

Abstract
Dcp1/2 is the major eukaryotic RNA decapping complex, comprised of the enzyme Dcp2 and activator Dcp1, which removes the 5′ m7G cap from mRNA, committing the transcript to degradation. Dcp1/2 activity is crucial for RNA quality control and turnover, and deregulation of these processes may lead to disease development. The molecular details of Dcp1/2 catalysis remain elusive, in part because both cap substrate (m7GpppN) and m7GDP product are bound by Dcp1/2 with weak (mM) affinity. In order to find inhibitors to use in elucidating the catalytic mechanism of Dcp2, we screened a small library of synthetic m7G nucleotides (cap analogs) bearing modifications in the oligophosphate chain. One of the most potent cap analogs, m7GpSpppSm7G, inhibited Dcp1/2 20 times more efficiently than m7GpppN or m7GDP. NMR experiments revealed that the compound interacts with specific surfaces of both regulatory and catalytic domains of Dcp2 with submillimolar affinities. Kinetics analysis revealed that m7GpSpppSm7G is a mixed inhibitor that competes for the Dcp2 active site with micromolar affinity. m7GpSpppSm7G-capped RNA undergoes rapid decapping, suggesting that the compound may act as a tightly bound cap mimic. Our identification of the first small molecule inhibitor of Dcp2 should be instrumental in future studies aimed at understanding the structural basis of RNA decapping and may provide insight toward the development of novel therapeutically relevant decapping inhibitors.
Keywords: Dcp1/Dcp2, RNA metabolism, decapping enzymes, inhibitors, mRNA cap
INTRODUCTION
Regulation of mRNA stability plays a vital role in development (Alonso 2012; Barckmann and Simonelig 2013), cell proliferation (Mazzoni and Falcone 2011), immune system adaptation (Carpenter et al. 2014), stress response (Romero-Santacreu et al. 2009), transcript localization (Walters and Parker 2014), and quality control (Inada 2013). The two major cellular RNA degradation pathways that are responsible for mRNA stability are 5′-to-3′ decay and 3′-to-5′ decay. In 5′-to-3′ RNA decay, hydrolysis of the 5′ m7G RNA cap structure (Supplemental Fig. S1A) is an irreversible step that commits the transcript to degradation by the conserved 5′-to-3′ exonuclease Xrn1 (Hsu and Stevens 1993). Dcp2 is the major RNA decapping enzyme in eukaryotes, and Dcp2-mediated cap hydrolysis is important in numerous 5′-to-3′ mRNA decay pathways, including bulk 5′-to-3′ decay (Wang et al. 2002; Balagopal et al. 2012), ARE-mediated decay (Fenger-Gron et al. 2005; von Roretz et al. 2011), nonsense-mediated decay (NMD) (Kervestin and Jacobson 2012; Popp and Maquat 2013), miRNA-induced decay (Guo et al. 2010; Wilczynska and Bushell 2015), and 3′-poly(U)-triggered decay (Rissland and Norbury 2009; Su et al. 2013; Lee et al. 2014). Efficient decapping by Dcp2 requires the essential activator Dcp1 and in metazoans the scaffolding protein Edc4 (Fenger-Gron et al. 2005; She et al. 2006; Eulalio et al. 2007; Chang et al. 2014). Coactivator proteins that further activate decapping and may target the decapping complex to specific RNA decay pathways have also recently been characterized, including the enhancer of decapping proteins (Edc1-3) and PNRC2 (Borja et al. 2010; Fromm et al. 2011; Lai et al. 2012). The Dcp1/2 decapping complex functions as part of a highly evolved network of protein–protein interactions that regulates cap hydrolysis and the degradation of mRNA.
In addition to Dcp2, several other classes of cap-degrading enzymes have been described. Scavenger decapping enzymes, including DcpS and Aph1/FHIT, are members of the HIT pyrophosphatase family that can degrade free cap molecules (m7GpppN) or short capped oligonucleotides that are remnants of the 3′-to-5′ mRNA decay pathways (Liu et al. 2002, 2004; Wang et al. 2002; Taverniti and Seraphin 2015). There are no reports indicating that scavenger decapping enzymes are able to hydrolyze the m7G cap structure attached to an RNA chain longer than several nucleotides (Milac et al. 2014). Recent evidence suggests that in mammals, many Nudix hydrolase family enzymes (Nudt2, 3, 12, 15, 16, 17) may have decapping activity in addition to Dcp2 (Taylor and Peculis 2008; Song et al. 2010, 2013; Li and Kiledjian 2011; Lu et al. 2011). Finally, the DXO family of enzymes possess pyrophosphohydrolase, decapping, and 5′-3′ exonuclease activities, and participates in quality control of pre-mRNA in the nucleus (Chang et al. 2012; Jiao et al. 2013; Jurado et al. 2014).
Dcp2 is a bilobed enzyme composed of an evolutionarily conserved N-terminal portion containing a regulatory domain (residues 1–94 in Schizosaccharomyces pombe), which binds the activator Dcp1, and a catalytic domain (residues 95–243) belonging to the Nudix hydrolase superfamily, which carries out cap hydrolysis and contains a positively charged channel for RNA binding (Mildvan et al. 2005; She et al. 2006, 2008; Deshmukh et al. 2008). The extended C-terminal portion of Dcp2 (residues 244–741) is highly divergent, largely disordered, and appears to contain protein–protein interaction modules that can recruit additional coactivators of decapping (Fromm et al. 2011; He and Jacobson 2015). Structural and kinetic studies suggest that Dcp2 uses a composite active site in which residues on both the regulatory and catalytic domains bind the 5′ cap structure and contribute to the catalysis of cap hydrolysis (Floor et al. 2010, 2012). These observations, along with dynamics studies of Dcp2, suggest that conformational transitions in the decapping complex play an important role in catalysis and may be used to regulate Dcp2 activity. Despite extensive structural and biochemical studies, it is unclear how the domains of Dcp2 come together to form the composite active site and bind and hydrolyze the 5′-terminal m7G cap.
A major obstacle to understanding cap recognition by Dcp2 is that all previously studied m7GpppN cap analogs bind the enzyme with very weak (millimolar) affinities. We have previously identified several phosphate-modified cap analogs which, after incorporation at the 5′ terminus of mRNAs, make them more resistant to Dcp2-mediated decapping (Grudzien-Nogalska et al. 2013; Ziemniak et al. 2013a). The modifications include bridging substitution at the α/β-position of the cap (O-to-CH2 or methylenebisphosphonate; O-to-NH or imidodiphosphate) (Grudzien et al. 2006; Su et al. 2011) or nonbridging substitution at the β-phosphate (O-to-S or phosphorothioate; O-to-BH3 or boranophosphate) (Supplemental Fig. S1B; Li et al. 2007; Su et al. 2011). The Dcp2-resistant transcripts are more stable than transcripts with corresponding unmodified caps both in vitro and in vivo. Some of the analogs that made mRNA resistant to Dcp2 also bound the translational cap-binding protein eIF4E with higher affinity and, consequently, produced mRNA with both increased cellular half-life and increased translational efficiency. These cap-modified transcripts have found application in experimental mRNA-based therapies (Kuhn et al. 2010; Ziemniak et al. 2013a; Sahin et al. 2014).
In the current work, we sought cap analogs that would bind Dcp2 tightly and inhibit decapping activity without the need for incorporation into an RNA transcript. Identification of such compounds would not only be useful in structural and biochemical studies on Dcp2 activity and cap binding, but would also potentially allow identification of inhibitor binding sites and pave the way for the development of novel Dcp2 inhibitors. Recent studies link Dcp2 activity to spinal muscular atrophy (Shukla and Parker 2014) and the interferon response (Li et al. 2012) in mammalian cells, suggesting that development of selective Dcp2 inhibitors may be medically useful.
