

Abstract
Background
Aspirin resistance established by different laboratory methods is still a debated problem. Using COX1 specific methods no aspirin resistance was detected among healthy volunteers. Here we tested the effect of chronic aspirin treatment on platelets from patients with stable coronary artery disease. The expression of COX2 mRNA in platelets and its influences on the effect of aspirin was also investigated.
Methods
One hundred and forty four patients were enrolled in the study. The direct measurement of COX1 acetylation was carried out by monoclonal antibodies specific to acetylated and non-acetylated COX1 (acCOX1 and nacCOX1) using Western blotting technique. Arachidonic acid (AA) induced TXB2 production by platelets was measured by competitive immunoassay. AA induced platelet aggregation, ATP secretion and VerifyNow Aspirin Assay were also performed. COX2 and COX1 mRNA expression in platelets were measured in 56 patients by RT-qPCR.
Results
In 138 patients only acCOX1 was detected, in the remaining six patients nacCOX1 disappeared after a compliance period. AA induced TXB2 production by platelets was very low in all patients including the 6 patients after compliance. AA induced platelet aggregation, secretion and with a few exceptions the VerifyNow Assay also demonstrated the effect of aspirin. Smoking, diabetes mellitus and inflammatory conditions did not influence the results. The very low amount of COX2 mRNA detected in 39 % of the investigated platelets did not influence the effect of aspirin.
Conclusions
No aspirin resistance was detected among patients with stable coronary artery disease. COX2 expression in platelets did not influence the effect of aspirin.
Keywords: Aspirin, Coronary artery disease, Cyclooxygenase-1, Cyclooxygenase-2, Platelet, Thromboxane B2
Background
Since the middle of the last century low dose aspirin (acetylsalicylic acid) has been effectively used in the prevention of acute atherothrombotic complications, like myocardial infarction and atherothrombotic ischemic stroke [1–5]. It prevents vascular death and nonfatal vascular events by approximately 15 and 30 %, respectively [1–3, 6]. The antithrombotic effect of aspirin is primarily due to its anti-platelet action. Aspirin as an acetylating agent acetylates the Ser529 residue of cyclooxygenase-1 (COX1), which is located in the wall of the enzyme’s active-site pocket [6–8]. Arachidonic acid (AA) released from membrane phospholipids as the result of signalization by platelet agonists, is transformed by COX1, exerting both cyclooxygenase and peroxidase activities, into cyclic endoperoxides, prostaglandin G2 and H2. The latter is the precursor of the powerful platelet activating prostaglandin derivative, thromboxane A2 (TXA2). The covalent modification of Ser529 prevents the access of AA to the active site of COX-1 and blocks TXA2 production by platelet throughout its lifespan.
In certain patients low dose aspirin therapy is not effective in preventing acute atherothrombotic complications, and for this reason the term “aspirin resistance” was introduced. In the PubMed browsing this term resulted in over 2000 publications. True aspirin resistance is the inability of aspirin to acetylate Ser259 residue in platelet COX1. The ineffectiveness of aspirin in protecting an individual from acute vascular events is often considered “aspirin resistance” although such cases are very likely related to the fact that aspirin inhibits effectively and selectively only one out of several pathways leading to platelet activation [9, 10]. It fails to inhibit platelet activation induced by strong agonists, like thrombin or high dose of collagen and only partially inhibits ADP induced platelet aggregation. The involvement of such stimuli in platelet activation could be prevalent in atherosclerotic vessel. In many studies non-compliance was not adequately controlled and was not separated from aspirin resistance. Further problem was the lack of laboratory tests that specifically detect the acetylation of COX1. Non-responsiveness to aspirin as measured by laboratory tests non-specific for detecting the effect of aspirin was also confused with aspirin resistance. In a recent study we developed monoclonal antibodies reacting with acetylated and non-acetylated COX1 (acCOX1 and nacCOX1, respectively) for the first time, and utilizing these antibodies a highly specific and sensitive method that detects 2.5 % of platelet nacCOX1 was developed [11]. Using this method that measures COX1 acetylation by aspirin directly, no aspirin resistance was found among 108 healthy volunteers taking 100 mg enteric-coated aspirin daily for 7 days. The same results were obtained by COX1-dependent functional tests, like AA-induced TXB2 formation in platelet rich plasma (PRP), AA-induced platelet aggregation and the VerifyNow Aspirin Assay [11, 12]. Practically complete inhibition of TXB2 formation during coagulation of whole blood was also observed in 24 healthy volunteers after 7 days of aspirin treatment [13].
However, in patients with coronary artery disease (CAD) the situation might be different from that observed in healthy volunteers. There could be conditions related to this disease, which might influence the effect of aspirin. The higher turnover of platelets and the expression/up-regulation of COX2, insensitive to low dose aspirin, have been claimed to contribute to impaired aspirin effect in diabetic patients and in inflammatory conditions [9, 14–17]. To explore the frequency of true aspirin resistance among patients with CAD 144 such patients being on long-term aspirin monotherapy were investigated for COX1 acetylation and tested for COX1 dependent functional assays. The expression of COX2 in this patient population was also investigated.
