

Abstract
Innate sensing of nucleic acids lies at the heart of antiviral immunity. During viral infection, dying cells may also release nucleic acids into the tissue microenvironment. It is presently unknown what effect such host signals have on the quality or duration of the immune response to viruses. Here we uncovered an immune-regulatory pathway that tempers the intensity of the host-response to influenza A virus (IAV) infection. We found that host-derived DNA accumulates in the lung microenvironment during IAV infection. Ablation of DNA in the lung resulted in increased mortality, increased cellular recruitment, and increased inflammation following IAV challenge. The released DNA in turn was sensed by the DNA receptor Absent in Melanoma (AIM2). Aim2−/− mice showed similarly exaggerated immune responses to IAV. Taken together, our results identify a novel mechanism of cross-talk between PAMP and DAMP sensing pathways, where sensing of host-derived DNA limits immune-mediated damage to infected tissues.
Introduction
Nucleic acid recognition is a primary mechanism driving protective host-defenses during infection with a wide variety of microbial pathogens. In contrast to microbial products, such as LPS, that are unique to bacteria and not found in mammalian hosts, nucleic acids are common to both the pathogen and the host cell they infect. During the course of infection when viral pathogens replicate, nucleic acids accumulate within cells and distinct families of PRRs including RIG-I-like receptors (RLR), Nod-like receptors (NLR), Toll-like receptors (TLR), and PYHIN proteins detect these molecules, and mobilize intracellular signaling pathways leading to anti-viral gene expression (1, 2).
Under normal circumstances, host nucleic acids are segregated from endosomal or cytosolic pattern recognition receptors (PRR), and DNA located in the nucleus and mitochondria of healthy cells, fails to be recognized by the innate immune system. During viral infection endogenous danger signals including DNA can also be released from damaged or dying cells. It is presently unclear if these danger signals are detected and if they in turn alter the host’s response to the pathogen. We set out to address this question using influenza A virus (IAV). IAV is an orthomyxovirus with a (−) ssRNA genome and the viral life cycle involves the creation RNA species only. IAV is sensed by RIG-I, TLR7, and TLR3 (3), all of which mobilize anti-viral defenses that curb viral replication, prevent viral spread and activate adaptive immunity.
Here we report the abundant release of endogenous DNA into the lung microenvironment which is protective during infection with IAV. We identified an important role for the DNA sensor AIM2 in sensing this host DNA. Mice lacking AIM2 were hyper susceptible to IAV infection. Although AIM2 contributes to the production of IL-1β early during IAV infection, the enhanced susceptibility of AIM2-deficient mice was not due to a failure in IAV-specific T cell responses. Rather, AIM2 appears to dampen inflammatory responses that would otherwise lead to excessive immunopathology. Thus our study uncovers previously unappreciated cross-talk between PAMP and DAMP sensing in controlling the magnitude and duration of the host response to virus infection.
Materials and Methods
Animals
Aim2−/− mice were generated as described (4, 5) and backcrossed to C57BL/6. WT and fully backcrossed Aim2−/− mice were maintained and caged separately. All procedures used in this study complied with federal guidelines and were approved by the University of Massachusetts Medical School Institutional Animal Care and Use Committee.
Influenza virus infection and viral titers
Influenza virus A/Puerto Rico/8/1934 H1N1 (PR8) grown in chicken eggs was purchased from Charles River Laboratories (Wilmington, MA). Mice were first anesthetized with isofluorane and inoculated via intranasal route with 4 × 104 pfu in 30 uL PBS. Viral titers in lung homogenates were measured by immunoplaque assay with MDCK cells. Monolayers were fixed, permeablized and stained with anti-IAV NP followed by secondary goat anti-mouse-HRP. Plaques were visualized using DAB substrate.
Quantification of DNA and albumin in BAL
BAL was harvested using 1 mL PBS, spun at 5,000 × g for 10 min and supernatants kept as cell-depleted BAL. dsDNA in BAL supernatants was quantitated by PicoGreen assay (Life Technologies). Albumin levels in the BAL were quantified by ELISA (GenWay).