Accordingly, we screened a library of synthetic, modified cap analogs for inhibition of Dcp1/2 decapping and identified several compounds with inhibitory activity at micromolar concentrations. The most potent of these, m7GpSpppSm7G (12b), was further characterized in biochemical and structural studies and found to bind with 20-fold tighter affinity than native m7GpppN cap to specific cap-binding surfaces on both the regulatory and catalytic domains of Dcp2. We propose a model of inhibition where 12b acts as a cap-mimic and binds tightly to the composite active site of Dcp2.
RESULTS
Screening a synthetic nucleotide library identifies Dcp1/2 inhibitors
A library consisting of 29 m7G nucleotide derivatives was screened in order to find potential inhibitors of the Dcp1/2 decapping complex (Fig. 1). The library contained several classes of phosphate-modified cap analogs, namely m7GDP (compounds 1–5, Fig. 1) and m7GTP derivatives (compounds 6–11) as well as tri- and tetraphosphate dinucleotide cap analogs (compounds 12–17). Several phosphate-unmodified parent compounds (1, 6, 16–17) as well as the common nucleotides ATP (18) and GTP (19), were also included in the inhibitor screen for comparison. The synthetic mononucleotide derivatives 1–11 possess either single or multiple modifications at bridging (CH2, NH) or nonbridging (S, BH3, F) positions of the phosphate chain. Dinucleotide derivatives, or “two-headed” cap analogs, 12–15 possess two m7G moieties, a lengthened tetraphosphate bridge, phosphorothioate, or boranophosphate substitution at the α and δ positions in the phosphate chain, and some have a methylene bridge between the β and γ phosphate groups. Previous studies on cap-interacting proteins including eIF4E (Grudzien et al. 2006; Grudzien-Nogalska et al. 2007), DcpS (Kalek et al. 2006; Kowalska et al. 2008), and Dcp2 itself (Grudzien-Nogalska et al. 2007; Su et al. 2011), suggest that introduction of these chemical modifications into the phosphate chain can significantly alter cap–protein interactions (Jemielity et al. 2010; Ziemniak et al. 2013b).
FIGURE 1.

Cap analogs included in the screened library. Section (§) indicates compounds containing a single stereogenic center on phosphorus atom, which consequently exist as a pair of diastereomers. Double section (§§) indicates compounds containing two stereogenic phosphorus atoms, which exist as a mixture of three diastereomers.
Incorporation of phosphorothioate and boranophosphate groups into the phosphate chain results in cap analogs which are isostructural, (pseudo)isoelectronic and retain the same negative net charge as unmodified counterparts. Both modifications lead to a slightly longer P–X bond as compared to the native P–O bond. The O-to-S substitution results in a higher preference for soft divalent metal cations, whereas the O-to-BH3 substitution generally decreases affinity to metal cations and reduces H-bond acceptor properties (Stivers and Nagarajan 2006; Li et al. 2007; Guga and Koziolkiewicz 2011). The O-to-F substitution at the terminal phosphate results in a smaller net negative charge and impairs H-bonding (Baranowski et al. 2015). The bridging O-to-CH2 substitution provides resistance to hydrolysis since the P–C bonds are remarkably stable, however, the inability to form H-bonds can deteriorate affinity to some proteins (Klein et al. 2002; Grudzien et al. 2006). The bridging imidodiphosphate modification is also highly resistant to hydrolysis by pyrophosphatases and better emulates the electronic properties of unmodified phosphatases, such as their charge distribution and H-bond accepting ability due to the higher electronegativity of nitrogen (Yount et al. 1971).
We used a radioactivity-based RNA-decapping assay to screen the synthetic nucleotide library for inhibition of Dcp1/2 activity (Supplemental Fig. S2A). A short (48 nt) radiolabeled RNA oligomer bearing an anti-reverse cap analog (ARCA), m27,3′-OGpppN at the 5′ end was incubated with S. pombe Dcp1/2 for either 15 or 30 min in the presence of 200 μM nucleotide analog. Capped (m27,3′-O GpppRNA) and decapped (pRNA) transcripts were resolved by high-resolution SDS-PAGE and quantified by autoradiography. During the reaction, decapped transcripts accumulate and lead to an increase in intensity of the gel band that contains both decapped and uncapped RNA (pppRNA; Fig. 2A; Supplemental Fig. S2B).
FIGURE 2.

Decapping inhibition screening assay. (A) Analysis of representative sequencing gels. In the absence of inhibitors, RNA transcripts are rapidly hydrolyzed by SpDcp1/2, only transcripts containing m27,2′-OGppCH2pG are resistant to degradation. In the presence of cap analogs 12a-b or 15a-c, a significant fraction of capped oligonucleotides is intact after 30 min treatment by the enzyme. (B) Inhibition properties of tested cap analogs (see Supplemental Table S1 for details). Phosphorothioate analogs of m7Gppppm7G (12a-b) are significantly more potent inhibitors than either m7GDP (1) (the product of Dcp2-mediated decapping reaction) or unmodified cap analog m27,3′-OGpppG (16). The data represent mean values from triplicate experiments ±SEM.
Uncapped mRNA represents the ∼10% of mRNAs for which a cap dinucleotide is not incorporated during in vitro transcription (Grudzien et al. 2013).
The results of the inhibition screen are given in Figure 2B and Supplemental Table S1, from which we can draw several broad conclusions about the inhibitory properties of the cap analogs tested. Cap analogs without any modifications, such as m7GDP (1), m7GTP (6), and m27,3′-OGpppG (16), as well as ATP (18) and GTP (19), were found to be poor Dcp1/2 inhibitors (Fig. 2B). The inhibitory effect was also minimal if the phosphate chain was simply elongated to tetraphosphate (m7GppppG, 17). These results are consistent with an earlier NMR-based study in which Kd values for several nucleotide–Dcp2 complexes were found to be in the range of 10–30 mM, indicating weak interaction of nucleotides with Dcp2 (Floor et al. 2010; see Supplemental Table S2 for selected data from this publication). Moreover, we found that there are no statistically significant differences between two previously studied cap analogs, m7GDP and m27,3′-OGpppG, with regard to their inhibitory potential (Student's t-test, P = 0.659). On the other hand, two-headed cap analogs (compounds 12–15) were generally found to be more potent Dcp2 inhibitors (Fig. 2). In particular, the diastereomers of m7GpSppppSm7G, compounds 12a (a mixture of D1 and D2 diastereomers) and 12b (D3 diastereomer), were found to be among the strongest inhibitors within the library (Figs. 2, 3). The greater inhibitory potency of m7GpS ppppSm7G (D3 diastereomer) (12b) compared to m7GDP (1) or m27,3′-O GpppG (16) is highly significant (Fig. 2B; P-values 0.003 and <0.001, respectively). Introduction of a methylene bridge, to either S- or BH3-modified two-headed cap analogs (13 and 15), resulted in no apparent enhancement of their inhibitory properties. We chose one of the most potent inhibitors of Dcp1/2 decapping activity identified in the inhibitor screen, cap analog 12b, for more detailed kinetic and structural studies aimed at verifying the potency of inhibition and understanding the mechanism of Dcp2 inhibition.
FIGURE 3.
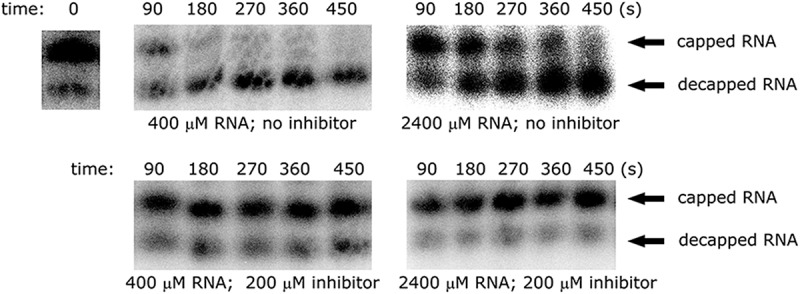
Representative sequencing gels showing the time course of in vitro decapping at two RNA concentrations in the presence or absence of 12b. In the first case, the decapping reaction is rapid—the substrate is degraded within several minutes. Addition of 200 μM 12b to the mixture blocks the decapping reaction.