Methods
Subjects
Patients who had undergone percutan coronary angioplasty or coronary artery bypass grafting surgery being on aspirin monotherapy for secondary prevention were enrolled in the study. The patients were regularly admitted at the outpatient service of the Department of Cardiology, University of Debrecen, Debrecen Hungary; study participants were enrolled within a 1.5-year period. They were taking 100 mg enteric-coated aspirin daily for at least 1 month before investigation. Exclusion criteria were: acute coronary syndrome or any other major clinical event or infection in the previous 3 months, malignant disorder, hyperthyroidism, autoimmune disease and chronic renal insufficiency, intake of antiplatelet drugs other than aspirin in the previous 2 weeks, known bleeding diathesis, anemia with hemoglobin value below 100 g/L and platelet count below 100 G/L. Finally, 144 patients were eligible for being involved in the study. The Jeffrey method was used for sample size calculation as described by Brown et al. [18]. Apparently healthy controls (n = 108) recruited for the study have been characterized in a previous publication [11]. All individuals enrolled in the study gave written informed consent prior to their inclusion in the study. Ethical approval was obtained from the Ethics Committee of the Medical Faculty, University of Debrecen, Hungary and the study was performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments.
Sample preparation
Blood samples were collected into Vacutainer tubes containing 0.109 mol/L trisodium citrate (Becton–Dickinson, Franklin Lakes, NJ) after overnight fasting. Anticoagulated blood was directly used for the VerifyNow® (VN) Aspirin assay (Accumetrics, San Diego, CA). Platelet rich plasma (PRP) separated by centrifugation (120g, 37 °C, 15 min) was used for AA induced platelet aggregation tests. Platelet depleted plasma (PDP), was obtained by two consecutive centrifugations (1500g, 25 °C, 20 min).
Methods used for the detection of aspirin effect
In a previous study we developed new methods for the direct and indirect detection of COX-1 acetylation by aspirin [11]. The first method is based on two monoclonal antibodies raised against the acetylated and non-acetylated nonapeptide that corresponded to human COX-1 525-533 residues (H-Gly-Ala-Pro-Phe-Ser-Leu-Lys-Gly-Leu-OH). Using the two purified antibodies nacCOX1 and acCOX-1 could be clearly distinguished in the lysate of washed platelets by Western blotting technique.
The indirect method measures 0.25 mg/mL AA-induced TXB2 generation in PRP diluted to 30 × 109/L by PDP. The produced TXB2 was separated from AA and from other interfering substances by sequential solid phase extraction [11]. In the final eluted sample TXB2 concentration was measured by competitive immunoassay (Assay Designs, Ann Arbor, MI) and it was expressed as pg TXB2 produced by 106 platelets.
As among the generally used routine laboratory tests used for the detection of aspirin effect AA-induced platelet aggregation, ATP secretion and the VN Asprin assay were proven to be the most reliable tests [12], we also used these assays to detect the effect of aspirin on patients with CAD being on secondary prevention. VN Aspirin Assay was performed according to the manufacturer’s instructions and the results were expressed as Aspirin Reaction Units (ARU). Platelet aggregation and secretion in platelet rich plasma was induced by 500 µg/mL AA (Helena, Gateshead, UK) and was monitored in Chrono-Log 700 lumiaggregometer (Chrono-Log, Havertown, PA) for 8 min. Prior to the experiments the platelet count was adjusted to 260 × 109 platelet/L. Aggregation was expressed as the percentage of maximal change in light transmission (Δtransmission %). ATP secretion was quantitated by bioluminescence method using luciferin-luciferase reagent (Biothema AB, Handen, Sweden). Maximal ATP secretion was expressed as μmol ATP/1011 platelets.
