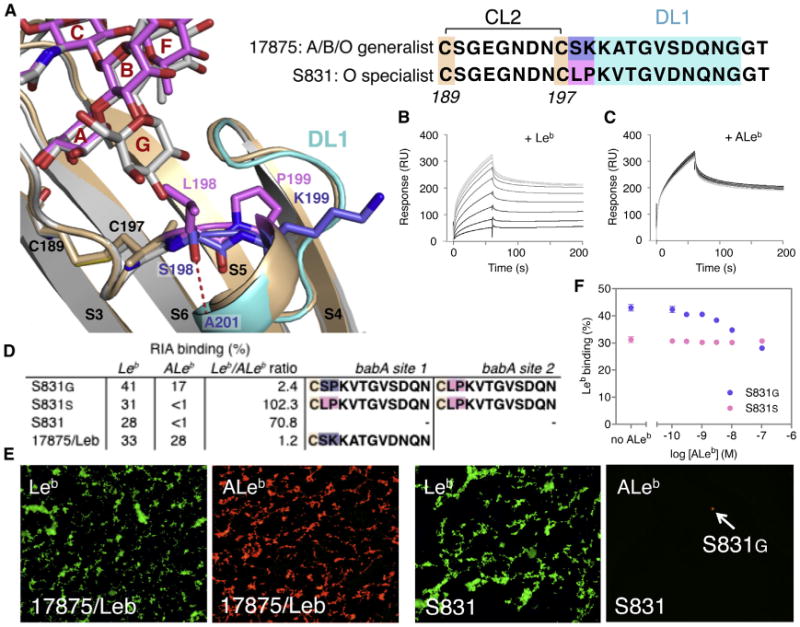
(A) Structure of the BabA S831 DL1 grafted hybrid (BabAAD;DL1-S831; tan, positions 198–199 in magenta) in complex with Leb6 (magenta), and wild-type 17875 BabAAD bound to BLeb7 (light gray, with DL1 region in cyan and positions 198–199 in blue). In the specialist hybrid the replacement of Lys199 with Pro199 results in the inward rotation of residue 198. Replacement of Ser198 of 17875 by Leu198 causes a steric occlusion of the Gal or GalNAc determinants in BLeb or ALeb.
(B and C) SPR sensorgrams of binding of BabAAD;DL1-S831 to a Leb-coated chip in the presence of competing soluble glycans: Leb5 (B) or ALeb5 (C), added in a 2-fold dilution series from 5 mM to 1 μM, colored gray to black.
(D) RIA of Leb versus ALeb binding of a generalist (S831G[D]) and specialist clone (S831S), and the control strains S831 and 17875/Leb. Amino acid sequence ofthe DL1 region (shown starting at Cys197) of the babA allele in the two loci of the corresponding strains. Binding and sequencing data are representative for two independent S831G(D) and S831S clones isolated.
(E) H. pylori 17875/Leb demonstrates the generalist phenotype and ability to bind both Alexa 488-labeled Leb (i) and Alexa 555-labeled ALeb (ii). Probing of theoriginal sweep population of the specialist H. pylori strain S831 by Alexa 555-labeled ALeb conjugate demonstrated by fluorescence microscopy the rarepresence of S831 bacterial cells of the generalist phenotype
(F) Competition RIA where Leb binding to a S831G(D) (blue) and S831S (magenta) clone is performed in competition with nonradiolabeled ALeb. Increasing concentrations of ALeb titrate out 34.6% of the Leb binding signal, demonstrating the concomitant expression of a generalist and a specialist babA variant. Data points show mean ± SD, n = 3. See also Figure S7.
Another adaptation in the bg preference site is seen in strain A730, where the α-helical turn in DL1 that shapes the bg A or B GalNAc or Gal binding pocket is stretched out into a hairpin loop (Figures 3B and S3). This results in up to five H-bond contacts with the bg derivative and increases affinity from 1.8 mM for Leb6 to 93 or 305 μM Kd for BLeb7 or A6-1, respectively. Thus, in A730, poor binding of the type 1 chain at the DL2 site can be compensated by improved binding of bg determinants at the DL1 site, effectively making the strain an A/B specialist (Figures S7C–S7E and S3).