Cap analog 12b is a mixed inhibitor of Dcp1/2
To determine the mechanism of inhibition for 12b we used a single-turnover kinetic assay with 32P-labeled native-capped RNA (29 nt) to measure decapping kinetics (kobs) at varying enzyme and inhibitor concentrations (Fig. 4A). Single-turnover conditions allow for direct measurement of the rate constant for the catalytic step (kmax) in response to inhibitor, in contrast to multiple-turnover conditions in which measured rates depend on both the rate of catalysis and product release. We used a synthetic, unstructured 29-nt RNA substrate bearing an unmodified 5′ m7G cap for these experiments because we have previously characterized kinetic constants for S. pombe Dcp2 and the Dcp1/2 complex with this substrate (Deshmukh et al. 2008; Floor et al. 2010). Fits of kobs versus enzyme concentration with different amounts of inhibitor revealed that the apparent KM increases by fivefold and the kmax for Dcp1/2 decapping decreases by threefold in the presence of 1 mM 12b inhibitor (Fig. 4A; Supplemental Fig. S3; Supplemental Tables S3, S4). The same trend in KMapp and kmax was found for SpDcp2 alone, in the absence of activator Dcp1 (Supplemental Fig. S4; Supplemental Table S5). This identifies 12b as a mixed inhibitor that can both (i) reversibly bind to the free enzyme and compete for RNA substrate binding (increasing KM) and also (ii) bind to the enzyme–substrate complex (decreasing kmax).
FIGURE 4.

Single-turnover inhibition kinetics of 12b with SpDcp1/2 and a 29-nt native-capped RNA substrate. (A) Fits to determine KMapp and kmax at different inhibitor concentrations; KMapp increases by fivefold and kmax decreases by threefold in the presence of 1 mM inhibitor, demonstrating that 12b is a mixed inhibitor of Dcp1/2 (see Supplemental Table S1). (B) Fits to Equation 1 of normalized rate data versus inhibitor concentration to determine KI under subsaturating enzyme concentrations. (C) Bar graph of determined KI values for 12b with Dcp1/2 or Dcp2 and m7GpppG with Dcp1/2; nucleotide 12b binds with ∼20-fold tighter affinity than m7GpppG.
In order to estimate the affinity of 12b for the free enzyme (KI), we measured kobs for decapping at different inhibitor concentrations while keeping the enzyme concentration below the KM for RNA substrate binding, where the vast majority of enzyme is not bound by substrate and inhibitor will be bound only to free enzyme. In this subsaturing enzyme regime, under single-turnover kinetics conditions, a normalized plot of kobs versus inhibitor concentration can be fit to Equation 1 to extract KI (Fig. 4B; Supplemental Figs. S5, S6; see Supplemental Information for derivation of Equation 1). For the Dcp1/2 complex, the KI for 12b is 65 ± 7 μM, and for Dcp2 alone, the KI for 12b is 121 ± 16 μM (Fig. 4C). Compound 12b binds 20-fold more tightly to Dcp1/2 than the unmodified m7GpppG, which has a KI of 1400 ± 300 μM.
| (1) |
Because 12b is a mixed inhibitor of Dcp1/2, the compound also has an independent affinity for the enzyme–substrate complex (KI′), which can in principle be measured by following kobs versus inhibitor concentration under saturating enzyme conditions where all substrate is bound as enzyme–substrate complex. However, under these conditions at high enzyme concentration, hydrolysis of 12b by Dcp1/2 occurs on a similar timescale as RNA decapping, preventing accurate determination of KI′ (Supplemental Fig. S7). No hydrolysis of 12b was observed under the conditions used to measure KI at subsaturating enzyme. In summary, decapping kinetics experiments with native-capped RNA show that the two-headed O-to-S modified cap analog 12b is a mixed inhibitor of Dcp2 decapping that binds reversibly with mid-micromolar affinity to the free enzyme. 12b binds ∼20-fold more tightly than cap analog m7GpppG, and is a significantly more potent inhibitor of Dcp2 than any previously tested cap analog or other nucleotide.
NMR titrations map binding of 12b to specific surfaces of Dcp2
To structurally map the interaction between 12b and Dcp2 we carried out NMR titrations on the isolated regulatory (residues 1–94) and catalytic (residues 95–243) domains of SpDcp2. Isolated domains were used in these experiments because conformational dynamics in the two domain Dcp2 construct (1–243) leads to significant peak broadening that allows only partial assignment of Dcp2 resonances, whereas the isolated domains have the majority of resonances assigned (Floor et al. 2010). Increasing amounts of 12b were added to either the 15N-labeled regulatory or catalytic domain of Dcp2 and 15N HSQC NMR spectra were recorded at 12b concentrations up to 2 mM. For both the regulatory and catalytic domains of Dcp2, we observed selective chemical shift perturbations and peak broadening upon addition of 12b (Fig. 5). NMR titrations of the regulatory domain indicate that 12b binds the same surface as m7GDP (Floor et al. 2010), strongly perturbing residues W43 and D47 on the conserved 31–52 helix of the regulatory domain (Fig. 5B–D; Supplemental Figs. S8, S9). Chemical shift versus cap analog concentration data are nicely fit by a two-state binding model to extract the Kd for ligand binding to the regulatory domain (Supplemental Fig. S10; Supplemental Table S2). Importantly, the affinity of 12b to the Dcp2 regulatory domain is about 0.8 mM, more than 10-fold tighter than any previously tested nucleotides (Floor et al. 2010), which bind with 10–40 mM affinity.
FIGURE 5.

Interaction of m7GpSpppSm7G (12b) with the regulatory and catalytic domains of SpDcp2. (A) Structure of the Dcp1/2 complex with regions strongly perturbed by 12b (PDB 2QKM); Dcp1 is yellow, Dcp2 regulatory domain (1–94) is purple, Dcp2 catalytic domain (95–243) is green with the catalytic Nudix helix in red. (B) Regulatory domain of Dcp2 colored by chemical shift perturbation upon addition of 2 mM 12b; aligned as in A. (C,D) Overlay of 15N-HSQC spectra for residue W43 (C) or D47 (D) during titration of 12b from 0 mM (red) to 2 mM (purple). (E) Catalytic domain of Dcp2 colored by chemical shift perturbation upon addition of 12b, resonances that undergo significant broadening (e.g., G122 as in G) are colored blue; aligned as in A. (F–I) Overlay of 15N-HSQC spectra for residue G116 (F), G122 (G), E192 (H), or E146 (I), during titration of 12b from 0 mM (red) to 2 mM (purple). NMR spectra for residues W117 and K118 are shown in Supplemental Figure S8.
