

Abstract
Coupling between transcription and RNA processing is key for gene regulation. Using live-cell photobleaching techniques, we investigated the factor TCERG1, which coordinates transcriptional elongation with splicing. We demonstrate that TCERG1 is highly mobile in the nucleoplasm and that this mobility is slightly decreased when it is associated with speckles. Dichloro-1-β-D-ribofuranosylbenzimidazole (DRB) but not α-amanitin treatment reduced the mobility of TCERG1, which suggests interaction with paused transcription elongation complexes. We found that TCERG1 mobility is rapid at the transcription site (TS) of a reporter that splices post-transcriptionally and that TCERG1 is recruited to the active TS independent of the CTD of RNAPII, thus excluding phosphorylated CTD as a requirement for recruiting this factor to the TS. Importantly, the mobility of TCERG1 is reduced when the reporter splices cotranscriptionally, which suggests that TCERG1 forms new macromolecular complexes when splicing occurs cotranscriptionally. In this condition, spliceostatin A has no effect, indicating that TCERG1 rapidly binds and dissociates from stalled spliceosomal complexes and that the mobility properties of TCERG1 do not depend on events occurring after the initial spliceosome formation. Taken together, these data suggest that TCERG1 binds independently to elongation and splicing complexes, thus performing their coupling by transient interactions rather than by stable association with one or the other complexes. This finding has conceptual implications for understanding the coupling between transcription and RNA processing.
Keywords: FRAP, transcription site, splicing, transcription–splicing coupling, RNAPII, HIV
INTRODUCTION
Mammalian cell nuclear proteins are concentrated within several stable compartments that are not defined by membranes (Mao et al. 2011). Although these compartments are apparently immobile structures, fluorescence recovery after photobleaching (FRAP) analysis has demonstrated that the proteins in these compartments are more mobile than previously assumed and are exchanged continuously between the compartments and the nucleoplasm (Phair and Misteli 2000; Snaar et al. 2000; Chen and Huang 2001; Rino et al. 2007). The absence of a defined membrane and the rapid dynamics of nuclear compartments are consistent with a self-organization model in which the structure of a body is determined by the global interactions among its constituents (Misteli 2001; Kaiser et al. 2008). One of the most dynamic nuclear compartments is the splicing factor-rich nuclear speckle (Lamond and Spector 2003). Pioneering time-lapse visualization studies revealed that nuclear speckles are markedly dynamic structures that respond to changes in gene expression (Misteli et al. 1997; Kruhlak et al. 2000). Subsequently, FRAP studies of nuclear speckle components revealed that these components move within the nucleus with kinetics ranging from milliseconds to seconds (Phair and Misteli 2000; Rino et al. 2008). This high mobility and the morphological changes that occur upon transcription inhibition (O'Keefe et al. 1994) indicate that speckles are formed by self-organization, which is achieved by direct interactions between their components, and that speckles then rearrange their structure upon expression of target RNAs.
The translocation of particles in the nucleus can be explained by a diffusion-like movement (Seksek et al. 1997; Gorisch et al. 2005). However, FRAP experiments have demonstrated that nuclear proteins exhibit a slower mobility than expected if only diffusion processes act on the proteins probably due to transient interactions with other nuclear components (Kruhlak et al. 2000; Pederson 2000; Phair and Misteli 2000; Shopland and Lawrence 2000). This reduced mobility may be dictated by interactions with quasi-immobile structures, such as the chromatin or nuclear matrix, or by integration into larger macromolecular complexes. For example, the mobilities of the splicing factors U2AF65, U2AF35, and SF1 are less than expected for the individual particles, which can be explained by the suggested presence of U2AF65–U2AF35–SF1 nucleoplasmic macromolecular complexes that determine their dynamic pattern (Rino et al. 2008). The study of other putative preassembled factor complexes, such as SR proteins with U2AF35 and U1-70K factors or snRNPs (Chusainow et al. 2005; Ellis et al. 2008; Huranova et al. 2010), reinforced the idea that the dynamic behavior of nuclear factors is largely determined by transient interactions. These aforementioned observations are consistent with a stochastic model in which nuclear proteins are permanently scanning the nuclear environment and searching for interaction partners using a combination of continuous diffusion-like movements and transient interactions. Consistent with this model and in the absence of transcription and splicing activity, nuclear factors move freely throughout the nucleoplasm, with higher dynamic rates due to the absence of pre-mRNA molecules or other factors engaged in pre-mRNA processing.
To date, many studies have reported that pre-mRNA processing, including splicing, is largely coupled with transcription (for review, see Bentley 2014). However, the in vivo spatial and dynamic properties of the functional coupling between transcriptional elongation and splicing have not been investigated in detail. Global analysis of nascent RNA in yeast has revealed that cotranscriptional splicing is associated with pauses of RNA polymerase II (RNAPII) at specific sites (Alexander et al. 2010; Carrillo Oesterreich et al. 2010; Wilhelm et al. 2011). Thus, a transcriptional pause has been proposed to be imposed by a regulatory checkpoint associated with cotranscriptional splicing (Alexander et al. 2010). This hypothesis implies that proteins acting at the interface of these processes serve as checkpoint factors to regulate cotranscriptional splicing. The identification of these regulators and their spatial properties in real time and in live cells within the context of the highly compartmentalized eukaryotic nucleus is essential for understanding the functional coordination of RNAPII transcription and splicing. One common model invoked for coupling factors is that these factors bind the transcription elongation complex, travel along the gene while associated with the polymerase, and mediate pausing and/or modification of splicing patterns when target introns are encountered.
TCERG1 is a human nuclear factor that was initially identified as a transcriptional cofactor regulating human immunodeficiency virus type 1 (HIV-1) gene expression (Suñé et al. 1997). Recently, we reported that TCERG1 functions as a positive regulator of HIV-1 transcriptional elongation by increasing the elongation rate of RNAPII (Coiras et al. 2013). Other data have demonstrated that TCERG1 interacts with transcription and splicing factors (Goldstrohm et al. 2001; Lin et al. 2004; Smith et al. 2004; Sánchez-Álvarez et al. 2006), localizes to the interface of nuclear speckles and presumed nearby transcription sites (TSs) (Sánchez-Álvarez et al. 2006; Sánchez-Hernández et al. 2012), and affects the transcriptional activity of CCAAT/enhancer-binding protein α (C/EBPα) (McFie et al. 2006) and the splicing of several minigene reporters (Lin et al. 2004; Cheng et al. 2007; Pearson et al. 2008; Montes et al. 2012b; Sánchez-Hernández et al. 2012). In addition, early mass analysis combined with a novel and precise computational approach provided the first evidence of a putative global role of TCERG1 in mRNA processing (Pearson et al. 2008). Given these data, TCERG1 has been suggested as a possible factor that coordinates transcription and splicing (Montes et al. 2012a), a hypothesis that we recently supported by demonstrating that TCERG1 regulates alternative splicing of the Bcl-x gene by modulating the rate of RNAPII transcription (Montes et al. 2012b).
Here, we investigated the dynamic properties of TCERG1 in real time in live cells. Our data do not support a situation where TCERG1 would be stably bound to the elongating polymerase but reveal a model where coupling is achieved by transient, rapid interactions with elongating and splicing complexes.
RESULTS
TCERG1 exhibits highly dynamic behavior in living cells
To study the dynamic properties of TCERG1 in living cells, we generated a GFP-tagged version of human full-length TCERG1 (GFP-TCERG1) and used confocal laser scanning microscopy to determine whether the subnuclear localization of this fusion protein was similar to that of the endogenous TCERG1, which localizes to nuclear speckles (Sánchez-Álvarez et al. 2006). GFP-TCERG1 was distributed throughout the nucleoplasm, with an increased signal in organized granule-like sites (Fig. 1A, panel a). Double-labeled immunofluorescence experiments using GFP-TCERG1 and antibodies against the essential splicing factor SRSF2, which is commonly used to define nuclear speckles, confirmed that the fusion protein was enriched in the speckle compartment (Fig. 1A, panels b–d). To confirm these data, the spatial relationship between GFP-TCERG1 relative to SRSF2 was assessed by quantitatively scanning specific nuclear regions containing speckles (Fig. 1A, panel e). These results indicate that the GFP-TCERG1 construct expresses a protein with the same nuclear distribution as that of endogenous TCERG1.