Flow cytometry and tetramer staining
Mice were perfused with PBS in the right ventricle. Cells suspensions from total lung tissue were stained for TCRβ, CD19, B220, CD11b, CD11c, Ly6G, Ly6C, NK1.1 (eBioscience), CD45, CD4, CD8α (BD Biosciences), and LD Blue (Life Technologies). For tetramer staining, cells were stained for TCRβ, CD8α, CD4, CD44 and APC-labeled tetramers (Kb;NP 366, PA 244, PB1 703) or I-Ab/NP 311 (Trudeau Institute, Saranac Lake, NY). Live cells were gated based on forward and side scatter, and Live/Dead Blue negative staining prior to subsequent gating. Data acquisition was performed on an LSRII (BD Biosciences) and data analyzed using FlowJo (TreeStar).
Cytokine quantification
Lung tissues were homogenized in 500 μL PBS, spun, and levels of IL-1β, IL-6, and TNFα in supernatants were quantified by ELISA (eBioscience).
Generation of recombinant adeno-associated virus
Briefly, mouse DNaseI was cloned into a rAAV vector plasmid carrying a vector genome with the expression cassette driven by CMV enhanced chicken β-actin promoter and flanked by AAV2 ITRs (17). The AAV-DnaseI plasmid was co-transfected into HEK 293 cells with an AAV9 packaging plasmid and adenovirus helper plasmid. The recombinant virus was purified by standard CsCl gradient sedimentation method and desalted by dialysis. Mice were first anesthetized with isofluorane and inoculated via intranasal route with 1010 pfu in 30 μL PBS at least two weeks prior to experiments.
Statistical Analysis
Data were analyzed using the two-tailed Student’s t test comparing means between groups. Kaplan-Meier survival curves were analyzed by Mantel-Cox log-rank test. Graphing and statistical analyses were done using GraphPad Prism.
Results and Discussion
Accumulated DNA in the lung microenvironment during IAV infection is protective
While examining the lungs of Influenza virus infected mice we noticed what appeared to be extracellular DNA extruded from necrotic cells primarily localized to the bronchi (Fig 1A). The source of this DNA is likely necrotic bronchiolar epithelial cells or neutrophil extracellular traps (NETs), both of which have previously been identified in IAV infected animals (7, 8). We formally quantified the levels of extracellular DNA released within tissues by obtaining bronchiolar lavage (BAL) fluid that we depleted of host cells. BAL collected from uninfected mice had low quantities of dsDNA (Fig 1B). Levels of dsDNA increased significantly as early as 1 dpi and continued to increase instep with the spread of infection at 2, 3, and 6 dpi (Fig 1B). Given that IAV is an orthomyxovirus with a negative-sense single-stranded RNA genome, known to only produce RNA intermediates during replication, host-derived DNA is the only plausible source of extracellular DNA in this model.
Figure 1.
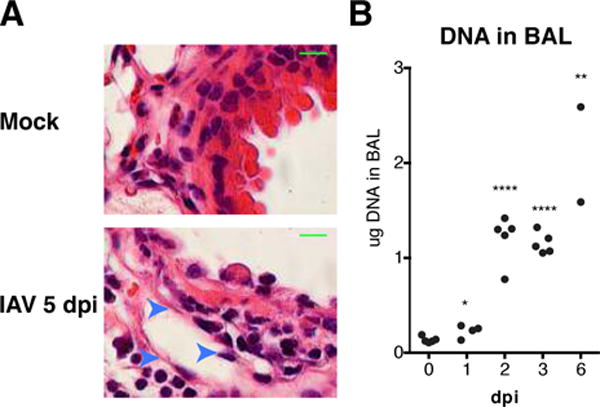
DNA accumulates in the lung microenvironment during IAV infection.
