

Abstract
The Hsp100/Clp protease complexes of Bacillus subtilis ClpXP and ClpCP are involved in the control of many interconnected developmental and stress response regulatory networks, including competence, redox stress response, and motility. Here we analyzed the role of regulatory proteolysis by ClpXP and ClpCP in motility development. We have demonstrated that ClpXP acts on the regulation of motility by controlling the levels of the oxidative and heat stress regulator Spx. We obtained evidence that upon oxidative stress Spx not only induces the thiol stress response, but also transiently represses the transcription of flagellar genes. Furthermore, we observed that in addition to the known impact of ClpCP via the ComK/FlgM-dependent pathway, ClpCP also affects flagellar gene expression via modulating the activity and levels of the global regulator DegU-P. This adds another layer to the intricate involvement of Clp mediated regulatory proteolysis in different gene expression programs, which may allow to integrate and coordinate different signals for a better-adjusted response to the changing environment of B. subtilis cells.
Keywords: AAA+ proteins, regulatory proteolysis, ClpC, ClpX, ClpP, motility, Bacillus subtilis
Introduction
Hsp100/Clp proteases are compartmentalized protein degradation machines, which consist of a peptidase component (i.e., ClpP) and an AAA+ ATPase (i.e., ClpC or ClpX). The peptidase subunits are arranged in a barrel-like double heptamer with the catalytic residues on the inside surface of the structure. Folded proteins are excluded from the catalytic sites because they are too large to fit through the opening of the pore and are thus protected from proteolysis. The AAA+ ATPases form a hexameric ring with a narrow pore, which associates with one or both sides of the peptidase barrel. Specific substrate proteins can be recognized by the N-terminal ATPase domain, often facilitated by adaptor proteins, and are unfolded and threaded through the pore by the AAA+ ATPase motor into the peptidase chamber, where they are degraded (Kirstein et al., 2009; Sauer and Baker, 2011).
Hsp100/Clp proteases participate in general and regulatory proteolysis in the bacterial cell. For example, the ClpCP complex in Bacillus subtilis acts in protein quality control by degradation of unfolded, misfolded or aggregated proteins, which accumulate under stress conditions such as heat shock (Krüger et al., 2000; Schlothauer et al., 2003). Interestingly, the same protein complex plays an important part in developmental processes by controlled degradation of transcription factors like the competence master regulator ComK (Turgay et al., 1998), the class III heat shock repressor CtsR (Derré et al., 1999; Krüger et al., 2001; Kirstein et al., 2007) and the anti-anti sigma factor SpoIIAB involved in sporulation (Pan et al., 2001). ClpCP may also play a role in the processing of SlrR, a newly identified regulator of biofilm formation (Chai et al., 2010). ClpE is homologous to ClpC, with the exception of the N-terminal domain, which is homologous to the N-terminal domain of ClpX (Kirstein et al., 2009). ClpE appears to be important under severe heat shock conditions (Miethke et al., 2006).
An important regulatory substrate of the third B. subtilis Hsp100/Clp ClpXP protease is the thiol and oxidative stress transcription factor Spx (Nakano M. M. et al., 2002; Nakano S. et al., 2002). Under non-stress conditions, Spx is very efficiently turned over by ClpXP aided by the adaptor protein YpbH, resulting in a low steady state concentration of the protein. When cells encounter oxidative or heat stress, spx transcription is up-regulated (Helmann et al., 2001; Leelakriangsak et al., 2007). More importantly, the Spx protein is stabilized either by oxidative inactivation (Garg et al., 2009) or heat-mediated sequestration (Engman and von Wachenfeldt, 2015) of the adaptor protein YjbH, leading to rapid accumulation of the active regulator (Zuber, 2009; Runde et al., 2014).
Spx is a transcriptional regulator, which forms a complex with the C-terminal domain of the RNA polymerase alpha subunit (alpha-CTD; Nakano et al., 2003b; Newberry et al., 2005). By enhancing RNA polymerase interaction with certain promoters, Spx can serve as an activator i.e., of genes encoding enzymes required to cope with thiol oxidative stress (Nakano et al., 2003a; Reyes and Zuber, 2008). Interestingly, Spx can act as a transcriptional repressor on another group of genes (Nakano et al., 2003a,b). According to the interference model, genes, which require an activator that binds to the RNA polymerase alpha-CTD, are repressed because Spx competes with binding of the activators to the alpha-CTD (Nakano et al., 2003b; Zhang et al., 2006).
Another interesting process, in which Clp proteases appear to be involved, is the regulation of swimming motility in B. subtilis (Mukherjee and Kearns, 2014). Already during their initial characterization, clpP, clpC, and clpX mutant strains were reported to be non-motile (Rashid et al., 1996; Liu and Zuber, 1998; Msadek et al., 1998). However, the mechanisms, by which Clp proteases affect swimming motility, are currently only partially understood. Swimming or swarming bacterial cells are propelled by flagella, rotating filamentous helical structures, which are powered by an intra-membrane revolving motor. Gene regulation of flagellar assembly is a hierarchical process as described for Escherichia coli (Chevance and Hughes, 2008) and B. subtilis (Mukherjee and Kearns, 2014). No obvious flagellar master regulator such as FlhDC of E. coli has been identified in the B. subtilis genome, instead, the early flagellar genes (class II genes) are located in a single large fla/che operon (Márquez-Magaña and Chamberlin, 1994). This operon is transcribed by the σA housekeeping sigma factor (Kearns and Losick, 2005) and is modulated by a number of transcription factors including DegU (Amati et al., 2004; Tsukahara and Ogura, 2008), CodY (Bergara et al., 2003), and SwrA (Kearns and Losick, 2005; Calvio et al., 2008). The sigD gene encoding the alternative sigma factor σD is positioned close to the 3′-end of the fla/che-operon (Márquez-Magaña and Chamberlin, 1994; Cozy and Kearns, 2010). The class III or late flagellar genes include hag, which encodes flagellin, the major structural subunit of the flagellum. They are organized in separate transcriptional units controlled by σD-dependent promoters (Márquez et al., 1990). σD is inhibited by its anti-sigma factor FlgM, which is an important morphogenetic checkpoint synchronizing gene expression with the assembly of the flagella (Mirel et al., 1994; Fredrick and Helmann, 1996; Bertero et al., 1999; Chevance and Hughes, 2008; Mukherjee and Kearns, 2014; Calvo and Kearns, 2015).
How could regulatory proteolysis by Hsp/100Clp proteins act on motility development? Liu et al. could demonstrate that high ComK concentrations in clpC or mecA mutant cells result in a transcriptional read-through from comFA into flgM. This leads to over-production of FlgM, which inhibits σD and represses hag transcription and thus motility development (Liu and Zuber, 1998). However, another study has proposed a second comK-independent effect of a clpC mutant on motility (Rashid et al., 1996). In addition, the proteolysis substrates responsible for the effect of clpX on swimming motility are unknown to date.
Here, we analyzed the influence of regulatory proteolysis on swimming motility in detail and identified two transcriptional regulators, which inhibit swimming motility and are affected by Clp proteases. We found that ClpCP, in addition to its control of the ComK mediated induction of FlgM expression (Liu and Zuber, 1998), also affects DegU~P mediated inhibition of motility. Most interestingly, we observed that Spx, a proteolysis substrate of ClpXP, negatively regulates motility genes by an unknown, probably indirect mechanism. Thereby heat or oxidative stress signals sensed by ClpXP/Spx can result in a halt of motility in B. subtilis cells.
Materials and methods
General methods
B. subtilis cells were cultured in Luria–Bertani (LB) medium (5 g/l yeast extract, 10 g/l tryptone–peptone, 5 g/l NaCl) at 37°C if not otherwise indicated. Overnight cultures were inoculated from freshly streaked colonies and grown in LB medium in the presence of appropriate antibiotics (10 μg/ml chloramphenicol, 1 μg/ml erythromycin + 25 μg/ml lincomycin, 10 μg/ml kanamycin, 10 μg/ml tetracycline, or 100 μg/ml spectinomycin). Standard DNA manipulation was carried out as described previously (Sambrook et al., 2001). Protein concentrations were determined using the Bradford method (Bradford, 1976).
Cloning
Cloning was performed in E. coli XL-1 blue cells (Stratagene). Phusion High Fidelity DNA Polymerase (New England Biolabs) was used for PCR amplifications. Chromosomal DNA was used as a template. Restriction enzymes and T4 DNA Ligase were obtained from Fermentas. Primer sequences are listed in Table S3, plasmids are listed in Table S2.
Plasmids pQE60-hag and pQE60-spx were constructed by amplification of the hag or spx genes using primers hagpQE60-for and hagpQE60-rev or spxpQE60-for and spxpQE60-rev, respectively and cloning into plasmid pQE60 (Qiagen) using the NcoI and BamHI restriction sites. Plasmids pX-hag1 and pX-hag4 were obtained by PCR amplification using primers hag1-for and hag1-rev or hag4-for and hag4-rev, respectively, BamHI digestion and ligation into BamHI-digested pX plasmid. For plasmid pflgB152, the flgB152 promoter fragment was amplified using primers flgB152-for and flgB152-rev and cloned into plasmid pDG268 using the EcoRI and BamHI restriction sites. All plasmids were sequenced.
Transformation/strain construction
All strains used in this study are described in Table S1. Transformation with chromosomal DNA or plasmid DNA was performed by a standard method (Anagnostopoulos and Spizizen, 1961). Strains BNM421 and BNM426 were constructed by transformation of strain BNM126 (Δhag) with plasmids pX-hag1 or pX-hag4, respectively. BNM421 expresses the hag gene from the xylose-inducible Pxyl promoter. In BNM426, a hag fragment, comprising 92 bases upstream and 32 bases downstream of the open reading frame, is under the control of Pxyl. Strain BNM426 complemented the swimming motility defect of the Δhag mutant in the presence of xylose.
BNM109 was constructed using the technique of long-flanking homology PCR as described previously (Wach, 1996) with the primers listed in Table S3. Strains BNM126 and BNM149 were constructed as described using plasmids pMADhag and pMADcomK as described previously (Arnaud et al., 2004; Blair et al., 2008). ΔclpP, ΔclpC, and ΔclpX mutants were obtained by transformation of the recipient strains with chromosomal DNA from strains BNM103, BNM105, and BNM106, respectively. Strains BNM350 and BNM351 were constructed by transformation of strain BNM111 (Δspx::kan) with plasmids pMMN521 or pSN56 (Nakano et al., 2003a), respectively. Strain BNM810 was acquired by transformation of the wild type strain with plasmid pSN56. The strains BNM1266, BNM1268, and BNM 1270 were constructed by transforming the B. subtilis168 swrA+ degQ+ (Gift of Nicola Stanley-Wall) with chromosomal DNA prepared from BNM103, 105, or 109 (Table S1).