12b is also recognized by a specific surface on the catalytic domain of Dcp2 with 0.5 mM affinity (Fig. 5E–H; Supplemental Fig. S8, S11). This is in contrast to all previously measured nucleotides, including m7GDP, that weakly bind the catalytic domain with 10–20 mM affinity (Floor et al. 2010), and appear to interact with many different surfaces of the domain nonspecifically. In the case of 12b the only significant chemical shift perturbations to the Dcp2 catalytic domain occur on clustered residues of adjacent loops (the 120s loop: residues G116, W117, K118, G122, are shifted and/or broadened and the 190s loop: residue E192 is shifted and broadened; Fig. 5A), indicating that the compound binds tightly and specifically to a single surface on the catalytic domain. Notably, unlike m7GDP, 12b does not significantly perturb E146, the catalytic glutamate on the Nudix helix of the catalytic domain of Dcp2 (Fig. 5I). Compound 12a, a mixture of the D1 and D2 diastereomers of m7GpS pppSm7G, is also recognized by the same surfaces of the Dcp2 regulatory and catalytic domains as the D3 diastereomer 12b (Supplemental Fig. S12, S13), suggesting that the stereochemistry of the phosphate modifications is not critical for 12 binding Dcp2.
m7GpSpppSm7G-capped RNA transcripts can be decapped by Dcp1/2
To investigate whether there is a correlation between the inhibitory properties of compounds 12–15 and their susceptibility to decapping, we synthesized 48-nt RNA transcripts incorporating these cap analogs at the 5′ end and tested the ability of Dcp1/2 to hydrolyze the cap-modified transcripts. Since bacterial or viral RNA polymerases are unable to discriminate between G and m7G for addition to the growing polynucleotide chain, 30%–50% of the RNAs are capped in the reverse orientation [i.e., Gpppm7GpNp(Np)n] if unmodified cap structures are used (Contreras et al. 1982). This can be prevented by replacement of conventional cap analogs with anti-reverse cap analogs (ARCAs) that contain modifications at either the 2′- or 3′-positions of m7G preventing them from being incorporated in the wrong orientation (Stepinski et al. 2001). Because 12–15 are composed of two symmetric m7G nucleotides, they can be attached to the 5′ end of RNA from either nucleotide, avoiding the need to incorporate additional 2′ or 3′ modifications. The efficiencies for incorporation of 12–15 into short (48 nt) RNA transcripts by T7 RNA polymerase were tested as described before (Ziemniak et al. 2013b), and ranged from 27%–80% for all cap analogs (Supplemental Table S6).
Decapping assays with analog 12–15-capped RNA were carried out as previously described, using recombinant S. pombe Dcp1/2 decapping complex (Ziemniak et al. 2013b). Transcripts containing analogs 12–15 at the 5′ terminus were incubated for 15 or 30 min with Dcp1/2, then the reaction was quenched, and capped versus decapped (and uncapped) transcripts were separated by gel electrophoresis then quantified by autoradiography. The percentage of capped RNA is calculated from the ratio of capped RNA to the total RNA, and decapping is expressed as the decrease in this percentage (Supplemental Table S6).
All RNA transcripts capped with diastereomers of compound 12 were degraded significantly faster than those capped with an unmodified ARCA analog (Fig. 6). This result was surprising since our previous studies on sulfur-modified cap analogs suggested that introduction of two nonbridging sulfur atoms in the phosphate chain should increase or have a neutral effect on cap analog stability (Grudzien-Nogalska et al. 2007). Cap analog 13, which introduces a methylene bridge into the O-to-S modified two-headed cap, is cleaved from RNA at a rate similar to the unmodified m27,3′-OGpppG. RNA capped with boranophosphate cap analogs with (15) and without (14) a bridging methylene substitution is cleaved at rates slower than unmodified ARCA. In particular, the D3 diastereomers 14c and 15c are strongly resistant to cleavage by Dcp1/2. The exception to this trend is 14b, a diastereomer of the boranophosphate-substituted two-headed cap analog, which is cleaved much faster than the other diastereomers of 14 or unmodified cap, similar to 12.
FIGURE 6.

(A) In vitro Dcp2-mediated hydrolysis of oligonucleotides capped by a standard ARCA cap (m27,3′-OGpppG) and analogs 12–15. After treatment with SpDcp1/2 for the indicated times, samples were loaded on a 16% RNA sequencing gel. Upper band represents capped transcripts whereas lower band represents a mixture of decapped and uncapped transcripts. (B) Susceptibility of cap analogs 12–15 to Dcp2-mediated degradation, compared to both a standard ARCA analog (m27,3′-OGpppG) and a Dcp2-resistant analog (m27,2′-OGppCH2pG). 12b is rapidly hydrolyzed by Dcp2 when attached to 5′ mRNA terminus.
Modified cap analogs 12–13 incorporated at the 5′ end of RNA transcripts, like the free dinucleotide 12b (see above and Supplemental Fig. S7), are recognized as good substrates by Dcp1/2 and hydrolyzed, whereas boranophosphate analogs 14–15, with the exception of 14b, are partially resistant to cleavage. These results indicate a lack of correlation between inhibitory properties of free cap analogs 12–15 and their susceptibility to decapping upon incorporation into RNA.
DISCUSSION
The decapping enzyme Dcp2 recognizes and cleaves the m7G cap at the 5′ end of mRNA, committing the transcript to degradation. Dcp2-mediated catalysis is a crucial step in many different 5′-to-3′ RNA decay pathways that control gene expression and transcript quality control in the cell. Despite several available crystal structures of Dcp2, and extensive biochemical and biophysical characterization, we still do not have a clear structural picture of how the enzyme binds and hydrolyzes substrate. This may in large part be due to the fact that Dcp2 weakly binds native cap structures (m7GpppN) with only mM affinity, making structural characterization of the Dcp2-cap interaction difficult. Here we have identified a chemically modified cap analog, m7GpSpppSm7G (12b), from a library of synthetic nucleotides that binds and inhibits Dcp2 ∼20-fold more strongly than native cap structures or any previously tested cap analogs. Our data suggest that 12b competes for the active site of Dcp2 and may act as a tightly bound mimic of the 5′ cap, paving the way for future structural studies and the development of potent, selective inhibitors of Dcp2.
Observations made during our initial screening of the synthetic nucleotide library for Dcp2 inhibition are consistent with earlier reports concerning the influences of structural alterations of nucleotide phosphate chains on cap–protein interactions (Kowalska et al. 2008, 2014; Rydzik et al. 2012). We find that the introduction of charge-retaining nonbridging modifications (S−, BH3−) into cap analogs seemed to generally have higher impact on Dcp2 decapping activity than the bridging (CH2 and NH) modifications. The more pronounced effect of nonbridging modifications in the phosphate chain on cap analog binding and inhibition of Dcp2 is also in line with detailed studies of other Nudix hydrolases, which have shown that nonbridging oxygen atoms in the phosphate chain often make important contacts with enzyme residues or metal ions (Mildvan et al. 2005).
NMR titrations of 12b with the isolated regulatory and catalytic domains of Dcp2 showed that the modified cap analog binds tightly to specific surfaces on each domain (Fig. 5). On the regulatory domain of Dcp2, 12b significantly perturbs residues on the 31–52 helix, particularly the catalytically important residues W43 and D47, similar to m7GDP (a product of mRNA decapping) or other native cap structures, but with ∼20-fold tighter affinity. On the catalytic domain of Dcp2, 12b selectively perturbs residues on the 120s and 190s loop, in contrast to m7GDP and ATP, which are bound much more weakly and appear to nonspecifically perturb many different surfaces of the catalytic domain at high concentrations. The importance of the 120s loop for catalysis has not been previously tested, however residues in this loop are perturbed by addition of RNA in NMR experiments (Deshmukh et al. 2008) and lie at the terminus of the predicted RNA binding channel on the catalytic domain of Dcp2, suggesting the 120s loop may play a role in binding nucleotides at the 5′ end of the RNA body. In the ATP-bound crystal structure of Dcp1/2 (She et al. 2008) the 120s loop forms part of the pocket occupied by ATP, suggesting that ATP may be mimicking a 5′ nucleotide of RNA and that one of the m7G moieties in 12b may do the same (Fig. 7; She et al. 2006).
FIGURE 7.