Given the apparent plasticity in bg preference site, we tested the possibility of changing generalist versus specialist preference profiles simply by spontaneous mutation. Glycan affinity-IF-microscopy of a specialist strain S831 population with Alexa 555-ALeb was used to identify the occurrence of generalist derivatives (Figure 7E). Colony screening identified several positive clones, designated S831G, able to bind both ALeb and Leb (Figure 7D). Like many strains, S831 contains babA genes at two different chromosomal loci. Each generalist derivative clone (S831G) contained one base substitution mutation in codon 198 (TCG to TTG), resulting in a Leu198 Ser (generalist) substitution, and the original TCG (Leu198) specialist codon at the other babA locus (Figure 7D). As comparison, two tested clones with original specialist phenotypes were unchanged in sequence at each babA locus (codon 198 TCG, Leu) (Figure 7D). A competition RIA showed cumulative binding of ALeb and Leb in the generalist-derivative phenotype, which corresponds to simultaneous expression of specialist and generalist BabA proteins (Figure 7F). Thus, H. pylori strains with adhesin gene duplications can coexpress both BabA generalist and specialist proteins from their duplicate and divergent babA genes.
Discussion
Our data show the glycan binding site of the virulence-associated adhesin BabA is structurally geared toward adaptation, here exemplified in the recognized generalist versus specialist ABO binding preferences. BabA's CBD colocalizes with two sequence regions (DL1 and DL2) that are high in positive selection of diversifying amino acid substitutions (Aspholm-Hurtig et al., 2004). Most structurally characterized blood antigen binding adhesins bind to small, di-, tri-, or tetrasaccharide epitopes at the terminal, nonreducing end of the glycan (Boraston et al., 2006; Choi et al., 2008; Gregg et al., 2008; Holmner et al., 2007). Instead, BabA employs a three-pronged binding site to bind the extended ABO/Leb glycans: (1) the main chain of the disulfide clasped loop CL2 wraps around the α1-2-linked secretor fucose and forms the structurally conserved anchor point in the BabA-Leb interaction; (2) the DL2 Asp-Ser-Ser triad binds the reducing end β1, 3GlcNAc-Gal-Glc moiety of the glycan, which is determinant of ABO type-1 chains; and (3) the DL1 region interacts with the bg A and B GalNAc or Gal substituents and determines the adhesins bg preference (Figures 3 and S7F). The DL2 region provides BabA with a specificity mechanism that selects type-1 chain ABO/Leb antigens and discriminates against type-2 chains antigens such as ABO-2/Ley antigens(Figure S6E). Together, the CL2 clasp, DL1, and the DL2 Asp-Ser-Ser triad cooperate to create the affinity/specificity balance that directs BabA binding toward ABO/Leb type-1 chain antigens and gives rise to the wide range of individual binding affinities among BabA clinical isolates (Aspholm-Hurtig et al., 2004). Type 1 chain selection reflects H. pylori's cell lineage tropism for the foveolar epithelium, which is glycosylated with both fucosylated type-1 and 2 chains, in contrast to the glandular (deeper located) region that mostly holds type-2 series ABO glycans (Mollicone et al., 1985). In this way, H. pylori can enter and invade the gastric mucosa without risk of detrimental entanglement with type-2 chain decorated cells and innate immunity decoys such as antimicrobial mucins (Kawakubo et al., 2004).