Measurement of COX1 and COX2 mRNA
COX1 and COX2 mRNA expression was determined in the platelets of 56 patients randomly selected out of the 144 subjects with CAD. RNA was isolated from leukocyte depleted platelets as described earlier [19]. The integrity of RNA samples was shown by determining the GAPDH 3ʹ:5ʹ signal ratio [20], the absence of RNA contamination from white blood cells was proven by RT-qPCR analysis of each sample for CD15 and HLA-DQβ mRNAs [21]. Reverse transcription and RT-qPCR reaction were carried out as described by Zsóri et al. [19]. Briefly, First Strand cDNA Synthesis Kit (Roche, Mannheim, Germany) was used for reverse transcription. The reaction solution was incubated at 42 °C for 60 min, and then at 94 °C for 5 min. No-template control and no-reverse transcriptase controls included in each run showed negative results. The absence of contaminating DNA was also demonstrated by melting curve analysis. RT-qPCR reactions were carried out on LightCycler 480 (Roche) in duplicates using SYBR Green I Master (Roche). PCRs were set up in a final volume of 20 µL consisting 10 µL Master Mix (2× concentration), 5 µL of cDNA template derived from reverse-transcribed RNA and 300 nM primers for COX1 and 400 nM primers for COX2. The primer sequences were tccatgttggtggactatgg (forward), gtggtggtccatgttcctg (reverse) for COX1, and cttcacgcatcagtttttcaag (forward), tcaccgtaaatatgatttaagtccac (reverse) for COX2. ACTB, GNAS and HDGF have been established as the most stable reference genes for the normalization of platelet mRNA expression in coronary artery disease; all three were amplified and used in the calculations [19]. The amplification program was: heating for 10 min at 95 °C, followed by 40 cycles of 10 s at 95 °C, 30 s at 60 °C and 1 s at 72 °C. Melting curve analysis was performed between 66 and 95 °C in 0.11°C/s increments with 5 acquisitions/ °C. CT values, corresponding to the number of cycles at which the fluorescence signal exceeds a threshold value, were used for the relative gene quantification based on the method of Livak and Schmittgen [22]. ΔCT,COX1 and ΔCT,COX2 were established against each of the three reference genes and the mean ΔΔCT,COX1,COX2 values were used for the calculation of COX1 mRNA:COX2 mRNA ratios.
Results
The acetylation of COX-1 by aspirin in patients with CAD
The characteristics of patients enrolled in the study are shown in Table 1. In the platelet lysate of 138 patients only acetylated COX1 was detected by Western blotting. These patients were represented by samples P1 and P2 in Fig. 1. Previously it has been shown that the method is highly sensitive and could detect as low as 2.5 % of the total platelet COX1 in non-acetylated form [11], i.e. more than 97.5 % of platelet COX-1 was acetylated as the consequence of long term aspirin treatment. It is to be noted that in platelets from these treated patients no nacCOX1 could be detected, which contradicts an earlier finding showing that only one monomer of the active dimeric enzyme is acetylated by aspirin [23]. In the case of six patients different amounts of non-acetylated COX1 remained in the platelet lysate. Lanes P3/1 and P4/1 show the two extremes; in sample P3/1 only a small amount of COX1 remained non-acetylated, while in a single sample (P4/1) no acetylation occurred. As noncompliance was assumed, these patients were contacted and their attention was drawn to the danger of not taking or irregularly taking the drug. After an additional two weeks the tests were repeated and full effectiveness of aspirin was demonstrated by the absence of non-acetylated COX1 and the presence of acetylated COX1 in the platelet lysate (see representative results on lanes P3/2 and P4/2, Fig. 1). According to sample size calculations the lack of aspirin resistance in all of the 144 patients indicated that aspirin is effective in more than 98 % of patients with stable coronary artery disease (confidence interval 95 %). It is to be noted that aspirin was fully effective in diabetic patients and also in smokers.
Table 1.
Characteristics of patient population
| Age (years) | 64 ± 10 (42–85) |
| Male gender | 67 % |
| BMI (kg/m2) | 30 (26–33; 17–51) |
| Current smoker | 10 % |
| Ever smoker | 61 % |
| Diabetes mellitus | 24 % |
| Triglyceride (mmol/L) | 1.54 (1.1–2.1; 0.6–6.5) |
| Total cholesterol (mmol/L) | 4.6 (3.8–5.4; 2.2–8.8) |
| HDL-C (mmol/L) | 1.2 (1.1–1.5; 0.7–2.5) |
| LDL-C (mmol/L) | 2.5 (2.0–3.3; 1.0–6.1) |
| Platelets (g/L) | 237 ± 58 (100–432) |
| Fibrinogen (g/L) | 3.7 (3.3–4.2; 2.0–7.1) |
| CRP (mg/L) | 2.1 (0.9–4.3; 0.5–20.5) |
| Serum creatinine (µmol/L) | 79 (68–90; 49–179) |
| GFR (mL/min/1.73 m2) | 86 (70–91; 23–91) |
Mean ± SD and total range in parenthesis are shown in the case of parameters with normal distribution and median with interquartile range and total range are given for parameters with non-normal distribution
BMI body mass index, HDL-C high-density lipoprotein cholesterol, LDL-C low-density lipoprotein cholesterol, CRP C-reactive protein, GFR glomerular filtration rate
Fig. 1.