To obtain strain BNM866, chromosomal DNA from strain ABH282, featuring a second copy of amyE at the ywrK gene locus (Camp and Losick, 2009), was first transformed into the wild type strain 168, resulting in strain BNM860. BNM860 was then transformed with plasmid pSN56 (Nakano et al., 2003a) selecting for spectinomycin resistance and chloramphenicol sensitivity, which indicates integration of the spxDD construct into the ywrK::amyE locus (BNM866). This strain produced Spx protein after isopropyl-β-D-thiogalactopyranoside (IPTG) induction, as verified by Western blot analysis (data not shown). Finally, to yield strains BNM878 and BNM1001, strain BNM866 was transformed with chromosomal DNA from strains BNM301 (amyE::PflgB-lacZ cat) or BNM328 (amyE::Phag-lacZ cat) selecting for chloramphenicol and spectinomycin resistance, indicating integration of the PflgB-lacZ or Phag-lacZ constructs at the original amyE locus, while the spxDD construct remains at the ywrK:amyE locus. We confirmed by Western blot analysis that the resulting strains produce Spx protein in response to IPTG induction (data not shown).
Pulse chase labeling and immunoprecipitation
Cells were grown in Belitsky minimal medium [50 mM Tris-(hydroxymethyl)-amino methane (Tris) -HCl pH 7, 5, 15 mM (NH4)2 SO4, 8 mM MgSO4, 27 mM KCl, 7 mM sodium citrate, 0.6 mM KH2PO4, 2 mM CaCl2, 160 μg/ml L-tryptophan, 10 μM MnSO4, 1 μM FeSO4, 4.5 mM potassium glutamate, 0, 2% glucose] to OD600 0.7 at 37°C. 3.5 ml bacteria were removed and pulse labeled with 30 μCi L-35S-methionine for 10 min at 37°C. Subsequently, cold L-methionine (0.3 M) was added in 30-fold excess and samples were taken after the indicated incubation times and mixed with trichloroacetic acid (TCA) to a final concentration of 10% w/v. TCA-precipitated samples were incubated on ice for 10 min and centrifuged for 15 min at 17,000 g and 4°C. The pellets were washed twice in 1 ml acetone, air-dried and resuspended in 20 μl lysis buffer (50 mM Tris-HCl pH 7.5, 5 mM EDTA, 1 mM PMSF, 4 mg/ml lysozyme). The samples were boiled for 3 min at 95°C. Two hundred and seventy microliters KI buffer (50 mM Tris-HCl pH 8, 150 mM NaCl, 0.5% Triton X-100, 1 mM PMSF) was added and the samples were incubated on ice for 15 min. Precipitate was separated by centrifugation for 15 min at 17,000 g at 4°C. Hundred microliters of the supernatant were mixed with 2 μl polyclonal anti-Hag antiserum and incubated over night at 4°C for immunoprecipitation. The next day, 8 μl Protein A Magnabeads (Thermo Scientific) were added to the solution and mixed. The magnetic beads were washed twice in 200 μl KI buffer. Subsequently, the magnetic beads were resuspended in SDS sample buffer, boiled for 3 min at 95°C and applied to 12.5% SDS PAGE gels. Electrophoresis was performed at 25 mA per gel for 1 h and the gels were vacuum dried on Whatman paper for 2 h at 85°C. The dried gels were placed on a phosphoimager screen for 24 h and screens were scanned using a Fla 2000 phosphoimager (Fujifilm, Japan).
Motility assay
Overnight cultures grown in LB medium at 37°C were diluted to OD600 2.0 in fresh LB medium and 3 μl were applied to tryptone agar plates containing 0.3% w/v agar (bacteriology grade, Carl Roth), 10 g/l tryptone/peptone and 5 g/l NaCl. The plates were incubated at 37°C for 8 h. The growth behavior of all examined strains in liquid culture with the tryptone salt medium (utilized for the swimming plates) was comparable to growth in LB (data not shown).
Protoplast preparation
Thirty milliliters of a growing B. subtilis culture were harvested by centrifugation and the pellets were washed twice in 1 ml STM (50 mM Tris-HCl pH 8, 50 mM NaCl, 5 mM MgCl2, 25% w/v sucrose). Subsequently, the pellets were re-suspended in 200 μl STM buffer + 0.3 mg/ml lysozyme and incubated for 30 min at 37°C to obtain protoplasts. The protoplasts were washed twice in 1 ml STM and then lysed by resuspension in 200 μl TM buffer without sucrose (50 mM Tris-HCl pH 8, 50 mM NaCl, 5 mM MgCl2) containing 10 μg/ml DNase I and 10 μg/ml RNase A and incubated on ice for 30 min. The lysate was centrifuged for 20 min at 17,000 g and 4°C and the supernatant was transferred to a fresh tube. Total protein concentration was determined by the Bradford assay.
Whole cell preparation
One milliliter of a growing B. subtilis culture was harvested by centrifugation and the pellet was washed twice in 1 ml STM (50 mM Tris-HCl pH 8.0, 50 mM NaCl, 5 mM MgCl2, 25% w/v sucrose) + 0.3 mg/ml lysozyme and incubated for 5 min at 37°C. 6x SDS sample buffer containing SDS and DTT was added and the samples were boiled at 95°C for 5 min. The samples were centrifuged for 5 min at 17,000 g prior to SDS–PAGE separation.
Western blot analysis
Polyclonal antibodies against Hag and Spx were produced in rabbits by inoculation with purified Hag-His6 or Spx-His6 (Pineda Antibody Services, Berlin, Germany). Rabbit-anti-Spx antibodies for initial experiments were kindly provided by Peter Zuber (University of Oregon). SigD antibodies from rabbit were kindly provided by John Helmann (Cornell University), rabbit-anti-CodY antibodies from Linc Sonenshein (Tufts University), and sheep-DegU antibodies from Nicola Stanley–Wall (University of Dundee).
Lysates from protoplasts were adjusted to equal concentration and 2.5–10 μg per lane total protein were loaded onto 12.5 or 15% SDS-gels and separated by electrophoresis. Gels were blotted onto PVDF membranes in 20 mM Tris-HCl pH 8.3, 150 mM glycine, 20% v/v methanol using a semi-dry blotting chamber. Blot membranes were blocked in TBS-M (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 5% w/v skim milk) and incubated with antisera diluted in TBS-M. Antisera were used at dilutions of 1:40,000 (anti-Hag), 1:5000 (anti-DegU, anti-SigD, anti-Spx), or 1:10,000 (anti-CodY). The blots were washed in TBS buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl) and incubated with secondary anti-rabbit (GE Healthcare) or anti-sheep (Sigma Aldrich) antibodies conjugated to alkaline phosphatase diluted 1:10,000 in TBS-M. The blot membranes were then equilibrated in AP buffer (100 mM Tris-HCl pH 9.5, 100 mM NaCl, 5 mM MgCl2) and developed using ECF Western Blotting Reagent (GE Healthcare). The fluorescence signals were scanned using a Fla 2000 phosphoimager (Fujifilm, Japan).
β-galactosidase assay
One to five milliliters samples of a growing B. subtilis carrying a lacZ fusion were collected and harvested by centrifugation for 5 min at 17,000 g and frozen at −20°C. For the β-galactosidase measurement, the cell pellets were thawed on ice and resuspended in 500 μl Z buffer (100 mM NaPO4 pH 7.0, 1 mM MgSO4, 100 mM β-mercaptoethanol). Ten microliters toluene was added and the samples were thoroughly mixed and incubated on ice for 30 min. For the assay, the samples were diluted 4-fold and transferred into a flat-bottom 96 well plate (final volume 200 μl). The reaction was started by addition of 50 μl ONPG (4 mg/ml in Z buffer) using an 8-channel multi-pipette (Eppendorf). Absorbance at 420 nm was measured every 60 s for 15 min at room temperature using a microplate reader (Tecan Instruments). The β-galactosidase activity (in Miller Units) was calculated from the linear slope of the absorbance at 420 nm over time correcting for the sample path length in the microplates. For comparison of β-galactosidase activities of strains exhibiting lag phases in growth (i.e., ΔclpP and ΔclpX mutants), the time axis was normalized to T0, the point of deviation from exponential growth.
SpxDD induction
Strains BNM351 (Δspx::kan amyE::PHy-spxDD) and BNM350 (Δspx::kan amyE::PHy-spx) were grown in LB medium at 37°C to OD600 0.3. Subsequently, the culture was split and expression of spxDD was induced by addition of 1 mM IPTG to one half of the culture. Samples were withdrawn before addition of ITPG (0 min), 30 and 60 min thereafter and total RNA or total protein were prepared as described above for Northern or Western blot analysis.
Thiol oxidative stress experiments
A growing culture was divided in early exponential phase and 1 mM N,N,N′,N′-Tetramethyl-azodicarboxamide (diamide) was added to half of the culture to induce thiol oxidative stress. Samples were removed before addition of diamide at the indicated time points and β-galactosidase activity was determined. Cell lysates of the same samples were analyzed by SDS–PAGE and Western blot against Spx.
Northern blot analysis
All buffers used for RNA work were treated with 0.1% diethyl pyrocarbonate (DEPC) and autoclaved (121°C, 20 min). RNA was prepared from 30 ml B. subtilis cultures using the FastRNA Pro Blue kit (MP Biochemicals). Lysis was performed by shaking in a Retsch mill for 10 min at 1800 rpm. The RNA was digested with RNase free DNase I (Roche Applied Sciences) to remove contaminating DNA and subsequently purified by phenol–chloroform extraction and ethanol precipitation. The RNA concentration was determined by absorbance measurement at 260 nm using a NanoDrop spectrophotometer (Peqlab).