(A) H&E staining of lung tissue from uninfected and infected WT mice at 5 dpi at 100× magnification. Arrows indicate necrotic cells associated with hematoxylin-rich strands. Scale bar = 10 μm (B) Concentration of DNA in cell-depleted BAL fluid from PR8 infected WT mice at indicated times. DNA quantified by PicoGreen assay. *, P 0.05; **, P 0.01; ****, P 0.001
It is presently unknown what affect host signals such as endogenous DNA have on the quality or duration of the immune response to viruses. To test the role of DNA in this context, we generated a recombinant adeno-associated virus (AAV) to ectopically express mouse DNaseI in the lungs of WT mice (Fig 2A). DNaseI is a secreted protein, normally absent from the lung with respect to its enzymatic activity and expression (9)(Fig 2B). Intranasal infection with AAV-DNaseI resulted in robust and stable expression of the transgene in the lung at least 2 weeks after transduction (Fig 2B). Importantly, the levels of DNA in the BAL fluid of IAV infected mice were significantly reduced in mice treated with AAV-DNaseI compared to GFP expressing control virus, confirming the product of the DNaseI transgene was secreted and enzymatically active (Fig 2C). In response to lethal PR8 challenge mice treated with AAV-DNaseI showed a significant decrease in survival (40%) compared to controls (79.6%) out to 14 dpi following challenge with an LD25 of PR8 (Fig 2D). Comparison of viral loads in the lungs of AAV-GFP and AAV-Dnase1 treated mice at 3, 6, and 9 dpi revealed similar viral loads (Fig 2E). FACS analysis of infiltrating leukocyte populations in the lungs of AAV-DnaseI revealed a significant increase in the number of CD4+ and CD8+ T cells compared to controls at 5 dpi (Fig 2F–H). Combined, these data suggest that DNA present within the lung microenvironment during IAV infection is protective, and may serve to limit the number of infiltrating T cells able to drive the development immune pathology.
Figure 2.
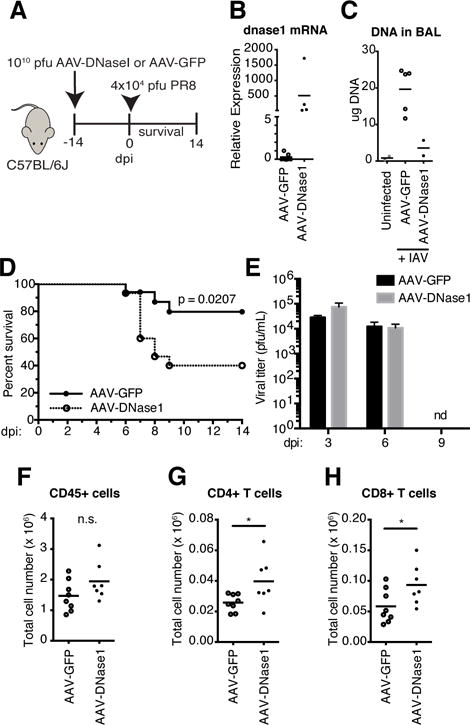
DNA in the lung is protective to IAV infection.