Analysis of 12b binding to the open and closed structures of Dcp1/2. Chemical shift perturbations (coloring as in Fig. 5B,E) of 12b binding to the isolated regulatory and catalytic domains of Dcp2 were mapped onto the structures of the open (A; PDB 2A6T) and closed, ATP-bound (B,C; PDB 2QKM) conformations of Dcp1/2. ATP is shown in red. Based on the location of observed chemical shift perturbations, it may be the case that one of the m7G moieties of 12b mimics cap and perturbs the 31–52 helix of the regulatory domain, which is known to be critical for cap binding and catalysis, while the other m7G moiety of 12b may bind the catalytic domain in a similar location as ATP in the closed conformation of Dcp1/2 (B,C), mimicking the 5′-most nucleotide of RNA.
On the 190s loop, E192, whose resonance is shifted and broadened upon addition of 12b to Dcp2, is a catalytically important residue that coordinates Mg2+ metal in the Dcp2 active site and has been shown to undergo pH-dependent motions that are likely coupled to the catalytic cycle (Aglietti et al. 2013). The detected perturbations of E192 cannot be the result of 12b interfering with metal binding in the active site however, since the NMR titrations are carried out with an excess of Mg2+ (5 mM, 20-fold excess over 12b) and the cap analog was co-titrated along with two equivalents of Mg2+. Additionally, NMR titrations adding only Mg2+ to the Dcp2 catalytic domain resulted in little to no visible chemical shift changes (data not shown). These data suggest that the dynamic, active site residue E192 is involved in binding of 12b. Alternatively, the chemical shift changes could result from a ligand induced conformational change near E192. Finally, E146, a catalytic glutamate on the Nudix helix involved in metal binding and catalysis that is significantly perturbed upon m7GDP addition (Floor et al. 2010), was not perturbed by 12b. One might interpret the lack of perturbations on the catalytic helix of Dcp2 as evidence that 12b binds to the active site of Dcp2 in a different, and catalytically incompetent mode than product m7GDP, however because 12b can be hydrolyzed by Dcp2 at high enzyme concentrations (Supplemental Fig. S7) and RNA transcripts capped with 12b undergo rapid decapping (Fig. 6), it is clear that the bound 12b can indeed access catalytically relevant conformations in solution.
Nucleotide analog 12b appears to act as a more tightly bound mimic of 5′ m7G cap based on the following observations: (i) single-turnover kinetic assays show 12b reversibly inhibits Dcp2 and competes with substrate binding to the active site, with a KI ∼20-fold tighter than m7GpppG, (ii) NMR titrations show that 12b binds tightly to specific surfaces of both the regulatory and catalytic domains of Dcp2 that are important for RNA binding and catalysis, and (iii) 12b is hydrolyzed by Dcp2 when bound as either the dinucleotide, or when incorporated at the 5′ end of RNA, demonstrating it is a viable substrate of the decapping enzyme that can bind the active site in a catalytically competent conformation. Dcp2 can adopt different conformations in solution (Fig. 7), and our combined data are consistent with a closed structure of the enzyme in which 12b contacts both domains of Dcp2. This might occur by one of the m7G moieties of 12b binding in the ATP binding site of the closed conformation of Dcp2, similar to the structure observed in PDB 2QKM (Fig. 7B), and the other m7G group extending back toward the 31–52 helix. However, we have previously shown that this conformation of Dcp2 is incompatible with catalysis (Floor et al. 2010) and thus favor a model of 12b binding in which one of the m7G moieties of 12b binds in a similar position on the catalytic domain of Dcp2 as ATP in Figure 7C, mimicking the 5′ nucleotide of RNA, and the other m7G group of 12b contacting the 31–52 helix of the regulatory domain in a different, closed conformation in which the regulatory domain positions W43 and the m7G cap closer to the catalytic helix of Dcp2. We favor the latter interpretation because our data show that 12b can bind to Dcp2 in a catalytically competent conformation both as the free nucleotide and when incorporated on the 5′ end of RNA; hydrolysis of 12b is inconsistent with the nucleotide analog binding Dcp2 in a closed conformation as depicted in Figure 7B, because the catalytic helix is positioned very far from the phosphate chain. Future structural studies will be aimed at using 12b as a tightly bound cap mimic to understand substrate recognition and catalysis by Dcp2.
Since analogs of all cap analogs, including m7Gppppm7G, could potentially interfere with translational machinery and DcpS-associated processes, their use for in vivo decapping studies may be limited (Ziemniak et al. 2015). Another obstacle to in vivo experiments is delivery of nucleotide analogs to the cytoplasm. Due to their negative charge, cap analogs are incapable of being transported across the cell membrane. However, some recent reports suggest that artificial delivery systems could overcome this encumbrance (Brudno et al. 2014; Howard et al. 2014; Zochowska et al. 2015). In spite of these difficulties the compounds described here can be further applied to biophysical and biochemical studies on Dcp1/2 and larger protein assemblies including coactivators of decapping. Ongoing structural studies involving X-ray crystallography are expected to unravel the detailed mechanism of binding, allowing the design of more efficient inhibitors of Dcp2. The structures of identified inhibitors may also serve as a starting point for the design of molecular probes to test Dcp2 binding or activity in high throughput format. Interestingly, recent studies have implicated Dcp2 in some diseases related to SMN assembly (Shukla and Parker 2014) and interferon response (Li et al. 2012). These findings suggest that small molecules targeting Dcp2 may also be interesting from a pharmacological point of view, and biophysical and biochemical insight into Dcp2 structure and function gained from studies using the tightly bound cap analogs described here may be important for the development of therapeutically relevant Dcp2 inhibitors.
MATERIALS AND METHODS
Compound library
mRNA cap analogs used for Dcp2 screening (Fig. 1) were synthesized as previously described: compound 2 (Kalek et al. 2005), compound 3 (Rydzik et al. 2012), compounds 4a, 4b (Strenkowska et al. 2012), compound 5 (Baranowski et al. 2015), compounds 9–11, 12–15 (Ziemniak et al. 2015), compounds 1, 6, 16, 17 (Jemielity et al. 2003).
Compounds 7 and 8 were prepared using the same methods as described in Strenkowska et al. (2012). The structure and purity for all compounds was confirmed by RP HPLC analysis, HRMS, and NMR before biological experiments. ATP and GTP were purchased from Sigma-Aldrich.
In vitro synthesis of RNA transcripts
Capped RNA transcripts were synthesized by in vitro transcription of plasmid pluc-A60 digested with NcoI, which yielded a capped RNA corresponding to the first 48 nt of firefly luciferase mRNA. All ribonucleotides were obtained in the presence of given cap analogs and 10 μCi/µL [α-32P] GTP (PerkinElmer) as described previously (Su et al. 2011). Reaction mixtures were extracted with phenol and chloroform; RNAs were separated from small-molecule impurities with NucAway spin columns (Ambion). The concentrations of RNAs were determined via measuring Cerenkov radiation in a scintillation counter (Beckman).
Dcp1/2 inhibitor screen
SpDcp1/2 was expressed and purified as described previously (Floor et al. 2010). Capped 32P-labeled 48-nt oligonucleotides were subjected to digestion with GST-SpDcp1/2 at 37°C for 30 min in a buffer composed of 50 mM Tris–HCl (pH 8 at 25°C), 50 mM NH4Cl, 0.01% NP-40, 1 mM DTT, and 5 mM MgCl2. In each case 200 μM of the synthetic nucleotide to be tested was added to the reaction. Reactions were quenched by adding two volumes of Precipitation/Inactivation Buffer III (Ambion). RNAs were precipitated at −20°C overnight, collected by centrifugation at 13,000g at 4°C for 20 min, purified by washing in 70% ethanol and collected again by centrifugation at 9000g at 4°C for 5 min. The quantity of RNA in the precipitated samples was determined via measurement of Cerenkov radiation in a scintillation counter (Beckman). The samples were resuspended in Sequencing Gel Loading Buffer (Ambion) and denatured at 95°C for 5 min. RNA sequencing gels (10% polyacrylamide) were run at 45–70 W for 3.5 h on a Base Runner Nucleic Acid Sequencer apparatus (International Biotechnologies). Gels were fixed in 5% acetic acid, 5% methanol for 10–15 min, dried onto Whatman 3MM filter paper (Fisher Scientific). Radioactivity in individual bands was quantified by analyzing scanned film using Quantity One program (Bio-Rad). Percentage of inhibition was defined as follows:
| (2) |
where RNAdeg indicates the fraction of decapped RNA in a given time point without inhibitor, and RNAdeg+Inh indicates the fraction of decapped RNA in a given time point when an inhibitor is present. Student's t-tests to compare inhibitory potential of cap analogs were calculated in SigmaPlot 11.0 software (Systat Software Inc.).