By exchanging DL1 sequences, we could exchange allele-specific binding properties such as receptor binding preference (Figures 7 and S7). In the canonical DL1 conformation seen in 17875 and S831 BabA, Pro199 in combination with a bulky amino acid in position 198 (such as Asn, Asp, or Leu) shifts generalist ABO binding to specialist bg O binding by steric interference with the bg A or B Gal derivatives. In Europe, with very few specialist strains, the Spanish S831 strain is exceptional by its remarkably strong preference for bg O antigens (Leb) (Aspholm-Hurtig et al., 2004). Nevertheless, we found that also this distinct phenotype can fluently shift into the generalist ABO binding preference, by merely a single nucleotide mutation and amino acid substitution in the determining aa198 position (Figures 7 and S7). Thus, H. pylori has the inherent ability for rapid dynamic adaptation to the host bg phenotype, yet panels of clinical isolates have shown no such individualized adaptation, even over a full lifetime of infection, but instead appear to reflect a population-level adaptation to the ABO prevalence (Aspholm-Hurtig et al., 2004). We speculate that the geographic specialist/generalist diversification stems from a transmission bottleneck, where in ABO-mixed populations, generalists can always adhere to a new individual regardless of bg phenotype. For most European generalists, a switch to specialist preference would require amino acid substitution in both the 198 (i.e., Ser to Asn, Asp, or Leu) and 199 (Glu or Lys to Pro) position (Figure S3), possibly explaining the lack of specialist adaptation within bgO individuals in this mixed ABO population. Notable exceptions are strains S855, S858, and S865, where the Ser-Pro 198-199 sequence is just one step away from a bg preference switch. In populations with no transmission bottleneck for bg O specialists, a consequence of convergence to specialist high-affinity binding may be efficient early life transmission as fetal gastric glycosylation slowly maturates into fucose containing glycan landscapes (Bry et al., 1996). Thus, high-affinity specialist strains may be better suited to establish infection at early infancy, and generalist strains may be lost from the population through attrition. bg O individuals have long been recognized to be at an increased risk for developing peptic and duodenal ulcer disease (Aird et al., 1954). Future analysis will need to show to what extent a stronger, specialist-type H. pylori binding to bg O/Leb antigens and associated vigorous inflammation responses maybe at the basis of these epidemiological observations, a scenario that could also contribute to the gastric cancer pandemic among South American Amerindians.
Structural stability in the BabA binding site has its basis in the CL2 disulfide that clasps a conserved nine-residue loop into a conformation poised to bind the glycan's α1-2 fucose residue. Loss of the CL2 clasp results in drastically lowered association (On-rate) in Leb-binding, whereas at least for strain 17875 BabA, dissociation (Off-rate) remains intact (Figure 4D), suggesting that the disulfide clasp acts to optimize conformational fit during receptor encounter. We show that chemical disruption of the CL2 disulfide reversibly attenuates Leb binding and interferes with H. pylori adherence to the gastric mucosa. Possibly, rapid changes in redox conditions of the gastric microenvironment, such as induced hypoxia and host or H. pylori-secreted oxido reductases (Hogg, 2003; Windle et al., 2000), could act as an adherence kill switch in response to rapid exacerbations in local inflammation activity. Of particular relevance, the sensitivities of BabA binding to reducing conditions are not uniform, but rather show that clinical isolates cluster into different susceptibility groups (Figure 4C), and suggests that the CL2 disulfide redox sensitivity is context dependent and possibly adaptive. Thus, H. pylori might modulate binding properties by amino acid substitutions both within the α1-2 fucose-binding site and in addition by contextual substitutions in the proximal domains.
Finally, the unique BabA redox sensitivity provides a target of translational potential. We demonstrate that treatment with N-acetylcysteine (NAC), a redox-active pharmaceutic, disrupts the CL2 disulfide, inactivates BabA's binding properties, and hence blocks BabA-mediated adherence of H. pylori to human gastric mucosa. In Leb-expressing mice infected with H. pylori, a 2 week oral NAC treatment resulted in close to 15-fold reduction in neutrophil recruitment (Figures 5 and S5). H. pylori eradication treatments with NAC supplementation have previously shown synergistic therapeutic potential, attributed to the mucolytic effects of NAC (Makipour and Friedenberg, 2011). However, in light of the BabA redox sensitivity, it is now tempting to propose that NAC augments efficacy of the eradication regime by inhibition and displacement of H. pylori mucosal adherence and hence destabilization of its replicative biofilm niche (Tan et al., 2011).
The redox-sensitive CL2 and two diversification hotspots in the BabA glycan-binding site emphasize BabA's extraordinary adaptive potential. The structural framework here provided will be important in establishing the relation between BabA polymorphisms and disease outcome as well as in devising immuno- or chemotherapeutic countermeasures.