(A) Schematic for transduced expression of DNaseI. (B) dnase1 mRNA measured by qPCR with total RNA isolated from lungs of AAV-GFP and AAV-DNaseI treated mice. (C) DNA in cell-depleted BAL fluid in uninfected controls compared to AAV-GFP and AAV-DNaseI treated mice at 5 dpi with PR8. (D) Survival comparison between AAV-GFP and AAV-DNaseI treated WT mice challenged with 4 ×104 pfu PR8. Data combined from two independent experiments (AAV-GFP, n = 17; AAV-DNaseI, n = 15). (E) Viral titers of lung homogenates from AAV-GFP and AAV-DNaseI treated mice at 3, 6, and 9 dpi with PR8. (F–H)) Total number of CD45+ leukocytes (F), CD4+ T cells (G), and CD8+ T cells (H) in the total lungs of AAV-GFP and AAV-DNaseI treated mice at 5 dpi with PR8. *, P 0.05
AIM2 regulates inflammatory responses in the lung during IAV infection
Upon finding that DNA accumulates in the lungs following IAV infection and that sensing of this DNA leads to protective responses, we thought it plausible that one, or more, DNA sensing receptors might be engaged in the lungs of IAV infected mice. To this end, we tested mice lacking Absent in Melanoma (AIM)-2, a well described cytosolic DNA receptor required for activation of the inflammasome complex in response to dsDNA (5, 10, 11). Previous studies have identified roles for inflammasomes in IAV infection. IAV infection has been shown to regulate IL-1β via the NLRP3 inflammasome (12, 13). Each component of the NLRP3 inflammasome, including ASC and caspase-1, have been shown to be required to drive the inflammatory response and leukocyte recruitment during IAV infection (13–15). To test the contribution of AIM2 during IAV infection, mice lacking AIM2 were infected with a lethal IAV challenge. While 75% of WT mice survived IAV infection, only 28% of AIM2−/− mice were surviving at 14 dpi (Fig 3A). There was no difference in viral load in the lungs of WT and Aim2−/− mice at 3, 6, and 9 dpi suggesting that although AIM2 was required for protection during lethal IAV challenge, AIM2 did not directly impact IAV replication (Fig 3B).
Figure 3.
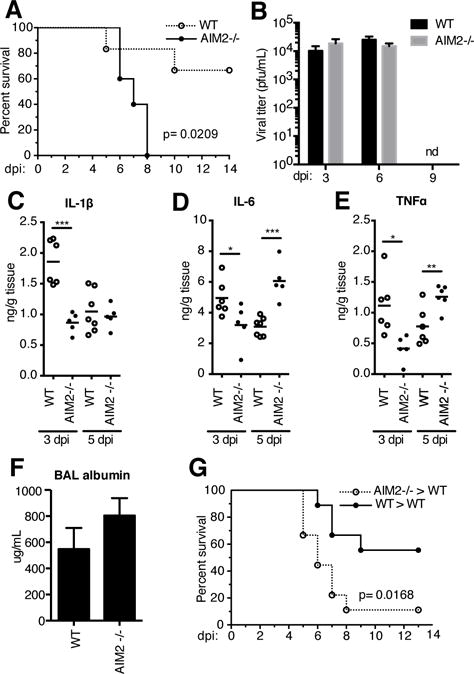
AIM2 is protective during IAV infection.
(A) Survival comparison of WT and Aim2−/− mice infected with 4 ×104 pfu PR8 (WT, n=6; Aim2−/−, n=5). (B) Viral titers of lung homogenates from WT and Aim2−/− at 3, 6, and 9 dpi with PR8. (C–F) Levels of IL-1β (D), IL-6 (E), and TNFα (F) protein in lung homogenates at 3 and 5 dpi.(G) Concentration of serum albumin in BAL washings of WT and Aim2−/− at 3 dpi. (G) Survival comparison of lethally irradiated WT mice engrafted with WT and Aim2−/− bone marrow infected with PR8 (WT>WT, n=10; Aim2−/−>WT, n=9)
We next assessed whether the susceptibility of Aim2−/− mice correlated with a defect in IL-1β production. Analysis of IL-1β, IL-1α and IL-1R antagonist (IL-1Ra) mRNA showed no defect in their induction between Aim2−/− and WT mice at 3 and 5 dpi (data not shown). However, the levels of IL-1β protein in lung homogenates were significantly lower at 3 dpi in AIM2-deficient mice but these levels increased by 5 dpi to match those of WT mice (Fig 3C). Similar to the effects we observed early for IL-1β levels, we found that IL-6 and TNFα were also decreased in lung homogenates of Aim2−/− mice at 3 dpi compared to WT controls (Fig 3D and E). However, by 5 dpi, the levels of both TNFα and IL-6 were significantly elevated in Aim2−/− mice (Fig 3D and E). The early effects of AIM2-deficiency on these cytokines may result from decreased IL-1β production and IL-1R dependent induction of these cytokines, or may reflect a greater number of leukocytes able to produce cytokines in the lungs of Aim2−/− mice. We observed a slight increase in albumin levels in the BAL of Aim2−/− mice at 3 dpi compared to WT controls suggesting that there was more extensive lung damage in the absence of AIM2 (Fig 3F). We also performed bone marrow transplants to elucidate whether expression of AIM2 in hematopoietic cells or stromal cells was required for protection from lethal IAV infection. Here, WT mice engrafted with Aim2−/− bone marrow were significantly more susceptible to IAV infection compared to those receiving WT bone marrow (Fig 3G). These data indicate that AIM2 in hematopoietic cells is responsible for the protective effects we observed.