RNA samples were diluted in DEPC treated H2O, mixed 1:1 with 2x RNA sample buffer (60 mM MOPS–NaOH pH 7.0, 0.02% w/v Bromphenol blue, 75% v/v formamide, 3.33% w/v formaldehyde, 3% w/v Ficoll 70) and heated to 65°C for 10 min. Ten microliters RNA Molecular Weight Marker III (Roche Applied Sciences) was applied to the gel as a size standard. The RNA was separated by electrophoresis on 1.2% w/v agarose gels in 40 mM MOPS–NaOH pH 7.0, 5 mM sodium acetate, 1 mM EDTA, 0.1% w/v diethyl pyrocarbonate (DEPC) and 37% v/v formaldehyde for 2 h and 45 min at 80 V. The gel was rinsed with 20x SSC (300 mM tri-sodium citrate pH 7.0, 3 M NaCl, 0.1% DEPC) and vacuum blotted onto a positively charged nylon membrane (Roche Applied Sciences) in 10x SSC (150 mM tri-sodium citrate pH 7.0, 1.5 M NaCl, 0.1% DEPC) for 1.5 h at 5 mm Hg pressure. UV crosslinking was performed for 10 min at 328 nm. Subsequently, the blot was stained in methylene blue solution (0.02% w/v methylene blue, 300 mM sodium acetate pH 5.5, 0.1% DEPC) for 5 min to visualize ribosomal RNAs as a control for equal sample application and blotting. The membrane was destained in Bleaching buffer (0.2x SSC 1% w/v SDS, 0.1% DEPC) and equilibrated in 2x SSC (30 mM tri-sodium citrate pH 7.0, 0.3 M NaCl, 0.1% DEPC).
Digoxigenin (DIG) labeled DNA probes were prepared by PCR using PCR DIG labeling mix containing DIG-dUTP (Roche Applied Sciences). PCR was performed with Phusion High Fidelity DNA polymerase (New England Biolabs) using primers hag-probe-for and hag1-rev (see Table S3). A first round of PCR was performed with chromosomal DNA as a template in the absence of DIG labeling mix. The product of this reaction was used as a template for a second round of PCR in the presence of DIG labeling mix. The PCR products were purified by gel extraction using the ZymoClean™ Gel DNA recovery kit (Hiss Diagnostics) and eluted in 20 μl DEPC treated H2O. The probes were denatured for 5 min at 95°C and cooled rapidly on ice.
The nylon membrane was transferred to a hybridization glass tube and incubated with 20 ml DIG Easy Hybridization solution (Roche Applied Sciences) for 1 h at 47°C with rotation in a hybridization oven. Twenty microliters DIG-labeled probe (100 ng/μl) was diluted in 20 ml DIG Easy Hybridization solution and the membrane applied to the blot over night at 47°C. The blot was washed twice with 20 ml wash buffer 1 (0.1% w/v SDS, 30 mM tri-sodium citrate pH 7.0, 0.3 M NaCl, 0.1% v/v DEPC) for 5 min at 47°C and twice with wash buffer 2 (0.1% w/v SDS, 1.5 mM tri-sodium citrate pH 7.0, 15 mM NaCl, 0.1% v/v DEPC) for 30 min at 47°C.
The blot membrane was blocked in Blocking buffer [100 mM maleic acid pH 7.5, 150 mM NaCl, 1% w/v Blocking reagent (Roche Applied Sciences)] for 30 min at room temperature. Anti-digoxigenin antibodies conjugated to alkaline phosphatase (Roche Applied Sciences) were diluted 1:5000 in Blocking buffer and applied to the blot for 1.5 h at room temperature. Subsequently, the blot was washed twice for 15 min in Detection buffer 1 (100 mM maleic acid pH 7.5, 150 mM NaCl) and equilibrated in Detection buffer 2 (100 mM Tris-HCl pH 9.5, 100 mM NaCl, 5 mM MgCl2) for 2 min. CDP Star solution (Roche Applied Sciences) was applied to the blot and the signal was detected using X-ray films.
Electrophoretic mobility shift assays
DNA probes were produced by PCR amplification from chromosomal DNA using the primer sets flgB (−209 to −6)-for/flgB (−209 to −6)-rev flgB (−106 to 98)-for/flgB (−106 to 98)-rev and flgB (−1 to 203)-for/flgB (−1 to 203)-rev (see Table S3) and purified by gel extraction.
Fifty nanograms of the DNA probes were mixed with purified Spx-His6 at 1.25, 2.5, 5 μM protein concentration in TSM buffer (10 mM Tris-HCl pH 7.5, 1 mM EDTA, 5% v/v glycerol, 1 mM MgCl2, 10 mM NaCl) in the presence of 1 μg poly-d(I–C; Roche Applied Sciences) and incubated for 20 min at room temperature. Subsequently, the samples were applied to 5% w/v polyacrylamide gels and electrophoresis was performed for 2 h at 80 V in TSM buffer. The gels were stained with ethidium bromide and bands were visualized by UV illumination.
Results
Clp proteases affect regulation of swimming motility
We examined the swimming motility of wild type and clp mutant B. subtilis cells and confirmed that clpP and clpC mutant strains exhibit a defect in swimming motility (Rashid et al., 1996; Liu and Zuber, 1998; Msadek et al., 1998). In addition we observed that a clpX mutant is non-motile, whereas a clpE mutant displayed similar motility to the wild type (Figure 1A).
Figure 1.
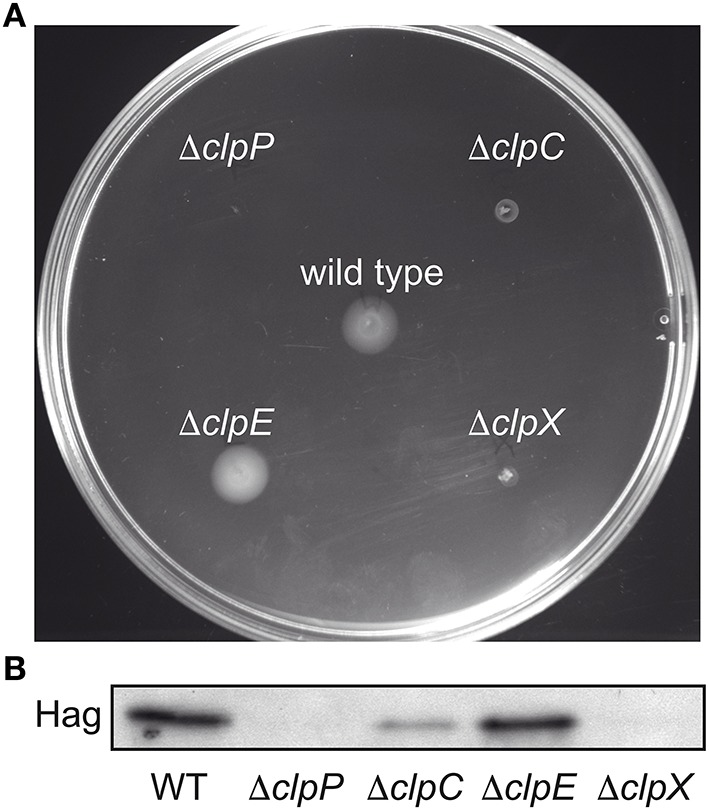
Clp proteases affect swimming motility and Hag protein levels in B. subtilis. (A) Motility assay of B. subtilis wild type and ΔclpP::spec (BNM103), ΔclpC::tet (BNM105), ΔclpE::spec (BNM106), and ΔclpX::kan (BNM107) mutants on 0.3% w/v agar plates. (B) Cells of the indicated strains (as in A) were grown to OD600 1.0 at 37°C. Cell lysates were analyzed by SDS–PAGE and Western blotting using anti-Hag antibodies.
We observed that the overall Hag protein level, the major Flagellin protein in B. subtilis, appears to be relatively stable, judging by in vivo by pulse chase experiments following immuno-precipitation in wild type B. subtilis cells (Figure S1). When we examined Hag levels in Clp mutant strains we observed very low Hag levels in clpC (Rashid et al., 1996) and no detectable Hag protein in clpX and clpP mutant strains (Figure 1B).
To investigate whether the observed effect of the mutants on Hag protein level is reflected in the mRNA levels of flagellar transcripts, we performed Northern blot experiments using a probe against the hag transcript. We observed that hag mRNA levels were strongly reduced in clpP, clpC, and clpX mutant strains (Figure S2). We noted an additional band of ~500 bases hybridizing with our probe in the clpC and hag mutants (Figure S2). This might be explained by an upregulation of the shorter B. subtilis yvzB transcript, which encodes a smaller homolog of Hag, in these mutants.
We conclude from these data that the absence of Clp proteases resulted in a swimming motility defect and strongly diminished Hag protein and transcript levels (Figure 1 and Figure S2), suggesting that ClpCP and ClpXP indirectly affect Hag protein levels, i.e., by regulatory proteolysis of transcriptional regulators controlling the synthesis of Hag.
Clp proteases regulate transcription from the flgB promoter
We first aimed to determine, which process in flagellar biogenesis is affected by the clp mutations. To this end, we performed reporter gene assays using transcriptional fusions of flagellar promoters to lacZ to elucidate whether transcription initiation from these promoters is altered in clp mutant strains. One construct (PflgB-lacZ) contains the upstream sequence of the fla/che operon from residues −479 to +47 relative to the transcriptional start site of the σA-dependent flgB promoter (PA) and is indicative of flagellar class II gene expression (Kearns and Losick, 2005). The minor σD-dependent promoter, which does not influence flagellar gene expression (Kearns and Losick, 2005), is also present in this sequence (PD3). To monitor the σD-dependent class III genes we used a transcriptional lacZ fusion to the hag promoter (Phag-lacZ) fusion (Kearns and Losick, 2005). All lacZ fusions were integrated into the ectopic amyE locus. We introduced clp mutations into these strains and determined β-galactosidase activities of samples along the growth curve.
In the wild type, both PflgB-lacZ and Phag-lacZ expression displayed the typical pattern of flagellar genes with a peak in post exponential phase (Mirel and Chamberlin, 1989; Figures 2A,B). Notably, in the clpP and clpX mutants, but not the clpC mutant, PflgB-lacZ activity was strongly reduced throughout growth (Figure 2A). The Phag-lacZ fusion was strongly down-regulated in the clpP, clpC, and clpX mutants (Figure 2B).
Figure 2.