Experimental Procedures
Bacterial Strains
17875/Leb is a spontaneous mutant from H. pylori CCUG17875 that binds Leb but not sialylated glycans (Mahdavi et al., 2002). Strain 17875babA1A2 has deletions of the two babA genes, replaced with kanamycin (A1) and chloramphenicol (A2) resistance markers (Ilver et al., 1998). The generalist strain J166, frequently used for Rhesus macaque challenges (Solnick et al., 2001), was used to make the Cys189Ala and Cys197Ala double mutation in CL2 (see Supplemental Experimental Procedures). The J166ΔbabA was used as a control. For SPR studies probing Leb binding of the CL2 mutant, H. pylori P1ΔbabA was used with 17875babA or its isogenic CL2 mutant (Cys189Ala/Cys197Ala) expressed from the pIB6 shuttle vector. Strain S831 is a specialist strain, and S831G(D) is a spontaneous generalist S831 derivative identified in this study. S831S cl-1 and S831G(D) cl-1 are clones obtained with colony membrane screening (described in the Supplemental Experimental Procedures).
Protein Production and Purification
Native BabA protein was purified from H. pylori CCUG 17875/Leb according to Bugaytsova et al. (personal communication). recBabA; 17875 BabAAD; its S831 DL1 loop mutant; and the P436, A730 isoforms are produced recombinantly in E. coli as outer membrane or periplasmically targeted proteins and purified by His-tag affinity purification. Cloning, production, and purification methodology is described in the Supplemental Experimental Procedures.
Structure Determination
The X-ray structure of the BabAAD fragment in complex with Nb-ER14 was determined by single anomalous dispersion using crystals with selenomethionine labeled BabAAD. All BabAAD structures in complex with Nb-ER19 were determined by molecular substitution with the 17875 BabAAD:Nb-ER14 structure. Ligand complexes were formed by cocrystallization with 1 mM of the various sugar ligands (Table S1). See Table S2 for data collection and refinement statistics and Supplemental Experimental Procedures for full details.
Binding Assays
Leb binding to various H. pylori strains was assessed by radioimmunoassay using 125I-labeled Leb-HSA glycoconjugate produced as described (Aspholm et al., 2006). In vitro binding properties of the BabA protein and the different BabA fragments and mutants were obtained by SPR and ITC. Expanded description is in the Supplemental Experimental Procedures.
In Vitro Inhibition and Detachment of H. pylori Adherence to Human Gastric Mucosa Tissue by NAC
Suspensions of FITC labeled H. pylori strain 17875/Leb were mixed with a series of N-acetylcysteine dilutions prepared in SIA blocking buffer (0,10,20, and 100 mg/mL). Bacteria were NAC-incubated at 37°C for 1 hr and then applied to human gastric mucosa histo tissue sections for 2 hr. Unspecific binding was removed by PBS-Tween, and slides were subjected to microscopy for digital quantification. In the “detachment” regime, adherent bacteria (in absence of NAC), applied and washed as above, were incubated for 1 hr at 37°C with a series of NAC dilutions in SIA blocking buffer (10, 20, 100, and 200 mg/mL). Slides were again washed in PBS-Tween and analyzed as described above. To assess the effect of NAC treatment on receptor availability in the human gastric mucosa sections, sections were preincubated with N-acetylcysteine dilutions prepared in SIA blocking buffer (0, 10, 20, 100, and 200 mg/mL NAC) for 2 hr at room temperature and then washed three times in PBS-Tween. Suspensions of FITC-labeled H. pylori strain 17875/Leb were applied and washed as above, and slides were subjected to microscopy.
Supplementary Material
Highlights.
Structural basis for Helicobacter pylori's polymorphic ABO/Leb glycan binding
A disulfide-clasped fucose binding loop directs BabA adherence
Redox-active pharmaceuticals block BabA-dependent binding and mucosal inflammation
Single amino acid substitutions can alter BabA's blood group binding preferences
Acknowledgments
We thank Ayla Debraekeleer and Joemar Taganna for assistance in recombinant protein production and figure preparation, respectively, and Lori M. Hansen for help in constructing H. pylori chromosomal mutant (JS). We acknowledge the use of the synchrotron-radiation facility at Diamond Light Source, Didcot, UK, under proposal MX9426 and the Soleil synchrotron, Gifsur-Yvette, France, under proposal 20131370; access support from the European Community's Seventh Framework Program (FP7/2007-2013) under Bio-Struct-X (grant agreement number 6601). This work is supported by project grant PRJ9 from the Flanders Institute of Biotechnology (VIB), the Odysseus program of the Flanders Science Foundation (FWO), and equipment grant UABR/09/005 from the Hercules Foundation. K.M. and S.S. are recipients of an FWO postdoc, and Erasmus Mundus PhD fellowship, respectively. T.B., L.H., and A.A. are supported by grants from Vetenskapsrådet/VR and Cancer-fonden, and T.B. is supported by the J.C. Kempe and Seth M. Kempe Memorial Foundation and the Knut and Alice Wallenberg Foundation (2012.0090); work was in part performed within the Umeå Centre for Microbial Research (UCMR) and the Biochemical Imaging Center Umeå (BICU).