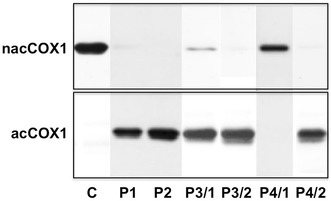
Detection of acetylated COX1 (acCOX1, lower panel) and non-acetylated COX1 (nacCOX1, upper panel) in platelet lysate of non-treated controls (C) and patients on aspirin treatment (P1–P4). Monoclonal antibodies specific to the acetylated and non-acetylated forms of COX1 were used in Western blotting detection system. In patients P3 and P4 non-compliance was presumed and the test was repeated after a 2 weeks period of compliance (P3/2 and P4/2). Below the band representing COX-1 another low intensity band cross-reacting with both antibodies is also apparent. It very likely represents COX-1 isoform 2, a 37 amino acids shorter transcript variant (http://www.ncbi.nlm.nih.gov/nuccore/18104968)
The effect of aspirin on arachidonic acid induced TXB2 production
Figure 2 demonstrates the AA induced TXB2 production of platelets in controls and in CAD patients being on long-term aspirin treatment. Aspirin drastically decreased the TXB2 generation, the median TXB2 production was only 1.2 % of that measured in the control group. Please, note that the y axis on the plot is broken and in the low range the scale is different. Here six patients also had higher TXB2 generation in the range of 43–835 pg TXB2/106 platelets, but at the second occasion their value returned to the range of the remaining 138 patients. In Fig. 2 their samples are represented only with the results of the second determinations.
Fig. 2.
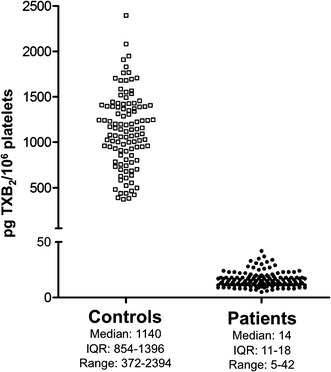
Arachidonic acid induced TXB2 generation of platelets from controls (open square) and from patients with coronary artery disease being on long-term aspirin treatment (filled circle). IQR interquartile range
The effect of aspirin on arachidonic acid induced platelet activation
In accordance with the considerably decreased TXB2 production, the extent of AA induced aggregation of platelets from aspirin treated patients was uniformly very low and no overlap between controls and patients was observed (Fig. 3). The median value of AA induced ATP release in PRP was 1.1 μmol ATP/1011 platelets (IQR: 0.9–1.4) in the control group, while in the PRP of all aspirin treated patients the amount of released ATP was below the limit of detection. In the case of VerifyNow Aspirin assay four ARU values from the patients group (2.8 %) overlapped with the total range of ARU values of the control group (Fig. 3). For this assay the manufacturer recommends 550 ARU as the cut off value for aspirin effect, however according to the results of the control group a cut off value of 585 ARU seems to be more appropriate. The reason for the slight difference between the other assays and the VerifyNow Aspirin Assay, the latter also uses AA as agonist, is not clear. Maybe the detection system used in the VerifyNow Aspirin Assay (aggregation of fibrinogen coated beads) is responsible for the few outliers in the patient’s group.
Fig. 3.
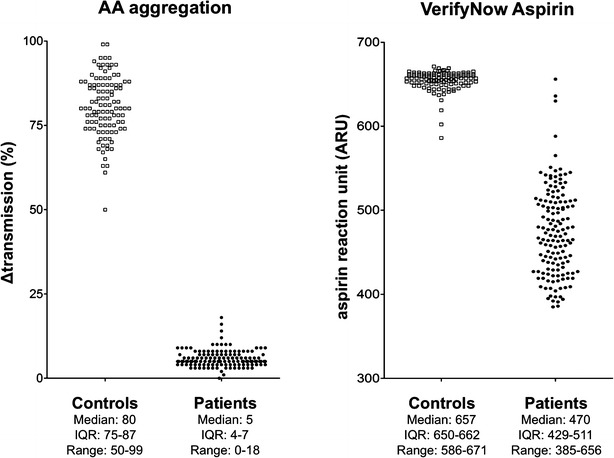
The results of arachidonic acid induced platelet aggregation and VerifyNow Aspirin Assay in controls (open square) and in patients with coronary artery disease being on long-term aspirin treatment (filled circle). IQR interquartile range
It is to be noted that 24 % of the study group were diabetic patients, the CRP level was above 5 mg/L in 21 % of the patients and in spite of the clinical condition 10 % of them was unable to quit smoking. In none of these patients was impaired aspirin effect measured by any of the methods used in the study.
The effect of aspirin on platelets with COX2 mRNA expression
Out of the 56 patients investigated for the expression of COX2 mRNA only in 22 cases was the amount above the limit of detection (Fig. 4). Even in these cases the relative amount of COX2 mRNA was less than 0.4 % of COX1 mRNA and with the exception of three patients this value was less than 0.05 %. None of the patients expressing COX2 mRNA in platelets showed aspirin resistance.
Fig. 4.