IL-1R signaling during IAV infection is required for eliciting a protective flu-specific T cell response (16). AIM2 is required for IL-1β production upon sensing DNA in the cytosol, thus we sought to determine whether AIM2 deficiency impaired the generation of flu-specific T cells. After infection with a sublethal dose of PR8, we performed tetramer staining in lung and spleen cell suspensions with four class I and one class II IAV epitopes to determine the frequency and number of flu-specific CD8+ and CD4+ T cells, respectively. Here, we found no significant difference in the frequency (Fig 4A) of NP 366-tetramer+ CD8+ T cells in the lungs at 9 dpi. Similarly, the numbers of CD8+ T cells positive for tetramers bearing three different IAV epitopes (NP 366, PA 224, PB1 703) were unaffected by AIM2-deficiency (Fig 4B). Similar results were found in both the lung and spleen for all class I and class II epitopes tested (data not shown). These data indicate that AIM2 plays a protective role during IAV infection and that the protective effect is unlikely to be due to the limited effects of AIM2 on early IL-1β production or the formation of flu-specific T cell responses all of which are unaffected by AIM2-deficiency.
Figure 4.
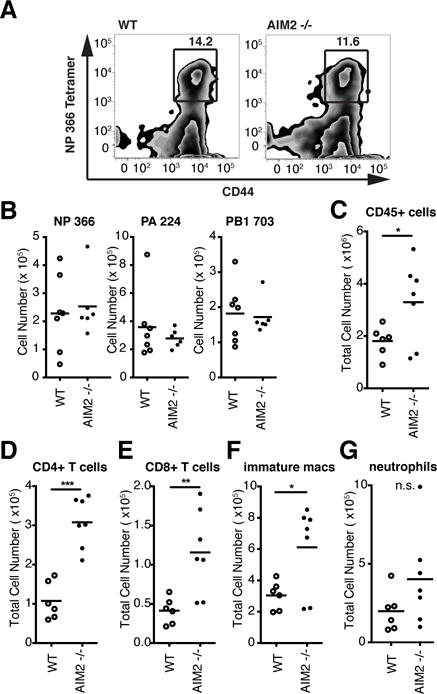
AIM2 dampens the inflammatory response to IAV infection.
(A) Frequency of NP366-tetramer+ CD8+ T cells and (B) total number tetramer+ CD8+ T cells in the lungs of WT and Aim2−/− mice at 9 dpi with 103 pfu PR8 using three epitopes (NP366, PA224, PB703). Populations were gated on CD45+ CD44hi CD8+ TCRβ+ cells.
(C–G) Total number of CD45+ cells (C), CD4+ T cells (D), CD8+ T cells (E), immature macrophages (F), and neutrophils (G) in the lung at 5 dpi with 4 ×104 pfu PR8. *, P 0.05; **, P 0.01; ***, P 0.005
We next used flow cytometric analysis to examine whether the absence of AIM2 altered leukocyte recruitment to the lungs of IAV infected mice. This analysis revealed a significant increase in CD45+ leukocytes in the lungs of Aim2−/− mice compared to WT controls in single-cell suspensions prepared from total lung at 5 dpi (Fig 4C). Looking at individual populations of immune cells within the CD45+ population, we found the increased numbers of CD4+ and CD8+ T cells, and immature macrophages in the lungs of Aim2−/− mice (Fig 4D–F). We observed no significant difference in either the frequency or number of neutrophils (Fig 4G), B cells, monocytes, or NK cells (data not shown).