Motility genes are down-regulated in B. subtilis clp mutant cells and the role of clpC and degSU. (A) β-galactosidase assays of the indicated strains carrying a PflgB-lacZ fusion. Circles: wild type 168 (BNM301), squares: ΔclpP::spec (BNM302), triangles: ΔclpC::tet (BNM303), diamonds: ΔclpX::kan (BNM305). Representative data from at least two independent experiments are shown. A schematic drawing of the promoter-lacZ fusion is depicted at the bottom. (B) Same as (A) for the hag promoter lacZ fusion. Circles: wild type 168 (BNM328), squares: ΔclpP::spec (BNM329), triangles: ΔclpC::tet (BNM330), diamonds: ΔclpX::kan (BNM332). (C) Cells of strains wild type, BNM103 (ΔclpP), BNM105 (ΔclpC), BNM107 (ΔclpX) were grown to OD600 1.0 at 37°C. Cell lysates were analyzed by SDS–PAGE and Western blotting using anti-SigD (D) β-galactosidase assays of the indicated strains carrying a PflgB152-lacZ fusion Circles: wild type 168 (BNM346), triangles: ΔclpC::tet (BNM347), diamonds: ΔdegSU::spec (BNM348), inverted triangles: ΔclpC::tet ΔdegSU::spec (BNM349). Representative data from at least two experiments are shown. A schematic drawing of the promoter-lacZ fusion is depicted at the bottom. (E) Same as (D) for the hag promoter lacZ fusion. Circles: wild type (BNM328), triangles: ΔclpC (BNM330), diamonds: ΔdegSU (BNM333) and inverted triangles: ΔclpC ΔdegSU (BNM338). (F) Cells of the wild type and strains BNM105 (ΔclpC), BNM138 (ΔdegSU), and BNM140 (ΔclpC ΔdegSU) were grown to OD600 1.0 at 37°C. Cell lysates were analyzed by SDS–PAGE and Western blotting using anti-Hag antibodies. (G) Cells were grown at 37°C to T0 (time of deviation from exponential growth) and cell lysates of the wild type and strains BNM103 (ΔclpP), BNM105 (ΔclpC), BNM106 (ΔclpE), BNM107 (ΔclpX), and BNM138 (ΔdegSU) were analyzed by SDS–PAGE followed by Western blotting using anti-DegU antibodies.
These results indicate that ClpXP affects transcription from the flgB promoter, whereas the lack of ClpC might affect hag promoter activity. However, as an additional control, we performed Western blots to determine the protein levels of the flagellar sigma factor σD, which is encoded in the fla/che operon and directly activates transcription from the hag promoter. As expected, clpP and clpX mutants exhibited lower σD protein levels, but the same was true for the clpC mutant (Figure 2C), even though clpC had no apparent effect on the activity of the tested PflgB-lacZ fusion (Figure 2A), suggesting that either clpC acts on σD post-transcriptionally or that additional elements near the fla/che promoter might be required for the observed down-regulation.
However, it was previously observed that the flgB promoter features two DegU binding sites, one located upstream of the promoter (BR1) and one downstream in the flgB coding region (BR2; Tsukahara and Ogura, 2008). The second BR2 element is not encoded in the flgB promoter LacZ fusion we used so far. Therefore, we constructed a longer lacZ fusion that included 17 additional bases upstream of the flgB start codon along with 88 bases of the flgB coding sequence (PflgB152-lacZ, see Materials and Methods and Figure 2D) including this additional DegU binding site (Tsukahara and Ogura, 2008). In the wild type background, this lacZ fusion displayed a similar expression pattern as the PflgB-lacZ construct, but peak expression was about three fold higher (Figure 2D). In addition, the clpC mutation had a strong negative effect on lacZ expression from this construct (Figure 2D). This suggests a possible role of DegU in the observed inhibition of swimming motility in the clpC mutant strain. In summary, our results suggest that transcription from the flgB promoter is strongly down-regulated in the clpP, clpC, and clpX mutants (Figures 2A,D), which results in lower protein levels of the flagellar sigma factor σD (Figure 2C). The lowered level of σD causes reduced transcription from the hag promoter (Figure 2B), lower Hag protein levels (Figure 1B) and reduced swimming motility (Figure 1A).
To further examine if the reduced Hag protein levels in clp mutants are solely a consequence of altered hag transcription, we uncoupled Hag production from σD regulation by placing a copy of hag downstream of the xylose-inducible Pxyl promoter at the ectopic amyE locus of a hag deletion mutant (Figure S3, see Section Materials and Methods). Interestingly, Hag levels were completely restored to wild type levels in the clpC background, implying that clpC acts on motility genes upstream of the hag promoter. In contrast, Hag levels were only partially restored in the clpP and clpX mutants, suggesting an additional effect of clpP and clpX on hag transcript or Hag protein levels downstream of transcription initiation from the hag promoter (Figure S3).
ClpC influences swimming motility through ComK and DegU
Controlled proteolytic degradation of a regulatory protein appears to be an important mechanism, by which Clp proteases can influence gene expression as demonstrated e.g., for the control of competence development (Kirstein et al., 2009; Battesti and Gottesman, 2013). Liu and Zuber previously described a pathway, by which ClpCP regulates swimming motility through ComK, which positively influences the transcription of FlgM. Briefly, ComK activates competence genes, among them comFA, which is located directly upstream of flgM on the chromosome. In the absence of clpC, more FlgM is produced by transcriptional readthrough. FlgM inhibits σD activity, leading to decreased hag expression and reduced motility (Liu and Zuber, 1998).
To confirm that the reduced swimming motility of the clpC mutant is due to raised ComK levels, we tested the motility and Hag protein levels of the clpC comK double mutant strain. As shown in Figure S4, the comK mutation partially suppressed the swimming motility and Hag production defect of the clpC mutant. These results indicate that part of the motility defect of a clpC mutant is due to higher levels of ComK and supports the read-through transcription of flgM as suggested by Liu and Zuber (Liu and Zuber, 1998). However, FlgM is unlikely to play a part in the down-regulation of flgB promoter activity because FlgM specifically inhibits σD (Caramori et al., 1996), whereas PflgB transcription is independent of σD (Kearns and Losick, 2005).
Therefore, we examined other known repressors of the fla/che operon. One candidate is DegU, which can act as a repressor of the fla/che operon in its phosphorylated form (Amati et al., 2004). In addition, the DNA element between positions +48 and +152 relative to the transcription start site of the flgB promoter, which is required for the clpC mediated down-regulation of fla/che transcription (Figure 2D), contains a DegU~P binding site (Tsukahara and Ogura, 2008) and DegU~P has been described as a possible ClpCP substrate (Ogura and Tsukahara, 2010).
We tested whether DegU levels can be elevated in a clpC mutant. To this end we performed DegU Western blots in wild type and clp mutant strains at different time points during growth. Notably, only between T0 and T2, at a time when cells are motile and expressing flagellar genes, we detected mildly increased levels of DegU in the clpC and to a lesser extent in the clpP mutants compared to the wild type (Figure 2G and Figure S5).
In order to test whether degU is responsible for the motility defect of the clpC mutant, we constructed a clpC degSU double mutant and tested swimming motility, Hag protein levels and motility gene expression of this strain. In this mutant, degU is deleted along with the degS gene, which encodes its cognate sensor kinase DegS. Indeed, the degSU mutation suppressed the swimming defect of the clpC mutant (Figure 3) and restored Hag production almost to wild type levels (Figure 2F), whereas mutation of degSU alone did not influence swimming and Hag concentration. Furthermore, β-galactosidase activity of the flgB-lacZ and to a little lesser extent of the hag-lacZ promoter reporter fusions was restored in the clpC degSU double mutant compared to the clpC mutant, but was significantly more similar to the wild type in the degSU mutant (Figures 2D,E). This suppression was specific to clpC, as the degSU mutation did not suppress down-regulation of the hag-lacZ promoter fusion and swimming motility in the clpX mutant (Figure S6). These results suggest that ClpC negatively influences DegU repressor activity.
Figure 3.

A degSU mutation suppresses the swimming motility defect of a B. subtilis clpC mutant strain. Motility assay of the wild type and strains BNM105 (ΔclpC), BNM138 (ΔdegSU), and BNM140 (ΔclpC ΔdegSU).
We observed a growth dependent mildly raised level of DegU in a clpC and to a lesser extent in a clpP mutant strain (Figure S5), which is consistent with the hypothesis that under specific conditions ClpC could inhibit or ClpCP could also degrade DegU-P (Ogura and Tsukahara, 2010) and that, in the absence of clpC, active DegU-P can accumulate and thereby represses transcription from the flgB promoter (Figure 2D).
Influence of the repressor cody on motility
The repressor CodY, which can sense GTP and branched chain amino acids, has been reported to bind to the flgB and hag promoters (Bergara et al., 2003; Ababneh and Herman, 2015). However, it was also observed that a codY deletion did not influence motility and fla/che expression (Amati et al., 2004) and a recent study, investigating the genome wide CodY binding sites did not detect CodY binding sites for controlling the flgB promoter (Belitsky and Sonenshein, 2013).
We could only detect small differences in motility of a codY mutant strain compared to wild type cells (Figure S7B) and we could not detect differences in the levels of cellular CodY protein in clpC, clpX, clpE, or clpP strains (Figure S7A). These results suggest that under our experimental conditions and in our strain background neither ClpCP nor ClpXP strongly influence motility via CodY.
ClpXP regulate swimming motility through Spx
According to the data presented above, motility genes are strongly down-regulated in the clpX and clpP mutants. Interestingly, these mutants are phenotypically distinct from the clpC mutant: for example, the shorter PflgB-lacZ fusion was down-regulated in clpX and clpP mutants, but not in the clpC mutant. Furthermore, our data indicate that clpX and clpP act on swimming motility independently of degU (Figure S6). Therefore, we assumed that distinct substrates of ClpCP and ClpXP regulate motility.
Both clpP and clpX mutants have a slow growth phenotype, which leads to frequent acquisition of second site suppressor mutants. We isolated such suppressor mutants, which could be easily identified by larger colony size on plates and loss of the characteristic lag phase during growth in liquid medium (Figure S8A). Interestingly, we noticed that this strain was only slightly less motile than the wild type (Figure S8B) and produced wild type levels of Hag protein (Figure S8C). One well-characterized suppressor mutation of clpX and clpP mutants is a loss of function mutation in the spx gene, which relieves the detrimental effect of raised levels of the ClpXP substrate Spx (Nakano et al., 2001). We analyzed Spx levels by Western blot using polyclonal Spx antibodies and detected only very low levels of Spx in the wild type strain and no Spx in the clpP suppressor mutant, whereas Spx accumulated to high levels in a freshly transformed clpP mutant (Figure S8C). This strongly suggested that our isolated suppressor mutant of clpP is phenotypically similar to a spx mutant.