FIGURE 1.

Characterization of the GFP-TCERG1 fusion protein. (A) Colocalization of GFP-TCERG1 with the essential splicing factor SRSF2. Dual labeling of HEK293T cells with EGFP-TCERG1 (a, green) and with the SRSF2 antibody (b, red) was performed. DAPI labeling was used to stain the chromatin (c, blue). Merge images are also presented (d). Line scans showing the local intensity distributions of GFP-TCERG1 in green, SRSF2 in red, and DAPI in blue are presented in panel e. A bar in panel d indicates the position of the line scans. Scale bar in panel d, 3 µm. (B) On the left is a schematic representation of the structure of the Bcl-x gene, with exons (boxes) and introns (lines). The two splice variants, the long antiapoptotic Bcl-xL and the short proapoptotic Bcl-xS, which are generated by alternative 5′ splice site selection, are depicted. The dotted lines indicate the alternative splicing events. The effect of TCERG1 and GFP-TCERG1 overexpression on the alternative splicing of the Bcl-x minigene is shown in the middle. HEK293T cells were transfected with the Bcl-x minigene together with empty plasmid (mock, lane 1), TCERG1 (lane 2), or GFP-TCERG1 expression plasmids (lane 3). Cells were harvested and processed for RT-PCR analysis. The graph on the right presents the densitometric analysis results as the ratio of Bcl-xL to Bcl-xS isoforms from three independent experiments (mean ± SEM). (**) P < 0.01; (***) P < 0.001.
To demonstrate that the fusion protein is functionally active, we performed an in vivo assay to test the effect of GFP-TCERG1 on Bcl-x alternative splicing. Human Bcl-x pre-mRNA undergoes alternative splicing at 5′ splice sites to produce the anti-apoptotic long (Bcl-xL) and the pro-apoptotic short (Bcl-xS) transcript isoforms (Fig. 1B, left panel). We reported previously that TCERG1 promotes splicing of the Bcl-xS isoform (Montes et al. 2012b). Total RNA was isolated from HEK293T cells transfected with the Bcl-x minigene under conditions of TCERG1 and GFP-TCERG1 overexpression, and splicing was analyzed using RT-PCR with specific primers to detect the Bcl-xL and Bcl-xS transcript isoforms. Consistent with our previous data (Montes et al. 2012b; Sánchez-Hernández et al. 2012), increased levels of TCERG1 resulted in a more efficient use of the Bcl-xS 5′ splice site, resulting in a decreased ratio of Bcl-xL to Bcl-xS (Fig. 1B, middle panel, lane 2). Similar results were obtained following GFP-TCERG1 overexpression; the levels of the Bcl-xS isoform were increased (Fig. 1B, middle panel, lane 3). Quantitative analysis of these data is also presented (Fig. 1B, right). Taken together, these results demonstrate that GFP-TCERG1 functions similar to TCERG1 in regulating Bcl-x alternative splicing. Thus, we concluded that the GFP-TCERG1 protein behaved analogously to its endogenous counterpart and was therefore suitable for further investigation.
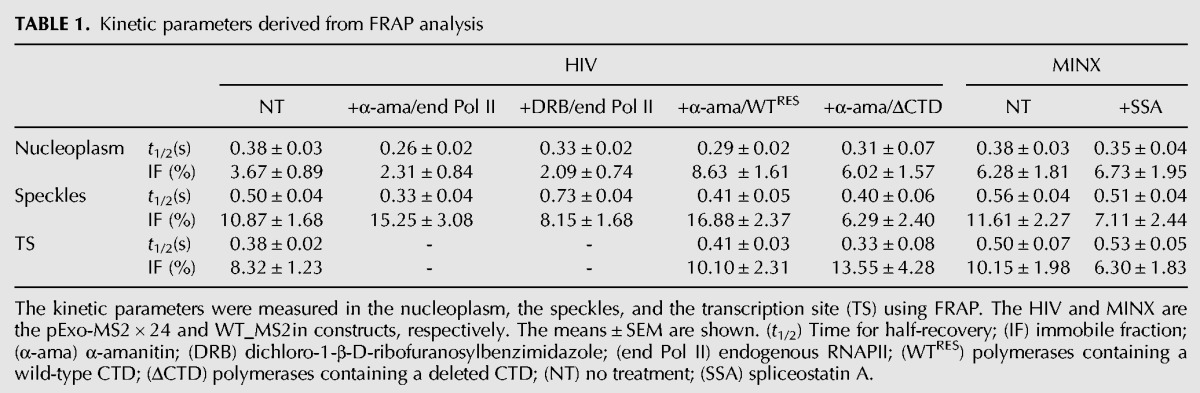
Next, we sought to analyze the mobility properties of GFP-TCERG1 in the nucleus using the well-established Exo1 cell line, which is a derivative of U2OS cells that express an MS2-tagged reporter RNA (Boireau et al. 2007) (see below). The FRAP method involves photobleaching a small area of the cell and monitoring the recovery of fluorescence intensity over a selected time interval (Fig. 2A). Fluorescence recovery indicates the movement of unbleached molecules into the bleached area. For these experiments, we selected cells with a low level of GFP fluorescence to avoid potential overexpression artifacts. The fluorescence of arbitrary areas in selected regions of the nucleoplasm and the speckles (located as far as possible from each other) were irreversibly photobleached, and the recovery was recorded simultaneously for 15 sec after bleaching. FRAP measurements revealed half times of 0.38 and 0.5 sec for the fluorescence of the nucleoplasm and the speckles, respectively (Table 1; Fig. 2B). These results indicate that TCERG1 is highly mobile in the nucleoplasm and speckles and that the dispersed nucleoplasmic population of TCERG1 molecules exhibited significantly faster mobility properties (P = 0.048) compared with those of the population of TCERG1 present at sites of enrichment. Analysis of the recovery rates after bleaching averaged over several nuclei revealed that the GFP-TCERG1 signal recovered after 10 sec in the nucleoplasm and the speckles (Fig. 2B). However, the fluorescent signal recovered only a fraction of its pre-bleach value in the nuclear speckle region, indicating the presence of an immobile fraction (IF) that might be bound to stable components of these nuclear compartments (Fig. 2B). Analysis of the FRAP curves revealed that IFs of ∼4% and 11% remained after the observation period of 15 sec in the nucleoplasm and the speckles, respectively (Table 1; Fig. 2B). Other studies of general splicing factors have reported ∼10% of immobile molecules in the splicing factor compartments (Rino et al. 2007; Huranova et al. 2010), which is comparable to our data. In summary, these experiments indicate that GFP-TCERG1 is a highly dynamic nuclear protein that exhibits lower mobility in speckles, which contain a significant population of immobile TCERG1 molecules that likely reflect an effect of its interaction with other RNA-related molecules.
FIGURE 2.