Single-turnover kinetic inhibition assays
General procedure. Single-turnover decapping assays were carried out as previously described (Deshmukh et al. 2008). SpDcp1/21-243 or SpDcp21-243 and inhibitor were combined at 3× concentration in the decapping reaction buffer (50 mM Tris–Cl [pH 8 at 25°C], 50 mM NH4Cl, 0.01% NP-40, 1 mM DTT, and 5 mM MgCl2). The decapping reaction was initiated by adding 30 µL capped 32P-labeled 29 nt RNA substrate at 1.5× concentration in decapping reaction buffer to 60 µL 3× protein + inhibitor at 4°C. Final protein concentrations ranged from 0.8–25 μM; final inhibitor concentrations ranged from 0.03–1 mM; final RNA concentration was <100 pM. Time points were quenched by addition of excess EDTA, RNA was separated from m7GDP product by thin layer chromatography, and the fraction of capped versus decapped transcripts was quantified using a GE Typhoon scanner and ImageQuant as previously described (Deshmukh et al. 2008). Plots of fraction m7GDP product versus time were fit to a first order exponential to extract kobs; plots of kobs versus enzyme concentration were fit to the Michaelis–Menten equation: kobs = kmax[E]/KM + [E], to extract kmax and KM.
Measuring kinetic constants with inhibitor 12b. KMapp and kmax were first measured for SpDcp1/2 and SpDcp2 in the presence of different concentrations of 12b, in order to determine the type of inhibition. Two different preparations of 29-nt RNA (denoted a and b) were used during these experiments, and these differed somewhat in their kinetic constants with Dcp1/2 and Dcp2 (For Dcp1/2, Fig. 4A; Supplemental Table S3 are preparation a; Supplemental Fig. S3; Supplemental Table S4 are preparation b). Importantly, both 29-nt RNA preparations showed the same trends in kinetic constants with added 12b, and gave the same KI for 12b (see above). For both SpDcp1/2 and SpDcp2, KMapp increases and kmax decreases in the presence of nucleotide analog 12b, identifying the compound as a mixed inhibitor of Dcp2 decapping.
15N-HSQC NMR titration experiments
The regulatory (1–94) and catalytic (95–243) domains of SpDcp2 were purified as described previously (Floor et al. 2010). NMR experiments were carried out in 150 mM NaCl, 2 mM MgCl2, 5 mM DTT, and 50 mM HEPES (pH 7.0) at 25°C. Titrations on the regulatory domain were performed at 250 μM protein in a volume of 500 μL by addition of increasing amounts of nucleotide analogs and the simultaneous addition of two equivalents of magnesium chloride. Titrations on the catalytic domain were identical except that 360 μM protein was used. All titration experiments were conducted on a Bruker Avance 800 MHz spectrometer outfitted with a cryogenic probe. The NMR data were processed using NMRPipe software package and visualized using Sparky (version 3.114). Composite chemical shift perturbations were calculated according to the equation:
| (3) |
Chemical shift perturbations were fit to a quadratic two-state binding model to extract Kdusing SigmaPlot 11.0 software (Systat Software Inc.):
| (4) |
where [L]0 is a ligand concentration, [P]0 is a protein concentration, and δmax is the maximal perturbation of the chemical shift.
In vitro synthesis of capped transcripts
Capped RNA transcripts containing modified cap analogs were synthesized by in vitro transcription of plasmid pluc-A60 digested with NcoI using T7 RNA polymerase, which yielded a capped RNA corresponding to the first 48-nt of firefly luciferase mRNA. All transcripts were obtained in the presence of given cap analogs and 10 µCi/µL [α-32P] GTP (PerkinElmer) as described previously (Su et al. 2011). Reaction mixtures were extracted with phenol and chloroform; RNAs were separated from small-molecule impurities with NucAway spin columns (Ambion). The concentrations of RNAs were determined via measuring Cerenkov radiation in a scintillation counter (Beckman).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
The authors are grateful to Ewa Grudzien-Nogalska, Sergey V. Slepenkov, and Wei Su for their help with Dcp2 screening assay and valuable discussions. This study was supported by the National Science Centre (Poland) (UMO-2011/01/N/NZ1/04326 and UMO-2014/12/T/NZ1/00528 to M.Z.; UMO-2013/09/B/ST5/01341 to J.J.) and the US National Institutes of Health (R01 GM078360 to J.D.G., NRSA fellowship F32 GM105313 to J.S.M., and R01 GM020818 to R.E.R.). M.Z. is supported by the Foundation for Polish Science International PhD Projects Programme, cofinanced by the EU European Regional Development Fund.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.055152.115.
REFERENCES
- Aglietti RA, Floor SN, McClendon CL, Jacobson MP, Gross JD. 2013. Active site conformational dynamics are coupled to catalysis in the mRNA decapping enzyme Dcp2. Structure 21: 1571–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso CR. 2012. A complex ‘mRNA degradation code’ controls gene expression during animal development. Trends Genet 28: 78–88. [DOI] [PubMed] [Google Scholar]
- Balagopal V, Fluch L, Nissan T. 2012. Ways and means of eukaryotic mRNA decay. Biochim Biophys Acta 1819: 593–603. [DOI] [PubMed] [Google Scholar]
- Baranowski MR, Nowicka A, Rydzik AM, Warminski M, Kasprzyk R, Wojtczak BA, Wojcik J, Claridge TD, Kowalska J, Jemielity J. 2015. Synthesis of fluorophosphate nucleotide analogues and their characterization as tools for 19F NMR studies. J Org Chem 80: 3982–3997. [DOI] [PubMed] [Google Scholar]
- Barckmann B, Simonelig M. 2013. Control of maternal mRNA stability in germ cells and early embryos. Biochim Biophys Acta 1829: 714–724. [DOI] [PubMed] [Google Scholar]
- Borja MS, Piotukh K, Freund C, Gross JD. 2010. Dcp1 links coactivators of mRNA decapping to Dcp2 by proline recognition. RNA 17: 278–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brudno Y, Silva EA, Kearney CJ, Lewin SA, Miller A, Martinick KD, Aizenberg M, Mooney DJ. 2014. Refilling drug delivery depots through the blood. Proc Natl Acad Sci 111: 12722–12727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter S, Ricci EP, Mercier BC, Moore MJ, Fitzgerald KA. 2014. Post-transcriptional regulation of gene expression in innate immunity. Nat Rev Immunol 14: 361–376. [DOI] [PubMed] [Google Scholar]
- Chang JH, Jiao X, Chiba K, Oh C, Martin CE, Kiledjian M, Tong L. 2012. Dxo1 is a new type of eukaryotic enzyme with both decapping and 5′-3′ exoribonuclease activity. Nat Struct Mol Biol 19: 1011–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CT, Bercovich N, Loh B, Jonas S, Izaurralde E. 2014. The activation of the decapping enzyme DCP2 by DCP1 occurs on the EDC4 scaffold and involves a conserved loop in DCP1. Nucleic Acids Res 42: 5217–5233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras R, Cheroutre H, Degrave W, Fiers W. 1982. Simple, efficient in vitro synthesis of capped RNA useful for direct expression of cloned eukaryotic genes. Nucleic Acids Res 10: 6353–6362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshmukh MV, Jones BN, Quang-Dang DU, Flinders J, Floor SN, Kim C, Jemielity J, Kalek M, Darzynkiewicz E, Gross JD. 2008. mRNA decapping is promoted by an RNA-binding channel in Dcp2. Mol Cell 29: 324–336. [DOI] [PubMed] [Google Scholar]