We therefore tested swimming motility and Hag levels in a clean clpX spx double deletion mutant. Interestingly, the spx mutation resulted in increased motility on swim plates, whereas cellular Hag levels were similar to the wild type strain in this mutant (Figures 4A,B). Furthermore, the spx mutant suppressed the swimming motility defect of the clpX mutant and restored Hag production to wild type levels (Figures 4A,B). In addition, the activity of the PflgB-lacZ and Phag-lacZ fusions was partially restored in the clpX spx double mutant (Figures 5A,B), suggesting that the flgB promoter is to a large extent regulated by clpX via spx. The spx single mutant was significantly more similar to the wild type in these reporter gene assays (Figures 5A,B). Interestingly, we observed that the Hag levels were also restored in clpX spx mutant in a strain with xylose-controlled hag expression (Figure S3D), implying that the observed additional posttranscriptional effect of clpX on hag is spx-dependent.
Figure 4.
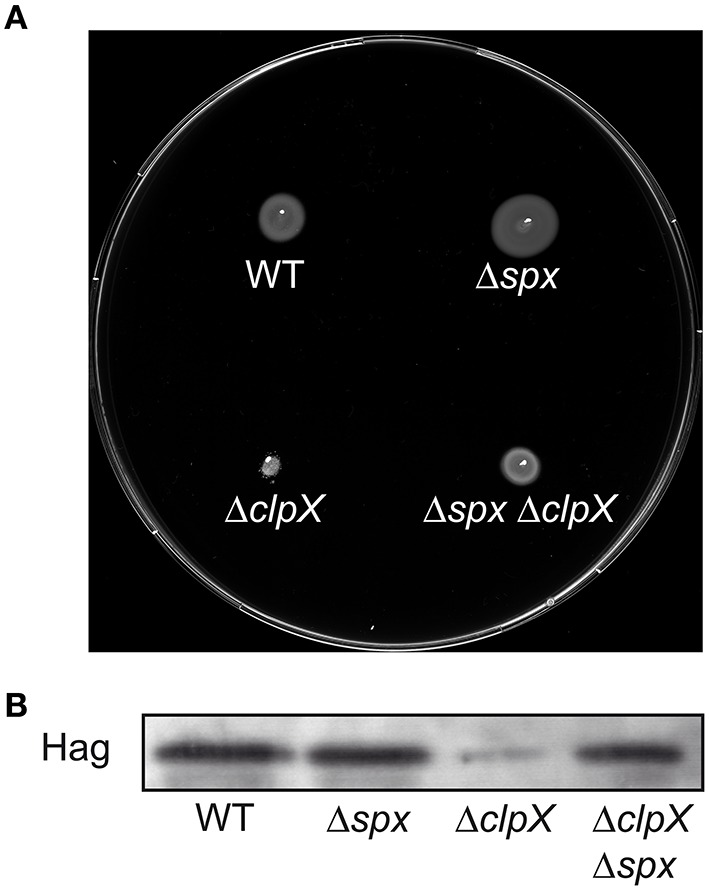
spx suppresses the swimming motility defect of a B. subtilis clpX mutant strain. (A) Motility assay of the wild type and strains BNM107 (ΔclpX), BNM111 (Δspx), and BNM112 (ΔclpX Δspx). (B) Cells of the indicated strains (as in A) were grown to OD600 1.0 at 37°C. Cell lysates were analyzed by SDS–PAGE and Western blotting using anti-Hag antibodies.
Figure 5.

clpX affects motility gene expression via spx. (A) β-galactosidase assays of the indicated strains carrying a PflgB-lacZ fusion. Circles: wild type 168, triangles: ΔclpX::kan (BNM305), diamonds: Δspx::kan (BNM307), inverted triangles: Δspx::kan ΔclpX::spec (BNM308). Representative data from 2 to 3 experiments are shown. (B) Same as (A) for the hag promoter lacZ fusion. Circles: wild type 168, triangles: ΔclpX::kan (BNM332), diamonds: Δspx::kan (BNM334), inverted triangles: Δspx::kan ΔclpX::spec (BNM335).
Spx negatively regulates motility genes
These results indicate that the reduced swimming motility of the clpX mutant might be caused by the presence of Spx, which negatively regulates the flgB promoter. To test whether Spx is able to inhibit motility also in a clpX+ background, we utilized a strain, in which a stabilized Spx variant (SpxDD) that can no longer be degraded by ClpXP, is encoded at the amyE locus under the control of an IPTG-inducible promoter (Nakano et al., 2003a). Notably, this strain was no more motile in the presence of IPTG, while an additional induction of wild type Spx had no effect on motility (Figures 6A,B), indicating that a raised level of Spx negatively regulates swimming motility.
Figure 6.

Spx acts as a negative regulator of motility genes. (A) Motility assay of the indicated strains [wild type: WT, BNM111: Δspx, BNM810: PHy-spxDD, BNM350 (Δspx PHy-spx), BNM351 (Δspx PHy-spxDD)]. Representative data from at least two experiments are shown. (B) Same as (A) using a swim agar plate containing 0.1 mM IPTG to induce spxDD. (C) A B. subtilis strain carrying both a transcriptional flgB promoter lacZ fusion at the ywrK locus and an IPTG-inducible copy of spxDD at the amyE locus (BNM878) was grown in LB medium at 37°C and induced with 0.1 mM IPTG in early exponential phase (OD600 0.2–0.3). β-galactosidase activity was measured at the indicated time points. Circles: control without IPTG, squares: induced with 0.1 mM IPTG, inverted triangles: OD600, no IPTG; triangles: OD600, induced with 0.1 mM IPTG. (D) Same as (C) using a strain with a hag promoter lacZ fusion at the ywrK locus combined with an IPTG-inducible copy of spxDD at the amyE locus (BNM1001).
In order to elucidate whether transcription from the flgB and hag promoters is regulated by spxDD induction, we inserted the PflgB-lacZ or Phag-lacZ reporter fusions at an additional ectopic locus (ywrK) of a strain carrying an IPTG-inducible copy of spxDD at the amyE locus (see Materials and Methods). We grew these strains to early exponential phase, induced spxDD by addition of IPTG and determined β-galactosidase activity of these strains (Figures 6C,D).
The activity of both promoters was strongly repressed compared to the un-induced control for a period of ~3 h after induction and subsequently increased (Figures 6C,D). As an additional control, we analyzed the hag mRNA levels by Northern blot analysis and Hag protein levels by Western blot analysis of a strain carrying an IPTG-inducible copy of spxDD at the amyE locus (Figure S9). The hag transcript level decreased below the detection limit of our Northern blot experiment after 30 min of spxDD induction Figure S9A). Hag protein levels also decreased after spxDD induction, however this effect was not as pronounced as for hag mRNA (Figure S9B), which is possibly due to the observed stability of Hag (Figure S1).
YjbH is an adapter protein, which specifically recognizes Spx and targets it for degradation by ClpXP. As an additional test to analyze the effect of increased Spx levels on motility, we assayed an yjbH deletion mutant for swimming motility and Hag levels. Similar to the clpX mutant, the yjbH mutant strain was unable to swim and displayed strongly decreased Hag levels. In contrast, an spx yjbH double mutant was highly motile and displayed wild type level of Hag protein (Figure S10).
Taken together, these data suggest that Spx acts as a negative regulator of swimming motility.
Motility genes are down-regulated in response to thiol oxidative stress
Spx is present at very low concentrations in growing, non-stressed cells due to regulatory proteolysis by ClpXP and repression of the spx gene. In response to oxidative stress, spx is transcriptionally de-repressed (Leelakriangsak et al., 2007) and Spx is stabilized (Zhang and Zuber, 2007; Garg et al., 2009).
Since our results suggest that Spx acts as a negative regulator of motility, it is conceivable that motility is repressed under conditions, when Spx accumulates in the cell, such as during thiol oxidative stress. To test this we subjected the PflgB-lacZ and Phag-lacZ reporter strains to oxidative stress by addition of 1 mM diamide, a strong inducer of Spx activity and collected samples for determination of β-galactosidase activity.
Whereas, the non-stressed control samples displayed a normal pattern of flagellar gene expression, PflgB-lacZ and Phag-lacZ activity strongly decreased for a period of 1–1.5 h after the application of oxidative stress (Figures 7A,B). Notably, Western blot analysis of the same samples with Spx-specific antibodies revealed that Spx protein was present in high amounts at the time points, at which flagellar gene expression was most strongly repressed (Figures 7A,B).
Figure 7.

Thiol oxidative stress results in transient down-regulation of motility genes in B. subtilis. (A) A growing culture of strain BNM301, carrying a transcriptional PflgB-lacZ fusion, was divided in early exponential phase and 1 mM diamide was added to half of the culture to induce thiol oxidative stress. Samples were removed before addition of diamide and at the indicated time points and β-galactosidase activity was determined. Cell lysates of the same samples were analyzed by SDS–PAGE and Western blot against Spx (lower panel). Representative data from at least two experiments are shown. (B) Same as (A) for strain BNM328, carrying a transcriptional Phag-lacZ fusion.
In summary, our data indicate that Spx acts as a negative regulator of the flgB promoter, which also affects σD levels and thus transcription from the hag promoter.
Spx regulates motility indirectly on both flgB and hag promoter
Our results clearly demonstrate that a raised cellular level of Spx results in a repression of the flgB promoter. We already demonstrated that Spx does not act on this promoter via DegU (Figure S6). Furthermore, we tested whether the defect of swimming motility in a clpX mutant is suppressed by a codY mutation. Swimming motility, Hag protein levels as well as flgB promoter activity were not increased in the clpX codY double mutant compared to the clpX single mutant (Figures S7B–E).
Interestingly, the hag promoter activity was increased especially at later time points in the double codY clpX mutant strain. These data suggest that CodY might be somehow indirectly involved in the Spx dependent repression of the hag promoter in the clpX mutant. The results presented earlier in Figure S3 already suggested an influence of Spx on the hag promoter independent of its influence on the flgB promoter (Figure S3). However, Spx-mediated down-regulation of the flgB promoter appears to be mostly independent of codY under our experimental conditions and in our strain background (Figure S7).
To test whether Spx directly binds to the flgB promoter, we performed electrophoretic mobility shift assays using flgB promoter DNA fragments and purified Spx protein (see SectionMaterials and Methods). In accordance with previously published results on Spx (Nakano et al., 2010), we did not observe DNA binding of Spx to the PflgB promotor region (Figure S11), suggesting that Spx indirectly regulates PflgB promoter activity.