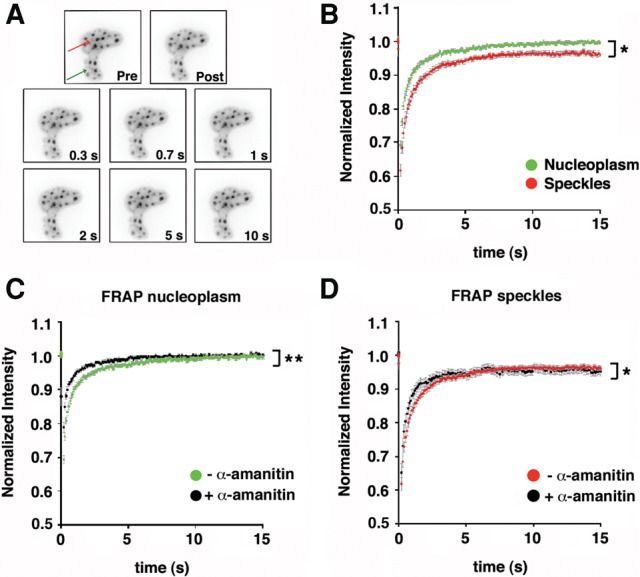
Analysis of GFP-TCERG1 dynamic properties in the Exo1 cells. (A) Time-lapse fluorescence microscopy of GFP-TCERG1 in nuclear speckles (the area of the bleach spot is indicated with the red arrow) and nucleoplasm (the area of the bleach spot is indicated with the green arrow) of the Exo1 cells. Images were taken at the time indicated in the bottom right-hand corner of the panels after the end of the bleach pulse. Pre, last pre-bleach pulse image; Post, first post-bleach pulse image. (B) Fluorescence recovery of GFP-TCERG1 was measured in the nucleoplasm (green) and nuclear speckles of Exo1 cells cotransfected with GFP-TCERG1, Tat and MS2-mcherry expression plasmids. The total number of analyzed cells was 34. The curves correspond to a pool of at least three independent experiments and the error bars represent the SEM. (C,D) Effect of the α-amanitin treatment on TCERG1 dynamics. Exo1 cells were transfected with GFP-TCERG1, Tat and MS2-mcherry and were then either untreated or treated with 0.1 µg/µL α-amanitin for 2 h and processed for FRAP experiments in the nucleoplasm and the nuclear speckle regions. FRAP curves in the nucleoplasm region in α-amanitin-treated (black; n = 23) and untreated (green; n = 34) cells are shown in panel C. FRAP curves in the nuclear speckles in α-amanitin-treated (black; n = 16) and untreated (red; n = 34) cells are shown in panel D. In all panels, the curves represent at least two independent experiments, and the error bars indicate the SEM. (*) P < 0.05; (**) P < 0.01.
TABLE 1.
Kinetic parameters derived from FRAP analysis
TCERG1 mobility in the nucleus upon transcriptional inhibition
Splicing factors are constantly moving into and out of nuclear speckles even in the absence of newly synthesized pre-mRNAs. Moreover, these proteins move more freely throughout the nucleus in the absence of transcription and splicing (Phair and Misteli 2000; Rino et al. 2007; Huranova et al. 2010), indicating that these proteins interact with pre-mRNA in these compartments. To test whether TCERG1 modifies its dynamic behavior in response to transcriptional inhibition, the cells were treated with α-amanitin, which strongly inhibits RNAPII-dependent transcription and leads to the destruction of its large subunit. We reported previously that α-amanitin treatment inhibited endogenous RNAPII (Montes et al. 2012b) and that TCERG1 moves from the nucleoplasm toward enlarged and rounded nuclear speckles upon transcription inhibition (Sánchez-Álvarez et al. 2006). We confirmed these results using the GFP-TCERG1 construct and observed the disappearance of TSs upon α-amanitin treatment (data not shown). Next, we quantitatively analyzed the recovery of TCERG1 in the nucleoplasmic and speckle regions of the cells with or without α-amanitin treatment. The half-recovery time under untreated conditions did not significantly differ for the nucleoplasm versus the speckles (Table 1; Fig. 2C,D). Slight and significant increases in the mobility of TCERG1 in the nucleoplasm (P = 0.004) and in the speckles (P = 0.014) upon inhibition of RNAPII transcription with α-amanitin were observed (Table 1; Fig. 2C,D), which are in agreement with the mobility of other splicing factors under these conditions (Phair and Misteli 2000). These observations likely reflected the absence of binding sites that depend on nascent transcripts in both the nucleoplasm and speckles.
RNAPII-mediated transcription can also be blocked by treatment with DRB, a nucleoside analog that inhibits RNAPII CTD phosphorylation by targeting the kinase activity of the P-TEFb complex (Chodosh et al. 1989; Marshall and Price 1992; Mancebo et al. 1997; Price 2000; Cheng and Price 2007). We previously used DRB to block HIV-1 gene transcription in vivo (Montes et al. 2012b). We further examined the dynamic properties of TCERG1 in DRB-treated cells. DRB treatment produced effects similar to those of the α-amanitin treatment on GFP-TCERG1 staining, with a significant increase in the GFP-associated signal in nuclear speckles due to the redistribution of the protein to enlarged and rounded speckles (Sánchez-Álvarez et al. 2006) (data not shown). Under these conditions, active TSs were undetectable, validating the transcription inhibition upon drug treatment (data not shown). Interestingly, we observed that the overall mobility of GFP-TCERG1 in nuclear speckles was delayed in the presence of DRB, with half-recovery times of ∼0.7 sec (P = 0.0002) (Table 1; Fig. 3A,B). Treatment with DRB had little effect on the dynamic behavior of GFP-TCERG1 in the nucleoplasm, with a half-recovery time of ∼0.3 sec (Table 1; Fig. 3A,B). These results indicate that a pool of GFP-TCERG1 molecules in speckles is delayed by DRB-dependent immobilization events, in contrast to the effects of α-amanitin treatment or the behavior of classical splicing factors upon DRB-treatment (Kruhlak et al. 2000; Rino et al. 2007; Huranova et al. 2010). After DRB treatment, an IF of ∼8% remained at the nuclear speckles, whereas ∼2% remained bound to the nucleoplasm. These results further indicate the presence of a fraction of TCERG1 bound to other molecules in speckles that is not dependent on active transcription.
FIGURE 3.
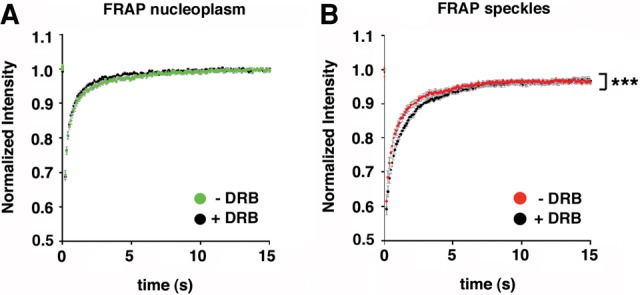
Effect of the DRB treatment on TCERG1 dynamics. The same experiment described for Figure 2C and D was performed with DRB. (A) FRAP curves in the nucleoplasm region in DRB-treated (black; n = 21) and untreated (green; n = 34) cells. (B) FRAP curves in the speckles in DRB-treated (black; n = 21) and -untreated (red; n = 34) cells. In all panels, the curves represent at least two independent experiments and the error bars indicate the SEM. (***) P < 0.001.
Dynamics of TCERG1 at the HIV-1 TS of a reporter that splices post-transcriptionally
The Exo1 cell line used in the present study expresses an HIV-1 reporter (termed pExo-MS2 × 24) that has been shown to splice post-transcriptionally (Boireau et al. 2007). In Exo1 cells, HIV-1 RNA transcript expression can be detected using fluorescence in situ hybridization (FISH) against the 24 MS2-binding sites present in the reporter gene or with the coexpression of a red fluorescent variant of MS2 (MS2-mCherry) (Fig. 4A). Because TCERG1 regulates HIV-1 transcription (Coiras et al. 2013), we took advantage of this system to address the dynamic properties of TCERG1 at the HIV-1 TS in addition to the nucleoplasm and the speckles. To evaluate the localization of TCERG1 to the HIV-1 TS, we cotransfected plasmids expressing GFP-TCERG1 and the HIV-1 transcriptional trans-activator Tat and performed FISH experiments using probes against the MS2 repeats within the HIV-1 reporter gene of Exo1 cells. As expected, GFP-TCERG1 exhibited a diffuse nucleoplasmic distribution with enrichment at the speckle region (Fig. 4B, panel a). After Tat induction, a bright spot was visible in the nucleoplasm, indicating the accumulation of labeled transcripts at the HIV-1 TS (Fig. 4B, panel b). Interestingly, we observed that TCERG1 accumulated at the HIV-1 TS in these cells (Fig. 4B, panel c), demonstrating an interaction between TCERG1 and the HIV-1 transcription machinery or nascent RNAs. Next, we wanted to examine the mobility of GFP-TCERG1 at the HIV-1 TS relative to the nucleoplasm and speckles using this construct. FRAP measurements revealed that GFP-TCERG1 exhibited similar fast mobility properties in the nucleoplasm and TS and reduced mobility in speckles (P = 0.0357) (Table 1; Fig. 4C), indicating that GFP-TCERG1 primarily establishes transient interactions with transcription and processing complexes.