Overall, our studies demonstrate that host-derived DNA accumulates in lung tissue damaged by IAV infection and has the capacity to modulate the intensity of the host response. Using a mouse influenza model, we found levels of host-derived DNA released from necrotic cells accumulates in the lung and continues to rise as the infection spreads throughout the tissue. Ablation of extracellular DNA using DNaseI (delivered via AAV) and genetic deletion of AIM2, a cytosolic sensor primarily known for its role in controlling inflammasome activation in response to cytosolic DNA, increased the susceptibility to IAV infection. Although AIM2 impacted the magnitude of the IAV induced IL-1β response in vivo early during infection, this response was indistinguishable from that of WT mice at later time points. The increase in cellular infiltration and inflammatory cytokines observed in Aim2−/− mice were not observed in prior studies that characterized the role of NLRP3, ASC, and caspase-1 during IAV infection (13–15). Our findings suggest the levels of IL-1β in Aim2−/− mice are sufficient for mounting an immune response and the observations reported here reflect an inflammasome-independent role for AIM2 in this context. Consistent with a largely intact IL-1β response, Aim2−/− mice were able to mount normal flu-specific adaptive responses, which are known to require IL-1R signaling (16). AIM2-deficient mice had similar viral loads to those seen in wild type mice, indicating that the effect of AIM2 in vivo was also not due to a failure to curb viral replication. Numbers of infiltrating leukocytes were increased however in the lungs of Aim2−/− mice. Moreover, levels of TNFα and IL-6, albeit slightly decreased early during infection, were also produced by Aim2−/− at levels greater than WT mice at 5 dpi likely due to the increased number of leukocytes in the lungs of Aim2−/− mice. Consistent with our findings that a DNA sensor is able to regulate inflammation and morbidity during infection with an RNA virus, we also found that ablation of DNA in the lung microenvironment by ectopic DNaseI expression yielded a similar effect to that observed in Aim2−/− mice. Collectively, these findings suggest that accumulation of DNA in tissues damaged by infection may provide a mechanism for alerting the immune system to the extent of tissue damage and functions as a signal to limit excessive immune pathology. It remains to be seen if the mechanism we have identified here is specific to IAV infection however or more generalizable. We predict that similar mechanisms may be at play in other viral infections.
The identification of AIM2 as a regulator of tissue inflammation adds to our understanding of the cross-talk that exists between innate immune pathways. Further study of endogenous DAMPs and their role in shaping antiviral immunity, repair and regenerative responses will also lead to improved understanding of viral pathogenesis and could also yield new insights for vaccine developments.
Acknowledgments
We would like to thank Dr. Zhaozhao Jiang and Kelly Army for their help with animal husbandry as well as Dr. Vijay Rathinam, Dr. Kai McInstry and Dr. Gabriel Hendricks for technical assistance.
Footnotes
This work is supported by grants from the NIH (AI093752 and AI083713 to K.A.F) and by an NIH T32 training grant (AI095213) to S.A.S.