Discussion
Regulatory and general proteolysis in swimming motility of B. subtilis
In this work, we present a detailed analysis of the impact of Clp proteases on swimming motility in the model organism B. subtilis. We found that regulatory proteolysis of the transcription factors DegU and Spx by ClpCP and ClpXP, respectively, is an important mechanism to facilitate and control swimming motility. In the absence of these proteases, active DegU-P or Spx can accumulate and negatively regulate expression of the fla/che operon, resulting in low σD levels, lower expression of late flagellar genes and a loss of swimming motility. As already suggested for B. subtilis mutated in clpC (Rashid et al., 1996), these results may also explain the previously observed increased cell chaining in clpP and clpX mutants (Gerth et al., 1998; Msadek et al., 1998), since autolysin genes, such as lytC and lytD, which are required for cell separation during division, are controlled by σD.
Regulation of flagellar assembly is also strongly influenced by regulatory proteolysis in E. coli or Salmonella, where the stability of the master regulator FlhDC and the flagellar sigma factor FliA are controlled by ClpXP (Tomoyasu et al., 2003; Barembruch and Hengge, 2007; Kitagawa et al., 2011; Takaya et al., 2012).
A master regulator and activator of motility, such as FlhDC in E. coli has not been identified in B. subtilis and the two proteins with a described activator function, SwrA (Kearns and Losick, 2005) and DegU (Tsukahara and Ogura, 2008), are both dispensable for normal expression of the fla/che operon for swimming motility in B. subtilis 168 strains. In most laboratory strains, such as B. subtilis 168, SwrA is encoded as a cryptic gene and not synthesized. In less domesticated B. subtilis strains, such as the biofilm forming NCIB 3610 B. subtilis strain, SwrA, like a small number of other regulatory proteins, is present and active as an activator of swimming motility even enabling swarming motility (Kearns et al., 2004; McLoon et al., 2011). It was suggested that SwrA acts in conjunction and interacting with DegU-P, switching it from a repressor to an activator of the flgB promoter (Ogura and Tsukahara, 2012; Mordini et al., 2013). Interestingly, when we tested the effect of clpC and clpX mutations on a B. subtilis 168 strain complemented with swrA+ and degQ+ alleles or the B. subtilis NCIB 3610 strain encoding SwrA, we still observed a negative effect on swimming motility (Figure S12).
ClpCP influences motility by controlling the activity of DegU~P
We demonstrate here that ClpCP regulates swimming motility not only by proteolysis of ComK, as previously reported (Liu and Zuber, 1998), but also by controlling the activity or stability of DegU, which was recently identified as a proteolysis target of ClpCP (Ogura and Tsukahara, 2010). In accordance with these observations, we observed that both the flgB and hag promotors were down-regulated in the clpC mutant in a degU-dependent manner and the σD and Hag levels were strongly decreased, rendering the bacteria non-motile.
The function of DegU in motility development is complex and has been controversially discussed in the literature. Presumably, DegU can act both as an activator (Tsukahara and Ogura, 2008) and in its phosphorylated form as a repressor (Amati et al., 2004) of the flgB promoter. However, it has been demonstrated that degU is required for swarming motility, whereas the gene is dispensable for swimming motility (Kobayashi, 2007; Verhamme et al., 2007). Our data are consistent with this hypothesis, as degSU mutants were motile and producing Hag and the degSU mutation had only a minor effect on transcription of motility genes in our hands. Therefore, we conclude that under our conditions and in our strain background only the repressor function of DegU in its phosphorylated form is relevant for swimming motility and influenced by ClpC.
The flgB promoter features two DegU binding sites, one located upstream of the promoter (BR1) and one downstream in the flgB coding region (BR2; Tsukahara and Ogura, 2008). The results presented here suggest that the downstream BR2 binding site is required for repression of the flgB promoter by DegU, since the longer lacZ fusion that incorporates the BR2 site was down-regulated in the clpC mutant in a degU-dependent manner whereas the shorter fusion was not affected by the clpC mutation (Figure 2). These data are in accordance with those of Tsukahara et al., who reported that the BR2 binding site is required for PflgB repression in a B. subtilis strain carrying a degU32 point mutant, which results in hyperphosphorylated DegU (Tsukahara and Ogura, 2008).
The long distance of the BR2 site from the core promoter suggests that DegU most probably does not repress flgB transcription by restricting access of RNA polymerase to the promoter. A similar mechanism was demonstrated for the repressor CodY, which binds to a site downstream of the ybgE transcription start and negatively regulates transcription elongation by a roadblock mechanism, resulting in a short terminated mRNA fragment (Belitsky and Sonenshein, 2011).
Interestingly, it was reported that phosphorylated DegU acts as a negative regulator of motility by transcriptional activation of flgM (Hsueh et al., 2011). This effect provides an additional explanation for the observed down-regulation of the hag promoter, which is σD-dependent and therefore negatively regulated by FlgM, in the clpC mutant.
DegU has been described as a cellular rheostat that allows adequate expression of different groups of genes during transition to stationary phase to allow processes such as competence, biofilm formation, and motility. It was proposed that DegU degradation by ClpCP plays a part in fine-tuning of DegU auto-activation (Veening et al., 2008; Ogura and Tsukahara, 2010). The results presented here suggest that DegU activity and stability is also important for swimming motility. We assume that ClpCP-mediated inhibition and proteolysis ensures that active DegU concentration in the cell is kept below a threshold level, such that the flgB promoter is de-repressed, but high enough to allow expression of other DegU-activated genes.
Most studies suggest that DegU acts as a PflgB repressor primarily in its phosphorylated form (Verhamme et al., 2007; Tsukahara and Ogura, 2008) and the experiments of Ogura and colleagues suggest that only phosphorylated DegU is targeted for ClpCP-mediated degradation (Ogura and Tsukahara, 2010). However, we observed in our strain and growth conditions some elevated DegU levels in vivo, but not to the extent observed before. Nevertheless, our experimental data are consistent with the role of DegU~P as a repressor of motility, which is specifically inhibited by ClpC. It should be noted that ClpC alone could be sufficient to repress DegU-P activity by unfolding DegU-P without targeting it to ClpP. It would be very interesting to explore by what mechanism phosphorylated DegU is recognized by ClpC and under what conditions it is targeted for degradation to ClpCP.
Regulatory proteolysis of Spx by ClpXP influences swimming motility
The second principal finding of this paper is the observation that swimming motility is inhibited in a clpX mutant via the stabilization of the ClpXP substrate Spx, which acts as a negative regulator of motility. We have demonstrated that an spx mutant suppresses the decreased swimming motility observed in a clpX mutant (Figure 4) and that raised levels of SpxDD in a clpX+ background inhibit motility (Figure 5). Our results suggest that Spx inhibits motility at the level of the flgB promoter (Figures 5, 6) and can in addition also influence the hag promoter (Figure S3). Furthermore, we could demonstrate that motility gene expression is transiently inhibited during thiol oxidative stress, which activates and stabilizes Spx (Figure 7).
Spx-mediated down-regulation of motility provides a connection between motility and a stress response pathway. The oxidative stress response requires a restructuring of the proteome redox enzymes and chaperones (Zuber, 2009). Likewise, swimming motility requires a substantial effort both for production of the flagellar proteins and their assembly (Chevance and Hughes, 2008). Exerting both programs at the same time could be detrimental for these cells. Our observations suggest that upon oxidative stress or induction of SpxDD the cells give priority to the stress response. However, they can resume motility development after oxidative stress has been alleviated (Figures 6, 7).
Importantly, this does not necessarily require that individual cells are non-motile during the stress response, since only flagellar gene expression is down-regulated. Already existing flagella could continue to function, which might even be an advantage for cells, enabling them to escape from the source of the stress by chemotaxis and swimming motility.
Possible mechanisms of motility regulation by Spx
Spx is a transcription factor, which interacts with the alpha subunit of RNA polymerase and enhances polymerase binding to certain promoters (Reyes and Zuber, 2008; Nakano et al., 2010). This mechanism is shared by a number of transcriptional activators, including response regulators such as ComA (Nakano et al., 2003b). According to the interference model, Spx does not directly act as a repressor, but restricts access of other transcription factors to the RNA polymerase alpha subunit when present at high concentrations. By this mechanism for example are competence genes repressed in the presence of Spx, because phosphorylated ComA can no longer bind to RNA polymerase (Nakano et al., 2003b). As already mentioned, an activator of transcription of the flgB promoter in B. subtilis 168 is not known, therefore it is very unlikely that Spx could act as a repressor of PflgB by interfering with an activator.
It was recently observed that Spx can activate the transcription of degSU (Shiwa et al., 2015), which could potentially effect the regulation of motility. However, we observed that a deletion of degSU did not interfere with the Spx mediated inhibition of motility (Figure S6) and no elevated DegU levels were observed in a clpX mutant (Figure 2G). However, the implications of raised levels of DegU and DegS on regulation of motility should be investigated in more detail.
DNA binding of Spx in the absence of RNA polymerase alpha CTD has never been observed and we have shown that Spx does not bind to the flgB promoter fragment in vitro (Figure S11). An indirect regulation of motility by Spx is also supported by a study, in which the Spx regulon was analyzed by tiling arrays and a genome wide characterization of Spx binding sites was accomplished. Spx-dependent repression of a number of motility and chemotaxis genes was observed in these experiments, but since no relevant Spx binding sites e.g., near the flgB and hag promoter were identified, this was considered an indirect Spx-mediated effect (Rochat et al., 2012). This suggests that Spx rather indirectly influences the flgB promoter and fla/che expression, for example by transcriptional activation of a repressor or other not yet identified intracellular signal transduction mechanisms. More experiments will be necessary to understand and elucidate the mechanism by which Spx influences motility via the flgB and hag promoters in B. subtilis.
In summary, we have uncovered two additional pathways, by which regulatory proteolysis affects swimming motility in B. subtilis. We could demonstrate that ClpCP contributes to motility development by controlling the stability of the response regulator DegU, which can act as a repressor of the fla/che operon. In turn, ClpXP facilitates swimming motility by proteolysis of its substrate Spx. We could show that the oxidative stress regulator Spx acts as a negative regulator of motility on the flgB promoter and the hag promoter. The additionally observed Spx-mediated repression of hag might also be facilitated by a yet unknown posttranscriptional process. Importantly, the influence of Spx on motility could also be observed in wild type cells during oxidative stress and can therefore be considered as a biologically relevant stress response mechanism.