- Eulalio A, Rehwinkel J, Stricker M, Huntzinger E, Yang SF, Doerks T, Dorner S, Bork P, Boutros M, Izaurralde E. 2007. Target-specific requirements for enhancers of decapping in miRNA-mediated gene silencing. Genes Dev 21: 2558–2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenger-Gron M, Fillman C, Norrild B, Lykke-Andersen J. 2005. Multiple processing body factors and the ARE binding protein TTP activate mRNA decapping. Mol Cell 20: 905–915. [DOI] [PubMed] [Google Scholar]
- Floor SN, Jones BN, Hernandez GA, Gross JD. 2010. A split active site couples cap recognition by Dcp2 to activation. Nat Struct Mol Biol 17: 1096–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floor SN, Borja MS, Gross JD. 2012. Interdomain dynamics and coactivation of the mRNA decapping enzyme Dcp2 are mediated by a gatekeeper tryptophan. Proc Natl Acad Sci 109: 2872–2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromm SA, Truffault V, Kamenz J, Braun JE, Hoffmann NA, Izaurralde E, Sprangers R. 2011. The structural basis of Edc3- and Scd6-mediated activation of the Dcp1:Dcp2 mRNA decapping complex. EMBO J 31: 279–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grudzien E, Kalek M, Jemielity J, Darzynkiewicz E, Rhoads RE. 2006. Differential inhibition of mRNA degradation pathways by novel cap analogs. J Biol Chem 281: 1857–1867. [DOI] [PubMed] [Google Scholar]
- Grudzien-Nogalska E, Jemielity J, Kowalska J, Darzynkiewicz E, Rhoads RE. 2007. Phosphorothioate cap analogs stabilize mRNA and increase translational efficiency in mammalian cells. RNA 13: 1745–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grudzien-Nogalska E, Kowalska J, Su W, Kuhn AN, Slepenkov SV, Darzynkiewicz E, Sahin U, Jemielity J, Rhoads RE. 2013. Synthetic mRNAs with superior translation and stability properties. Methods Mol Biol 969: 55–72. [DOI] [PubMed] [Google Scholar]
- Guga P, Koziolkiewicz M. 2011. Phosphorothioate nucleotides and oligonucleotides—recent progress in synthesis and application. Chem Biodivers 8: 1642–1681. [DOI] [PubMed] [Google Scholar]
- Guo H, Ingolia NT, Weissman JS, Bartel DP. 2010. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 466: 835–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He F, Jacobson A. 2015. Control of mRNA decapping by positive and negative regulatory elements in the Dcp2 C-terminal domain. RNA 21: 1633–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard JD, Sarojini H, Wan R, Chien S. 2014. Rapid granulation tissue regeneration by intracellular ATP delivery—a comparison with Regranex. PLoS One 9: e91787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu CL, Stevens A. 1993. Yeast cells lacking 5′-->3′ exoribonuclease 1 contain mRNA species that are poly(A) deficient and partially lack the 5′ cap structure. Mol Cell Biol 13: 4826–4835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inada T. 2013. Quality control systems for aberrant mRNAs induced by aberrant translation elongation and termination. Biochim Biophys Acta 1829: 634–642. [DOI] [PubMed] [Google Scholar]
- Jemielity J, Fowler T, Zuberek J, Stepinski J, Lewdorowicz M, Niedzwiecka A, Stolarski R, Darzynkiewicz E, Rhoads RE. 2003. Novel “anti-reverse” cap analogs with superior translational properties. RNA 9: 1108–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jemielity J, Kowalska J, Rydzik AM, Darzynkiewicz E. 2010. Synthetic mRNA cap analogs with a modified triphosphate bridge—synthesis, applications and prospects. New J Chem 34: 829–844. [Google Scholar]
- Jiao X, Chang JH, Kilic T, Tong L, Kiledjian M. 2013. A mammalian pre-mRNA 5′ end capping quality control mechanism and an unexpected link of capping to pre-mRNA processing. Mol Cell 50: 104–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurado AR, Tan D, Jiao X, Kiledjian M, Tong L. 2014. Structure and function of pre-mRNA 5′-end capping quality control and 3′-end processing. Biochemistry 53: 1882–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalek M, Jemielity J, Stepinski J, Stolarski R, Darzynkiewicz E. 2005. A direct method for the synthesis of nucleoside 5′-methylenebis(phosphonate)s from nucleosides. Tetrahedron Lett 46: 2417–2421. [Google Scholar]
- Kalek M, Jemielity J, Darzynkiewicz ZM, Bojarska E, Stepinski J, Stolarski R, Davis RE, Darzynkiewicz E. 2006. Enzymatically stable 5′ mRNA cap analogs: synthesis and binding studies with human DcpS decapping enzyme. Bioorg Med Chem 14: 3223–3230. [DOI] [PubMed] [Google Scholar]
- Kervestin S, Jacobson A. 2012. NMD: a multifaceted response to premature translational termination. Nat Rev Mol Cell Biol 13: 700–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein E, Mons S, Valleix A, Mioskowski C, Lebeau L. 2002. Synthesis of enzymatically and chemically non-hydrolyzable analogues of dinucleoside triphosphates Ap3A and Gp3G. J Org Chem 67: 146–153. [DOI] [PubMed] [Google Scholar]
- Kowalska J, Lewdorowicz M, Zuberek J, Grudzien-Nogalska E, Bojarska E, Stepinski J, Rhoads RE, Darzynkiewicz E, Davis RE, Jemielity J. 2008. Synthesis and characterization of mRNA cap analogs containing phosphorothioate substitutions that bind tightly to eIF4E and are resistant to the decapping pyrophosphatase DcpS. RNA 14: 1119–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalska J, Wypijewska Del Nogal A, Darzynkiewicz ZM, Buck J, Nicola C, Kuhn AN, Lukaszewicz M, Zuberek J, Strenkowska M, Ziemniak M, et al. 2014. Synthesis, properties, and biological activity of boranophosphate analogs of the mRNA cap: versatile tools for manipulation of therapeutically relevant cap-dependent processes. Nucleic Acids Res 42: 10245–10264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn AN, Diken M, Kreiter S, Selmi A, Kowalska J, Jemielity J, Darzynkiewicz E, Huber C, Tureci O, Sahin U. 2010. Phosphorothioate cap analogs increase stability and translational efficiency of RNA vaccines in immature dendritic cells and induce superior immune responses in vivo. Gene Ther 17: 961–971. [DOI] [PubMed] [Google Scholar]
- Lai T, Cho H, Liu Z, Bowler MW, Piao S, Parker R, Kim YK, Song H. 2012. Structural basis of the PNRC2-mediated link between mrna surveillance and decapping. Structure 20: 2025–2037. [DOI] [PubMed] [Google Scholar]
- Lee M, Kim B, Kim VN. 2014. Emerging roles of RNA modification: m6A and U-tail. Cell 158: 980–987. [DOI] [PubMed] [Google Scholar]
- Li Y, Kiledjian M. 2011. Regulation of mRNA decapping. Wiley Interdiscip Rev RNA 1: 253–265. [DOI] [PubMed] [Google Scholar]
- Li P, Sergueeva ZA, Dobrikov M, Shaw BR. 2007. Nucleoside and oligonucleoside boranophosphates: chemistry and properties. Chem Rev 107: 4746–4796. [DOI] [PubMed] [Google Scholar]