FIGURE 4.

Dynamics of TCERG1 at the HIV-1 TS in the Exo1 cells. (A) Schematic representation of the HIV-1 reporter (pExo-MS2 × 24, HIV) stably integrated into U2OS cells (Exo1 cells). The reporter contains the two HIV-1 LTR sequences, the packaging sequence (ψ), the splice donor (SD1), the Rev-responsive element (RRE), the splice acceptor (SA7), and 24 MS2-binding sites in the 3′untranslated regions for detection in live and fixed cells. (B) Exo1 cells were transfected with GFP-TCERG1 and Tat expression vectors and processed for fluorescence in situ hybridization. GFP-TCERG1 was visualized using immunofluorescence (a), and HIV-1 RNAs were detected using probes against the MS2 repeats (b). Merge images present GFP-TCERG1 in green and RNAs in red (c). Scale bar, 3 µm. (C) Fluorescence recovery of GFP-TCERG1 was measured in the nucleoplasm (green; n = 34), nuclear speckles (red; n = 34), and HIV-1 TS (blue; n = 32) of Exo1 cells cotransfected with GFP-TCERG1, Tat and MS2-mcherry expression plasmids. The curves correspond to a pool of at least three independent experiments, and the error bars represent the SEM. (D) FRAP of Exo1 cells transfected with GFP-TCERG1, Tat, MS2-mcherry, and the α-amanitin-resistant wild-type (WTRES) RNAPII expression plasmids. Cells were treated with 0.1 µg/µL α-amanitin for 2 h before FRAP analysis. Fluorescence was measured at the HIV-1 TS (green; n = 21). The same analysis was also performed for the endogenous polymerase (red; n = 32). (E) The same experiment described for panel D was performed with the α-amanitin-resistant wild-type (green; n = 21) and ΔCTD (ΔCTDRES) (blue; n = 11) polymerases. In both panels, the curves represent at least two independent experiments, and the error bars indicate the SEM. (*) P < 0.05.
The CTD of the largest subunit of RNAPII is believed to play a critical role in RNA biogenesis by recruiting distinct sets of factors to the nascent transcript depending on its phosphorylation status (Buratowski 2003; de Almeida and Carmo-Fonseca 2008; Bartkowiak et al. 2011; Egloff et al. 2012). Importantly, truncation of the CTD of RNAPII prevents the accumulation of SR protein splicing factors as well as snRNPs at TSs in vivo (Misteli and Spector 1999). Given these data and our previous results, we sought to investigate the role of the CTD in the dynamic properties of GFP-TCERG1 at the TS. Exo1 cells were cotransfected with the GFP-TCERG1 vector and plasmids that expressed polymerases containing a wild-type CTD (WTRES) or a completely deleted CTD (ΔCTDRES). The transfected polymerases carry a point mutation that confers α-amanitin resistance, allowing inhibition of the endogenous RNAPII without affecting the activity of the transiently transfected polymerases (de la Mata and Kornblihtt 2006; Montes et al. 2012b). Inhibition of transcriptional elongation of pExo-MS2 ×24 by ΔCTDRES polymerase has been reported previously (Boireau et al. 2007). Interestingly, GFP-TCERG1 was detected at the HIV-1 TS upon complete deletion of the CTD (data not shown), indicating that the recruitment of TCERG1 to the HIV-1 TS is independent of the CTD of RNAPII and, therefore, of its phosphorylation status. Next, we compared the dynamics of TCERG1 at the TS of cells expressing WT and variant RNAPII. With WTRES, FRAP measurements in the α-amanitin-treated cells revealed fast fluorescence recovery similar to that observed with endogenous RNAPII transcription (Table 1; Fig. 4D). FRAP analysis of the GFP-TCERG1 kinetics in cells with WTRES and ΔCTDRES produced similar results (Table 1; Fig. 4E).
Dynamics of TCERG1 at the HIV-1 TS of a reporter that splices cotranscriptionally
To address the role of the spliceosome assembly in the kinetic properties of TCERG1, we used the previously described MINX reporter, which is derived from the major late transcript of adenovirus-2, contains strong splicing signals and splices entirely cotranscriptionally (Schmidt et al. 2011; Nawroth et al. 2014). The MINX reporter is under the control of the HIV-1 LTR and contains a downstream copy of the LacZ gene and cleavage and polyadenylation sequences of the bovine growth hormone gene (bGH) (Fig. 5A). The transcripts can be monitored in live cells using 4 MS2-binding sites located within the MINX intron (WT_MS2in). WT_MS2in cells were transiently cotransfected with Tat and GFP-TCERG1. TCERG1 was detected by immunofluorescence and the MINX TS was observed by detecting the resulting RNAs using FISH. GFP-TCERG1 colocalized with the nuclear MINX RNA (Fig. 5B). Next, we examined the mobility of GFP-TCERG1 at the TS relative to the nucleoplasm and speckles using the MINX construct. The FRAP curves revealed fast initial recovery at the TS during the first seconds of photobleaching (Fig. 5C). As for Exo1 cells, quantification of the FRAP data indicated slightly reduced mobility of the fluorescent protein at the TS and speckles compared with the nucleoplasm (P = 0.008) (Table 1). The mobility of GFP-TCERG1 in the speckles and at the TS was also reduced in the cells with the MINX reporter construct compared with that observed in cells with the pExo-MS2 × 24 reporter construct (Table 1; Fig. 5D,E), which could reflect the presence of new interactions that slowed the mobility of the fusion protein and that are dependent on cotranscriptional splicing. Most of the fluorescence at the TS was recovered during the experimental interval, but a residual IF of ∼10% persisted, indicating a stable interaction with slow moving molecules that was also observed in the speckle compartment (Table 1).
FIGURE 5.

Analysis of GFP-TCERG1 dynamic properties in the WT_MS2in cells. (A) Schematic representation of the MINX reporter containing 4 MS2-binding sites within the MINX intron (WT_MS2in). The reporter contains the HIV-1 LTR, the LacZ gene, and the cleavage and polyadenylation sequence of the bovine growth hormone gene (bGH). (B) WT_MS2in cells were transfected with the GFP-TCERG1 and Tat expression vectors. TCERG1 was visualized using immunofluorescence (a) and RNAs were visualized by FISH with probes against the MS2 repeats (b) 24 h after transfection. Merge images present GFP-TCERG1 in green and RNAs in red (c). Scale bar, 3 µm. (C) FRAP of WT_MS2in cells transfected with the GFP-TCERG1, Tat, and MS2-mcherry expression plasmids. Fluorescence was measured in the nucleoplasm (green; n = 16), at the HIV-1 TS (blue; n = 14), and in the nuclear speckles (red; n = 16). The curves represent a pool of at least three independent experiments, and the error bars indicate the SEM. (D,E) FRAP curves for GFP-TCERG1 at the speckles (D) and TS (E) of the Exo1 (HIV) (red; n = 32) and the WT_MS2in (MINX) (green; n = 14) cells are presented. The curves represent at least two independent experiments, and the error bars indicate the SEM. (**) P < 0.01.