These results highlight the complex involvement of controlled proteolysis in the regulation of motility and its intricate connections to stress response pathways such as the Spx controlled thiol stress response (Zuber, 2004, 2009) or heat shock response (Runde et al., 2014) and the various processes (such as e.g., biofilm formation) controlled by the master regulator DegU (Murray et al., 2009).
Author contributions
NM designed research, performed experiments, analyzed results, and wrote the paper. JH designed research, performed experiments and analyzed data. HS designed research, performed experiments and analyzed data KT designed research, analyzed results, and wrote the paper.
Funding
We acknowledge support by Deutsche Forschungsgemainschaft (Tu106/6, Tu106/7) and Open Access Publishing Fund of Leibniz Universität Hannover.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We want to thank Peter Zuber (University of Oregon), Dan Kearns (Indiana University), Ulf Gerth and Michael Hecker (University of Greifswald), Linc Sonenshein (Tufts University), Nicola Stanley-Wall (University of Dundee), Claes von Wachenfeldt (Lund University), and John Helmann (Cornell University) for the gift of antibodies, strains, or plasmids. We are grateful to Ilka Slosarek, Sabine Kretschmer (FU Berlin), and Armgard Janczikowski (Leibniz Universität Hannover) for excellent technical assistance. In addition, we thank Alex Elsholz (MPI Marburg) and Hendrik Osadnik (UC San Francisco) for critical reading of the manuscript.
Supplementary material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2016.00315
References
- Ababneh Q. O., Herman J. K. (2015). CodY regulates SigD levels and activity by binding to three sites in the fla/che operon. J. Bacteriol. 197, 2999–3006. 10.1128/JB.00288-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amati G., Bisicchia P., Galizzi A. (2004). DegU-P represses expression of the motility fla-che operon in Bacillus subtilis. J. Bacteriol. 186, 6003–6014. 10.1128/JB.186.18.6003-6014.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anagnostopoulos C., Spizizen J. (1961). Requirements for Transformation in Bacillus subtilis. J. Bacteriol. 81, 741–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnaud M., Chastanet A., Débarbouillé M. (2004). New vector for efficient allelic replacement in naturally nontransformable, low-GC-content, gram-positive bacteria. Appl. Environ. Microbiol. 70, 6887–6891. 10.1128/AEM.70.11.6887-6891.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barembruch C., Hengge R. (2007). Cellular levels and activity of the flagellar sigma factor FliA of Escherichia coli are controlled by FlgM-modulated proteolysis. Mol. Microbiol. 65, 76–89. 10.1111/j.1365-2958.2007.05770.x [DOI] [PubMed] [Google Scholar]
- Battesti A., Gottesman S. (2013). Roles of adaptor proteins in regulation of bacterial proteolysis. Curr. Opin. Microbiol. 16, 140–147. 10.1016/j.mib.2013.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belitsky B. R., Sonenshein A. L. (2011). Roadblock repression of transcription by Bacillus subtilis CodY. J. Mol. Biol. 411, 729–743. 10.1016/j.jmb.2011.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belitsky B. R., Sonenshein A. L. (2013). Genome-wide identification of Bacillus subtilis CodY-binding sites at single-nucleotide resolution. Proc. Natl. Acad. Sci. U.S.A. 110, 7026–7031. 10.1073/pnas.1300428110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergara F., Ibarra C., Iwamasa J., Patarroyo J. C., Aguilera R., Márquez-Magaña L. M. (2003). CodY is a nutritional repressor of flagellar gene expression in Bacillus subtilis. J. Bacteriol. 185, 3118–3126. 10.1128/JB.185.10.3118-3126.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertero M. G., Gonzales B., Tarricone C., Ceciliani F., Galizzi A. (1999). Overproduction and characterization of the Bacillus subtilis anti-sigma factor FlgM. J. Biol. Chem. 274, 12103–12107. 10.1074/jbc.274.17.12103 [DOI] [PubMed] [Google Scholar]
- Blair K. M., Turner L., Winkelman J. T., Berg H. C., Kearns D. B. (2008). A molecular clutch disables flagella in the Bacillus subtilis biofilm. Science 320, 1636–1638. 10.1126/science.1157877 [DOI] [PubMed] [Google Scholar]
- Bradford M. M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254. 10.1016/0003-2697(76)90527-3 [DOI] [PubMed] [Google Scholar]
- Calvio C., Osera C., Amati G., Galizzi A. (2008). Autoregulation of swrAA and motility in Bacillus subtilis. J. Bacteriol. 190, 5720–5728. 10.1128/JB.00455-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo R. A., Kearns D. B. (2015). FlgM is secreted by the flagellar export apparatus in Bacillus subtilis. J. Bacteriol. 197, 81–91. 10.1128/JB.02324-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camp A. H., Losick R. (2009). A feeding tube model for activation of a cell-specific transcription factor during sporulation in Bacillus subtilis. Genes Dev. 23, 1014–1024. 10.1101/gad.1781709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caramori T., Barilla D., Nessi C., Sacchi L., Galizzi A. (1996). Role of FlgM in sigma D-dependent gene expression in Bacillus subtilis. J. Bacteriol. 178, 3113–3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai Y., Kolter R., Losick R. (2010). Reversal of an epigenetic switch governing cell chaining in Bacillus subtilis by protein instability. Mol. Microbiol. 78, 218–229. 10.1111/j.1365-2958.2010.07335.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevance F. F. V., Hughes K. T. (2008). Coordinating assembly of a bacterial macromolecular machine. Nat. Rev. Microbiol. 6, 455–465. 10.1038/nrmicro1887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cozy L. M., Kearns D. B. (2010). Gene position in a long operon governs motility development in Bacillus subtilis. Mol. Microbiol. 76, 273–285. 10.1111/j.1365-2958.2010.07112.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derré I., Rapoport G., Msadek T. (1999). CtsR, a novel regulator of stress and heat shock response, controls clp and molecular chaperone gene expression in gram-positive bacteria. Mol. Microbiol. 31, 117–131. 10.1046/j.1365-2958.1999.01152.x [DOI] [PubMed] [Google Scholar]
- Engman J., von Wachenfeldt C. (2015). Regulated protein aggregation: a mechanism to control the activity of the ClpXP adaptor protein YjbH. Mol. Microbiol. 95, 51–63. 10.1111/mmi.12842 [DOI] [PubMed] [Google Scholar]
- Fredrick K., Helmann J. D. (1996). FlgM is a primary regulator of sigmaD activity, and its absence restores motility to a sinR mutant. J. Bacteriol. 178, 7010–7013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg S. K., Kommineni S., Henslee L., Zhang Y., Zuber P. (2009). The YjbH protein of Bacillus subtilis enhances ClpXP-catalyzed proteolysis of Spx. J. Bacteriol. 191, 1268–1277. 10.1128/JB.01289-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerth U., Krüger E., Derré I., Msadek T., Hecker M. (1998). Stress induction of the Bacillus subtilis clpP gene encoding a homologue of the proteolytic component of the Clp protease and the involvement of ClpP and ClpX in stress tolerance. Mol. Microbiol. 28, 787–802. 10.1046/j.1365-2958.1998.00840.x [DOI] [PubMed] [Google Scholar]
- Helmann J. D., Wu M. F., Kobel P. A., Gamo F. J., Wilson M., Morshedi M. M., et al. (2001). Global transcriptional response of Bacillus subtilis to heat shock. J. Bacteriol. 183, 7318–7328. 10.1128/JB.183.24.7318-7328.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsueh Y.-H., Cozy L. M., Sham L.-T., Calvo R. A., Gutu A. D., Winkler M. E., et al. (2011). DegU-phosphate activates expression of the anti-sigma factor FlgM in Bacillus subtilis. Mol. Microbiol. 81, 1092–1108. 10.1111/j.1365-2958.2011.07755.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearns D. B., Chu F., Rudner R., Losick R. (2004). Genes governing swarming in Bacillus subtilis and evidence for a phase variation mechanism controlling surface motility. Mol. Microbiol. 52, 357–369. 10.1111/j.1365-2958.2004.03996.x [DOI] [PubMed] [Google Scholar]
- Kearns D. B., Losick R. (2005). Cell population heterogeneity during growth of Bacillus subtilis. Genes Dev. 19, 3083–3094. 10.1101/gad.1373905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirstein J., Dougan D. A., Gerth U., Hecker M., Turgay K. (2007). The tyrosine kinase McsB is a regulated adaptor protein for ClpCP. EMBO J. 26, 2061–2070. 10.1038/sj.emboj.7601655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirstein J., Molière N., Dougan D. A., Turgay K. (2009). Adapting the machine: adaptor proteins for Hsp100/Clp and AAA+ proteases. Nat. Rev. Microbiol. 7, 589–599. 10.1038/nrmicro2185 [DOI] [PubMed] [Google Scholar]
- Kitagawa R., Takaya A., Yamamoto T. (2011). Dual regulatory pathways of flagellar gene expression by ClpXP protease in enterohaemorrhagic Escherichia coli. Microbiology 157, 3094–3103. 10.1099/mic.0.051151-0 [DOI] [PubMed] [Google Scholar]
- Kobayashi K. (2007). Gradual activation of the response regulator DegU controls serial expression of genes for flagellum formation and biofilm formation in Bacillus subtilis. Mol. Microbiol. 66, 395–409. 10.1111/j.1365-2958.2007.05923.x [DOI] [PubMed] [Google Scholar]