Footnotes
Supplemental Information: Supplemental Information includes Supplemental Experimental Procedures, seven figures, and three tables and can be found with this article online at http://dx.doi.org/10.1016/j.chom.2015.12.004.
Author Contributions: K.M. and S.S. produced the recombinant BabA ectodomains, solved their X-ray structures, and performed binding assays. E.R., S.M., and L.H. generated BabA-specific Nbs. P.G. performed mutagenesis, characterization, and binding studies on CL2 mutants and H. pylori strains. P.G., M.F., and L.R. performed NAC experiments. G.C. performed binding studies on recombinant FL BabA. A.L. produced expression constructs. J.B. purified native BabA, and performed IF-microscopy and crosslinking experiments. A.S. performed RIA experiments. M.M. and J.N. isolated and characterized S831 derivatives. K.B. performed ProteOn experiments. G.V. performed MS analysis of NAC titration. A.A., J.S., and T.B. supervised P.G., and T.N. supervised M.F. S.O. and M.L. produced hepta-BLeb and Leb-HSA. T.B. and H.R. conceived and supervised the study, with contributions from all authors. K.M., P.G., D.B., J.B., T.B., and H.R. wrote the paper.
References
- Aird I, Bentall HH, Mehigan JA, Roberts JAF. The blood groups in relation to peptic ulceration and carcinoma of colon, rectum, breast, and bronchus; an association between the ABO groups and peptic ulceration. BMJ. 1954;2:315–321. doi: 10.1136/bmj.2.4883.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alm RA, Bina J, Andrews BM, Doig P, Hancock RE, Trust TJ. Comparative genomics of Helicobacter pylori: analysis of the outer membrane protein families. Infect Immun. 2000;68:4155–4168. doi: 10.1128/iai.68.7.4155-4168.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aspholm M, Kalia A, Ruhl S, Schedin S, Arnqvist A, Lindén S, Sjöström R, Gerhard M, Semino-Mora C, Dubois A, et al. Helicobacter pylori adhesion to carbohydrates. Methods Enzymol. 2006;417:293–339. doi: 10.1016/S0076-6879(06)17020-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aspholm-Hurtig M, Dailide G, Lahmann M, Kalia A, Ilver D, Roche N, Vikström S, Sjöström R, Lindén S, Bäckström A, et al. Functional adaptation of BabA, the H. pylori ABO blood group antigen binding adhesin. Science. 2004;305:519–522. doi: 10.1126/science.1098801. [DOI] [PubMed] [Google Scholar]
- Boraston AB, Wang D, Burke RD. Blood group antigen recognition by a Streptococcus pneumoniae virulence factor. J Biol Chem. 2006;281:35263–35271. doi: 10.1074/jbc.M607620200. [DOI] [PubMed] [Google Scholar]
- Borén T, Falk P, Roth KA, Larson G, Normark S. Attachment of Helicobacter pylori to human gastric epithelium mediated by blood group antigens. Science. 1993;262:1892–1895. doi: 10.1126/science.8018146. [DOI] [PubMed] [Google Scholar]
- Bry L, Falk PG, Midtvedt T, Gordon JI. A model of host-microbial interactions in an open mammalian ecosystem. Science. 1996;273:1380–1383. doi: 10.1126/science.273.5280.1380. [DOI] [PubMed] [Google Scholar]
- Cammarota G, Branca G, Ardito F, Sanguinetti M, Ianiro G, Cianci R, Torelli R, Masala G, Gasbarrini A, Fadda G, et al. Biofilm demolition and antibiotic treatment to eradicate resistant Helicobacter pylori: a clinical trial. Clin Gastroenterol Hepatol. 2010;8:817–820. doi: 10.1016/j.cgh.2010.05.006. [DOI] [PubMed] [Google Scholar]