The kinetics properties of transcriptional elongation by RNAPII are known to influence splicing (de la Mata et al. 2003); however, less information is available on how splicing events that occur cotranscriptionally on the pre-mRNA can influence transcriptional elongation. To gain further insight into the role of the spliceosome assembly process in the recruitment and kinetic properties of TCERG1, we repeated this experiment in the presence of spliceostatin A (SSA), a potent antitumor compound that targets the SF3b subunit of the U2 snRNP particle and blocks the formation of a catalytic spliceosome after U2 snRNP recruitment (Kaida et al. 2007; Roybal and Jurica 2010; Corrionero et al. 2011). The inhibition of cotranscriptional splicing of the MINX reporter with SSA has been previously reported (Schmidt et al. 2011). Treatment of cells with SSA did not abolish the localization of TCERG1 to the TS (Fig. 6A), confirming that TCERG1 is recruited during an early step of the transcription cycle and before U2 snRNP is incorporated onto the RNA. The dynamic behavior of GFP-TCERG1 in the nucleoplasm, the speckles, and the TS upon SSA treatment was assessed using FRAP analysis and quantification (Table 1; Fig. 6B–E). GFP-TCERG1 moved rapidly in the nucleus similar to the behavior observed in cells without SSA treatment, and the fluorescent protein exhibited reduced mobility in the speckles (P = 0.0034) and the TS (P = 0.011) compared with the nucleoplasm, which might reflect the interaction of TCERG1 with pre-mRNA factors. These results indicate that the mobility properties of TCERG1 do not depend on events occurring after the initial spliceosome formation.
FIGURE 6.

Effect of SSA on GFP-TCERG1 dynamics. WT_MS2in cells were transfected with the GFP-TCERG1, Tat and MS2-mcherry expression plasmids and treated with 100 ng/mL spliceostatin A (SSA) for 3 h before processing and visualization. (A) GFP (a) and mCherry (b) signals were visualized using immunofluorescence. Merge images (c) present the GFP signal in green and the mCherry signal in red. Scale bar, 3 µm (B) FRAP curves for GFP-TCERG1 in the nucleoplasmic regions of untreated (green; n = 16) and SSA-treated (black; n = 13) cells. (C) FRAP curves for GFP-TCERG1 in the nuclear speckles of SSA-untreated (red; n = 16) and -treated (black; n = 13) cells. (D) FRAP curves for GFP-TCERG1 at the TS of untreated (blue; n = 14) and SSA-treated (black; n = 12) cells. (E) FRAP of WT_MS2in cells treated with SSA and transfected with the GFP-TCERG1, Tat and MS2-mcherry expression plasmids. Fluorescence was measured in the nucleoplasm (green; n = 13), the HIV-1 TS (blue; n = 12) and the nuclear speckles (red; n = 13). In all panels, the curves represent at least two independent experiments, and the error bars indicate the SEM. (*) P < 0.05; (**) P < 0.01.
DISCUSSION
Evidence gathered in recent years has established that transcription and alternative splicing are coupled physically and functionally and this coupling is emerging as an essential component of gene expression regulation. The functional coupling of transcription and pre-mRNA processing in the context of the highly compartmentalized eukaryotic nucleus remains to be characterized. Here, we have measured the cell nuclear dynamics of TCERG1, a human protein that has been proposed to act as a checkpoint regulator to promote cotranscriptional splicing by regulating RNAPII processivity (Montes et al. 2012b). Previous studies have found that the splicing proteins are very mobile in the nucleoplasm and speckles, with a small IF in this latter compartment (Kruhlak et al. 2000; Phair and Misteli 2000; Rino et al. 2007, 2008). Similarly, we found that TCERG1 is a highly dynamic nuclear protein that exhibits similar behavior in the nucleoplasm, speckles, and TS using an HIV-1 reporter that has been shown to splice post-transcriptionally (Boireau et al. 2007). The half-time fluorescence recovery of TCERG1 was ∼0.4–0.5 sec, with a small population of molecules that were immobile in the speckles and in the TS.
Surprisingly, the mobility of TCERG1 is reduced by DRB but not α-amanitin treatment. This paradoxical effect could be explained by the distinct mechanism of action of the transcriptional inhibitors and might provide clues regarding the dynamics of TCERG1. Transcriptional inhibition by α-amanitin is an irreversible process because it triggers degradation of the largest subunit of endogenous RNAPII (Nguyen et al. 1996). In contrast, DRB acts reversibly on transcription by inhibiting phosphorylation of the CTD of RNAPII, thereby inhibiting elongation (Dubois et al. 1994). Unlike α-amanitin, DRB does not cause RNAPII degradation (Nguyen et al. 1996). Here, we demonstrated that TCERG1 could be recruited at the TS in the absence of splicing. Moreover, we previously reported that TCERG1 is present in the RNAPII complexes assembled onto the HIV-1 promoter in vitro and in vivo (Coiras et al. 2013). Thus, TCERG1 may be stalled because the RNAPII complexes are unable to proceed with transcription due to the action of DRB.
In the recruitment model that explains the mechanism underlying the transcriptional control of alternative splicing, the CTD of RNAPII functions as a “landing pad” (Greenleaf 1993) that recruits factors involved in pre-mRNA processing to the transcription elongation complex, depending on the phosphorylation status of the CTD. Thus, the ability to specifically recognize hyperphosphorylated forms of the CTD is currently regarded as a significant feature indicating the putative role of a given factor in cotranscriptional pre-mRNA processing. TCERG1 binds to the CTD (Carty et al. 2000), and this interaction requires the phosphorylation of Ser7 within the CTD (Liu et al. 2013), which is specifically enriched over introns (Kim et al. 2010). Based on these data, this specific CTD modification of Ser7 has been suggested to be involved in the recruitment of adaptor proteins, such as TCERG1, which couple the transcribing RNAPII with spliceosomes to regulate cotranscriptional splicing events (Liu et al. 2013). Our results demonstrate that TCERG1 is recruited to the active TS independent of the CTD of RNAPII. These data, together with our previous results demonstrating that TCERG1 is found in preinitiation complexes assembled both in vitro and in vivo over promoter sequences (Coiras et al. 2013), exclude phosphorylated CTD as a requirement for the recruitment of this factor to the TS. This finding indicates that TCERG1 interacts with the phosphorylated CTD later during the transcription process.
We found that the mobility of TCERG1 slows down at the TS when using a construct that splices cotranscriptionally. This result suggests that TCERG1 forms new macromolecular complexes when splicing occurs cotranscriptionally. Interestingly, a similar recovery rate was observed following splicing inhibition with SSA, which blocks spliceosome assembly at the U2snRNP recruitment step. This finding indicates that TCERG1 interacts with stalled spliceosomal complexes only transiently. Given that TCERG1 interacts with SF1 and U2AF65 (Goldstrohm et al. 2001; Sánchez-Álvarez et al. 2006), these splicing factors may be responsible for slowing down the mobility of TCERG1 at splice sites. In turn, interaction of TCERG1 with splicing factors such as SF1 may stabilize them long enough to stimulate spliceosome assembly.
Cotranscriptional splicing is associated with RNAPII pausing at specific sites (Alexander et al. 2010; Carrillo Oesterreich et al. 2010; Wilhelm et al. 2011). We previously showed that TCERG1 promotes the splicing of the short isoform of Bcl-x through a regulatory element located in the first half of exon 2, which contained a higher level of polymerase molecules (Montes et al. 2012b), consistent with the existence of an RNAPII pause site at this position. On the basis of these observations and our present work, an exciting possibility remains that TCERG1 interacts with transcription elongation complexes to stimulate spliceosome assembly. Given that transcription elongation and splicing occur in minutes (Boireau et al. 2007; Schmidt et al. 2011), and that TCERG1 recovery occurs in seconds, our data clearly rule out a model in which TCERG1 would travel along the gene while associated with RNAPII. Instead, TCERG1 appears to interact only very transiently with elongation and splicing complexes, thus likely acting by a hit-and-run type of mechanism.