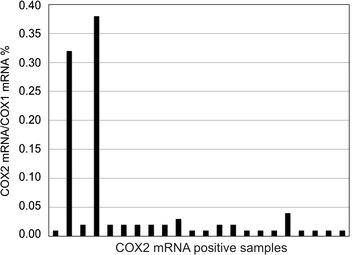
COX2:COX1 mRNA ratios in the platelets of patients with detectable COX2 mRNA
Discussion
So far, the direct measurement of COX1 acetylation in healthy volunteers taking 100 mg aspirin has been carried out in two studies using different methodologies. Our former study measured nacCOX1 and acCOX1 by Western blotting technique using specific monoclonal antibodies [11], in another study platelet proteins were separated by nuPAGE then, in-gel enzymatic digestion was carried out and the COX1 peptide carrying the Ser529 residue was determined by mass spectrometry [13]. One hundred and eight and twenty-four individuals were enrolled in the studies, respectively and in none of the cases was true aspirin resistance revealed. It is to be noted that using AA-induced platelet aggregation and serum TXB2 measurement Grosser et al. [24] also failed to detect specific phenotype of true aspirin resistance among 400 healthy volunteers. As in patients with CAD the occurrence of aspirin resistance has not been explored by detecting the acetylation of COX1 in platelets, our method developed as part of basic research was translated to address this question in clinical setting. In this study none of the 144 patients enrolled in the study showed impaired acetylation of platelet COX1, TXB2 generation, AA-induced platelet aggregation and ATP release were profoundly inhibited by aspirin in all patients. Neither diabetes nor smoking influenced the effectiveness of aspirin and inflammatory condition shown by elevated CRP also failed to influence COX1 acetylation. This finding suggests that in patients with coronary artery disease, just like in healthy individuals aspirin resistance, if it exists at all, is a rarity.
Several factors have been suggested to influence the effect of aspirin in pathological conditions. Increased platelet turnover was presumed to release newly formed platelets with nacCOX1 into the circulation and produce enough TXA2 to activate platelets, even platelets with acCOX1 [14, 15, 25, 26]. This problem was thought particularly important in the case of enteric-coated aspirin administered in a single daily dose. However, important component of the inhibitory effect of aspirin occurs in the presystemic circulation [13, 27]. Furthermore, COX1 of megakaryocytes is acetylated by aspirin and platelets released from megakaryocytes contain acCOX1 [28]. Furthermore, the finding of a persistent level of acCOX1 throughout a 7 days interval of treatment with once daily 100 mg enteric-coated aspirin provides direct evidence that acetylated platelets entered into the circulation during this time-frame [13]. Our present finding demonstrating that patients on long-term aspirin contain more than 97.5 % COX1 in acetylated form also support the cumulative nature of platelet COX1 inhibition upon repeated daily dosing [29]. The reports on the increased level of COX1 acetylation 24 h after the sixth daily dose of aspirin as compared to the level measured 24 h after a single aspirin dose also indicate cumulative saturable acetylation of platelet COX1 [11, 13].
COX2, an isoenzyme of COX1, is also sensitive to the inhibitory effect of aspirin [8, 30]. However a much higher dose of aspirin, well above the range reached during the prophylactic low dose aspirin therapy, is needed to acetylate the respective serine residue (in this case Ser516) [16]. This enzyme is present in megakaryocytes, it is expressed in young platelets [31] and its up-regulation has been described in diabetes mellitus and inflammatory conditions [17, 32, 33]. It has been proposed that COX2 expression in platelets might result in the suppression of aspirin effect. However, Riondino et al. [34] demonstrated on 100 patients being on chronic aspirin treatment by immunoblot analysis that COX2 could be detected only in 46 % of patients and its amount was markedly lower than that of COX1. By using COX2 inhibitor CAY10404 and aspirin they also demonstrated that COX2 dependent TXA2 production is less than 2 %. Our results are in line with the latter observations. We found detectable amount of platelet COX2 mRNA only in 39 % of patients with stable coronary artery disease. Even in those cases in which COX2 mRNA was detected, its amount was incomparably lower than that of COX1 mRNA and platelets from these patients produced a very low amount of TXB2 comparable to the rest of the aspirin treated patients. These findings do not support the role of COX2 expressed in platelets in the diminished response to low dose aspirin therapy.
In the light of the above findings it is a question if aspirin therapy should be monitored at all. In theory, mutations in the COX1 gene may result in a situation in which aspirin is unable to acetylate the enzyme, however such a situation has not been reported. The results suggest that the effect of aspirin does not need to be controlled in patients with coronary artery disease, unless non-compliance or drug interference is suspected. The main reason for the ineffectiveness of aspirin as detected by laboratory tests is non-compliance [35–37], testing of patients on aspirin therapy could provide information on their compliance. Non-steroid anti-inflammatory drugs may interfere with the effect of aspirin [38–40] and insufficient aspirin effect measured by adequate laboratory tests could draw the attention to such drug interference. It is to be emphasized that for the above purposes only methods specific to the effect of COX1 acetylation should be used. Among the routinely used methods these include AA induced platelet aggregation and secretion, the VerifyNow Aspirin Assay and the measurement of serum TXB2 [41]. This statement is valid only for aspirin monotherapy, its combination with P2Y12 receptor inhibitors (dual antiplatelet therapy) might influence the test results [42]. A limitation of the study is the patient population that comprised of individuals with stable coronary disease. It is to be seen if the results could be extended to patients with acute coronary syndrome.