- Li Y, Dai J, Song M, Fitzgerald-Bocarsly P, Kiledjian M. 2012. Dcp2 decapping protein modulates mRNA stability of the critical interferon regulatory factor (IRF) IRF-7. Mol Cell Biol 32: 1164–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Rodgers ND, Jiao X, Kiledjian M. 2002. The scavenger mRNA decapping enzyme DcpS is a member of the HIT family of pyrophosphatases. EMBO J 21: 4699–4708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SW, Jiao X, Liu H, Gu M, Lima CD, Kiledjian M. 2004. Functional analysis of mRNA scavenger decapping enzymes. RNA 10: 1412–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu G, Zhang J, Li Y, Li Z, Zhang N, Xu X, Wang T, Guan Z, Gao GF, Yan J. 2011. hNUDT16: a universal decapping enzyme for small nucleolar RNA and cytoplasmic mRNA. Protein Cell 2: 64–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzoni C, Falcone C. 2011. mRNA stability and control of cell proliferation. Biochem Soc Trans 39: 1461–1465. [DOI] [PubMed] [Google Scholar]
- Milac AL, Bojarska E, Wypijewska del Nogal A. 2014. Decapping Scavenger (DcpS) enzyme: advances in its structure, activity and roles in the cap-dependent mRNA metabolism. Biochim Biophys Acta 1839: 452–462. [DOI] [PubMed] [Google Scholar]
- Mildvan AS, Xia Z, Azurmendi HF, Saraswat V, Legler PM, Massiah MA, Gabelli SB, Bianchet MA, Kang LW, Amzel LM. 2005. Structures and mechanisms of Nudix hydrolases. Arch Biochem Biophys 433: 129–143. [DOI] [PubMed] [Google Scholar]
- Popp MW, Maquat LE. 2013. Organizing principles of mammalian nonsense-mediated mRNA decay. Annu Genet 47: 139–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rissland OS, Norbury CJ. 2009. Decapping is preceded by 3′ uridylation in a novel pathway of bulk mRNA turnover. Nat Struct Mol Biol 16: 616–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero-Santacreu L, Moreno J, Perez-Ortin JE, Alepuz P. 2009. Specific and global regulation of mRNA stability during osmotic stress in Saccharomyces cerevisiae. RNA 15: 1110–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rydzik AM, Kulis M, Lukaszewicz M, Kowalska J, Zuberek J, Darzynkiewicz ZM, Darzynkiewicz E, Jemielity J. 2012. Synthesis and properties of mRNA cap analogs containing imidodiphosphate moiety—fairly mimicking natural cap structure, yet resistant to enzymatic hydrolysis. Bioorg Med Chem 20: 1699–1710. [DOI] [PubMed] [Google Scholar]
- Sahin U, Kariko K, Tureci O. 2014. mRNA-based therapeutics—developing a new class of drugs. Nat Rev Drug Discov 13: 759–780. [DOI] [PubMed] [Google Scholar]
- She M, Decker CJ, Chen N, Tumati S, Parker R, Song H. 2006. Crystal structure and functional analysis of Dcp2p from Schizosaccharomyces pombe. Nat Struct Mol Biol 13: 63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- She M, Decker CJ, Svergun DI, Round A, Chen N, Muhlrad D, Parker R, Song H. 2008. Structural basis of dcp2 recognition and activation by dcp1. Mol Cell 29: 337–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla S, Parker R. 2014. Quality control of assembly-defective U1 snRNAs by decapping and 5′-to-3′ exonucleolytic digestion. Proc Natl Acad Sci 111: E3277–3286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song MG, Li Y, Kiledjian M. 2010. Multiple mRNA decapping enzymes in mammalian cells. Mol Cell 40: 423–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song M-G, Bail S, Kiledjian M. 2013. Multiple Nudix family proteins possess mRNA decapping activity. RNA 19: 390–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepinski J, Waddell C, Stolarski R, Darzynkiewicz E, Rhoads RE. 2001. Synthesis and properties of mRNAs containing the novel “anti-reverse” cap analogs 7-methyl(3′-O-methyl)GpppG and 7-methyl(3′-deoxy)GpppG. RNA 7: 1486–1495. [PMC free article] [PubMed] [Google Scholar]
- Stivers JT, Nagarajan R. 2006. Probing enzyme phosphoester interactions by combining mutagenesis and chemical modification of phosphate ester oxygens. Chem Rev 106: 3443–3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strenkowska M, Wanat P, Ziemniak M, Jemielity J, Kowalska J. 2012. Preparation of synthetically challenging nucleotides using cyanoethyl P-imidazolides and microwaves. Org Lett 14: 4782–4785. [DOI] [PubMed] [Google Scholar]
- Su W, Slepenkov S, Grudzien-Nogalska E, Kowalska J, Kulis M, Zuberek J, Lukaszewicz M, Darzynkiewicz E, Jemielity J, Rhoads RE. 2011. Translation, stability, and resistance to decapping of mRNAs containing caps substituted in the triphosphate chain with BH3, Se, and NH. RNA 17: 978–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su W, Slepenkov SV, Slevin MK, Lyons SM, Ziemniak M, Kowalska J, Darzynkiewicz E, Jemielity J, Marzluff WF, Rhoads RE. 2013. mRNAs containing the histone 3′ stem-loop are degraded primarily by decapping mediated by oligouridylation of the 3′ end. RNA 19: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taverniti V, Seraphin B. 2015. Elimination of cap structures generated by mRNA decay involves the new scavenger mRNA decapping enzyme Aph1/FHIT together with DcpS. Nucleic Acids Res 43: 482–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MJ, Peculis BA. 2008. Evolutionary conservation supports ancient origin for Nudt16, a nuclear-localized, RNA-binding, RNA-decapping enzyme. Nucleic Acids Res 36: 6021–6034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Roretz C, Di Marco S, Mazroui R, Gallouzi IE. 2011. Turnover of AU-rich-containing mRNAs during stress: a matter of survival. Wiley Interdiscip Rev RNA 2: 336–347. [DOI] [PubMed] [Google Scholar]
- Walters R, Parker R. 2014. Quality control: is there quality control of localized mRNAs? J Cell Biol 204: 863–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Jiao X, Carr-Schmid A, Kiledjian M. 2002. The hDcp2 protein is a mammalian mRNA decapping enzyme. Proc Natl Acad Sci 99: 12663–12668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilczynska A, Bushell M. 2015. The complexity of miRNA-mediated repression. Cell Death Differ 22: 22–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yount RG, Babcock D, Ballantyne W, Ojala D. 1971. Adenylyl imidodiphosphate, an adenosine triphosphate analog containing a P–N–P linkage. Biochemistry 10: 2484–2489. [DOI] [PubMed] [Google Scholar]
- Ziemniak M, Strenkowska M, Kowalska J, Jemielity J. 2013a. Potential therapeutic applications of RNA cap analogs. Future Med Chem 5: 1141–1172. [DOI] [PubMed] [Google Scholar]
- Ziemniak M, Szabelski M, Lukaszewicz M, Nowicka A, Darzynkiewicz E, Rhoads RE, Wieczorek Z, Jemielity J. 2013b. Synthesis and evaluation of fluorescent cap analogues for mRNA labelling. RSC Adv 3: 20943–20958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziemniak M, Kowalska J, Lukaszewicz M, Zuberek J, Wnek K, Darzynkiewicz E, Jemielity J. 2015. Phosphate-modified analogues of m7GTP and m7Gppppm7G—synthesis and biochemical properties. Bioorg Med Chem 23: 5369–5381. [DOI] [PubMed] [Google Scholar]
- Zochowska M, Piguet AC, Jemielity J, Kowalska J, Szolajska E, Dufour JF, Chroboczek J. 2015. Virus-like particle-mediated intracellular delivery of mRNA cap analog with in vivo activity against hepatocellular carcinoma. Nanomedicine 11: 67–76. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.