- Choi JM, Hutson AM, Estes MK, Prasad BVV. Atomic resolution structural characterization of recognition of histo-blood group antigens by Norwalk virus. Proc Natl Acad Sci USA. 2008;105:9175–9180. doi: 10.1073/pnas.0803275105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk PG, Bry L, Holgersson J, Gordon JI. Expression of a human alpha-1,3/4-fucosyltransferase in the pit cell lineage of FVB/N mouse stomach results in production of Leb-containing glycoconjugates: a potential transgenic mouse model for studying Helicobacter pylori infection. Proc Natl Acad Sci USA. 1995;92:1515–1519. doi: 10.1073/pnas.92.5.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhard M, Lehn N, Neumayer N, Bore´n T, Rad R, Schepp W, Miehlke S, Classen M, Prinz C. Clinical relevance of the Helicobacter pylori gene for blood-group antigen-binding adhesin. Proc Natl Acad Sci USA. 1999;96:12778–12783. doi: 10.1073/pnas.96.22.12778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregg KJ, Finn R, Abbott DW, Boraston AB. Divergent modes of glycan recognition by a new family of carbohydrate-binding modules. J Biol Chem. 2008;283:12604–12613. doi: 10.1074/jbc.M709865200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guruge JL, Falk PG, Lorenz RG, Dans M, Wirth HP, Blaser MJ, Berg DE, Gordon JI. Epithelial attachment alters the outcome of Helicobacter pylori infection. Proc Natl Acad Sci USA. 1998;95:3925–3930. doi: 10.1073/pnas.95.7.3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hage N, Howard T, Phillips C, Brassington C, Overman R, Debreczeni J, Gellert P, Stolnik S, Winkler GS, Falcone FH. Structural basis of Lewis(b) antigen binding by the Helicobacter pylori adhesin BabA. Sci Adv. 2015;1:e1500315. doi: 10.1126/sciadv.1500315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatakeyama M, Higashi H. Helicobacter pylori CagA: a new paradigm for bacterial carcinogenesis. Cancer Sci. 2005;96:835–843. doi: 10.1111/j.1349-7006.2005.00130.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogg PJ. Disulfide bonds as switches for protein function. Trends Biochem Sci. 2003;28:210–214. doi: 10.1016/S0968-0004(03)00057-4. [DOI] [PubMed] [Google Scholar]
- Holmner A, Askarieh G, ökvist M, Krengel U. Blood group antigen recognition by Escherichia coli heat-labile enterotoxin. J Mol Biol. 2007;371:754–764. doi: 10.1016/j.jmb.2007.05.064. [DOI] [PubMed] [Google Scholar]
- Ilver D, Arnqvist A, Ogren J, Frick IM, Kersulyte D, Incecik ET, Berg DE, Covacci A, Engstrand L, Bore´n T. Helicobacter pylori adhesin binding fucosylated histo-blood group antigens revealed by retagging. Science. 1998;279:373–377. doi: 10.1126/science.279.5349.373. [DOI] [PubMed] [Google Scholar]
- Ishijima N, Suzuki M, Ashida H, Ichikawa Y, Kanegae Y, Saito I, Borén T, Haas R, Sasakawa C, Mimuro H. BabA-mediated adherence is a potentiator of the Helicobacter pylori type IV secretion system activity. J Biol Chem. 2011;286:25256–25264. doi: 10.1074/jbc.M111.233601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang J, Blaser MJ. Bacterial populations as perfect gases: genomic integrity and diversification tensions in Helicobacter pylori. Nat Rev Microbiol. 2006;4:826–836. doi: 10.1038/nrmicro1528. [DOI] [PubMed] [Google Scholar]
- Kawakubo M, Ito Y, Okimura Y, Kobayashi M, Sakura K, Kasama S, Fukuda MN, Fukuda M, Katsuyama T, Nakayama J. Natural antibiotic function of a human gastric mucin against Helicobacter pylori infection. Science. 2004;305:1003–1006. doi: 10.1126/science.1099250. [DOI] [PubMed] [Google Scholar]