Conclusions
The acetylation of platelet COX1 was tested in 144 patients with stable coronary artery disease. A direct method based on monoclonal antibodies against nacCOX1 and acCOX1 failed to detect aspirin resistance among the patients. Similar results were obtained with the measurement of AA induced TXB2 production of platelets, with AA induced platelet aggregation and ATP secretion and the VerifyNow Aspirin assay. These methods are useful in detecting non-compliance and very likely also drug interference. COX2 mRNA was expressed in 39 % of 56 investigated patients in very low amount, but it was not related to impaired aspirin effect.
Authors’ contributions
NH, EGK, RGK, IÉ, LM participated in the study design. NH, SL, LB, ZB, RGK, IÉ were involved in enrolling then patients, collection and evaluation of patient’s data. ÉK, produced the monoclonal antibodies for the Western blot assay. ÉK, GH, performed the Western blot assays. EGK, GH, HP, GS were responsible for the TXB2 assays. NH, EGK, SL, GS, HP performed platelet aggregation and ATP release determinations. AHS, LM carried out RT qPCR assays. ZB, RGK, IÉ, LM evaluated the results. NH, EGK, LM wrote the manuscript. All authors read and approved the final manuscript.
Acknowledgements
The authors are indebted to Associate Professor János Sándor (Ph.D.) for his advice in the statistical calculation. Support from the National Office of Research and Technology, Hungary (Jedlik Ányos Grant, NKFP-07-A1-2008-0127), from the Hungarian Academy of Science (MTA11003, TKI417), from the National Development Agency, Hungary (TÁMOP 4.2.2.A-11/1/KONV-2012-0045) and from the Hungarian National Research Fund (OTKA K113097) is acknowledged.
Competing interests
The authors declare that they have no competing interests.
Contributor Information
Nóra Homoródi, Email: homorodin@yahoo.com.
Emese G. Kovács, Email: emesekovacsmd@gmail.com
Sarolta Leé, Email: leesaci@gmail.com.
Éva Katona, Email: ekatona@med.unideb.hu.
Amir H. Shemirani, Email: amir@med.unideb.hu
Gizella Haramura, Email: haramura@med.unideb.hu.
László Balogh, Email: laszlobalogh76@yahoo.com.
Zsuzsanna Bereczky, Email: zsbereczky@med.unideb.hu.
Gabriella Szőke, Email: szokegaby@gmail.com.
Hajna Péterfy, Email: peterfyhajna@diagnosticum.hu.
Róbert G. Kiss, Email: robertgaborkiss@gmail.com
István Édes, Email: edes@med.unideb.hu.
László Muszbek, Email: muszbek@med.unideb.hu.
References
- 1.Eikelboom JW, Hirsh J, Spencer FA, Baglin TP, Weitz JI. Antiplatelet drugs: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest. 2012;141:e89S–e119S. doi: 10.1378/chest.11-2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Patrono C, Rocca B. Aspirin, 110 years later. J Thromb Haemost. 2009;7(Suppl 1):258–261. doi: 10.1111/j.1538-7836.2009.03391.x. [DOI] [PubMed] [Google Scholar]
- 3.Antithrombotic Trialists’ Collaboration Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ. 2002;324:71–86. doi: 10.1136/bmj.324.7329.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baigent C, Blackwell L, Collins R, Emberson J, Godwin J, Peto R, et al. Aspirin in the primary and secondary prevention of vascular disease: collaborative meta-analysis of individual participant data from randomised trials. Lancet. 2009;373:1849–1860. doi: 10.1016/S0140-6736(09)60503-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vandvik PO, Lincoff AM, Gore JM, Gutterman DD, Sonnenberg FA, Alonso-Coello P, et al. Primary and secondary prevention of cardiovascular disease: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest. 2012;141:e637S–e668S. doi: 10.1378/chest.11-2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Patrono C, Garcia Rodriguez LA, Landolfi R, Baigent C. Low-dose aspirin for the prevention of atherothrombosis. N Engl J Med. 2005;353:2373–2383. doi: 10.1056/NEJMra052717. [DOI] [PubMed] [Google Scholar]
- 7.Kulmacz RJ, van der Donk WA, Tsai AL. Comparison of the properties of prostaglandin H synthase-1 and -2. Prog Lipid Res. 2003;42:377–404. doi: 10.1016/S0163-7827(03)00023-7. [DOI] [PubMed] [Google Scholar]
- 8.Simmons DL, Botting RM, Hla T. Cyclooxygenase isozymes: the biology of prostaglandin synthesis and inhibition. Pharmacol Rev. 2004;56:387–437. doi: 10.1124/pr.56.3.3. [DOI] [PubMed] [Google Scholar]