MATERIALS AND METHODS
Plasmids and alternative splicing reporter minigenes
The intermediate vector pEFBOS/GFP was obtained by amplifying the enhanced green fluorescent protein (EGFP) fragment from the pEGFP-C1 plasmid (Invitrogen) by PCR and inserting the fragment into the EcoRI-digested pEFBOST7 vector. pEFBOS/GFP/T7-TCERG1[1-1098] was generated using the EcoRI fragment obtained from the pEFBOS-T7-TCERG1 [1-1098] parental vector (Suñé and Garcia-Blanco 1999) using standard cloning procedures. Benoit Chabot (University of Sherbrooke, Canada) kindly provided the HIV-X2 reporter minigene. The plasmids expressing Tat and MS2-cherry have been previously described (Boireau et al. 2007; Molle et al. 2007). Alberto Kornblihtt (Universidad de Buenos Aires, Argentina) kindly provided the expression vectors for the α-amanitin-resistant variants of the human RNAPII (WTRES and ΔCTDRES), which have been described previously (de la Mata et al. 2003).
Cell culture and transfection assays
HEK293T cells were used for alternative splicing, immunofluorescence and RNA interference experiments. Cells were grown and maintained as previously described (Sánchez-Álvarez et al. 2010). The cells were grown to ∼60% to 70% confluence and then transfected with the appropriate amounts of the indicated constructs using calcium phosphate. U2OS cells stably expressing the HIV-1 reporter pExo-MS2 × 24 (Exo1 cells) and the construct based on the MINX intron WT_MS2in have been described previously (Boireau et al. 2007; Schmidt et al. 2011). The cells were grown and maintained in Dulbecco's modified Eagle medium (Invitrogen) supplemented with 10% fetal bovine serum, penicillin–streptomycin 100 U and 100 µg/mL, respectively, (Invitrogen), and 132 µg/mL of hygromycin. For the FISH experiments, the cells were grown on coverslips. For the live-cell experiments, the medium was replaced by medium that was identical except that it lacked riboflavin and phenol red. The cells were grown to ∼70% to 80% confluence and transfected using Gene Juice reagent (Merck Millipore) according to the manufacturer's protocols. The transcription inhibitor treatments were performed by incubating the cells with 0.1 µg/µL α-amanitin (Sigma) for 2 h at 37°C and with 100 µM DRB for 2 h at 37°C before live-cell imaging. To inhibit splicing, the cells were treated with 100 ng/mL SSA (Kaida et al. 2007) for 3 h, and the samples were processed for FRAP analysis.
RNA extraction and RT-PCR analysis
HEK293T cells were transfected with 0.6 µg of Bcl-X2 minigene reporter and 1 µg of pEFBOS/GFP/T7-TCERG1 [1-1098], pEFBOS-T7-TCERG1 [1-1098] parental vector or empty vector as indicated in the legend of Figure 1B. After 48 h, total RNA was isolated from transfected cells using TRIzol reagent (Invitrogen) and digested with 10 U RNase-free DNase I (Roche) for 30 min at 37°C. After DNase inactivation at 70°C for 5 min, 400 ng of RNA was reverse-transcribed using the Moloney murine leukemia virus RT (Invitrogen) for 1 h at 37°C with the primer RT-Sveda (5′-GGGAAGCTAGAGTAAGTAG-3′). One-tenth of the resulting cDNA was amplified by PCR using the oligonucleotides X34 (5′-AGGGAGGCAGGCGACGGCGACGAGTTT-3′) and XAge1R (5′-GTGGATCCCCCGGGCTGCAGGAATTCGAT-3′). The PCR products were analyzed on a 2% agarose gel. The intensity of the bands was quantified using Quantity One 4.5.0 software (BioRad).
Antibodies
Antibodies against the splicing factor SRSF2 (catalog no. S4045, Sigma) were used at a dilution of 1:4000 for immunofluorescence. Primary antibodies were detected using Alexa Fluor 647-conjugated goat anti-mouse (Molecular Probes) diluted 1:500 for immunofluorescence.
Immunofluorescence
Cells were grown on coverslips. Approximately 24 h after transfection, cells were fixed with 3.5% paraformaldehyde in PBS buffer (pH 7.4) for 45 min on ice. Cells were washed three times with PBS, permeabilized in PBS containing 0.1% Triton X-100 (Sigma) for 5 min at room temperature, and blocked in PBS containing 2.5% BSA overnight at 4°C. Cells were incubated with primary antibodies at appropriate dilutions in PBS containing 0.1% BSA for 1 h at room temperature (humidity chamber), then washed with 0.1% BSA in PBS and incubated with the appropriate secondary antibodies under the conditions described previously. DAPI was added to the cells at 1 µg/mL in PBS for 5 min. After the cells were stained, they were rinsed extensively with 0.1% BSA in PBS and then with PBS. Coverslips were then mounted onto the glass slides using ProLong Gold Antifade reagent (Molecular Probes).
Fluorescence in situ hybridization
Approximately 24 h after transfection, cells were fixed with 4% paraformaldehyde in PBS for 30 min at room temperature, washed twice with PBS, and permeabilized in 70% ethanol overnight at 4°C. The cells were rehydrated in PBS and in SSC buffer containing 20% formamide (Sigma) and incubated overnight at 37°C with hybridization mix (1× SSC, 20% formamide, 10% dextran sulfate, 0.01 µg/µL RNA grade BSA, 2 mM Vanadyl-R-C, 20 µg of tRNA, and 20 ng of MS2-Cy3 probe). The probe was briefly denaturated at 90°C before being added to the hybridization mix. Post-hybridization washes were performed twice in SSC containing 20% formamide for 30 min each at 37°C. Finally, the cells were washed in PBS for 2 min and then mounted using a mix containing 90% glycerol, 1 mg/mL p-phenylenediamine and 0.1 µg/mL DAPI. The following MS2 probe was used (X stands for amino-allyl-T): MS2 transcribed strand 5′-AXGTCGACCTGCAGACAXGGGTGATCCTCAXGTTTTCTAGGCAATXA-3′.
Microscopy and imaging processing
Immunofluorescence images were captured on a Leica SP5 confocal microscope with an HCX PL APO 63×/1.4 oil objective. GFP and Alexa Fluor 647 signals were collected simultaneously and excited with the 488-nm line of an argon laser and with the 633-nm line of a HeNe laser, respectively. The DAPI signal was acquired sequentially and excited at 405 nm. Images from the FISH experiments were captured on a Leica DM6000 M wide-field microscope driven by MetaMorph software (Universal Imaging Corp) using a 100× PL APO 1.4–0.7 oil objective. Images were prepared using ImageJ and Adobe Photoshop CS3 extended v10.0 software.
FRAP experiments
FRAP was performed on a Nikon TE200 microscope with a 100× NA 1.45 objective. Twenty pre-bleach images were acquired at an exposure time of 100 msec. The GFP signals at the TS, nuclear speckles and nucleoplasm were simultaneously bleached at 488 nm in a region of interest (ROI) 1 µm in diameter at a 56% laser power for 300 msec, and 300 post-bleach frames were collected every 100 msec for 20 sec. Signal recovery was recorded at ≤1% of the power 488-nm laser line using MetaMorph software (Universal Imaging Corp.). Images were analyzed by recording the fluorescence of the bleached region. The background was removed, the intensities at each time point were corrected for bleaching by dividing them by the total cell fluorescence, and these values were finally normalized by dividing them by the fluorescent intensity before bleaching. Half-time values were obtained from recovery curves in which the first pre-bleach value was normalized to 0. The IFs were calculated according to the following equation: 1 − IE where 1 is the total amount of the protein and IE represents the end value of the recovered fluorescence intensity. The data shown in Table 1 correspond to the mean ± SEM of individual cells. P values were calculated using a non-parametric Mann–Whitney test.