References
- 1.Barbalat R, Ewald SE, Mouchess ML, Barton GM. Nucleic acid recognition by the innate immune system. Annu Rev Immunol. 2011;29:185–214. doi: 10.1146/annurev-immunol-031210-101340. [DOI] [PubMed] [Google Scholar]
- 2.Schattgen SA, Fitzgerald KA. The PYHIN protein family as mediators of host defenses. Immunol Rev. 2011;243:109–118. doi: 10.1111/j.1600-065X.2011.01053.x. [DOI] [PubMed] [Google Scholar]
- 3.Iwasaki A, Pillai PS. Innate immunity to influenza virusinfection. Nat Rev Immunol. 2014;14:315–328. doi: 10.1038/nri3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jones JW, Kayagaki N, Broz P, Henry T, Newton K, O’Rourke K, Chan S, Dong J, Qu Y, Roose-Girma M, Dixit VM, Monack DM. Absent in melanoma 2 is required for innate immune recognition of Francisella tularensis. Proc Natl Acad Sci. 2010;107:9771–9776. doi: 10.1073/pnas.1003738107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rathinam VAK, Jiang Z, Waggoner SN, Sharma S, Cole LE, Waggoner L, Vanaja SK, Monks BG, Ganesan S, Latz E, Hornung V, Vogel SN, Szomolanyi-Tsuda E, Fitzgerald KA. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat Immunol. 2010;11:395–402. doi: 10.1038/ni.1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McKinstry KK, Strutt TM, Buck A, Curtis JD, Dibble JP, Huston G, Tighe M, Hamada H, Sell S, Dutton RW, Swain SL. IL-10 Deficiency Unleashes an Influenza-Specific Th17 Response and Enhances Survival against High-Dose Challenge. J Immunol. 2009;182:7353–7363. doi: 10.4049/jimmunol.0900657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Capelozzi VL, Parra ER, Ximenes M, Bammann RH, Barbas CSV, Duarte MIS. Pathological and ultrastructural analysis of surgical lung biopsies in patients with swine-origin influenza type A/H1N1 and acute respiratory failure. Clinics. 2010;65:1229–1237. doi: 10.1590/S1807-59322010001200003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Narasaraju T, Yang E, Samy RP, Ng HH, Poh WP, Liew AA, Phoon MC, van Rooijen N, Chow VT. Excessive Neutrophils and Neutrophil Extracellular Traps Contribute to Acute Lung Injury of Influenza Pneumonitis. Am J Pathol. 2011;179:199–210. doi: 10.1016/j.ajpath.2011.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Napirei M, Ricken A, Eulitz D, Knoop H, Mannherz HG. Expression pattern of the deoxyribonuclease 1 gene: lessons from the Dnase1 knockout mouse. Biochem J. 2004;380:929–937. doi: 10.1042/BJ20040046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, Latz E, Fitzgerald KA. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. 2009;458:514–518. doi: 10.1038/nature07725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fernandes-Alnemri T, Yu J-W, Datta P, Wu J, Alnemri ES. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature. 2009;458:509–513. doi: 10.1038/nature07710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ichinohe T, Pang IK, Iwasaki A. Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat Immunol. 2010;11:404–410. doi: 10.1038/ni.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Allen IC, Scull MA, Moore CB, Holl EK, Mcelvania-Tekippe E, Taxman DJ, Guthrie EH, Pickles RJ, Ting JP-Y. The NLRP3 Inflammasome Mediates In Vivo Innate Immunity to Influenza A Virus through Recognition of Viral RNA. Immunity. 2009;30:556–565. doi: 10.1016/j.immuni.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ichinohe T, Lee HK, Ogura Y, Flavell R, Iwasaki A. Inflammasome recognition of influenza virus is essential for adaptive immune responses. Journal of Experimental Medicine. 2009;206:79–87. doi: 10.1084/jem.20081667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thomas PG, Dash P, Aldridge JR, Jr, Ellebedy AH, Reynolds C, Funk AJ, Martin WJ, Lamkanfi M, Webby RJ, Boyd KL. The Intracellular Sensor NLRP3 Mediates Key Innate and Healing Responses to Influenza A Virus via the Regulation of Caspase-1. Immunity. 2009;30:566–575. doi: 10.1016/j.immuni.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pang IK, Ichinohe T, Iwasaki A. IL-1R signaling in dendritic cells replaces pattern-recognition receptors in promoting CD8+ T cell responses to influenza A virus. Nat Immunol. 2013:1–9. doi: 10.1038/ni.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gao G, Sena-Esteves M. Introducing Genes into Mammalian Cells: Viral Vectors. In: Green MR, Sambrook J, editors. Molecular Cloning, Vol 2: A Laboratory Manual. Cold Spring Harbor Laboratory Press; New York: 2012. pp. 1209–1313. [DOI] [PubMed] [Google Scholar]