- 9.Pettersen AA, Arnesen H, Seljeflot I. A brief review on high on-aspirin residual platelet reactivity. Vascul Pharmacol. 2015;67–69:6–9. doi: 10.1016/j.vph.2015.03.018. [DOI] [PubMed] [Google Scholar]
- 10.Angiolillo DJ, Capodanno D, Goto S. Platelet thrombin receptor antagonism and atherothrombosis. Eur Heart J. 2010;31:17–28. doi: 10.1093/eurheartj/ehp504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kovacs EG, Katona E, Bereczky Z, Homorodi N, Balogh L, Toth E, et al. New direct and indirect methods for the detection of cyclooxygenase 1 acetylation by aspirin; the lack of aspirin resistance among healthy individuals. Thromb Res. 2013;131:320–324. doi: 10.1016/j.thromres.2013.01.033. [DOI] [PubMed] [Google Scholar]
- 12.Kovacs EG, Katona E, Bereczky Z, Homorodi N, Balogh L, Toth E, et al. Evaluation of laboratory methods routinely used to detect the effect of aspirin against new reference methods. Thromb Res. 2014;133:811–816. doi: 10.1016/j.thromres.2013.10.008. [DOI] [PubMed] [Google Scholar]
- 13.Patrignani P, Tacconelli S, Piazuelo E, Di Francesco L, Dovizio M, Sostres C, et al. Reappraisal of the clinical pharmacology of low-dose aspirin by comparing novel direct and traditional indirect biomarkers of drug action. J Thromb Haemost. 2014;12:1320–1330. doi: 10.1111/jth.12637. [DOI] [PubMed] [Google Scholar]
- 14.Grove EL, Hvas AM, Kristensen SD. Immature platelets in patients with acute coronary syndromes. Thromb Haemost. 2009;101:151–156. [PubMed] [Google Scholar]
- 15.Guthikonda S, Lev EI, Patel R, DeLao T, Bergeron AL, Dong JF, et al. Reticulated platelets and uninhibited COX-1 and COX-2 decrease the antiplatelet effects of aspirin. J Thromb Haemost. 2007;5:490–496. doi: 10.1111/j.1538-7836.2007.02387.x. [DOI] [PubMed] [Google Scholar]
- 16.Rocca B, Secchiero P, Ciabattoni G, Ranelletti FO, Catani L, Guidotti L, et al. Cyclooxygenase-2 expression is induced during human megakaryopoiesis and characterizes newly formed platelets. Proc Natl Acad Sci USA. 2002;99:7634–7639. doi: 10.1073/pnas.112202999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Capodanno D, Patel A, Dharmashankar K, Ferreiro JL, Ueno M, Kodali M, et al. Pharmacodynamic effects of different aspirin dosing regimens in type 2 diabetes mellitus patients with coronary artery disease. Circ Cardiovasc Interv. 2011;4:180–187. doi: 10.1161/CIRCINTERVENTIONS.110.960187. [DOI] [PubMed] [Google Scholar]
- 18.Brown DL, Cai TT, DasGupta A. Interval estimation for a binomial proportion. Stat Sci. 2001;16:101–133. [Google Scholar]
- 19.Zsori KS, Muszbek L, Csiki Z, Shemirani AH. Validation of reference genes for the determination of platelet transcript level in healthy individuals and in patients with the history of myocardial infarction. Int J Mol Sci. 2013;14:3456–3466. doi: 10.3390/ijms14023456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nolan T, Hands RE, Bustin SA. Quantification of mRNA using real-time RT-PCR. Nat Protoc. 2006;1:1559–1582. doi: 10.1038/nprot.2006.236. [DOI] [PubMed] [Google Scholar]
- 21.Birschmann I, Mietner S, Dittrich M, Pfrang J, Dandekar T, Walter U. Use of functional highly purified human platelets for the identification of new proteins of the IPP signaling pathway. Thromb Res. 2008;122:59–68. doi: 10.1016/j.thromres.2007.08.022. [DOI] [PubMed] [Google Scholar]
- 22.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 23.Zou H, Yuan C, Dong L, Sidhu RS, Hong YH, Kuklev DV, et al. Human cyclooxygenase-1 activity and its responses to COX inhibitors are allosterically regulated by nonsubstrate fatty acids. J Lipid Res. 2012;53:1336–1347. doi: 10.1194/jlr.M026856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grosser T, Fries S, Lawson JA, Kapoor SC, Grant GR, FitzGerald GA. Drug resistance and pseudoresistance: an unintended consequence of enteric coating aspirin. Circulation. 2013;127:377–385. doi: 10.1161/CIRCULATIONAHA.112.117283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pascale S, Petrucci G, Dragani A, Habib A, Zaccardi F, Pagliaccia F, et al. Aspirin-insensitive thromboxane biosynthesis in essential thrombocythemia is explained by accelerated renewal of the drug target. Blood. 2012;119:3595–3603. doi: 10.1182/blood-2011-06-359224. [DOI] [PubMed] [Google Scholar]