ACKNOWLEDGMENTS
We are grateful to members of the laboratories for their helpful suggestions, critical discussions, and comments. This work was supported by grants from the Spanish Ministry of Economy and Competitiveness (grant numbers BFU2011-24577 and BFU2014-54660-R) and the Andalusian Government (Excellence Projects CVI-4626/2009 and BIO-2515/2012) to C.S.; the Spanish Ministry of Economy and Competitiveness (grant number BFU2013-44660-R) and the Andalusian Government (Excellence Project CTS-6587) to C.H.M. N.S.-H. was supported by a fellowship from the CSIC (JAE Program). Support from the European Region Development Fund, ERDF (FEDER), is also acknowledged.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.052795.115.
REFERENCES
- Alexander RD, Innocente SA, Barrass JD, Beggs JD. 2010. Splicing-dependent RNA polymerase pausing in yeast. Mol Cell 40: 582–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartkowiak B, Mackellar AL, Greenleaf AL. 2011. Updating the CTD story: from tail to epic. Genet Res Int 2011: 623718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley DL. 2014. Coupling mRNA processing with transcription in time and space. Nat Rev Genet 15: 163–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boireau S, Maiuri P, Basyuk E, de la Mata M, Knezevich A, Pradet-Balade B, Backer V, Kornblihtt A, Marcello A, Bertrand E. 2007. The transcriptional cycle of HIV-1 in real-time and live cells. J Cell Biol 179: 291–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buratowski S. 2003. The CTD code. Nat Struct Biol 10: 679–680. [DOI] [PubMed] [Google Scholar]
- Carrillo Oesterreich F, Preibisch S, Neugebauer KM. 2010. Global analysis of nascent RNA reveals transcriptional pausing in terminal exons. Mol Cell 40: 571–581. [DOI] [PubMed] [Google Scholar]
- Carty SM, Goldstrohm AC, Suñé C, Garcia-Blanco MA, Greenleaf AL. 2000. Protein-interaction modules that organize nuclear function: FF domains of CA150 bind the phosphoCTD of RNA polymerase II. Proc Natl Acad Sci 97: 9015–9020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Huang S. 2001. Nucleolar components involved in ribosome biogenesis cycle between the nucleolus and nucleoplasm in interphase cells. J Cell Biol 153: 169–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng B, Price DH. 2007. Properties of RNA polymerase II elongation complexes before and after the P-TEFb-mediated transition into productive elongation. J Biol Chem 282: 21901–21912. [DOI] [PubMed] [Google Scholar]
- Cheng D, Côté J, Shaaban S, Bedford MT. 2007. The arginine methyltransferase CARM1 regulates the coupling of transcription and mRNA processing. Mol Cell 25: 71–83. [DOI] [PubMed] [Google Scholar]
- Chodosh LA, Fire A, Samuels M, Sharp PA. 1989. 5,6-Dichloro-1-β-D-ribofuranosylbenzimidazole inhibits transcription elongation by RNA polymerase II in vitro. J Biol Chem 264: 2250–2257. [PubMed] [Google Scholar]
- Chusainow J, Ajuh PM, Trinkle-Mulcahy L, Sleeman JE, Ellenberg J, Lamond AI. 2005. FRET analyses of the U2AF complex localize the U2AF35/U2AF65 interaction in vivo and reveal a novel self-interaction of U2AF35. RNA 11: 1201–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coiras M, Montes M, Montanuy I, López-Huertas MR, Mateos E, Le Sommer C, Garcia-Blanco MA, Hernández-Munain C, Alcamí J, Suñé C. 2013. Transcription elongation regulator 1 (TCERG1) regulates competent RNA polymerase II-mediated elongation of HIV-1 transcription and facilitates efficient viral replication. Retrovirology 10: 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrionero A, Miñana B, Valcárcel J. 2011. Reduced fidelity of branch point recognition and alternative splicing induced by the anti-tumor drug spliceostatin A. Genes Dev 25: 445–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Almeida SF, Carmo-Fonseca M. 2008. The CTD role in cotranscriptional RNA processing and surveillance. FEBS Lett 582: 1971–1976. [DOI] [PubMed] [Google Scholar]
- de la Mata M, Kornblihtt AR. 2006. RNA polymerase II C-terminal domain mediates regulation of alternative splicing by SRp20. Nat Struct Mol Biol 13: 973–980. [DOI] [PubMed] [Google Scholar]
- de la Mata M, Alonso CR, Kadener S, Fededa JP, Blaustein M, Pelisch F, Cramer P, Bentley D, Kornblihtt AR. 2003. A slow RNA polymerase II affects alternative splicing in vivo. Mol Cell 12: 525–532. [DOI] [PubMed] [Google Scholar]
- Dubois MF, Nguyen VT, Bellier S, Bensaude O. 1994. Inhibitors of transcription such as 5,6-dichloro-1-β-D-ribofuranosylbenzimidazole and isoquinoline sulfonamide derivatives (H-8 and H-7) promote dephosphorylation of the carboxyl-terminal domain of RNA polymerase II largest subunit. J Biol Chem 269: 13331–13336. [PubMed] [Google Scholar]
- Egloff S, Dienstbier M, Murphy S. 2012. Updating the RNA polymerase CTD code: adding gene-specific layers. Trends Genet 28: 333–341. [DOI] [PubMed] [Google Scholar]
- Ellis JD, Llères D, Denegri M, Lamond AI, Cáceres JF. 2008. Spatial mapping of splicing factor complexes involved in exon and intron definition. J Cell Biol 181: 921–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstrohm AC, Albrecht TR, Suñé C, Bedford MT, Garcia-Blanco MA. 2001. The transcription elongation factor CA150 interacts with RNA polymerase II and the pre-mRNA splicing factor SF1. Mol Cell Biol 21: 7617–7628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorisch SM, Lichter P, Rippe K. 2005. Mobility of multi-subunit complexes in the nucleus: accessibility and dynamics of chromatin subcompartments. Histochem Cell Biol 123: 217–228. [DOI] [PubMed] [Google Scholar]
- Greenleaf AL. 1993. Positive patches and negative noodles: linking RNA processing to transcription? Trends Biochem Sci 18: 117–119. [DOI] [PubMed] [Google Scholar]
- Huranova M, Ivani I, Benda A, Poser I, Brody Y, Hof M, Shav-Tal Y, Neugebauer KM, Stanek D. 2010. The differential interaction of snRNPs with pre-mRNA reveals splicing kinetics in living cells. J Cell Biol 191: 75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaida D, Motoyoshi H, Tashiro E, Nojima T, Hagiwara M, Ishigami K, Watanabe H, Kitahara T, Yoshida T, Nakajima H, et al. 2007. Spliceostatin A targets SF3b and inhibits both splicing and nuclear retention of pre-mRNA. Nat Chem Biol 3: 576–583. [DOI] [PubMed] [Google Scholar]
- Kaiser TE, Intine RV, Dundr M. 2008. De novo formation of a subnuclear body. Science 322: 1713–1717. [DOI] [PubMed] [Google Scholar]
- Kim H, Erickson B, Luo W, Seward D, Graber JH, Pollock DD, Megee PC, Bentley DL. 2010. Gene-specific RNA polymerase II phosphorylation and the “CTD code”. Nat Struct Mol Biol 17: 1279–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruhlak MJ, Lever MA, Fischle W, Verdin E, Bazett-Jones DP, Hendzel MJ. 2000. Reduced mobility of the alternate splicing factor (ASF) through the nucleoplasm and steady state speckle compartments. J Cell Biol 150: 41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamond AI, Spector DL. 2003. Nuclear speckles: a model for nuclear organelles. Nat Rev Mol Cell Biol 4: 605–612. [DOI] [PubMed] [Google Scholar]