- Mahdavi J, Sonde´n B, Hurtig M, Olfat FO, Forsberg L, Roche N, Angstrom J, Larsson T, Teneberg S, Karlsson KA, et al. Helicobacter pylori SabA adhesin in persistent infection and chronic inflammation. Science. 2002;297:573–578. doi: 10.1126/science.1069076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makipour K, Friedenberg FK. The potential role of N-acetylcysteine for the treatment of Helicobacter pylori. J Clin Gastroenterol. 2011;45:841–843. doi: 10.1097/MCG.0b013e31822be4d6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollicone R, Bara J, Le Pendu J, Oriol R. Immunohistologic pattern of type 1 (Lea, Leb) and type 2 (X, Y, H) blood group-related antigens in the human pyloric and duodenal mucosae. Lab Invest. 1985;53:219–227. [PubMed] [Google Scholar]
- Pang SS, Nguyen STS, Perry AJ, Day CJ, Panjikar S, Tiralongo J, Whisstock JC, Kwok T. The three-dimensional structure of the extracellular adhesion domain of the sialic acid-binding adhesin SabA from Helicobacter pylori. J Biol Chem. 2014;289:6332–6340. doi: 10.1074/jbc.M113.513135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peek RM, Jr, Blaser MJ. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat Rev Cancer. 2002;2:28–37. doi: 10.1038/nrc703. [DOI] [PubMed] [Google Scholar]
- Polk DB, Peek RM., Jr Helicobacter pylori: gastric cancer and beyond. Nat Rev Cancer. 2010;10:403–414. doi: 10.1038/nrc2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posselt G, Backert S, Wessler S. The functional interplay of Helicobacter pylori factors with gastric epithelial cells induces a multi-step process in pathogenesis. Cell Commun Signal. 2013 doi: 10.1186/1478-811X-11-77. http://dx.doi.org/10.1186/1478-811X-11-77. [DOI] [PMC free article] [PubMed]
- Prinz C, Schöniger M, Rad R, Becker I, Keiditsch E, Wagenpfeil S, Classen M, Rösch T, Schepp W, Gerhard M. Key importance of the Helicobacter pylori adherence factor blood group antigen binding adhesin during chronic gastric inflammation. Cancer Res. 2001;61:1903–1909. [PubMed] [Google Scholar]
- Rautelin H, Blomberg B, Fredlund H, Järnerot G, Danielsson D. Incidence of Helicobacter pylori strains activating neutrophils in patients with peptic ulcer disease. Gut. 1993;34:599–603. doi: 10.1136/gut.34.5.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solnick JV, Hansen LM, Canfield DR, Parsonnet J. Determination of the infectious dose of Helicobacter pylori during primary and secondary infection in rhesus monkeys (Macaca mulatta) Infect Immun. 2001;69:6887–6892. doi: 10.1128/IAI.69.11.6887-6892.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subedi S, Moonens K, Romão E, Lo A, Vandenbussche G, Bugaytsova J, Muyldermans S, Borén T, Remaut H. Expression, purification and X-ray crystallographic analysis of the Helicobacter pylori blood group antigen-binding adhesin BabA. Acta Crystallogr F Struct Biol Commun. 2014;70:1631–1635. doi: 10.1107/S2053230X14023188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suerbaum S, Josenhans C. Helicobacter pylori evolution and phenotypic diversification in a changing host. Nat Rev Microbiol. 2007;5:441–452. doi: 10.1038/nrmicro1658. [DOI] [PubMed] [Google Scholar]
- Tan S, Noto JM, Romero-Gallo J, Peek RM, Jr, Amieva MR. Helicobacter pylori perturbs iron trafficking in the epithelium to grow on the cell surface. PLoS Pathog. 2011;7:e1002050. doi: 10.1371/journal.ppat.1002050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windle HJ, Fox A, Ní Eidhin D, Kelleher D. The thioredoxin system of Helicobacter pylori. J Biol Chem. 2000;275:5081–5089. doi: 10.1074/jbc.275.7.5081. [DOI] [PubMed] [Google Scholar]
- Younson J, O'Mahony R, Liu H, Basset C, Grant S, Campion C, Jennings L, Vaira D, Kelly CG, Roitt IM, Holton J. A human domain antibody and Lewis b glycoconjugate that inhibit binding of Helicobacter pylori to Lewis b receptor and adhesion to human gastric epithelium. J Infect Dis. 2009;200:1574–1582. doi: 10.1086/644596. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.