- 26.Rocca B, Santilli F, Pitocco D, Mucci L, Petrucci G, Vitacolonna E, et al. The recovery of platelet cyclooxygenase activity explains interindividual variability in responsiveness to low-dose aspirin in patients with and without diabetes. J Thromb Haemost. 2012;10:1220–1230. doi: 10.1111/j.1538-7836.2012.04723.x. [DOI] [PubMed] [Google Scholar]
- 27.Pedersen AK, FitzGerald GA. Dose-related kinetics of aspirin. Presystemic acetylation of platelet cyclooxygenase. N Engl J Med. 1984;311:1206–1211. doi: 10.1056/NEJM198411083111902. [DOI] [PubMed] [Google Scholar]
- 28.van Pampus EC, Huijgens PC, Zevenbergen A, Twaalfhoven H, van Kamp GJ, Langenhuijsen MM. Influence of aspirin on human megakaryocyte prostaglandin synthesis. Eur J Haematol. 1993;50:264–268. doi: 10.1111/j.1600-0609.1993.tb00160.x. [DOI] [PubMed] [Google Scholar]
- 29.Patrignani P, Filabozzi P, Patrono C. Selective cumulative inhibition of platelet thromboxane production by low-dose aspirin in healthy subjects. J Clin Invest. 1982;69:1366–1372. doi: 10.1172/JCI110576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Blobaum AL, Marnett LJ. Structural and functional basis of cyclooxygenase inhibition. J Med Chem. 2007;50:1425–1441. doi: 10.1021/jm0613166. [DOI] [PubMed] [Google Scholar]
- 31.Santilli F, Simeone P, Liani R, Davi G. Platelets and diabetes mellitus. Prostaglandins Other Lipid Mediat. 2015 doi: 10.1016/j.prostaglandins.2015.05.002. [DOI] [PubMed] [Google Scholar]
- 32.Halushka MK, Halushka PV. Why are some individuals resistant to the cardioprotective effects of aspirin? Could it be thromboxane A2? Circulation. 2002;105:1620–1622. doi: 10.1161/01.CIR.0000015422.86569.52. [DOI] [PubMed] [Google Scholar]
- 33.Weber AA, Zimmermann KC, Meyer-Kirchrath J, Schror K. Cyclooxygenase-2 in human platelets as a possible factor in aspirin resistance. Lancet. 1999;353:900. doi: 10.1016/S0140-6736(99)00498-5. [DOI] [PubMed] [Google Scholar]
- 34.Riondino S, Trifiro E, Principessa L, Mascioletti S, Di Renzo L, Gaudio C, et al. Lack of biological relevance of platelet cyclooxygenase-2 dependent thromboxane A2 production. Thromb Res. 2008;122:359–365. doi: 10.1016/j.thromres.2007.12.011. [DOI] [PubMed] [Google Scholar]
- 35.Schwartz KA, Schwartz DE, Barber K, Reeves M, De Franco AC. Non-compliance is the predominant cause of aspirin resistance in chronic coronary arterial disease patients. J Transl Med. 2008;6:46. doi: 10.1186/1479-5876-6-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schwartz KA, Schwartz DE, Ghosheh K, Reeves MJ, Barber K, DeFranco A. Compliance as a critical consideration in patients who appear to be resistant to aspirin after healing of myocardial infarction. Am J Cardiol. 2005;95:973–975. doi: 10.1016/j.amjcard.2004.12.038. [DOI] [PubMed] [Google Scholar]
- 37.Meen O, Brosstad F, Khiabani H, Gjertsen E, Lauritsen ME, Pedersen TM, et al. No case of COX-1-related aspirin resistance found in 289 patients with symptoms of stable CHD remitted for coronary angiography. Scand J Clin Lab Invest. 2008;68:185–191. doi: 10.1080/00365510701601673. [DOI] [PubMed] [Google Scholar]
- 38.Huber K, Schror K. High on-treatment platelet reactivity—why should we be concerned? Thromb Haemost. 2013;109:789–791. doi: 10.1160/TH13-04-0281. [DOI] [PubMed] [Google Scholar]
- 39.Fitzgerald R, Pirmohamed M. Aspirin resistance: effect of clinical, biochemical and genetic factors. Pharmacol Ther. 2011;130:213–225. doi: 10.1016/j.pharmthera.2011.01.011. [DOI] [PubMed] [Google Scholar]
- 40.Ho PM, Spertus JA, Masoudi FA, Reid KJ, Peterson ED, Magid DJ, et al. Impact of medication therapy discontinuation on mortality after myocardial infarction. Arch Intern Med. 2006;166:1842–1847. doi: 10.1001/archinte.166.17.1842. [DOI] [PubMed] [Google Scholar]
- 41.Patrono C, Ciabattoni G, Pinca E, Pugliese F, Castrucci G, De Salvo A, et al. Low dose aspirin and inhibition of thromboxane B2 production in healthy subjects. Thromb Res. 1980;17:317–327. doi: 10.1016/0049-3848(80)90066-3. [DOI] [PubMed] [Google Scholar]
- 42.Bagoly Z, Homorodi N, Kovacs EG, Sarkady F, Csiba L, Edes I, et al. How to test the effect of aspirin and clopidogrel in patients on dual antiplatelet therapy? Platelets. 2016;27:59–65. doi: 10.3109/09537104.2015.1031098. [DOI] [PubMed] [Google Scholar]