- Lin KT, Lu RM, Tarn WY. 2004. The WW domain-containing proteins interact with the early spliceosome and participate in pre-mRNA splicing in vivo. Mol Cell Biol 24: 9176–9185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Fan S, Lee CJ, Greenleaf AL, Zhou P. 2013. Specific interaction of the transcription elongation regulator TCERG1 with RNA polymerase II requires simultaneous phosphorylation at Ser2, Ser5, and Ser7 within the carboxyl-terminal domain repeat. J Biol Chem 288: 10890–10901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancebo HS, Lee G, Flygare J, Tomassini J, Luu P, Zhu Y, Peng J, Blau C, Hazuda D, Price D, et al. 1997. P-TEFb kinase is required for HIV Tat transcriptional activation in vivo and in vitro. Genes Dev 11: 2633–2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao YS, Zhang B, Spector DL. 2011. Biogenesis and function of nuclear bodies. Trends Genet 27: 295–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall NF, Price DH. 1992. Control of formation of two distinct classes of RNA polymerase II elongation complexes. Mol Cell Biol 12: 2078–2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFie PJ, Wang GL, Timchenko NA, Wilson HL, Hu X, Roesler WJ. 2006. Identification of a co-repressor that inhibits the transcriptional and growth-arrest activities of CCAAT/enhancer-binding protein α. J Biol Chem 281: 18069–18080. [DOI] [PubMed] [Google Scholar]
- Misteli T. 2001. Protein dynamics: implications for nuclear architecture and gene expression. Science 291: 843–847. [DOI] [PubMed] [Google Scholar]
- Misteli T, Spector DL. 1999. RNA polymerase II targets pre-mRNA splicing factors to transcription sites in vivo. Mol Cell 3: 697–705. [DOI] [PubMed] [Google Scholar]
- Misteli T, Caceres JF, Spector DL. 1997. The dynamics of a pre-mRNA splicing factor in living cells. Nature 387: 523–527. [DOI] [PubMed] [Google Scholar]
- Molle D, Maiuri P, Boireau S, Bertrand E, Knezevich A, Marcello A, Basyuk E. 2007. A real-time view of the TAR:Tat:P-TEFb complex at HIV-1 transcription sites. Retrovirology 4: 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montes M, Becerra S, Sánchez-Álvarez M, Suñé C. 2012a. Functional coupling of transcription and splicing. Gene 501: 104–117. [DOI] [PubMed] [Google Scholar]
- Montes M, Cloutier A, Sánchez-Hernández N, Michelle L, Lemieux B, Blanchette M, Hernández-Munain C, Chabot B, Suñé C. 2012b. TCERG1 regulates alternative splicing of the Bcl-x gene by modulating the rate of RNA polymerase II transcription. Mol Cell Biol 32: 751–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawroth I, Mueller F, Basyuk E, Beerens N, Rahbek UL, Darzacq X, Bertrand E, Kjems J, Schmidt U. 2014. Stable assembly of HIV-1 export complexes occurs cotranscriptionally. RNA 20: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen VT, Giannoni F, Dubois MF, Seo SJ, Vigneron M, Kedinger C, Bensaude O. 1996. In vivo degradation of RNA polymerase II largest subunit triggered by α-amanitin. Nucleic Acids Res 24: 2924–2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Keefe RT, Mayeda A, Sadowski CL, Krainer AR, Spector DL. 1994. Disruption of pre-mRNA splicing in vivo results in reorganization of splicing factors. J Cell Biol 124: 249–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson JL, Robinson TJ, Muñoz MJ, Kornblihtt AR, Garcia-Blanco MA. 2008. Identification of the cellular targets of the transcription factor TCERG1 reveals a prevalent role in mRNA processing. J Biol Chem 283: 7949–7961. [DOI] [PubMed] [Google Scholar]
- Pederson T. 2000. Diffusional protein transport within the nucleus: a message in the medium. Nat Cell Biol 2: E73–E74. [DOI] [PubMed] [Google Scholar]
- Phair RD, Misteli T. 2000. High mobility of proteins in the mammalian cell nucleus. Nature 404: 604–609. [DOI] [PubMed] [Google Scholar]
- Price DH. 2000. P-TEFb, a cyclin-dependent kinase controlling elongation by RNA polymerase II. Mol Cell Biol 20: 2629–2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rino J, Carvalho T, Braga J, Desterro JM, Luhrmann R, Carmo-Fonseca M. 2007. A stochastic view of spliceosome assembly and recycling in the nucleus. PLoS Comput Biol 3: 2019–2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rino J, Desterro JM, Pacheco TR, Gadella TW Jr, Carmo-Fonseca M. 2008. Splicing factors SF1 and U2AF associate in extraspliceosomal complexes. Mol Cell Biol 28: 3045–3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roybal GA, Jurica MS. 2010. Spliceostatin A inhibits spliceosome assembly subsequent to prespliceosome formation. Nucleic Acids Res 38: 6664–6672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez-Álvarez M, Goldstrohm AC, Garcia-Blanco MA, Suñé C. 2006. Human transcription elongation factor CA150 localizes to splicing factor-rich nuclear speckles and assembles transcription and splicing components into complexes through its amino and carboxyl regions. Mol Cell Biol 26: 4998–5014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez-Álvarez M, Montes M, Sánchez-Hernández N, Hernández-Munain C, Suñé C. 2010. Differential effects of sumoylation on transcription and alternative splicing by transcription elongation regulator 1 (TCERG1). J Biol Chem 285: 15220–15233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez-Hernández N, Ruiz L, Sánchez-Álvarez M, Montes M, Macias MJ, Hernández-Munain C, Suñé C. 2012. The FF4 and FF5 domains of transcription elongation regulator 1 (TCERG1) target proteins to the periphery of speckles. J Biol Chem 287: 17789–17800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt U, Basyuk E, Robert MC, Yoshida M, Villemin JP, Auboeuf D, Aitken S, Bertrand E. 2011. Real-time imaging of cotranscriptional splicing reveals a kinetic model that reduces noise: implications for alternative splicing regulation. J Cell Biol 193: 819–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seksek O, Biwersi J, Verkman AS. 1997. Translational diffusion of macromolecule-sized solutes in cytoplasm and nucleus. J Cell Biol 138: 131–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shopland LS, Lawrence JB. 2000. Seeking common ground in nuclear complexity. J Cell Biol 150: F1–F4. [DOI] [PubMed] [Google Scholar]
- Smith MJ, Kulkarni S, Pawson T. 2004. FF domains of CA150 bind transcription and splicing factors through multiple weak interactions. Mol Cell Biol 24: 9274–9285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snaar S, Wiesmeijer K, Jochemsen AG, Tanke HJ, Dirks RW. 2000. Mutational analysis of fibrillarin and its mobility in living human cells. J Cell Biol 151: 653–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suñé C, Garcia-Blanco MA. 1999. Transcriptional cofactor CA150 regulates RNA polymerase II elongation in a TATA-box-dependent manner. Mol Cell Biol 19: 4719–4728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suñé C, Hayashi T, Liu Y, Lane WS, Young RA, Garcia-Blanco MA. 1997. CA150, a nuclear protein associated with the RNA polymerase II holoenzyme, is involved in Tat-activated human immunodeficiency virus type 1 transcription. Mol Cell Biol 17: 6029–6039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm BT, Marguerat S, Aligianni S, Codlin S, Watt S, Bähler J. 2011. Differential patterns of intronic and exonic DNA regions with respect to RNA polymerase II occupancy, nucleosome density and H3K36me3 marking in fission yeast. Genome Biol 12: R82. [DOI] [PMC free article] [PubMed] [Google Scholar]