- Krüger E., Witt E., Ohlmeier S., Hanschke R., Hecker M. (2000). The Clp proteases of Bacillus subtilis are directly involved in degradation of misfolded proteins. J. Bacteriol. 182, 3259–3265. 10.1128/JB.182.11.3259-3265.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krüger E., Zühlke D., Witt E., Ludwig H., Hecker M. (2001). Clp-mediated proteolysis in Gram-positive bacteria is autoregulated by the stability of a repressor. EMBO J. 20, 852–863. 10.1093/emboj/20.4.852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leelakriangsak M., Kobayashi K., Zuber P. (2007). Dual negative control of spx transcription initiation from the P3 promoter by repressors PerR and YodB in Bacillus subtilis. J. Bacteriol. 189, 1736–1744. 10.1128/JB.01520-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Zuber P. (1998). A molecular switch controlling competence and motility: competence regulatory factors ComS, MecA, and ComK control sigmaD-dependent gene expression in Bacillus subtilis. J. Bacteriol. 180, 4243–4251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Márquez L. M., Helmann J. D., Ferrari E., Parker H. M., Ordal G. W., Chamberlin M. J. (1990). Studies of sigma D-dependent functions in Bacillus subtilis. J. Bacteriol. 172, 3435–3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Márquez-Magaña L. M., Chamberlin M. J. (1994). Characterization of the sigD transcription unit of Bacillus subtilis. J. Bacteriol. 176, 2427–2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLoon A. L., Guttenplan S. B., Kearns D. B., Kolter R., Losick R. (2011). Tracing the domestication of a biofilm-forming bacterium. J. Bacteriol. 193, 2027–2034. 10.1128/JB.01542-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miethke M., Hecker M., Gerth U. (2006). Involvement of Bacillus subtilis ClpE in CtsR degradation and protein quality control. J. Bacteriol. 188, 4610–4619. 10.1128/JB.00287-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirel D. B., Chamberlin M. J. (1989). The Bacillus subtilis flagellin gene (hag) is transcribed by the sigma 28 form of RNA polymerase. J. Bacteriol. 171, 3095–3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirel D. B., Lauer P., Chamberlin M. J. (1994). Identification of flagellar synthesis regulatory and structural genes in a Sigma D-dependent operon of Bacillus subtilis. J. Bacteriol. 176, 4492–4500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mordini S., Osera C., Marini S., Scavone F., Bellazzi R., Galizzi A., et al. (2013). The role of SwrA, DegU and P(D3) in fla/che expression in B. subtilis. PLoS ONE 8:e85065. 10.1371/journal.pone.0085065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Msadek T., Dartois V., Kunst F., Herbaud M. L., Denizot F., Rapoport G. (1998). ClpP of Bacillus subtilis is required for competence development, motility, degradative enzyme synthesis, growth at high temperature and sporulation. Mol. Microbiol. 27, 899–914. 10.1046/j.1365-2958.1998.00735.x [DOI] [PubMed] [Google Scholar]
- Mukherjee S., Kearns D. B. (2014). The structure and regulation of flagella in Bacillus subtilis. Annu. Rev. Genet. 48, 319–340. 10.1146/annurev-genet-120213-092406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray E. J., Kiley T. B., Stanley-Wall N. R. (2009). A pivotal role for the response regulator DegU in controlling multicellular behaviour. Microbiology 155, 1–8. 10.1099/mic.0.023903-0 [DOI] [PubMed] [Google Scholar]
- Nakano M. M., Hajarizadeh F., Zhu Y., Zuber P. (2001). Loss-of-function mutations in yjbD result in ClpX- and ClpP-independent competence development of Bacillus subtilis. Mol. Microbiol. 42, 383–394. 10.1046/j.1365-2958.2001.02639.x [DOI] [PubMed] [Google Scholar]
- Nakano M. M., Lin A., Zuber C. S., Newberry K. J., Brennan R. G., Zuber P. (2010). Promoter recognition by a complex of Spx and the C-terminal domain of the RNA polymerase alpha subunit. PLoS ONE 5:e8664. 10.1371/journal.pone.0008664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano M. M., Nakano S., Zuber P. (2002). Spx (YjbD), a negative effector of competence in Bacillus subtilis, enhances ClpC-MecA-ComK interaction. Mol. Microbiol. 44, 1341–1349. 10.1046/j.1365-2958.2002.02963.x [DOI] [PubMed] [Google Scholar]
- Nakano S., Küster-Schöck E., Grossman A. D., Zuber P. (2003a). Spx-dependent global transcriptional control is induced by thiol-specific oxidative stress in Bacillus subtilis. Proc. Natl. Acad. Sci. U.S.A. 100, 13603–13608. 10.1073/pnas.2235180100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano S., Nakano M. M., Zhang Y., Leelakriangsak M., Zuber P. (2003b). A regulatory protein that interferes with activator-stimulated transcription in bacteria. Proc. Natl. Acad. Sci. U.S.A. 100, 4233–4238. 10.1073/pnas.0637648100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano S., Zheng G., Nakano M. M., Zuber P. (2002). Multiple pathways of Spx (YjbD) proteolysis in Bacillus subtilis. J. Bacteriol. 184, 3664–3670. 10.1128/JB.184.13.3664-3670.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newberry K. J., Nakano S., Zuber P., Brennan R. G. (2005). Crystal structure of the Bacillus subtilis anti-alpha, global transcriptional regulator, Spx, in complex with the alpha C-terminal domain of RNA polymerase. Proc. Natl. Acad. Sci. U.S.A. 102, 15839–15844. 10.1073/pnas.0506592102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogura M., Tsukahara K. (2010). Autoregulation of the Bacillus subtilis response regulator gene degU is coupled with the proteolysis of DegU-P by ClpCP. Mol. Microbiol. 75, 1244–1259. 10.1111/j.1365-2958.2010.07047.x [DOI] [PubMed] [Google Scholar]
- Ogura M., Tsukahara K. (2012). SwrA regulates assembly of Bacillus subtilis DegU via its interaction with N-terminal domain of DegU. J. Biochem. 151, 643–655. 10.1093/jb/mvs036 [DOI] [PubMed] [Google Scholar]
- Pan Q., Garsin D. A., Losick R. (2001). Self-reinforcing activation of a cell-specific transcription factor by proteolysis of an anti-sigma factor in B. subtilis. Mol. Cell 8, 873–883. 10.1016/S1097-2765(01)00362-8 [DOI] [PubMed] [Google Scholar]
- Rashid M. H., Tamakoshi A., Sekiguchi J. (1996). Effects of mecA and mecB (clpC) mutations on expression of sigD, which encodes an alternative sigma factor, and autolysin operons and on flagellin synthesis in Bacillus subtilis. J. Bacteriol. 178, 4861–4869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes D. Y., Zuber P. (2008). Activation of transcription initiation by Spx: formation of transcription complex and identification of a Cis-acting element required for transcriptional activation. Mol. Microbiol. 69, 765–779. 10.1111/j.1365-2958.2008.06330.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochat T., Nicolas P., Delumeau O., Rabatinová A., Korelusová J., Leduc A., et al. (2012). Genome-wide identification of genes directly regulated by the pleiotropic transcription factor Spx in Bacillus subtilis. Nucleic Acids Res. 40, 9571–9583. 10.1093/nar/gks755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runde S., Molière N., Heinz A., Maisonneuve E., Janczikowski A., Elsholz A. K. W., et al. (2014). The role of thiol oxidative stress response in heat-induced protein aggregate formation during thermotolerance in Bacillus subtilis. Mol. Microbiol. 91, 1036–1052. 10.1111/mmi.12521 [DOI] [PubMed] [Google Scholar]
- Sambrook J., Russell D. W., Cold Spring Harbor Laboratory (2001). Molecular Cloning: A Laboratory Manual, 3rd Edn. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory. [Google Scholar]
- Sauer R. T., Baker T. A. (2011). AAA+ proteases: ATP-fueled machines of protein destruction. Annu. Rev. Biochem. 80, 587–612. 10.1146/annurev-biochem-060408-172623 [DOI] [PubMed] [Google Scholar]
- Schlothauer T., Mogk A., Dougan D. A., Bukau B., Turgay K. (2003). MecA, an adaptor protein necessary for ClpC chaperone activity. Proc. Natl. Acad. Sci. U.S.A. 100, 2306–2311. 10.1073/pnas.0535717100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiwa Y., Yoshikawa H., Tanaka T., Ogura M. (2015). Bacillus subtilis degSU operon is regulated by the ClpXP-Spx regulated proteolysis system. J. Biochem. 157, 321–330. 10.1093/jb/mvu076 [DOI] [PubMed] [Google Scholar]
- Takaya A., Erhardt M., Karata K., Winterberg K., Yamamoto T., Hughes K. T. (2012). YdiV: a dual function protein that targets FlhDC for ClpXP-dependent degradation by promoting release of DNA-bound FlhDC complex. Mol. Microbiol. 83, 1268–1284. 10.1111/j.1365-2958.2012.08007.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomoyasu T., Takaya A., Isogai E., Yamamoto T. (2003). Turnover of FlhD and FlhC, master regulator proteins for Salmonella flagellum biogenesis, by the ATP-dependent ClpXP protease. Mol. Microbiol. 48, 443–452. 10.1046/j.1365-2958.2003.03437.x [DOI] [PubMed] [Google Scholar]
- Tsukahara K., Ogura M. (2008). Promoter selectivity of the Bacillus subtilis response regulator DegU, a positive regulator of the fla/che operon and sacB. BMC Microbiol. 8:8. 10.1186/1471-2180-8-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turgay K., Hahn J., Burghoorn J., Dubnau D. (1998). Competence in Bacillus subtilis is controlled by regulated proteolysis of a transcription factor. EMBO J. 17, 6730–6738. 10.1093/emboj/17.22.6730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veening J.-W., Igoshin O. A., Eijlander R. T., Nijland R., Hamoen L. W., Kuipers O. P. (2008). Transient heterogeneity in extracellular protease production by Bacillus subtilis. Mol. Syst. Biol. 4, 184. 10.1038/msb.2008.18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhamme D. T., Kiley T. B., Stanley-Wall N. R. (2007). DegU co-ordinates multicellular behaviour exhibited by Bacillus subtilis. Mol. Microbiol. 65, 554–568. 10.1111/j.1365-2958.2007.05810.x [DOI] [PubMed] [Google Scholar]
- Wach A. (1996). PCR-synthesis of marker cassettes with long flanking homology regions for gene disruptions in S. cerevisiae. Yeast 12, 259–265. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Nakano S., Choi S.-Y., Zuber P. (2006). Mutational analysis of the Bacillus subtilis RNA polymerase alpha C-terminal domain supports the interference model of Spx-dependent repression. J. Bacteriol. 188, 4300–4311. 10.1128/JB.00220-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Zuber P. (2007). Requirement of the zinc-binding domain of ClpX for Spx proteolysis in Bacillus subtilis and effects of disulfide stress on ClpXP activity. J. Bacteriol. 189, 7669–7680. 10.1128/JB.00745-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuber P. (2004). Spx-RNA polymerase interaction and global transcriptional control during oxidative stress. J. Bacteriol. 186, 1911–1918. 10.1128/JB.186.7.1911-1918.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuber P. (2009). Management of oxidative stress in Bacillus. Annu. Rev. Microbiol. 63, 575–597. 10.1146/annurev.micro.091208.073241 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.