

Abstract
Human ether-á-go-go related gene (hERG, Kv11.1) potassium channels play a significant role in cardiac excitability. Like other Kv channels, hERG is activated by membrane voltage; however, distinct from other Kv channels, hERG channels have unusually slow kinetics of closing (deactivation). The mechanism for slow deactivation involves an N-terminal “eag domain” which comprises a PAS (Per-Arnt-Sim) domain and a short Cap domain. Here we review recent advances in understanding how the eag domain regulates deactivation, including several new Nuclear Magnetic Resonance (NMR) solution structures of the eag domain, and evidence showing that the eag domain makes a direct interaction with the C-terminal C-linker and Cyclic Nucleotide-Binding Homology Domain.
1. Introduction
The human ether-á-go-go related gene (hERG, Kv11.1) encodes a voltage-gated potassium (K+) channel which is expressed in a variety of tissues, including the heart and brain [1, 2]. The role of hERG in the heart is well-characterized: hERG encodes the IKr current [3, 4], which contributes to the repolarization of the ventricular action potential [5]. Mutations in hERG channels are linked to type II long QT syndrome (LQT2) [2], a disorder typified by a prolongation of the cardiac action potential, early after-depolarizations, and “torsades de pointe” tachycardias [6–9]. An acquired form of LQT is associated with drug inhibition of hERG channels [10]. As with other voltage-activated channels, hERG subunits have six transmembrane domains, large cytoplasmic N- and C-terminal regions (Fig. 1A) and four hERG subunits likely form a functional channel [1]. hERG and other members of the eag family of channels are structurally distinct from other Kv channels because they have a conserved “eag” domain encoded by amino acids 1–135 in the N-terminal region. The eag domain contains a Per-Arnt-Sim (PAS) homology domain that is encoded by amino acids 26–135 [11] (Fig. 1A). The hERG PAS domain is adjacent to a domain encoded by amino acids 1–25 that “caps” the PAS domain and that we term the hERG Cap domain (Fig. 1A). Cap domains are key regulators of PAS function in other proteins [12–15]. Evidence suggests that the hERG Cap domain directly regulates channel gating [16]. Each of the four hERG subunits (and other subunits in the eag family) contains a C-terminal C-linker region and cyclic nucleotide-binding homology domain (CNBHD) that are homologous to that of cyclic nucleotide-gated (CNG) and hyperpolarization-activated cyclic nucleotide-gated (HCN) channels [1, 17] (Fig 1A). However, unlike CNG and HCN channels, hERG is not directly regulated by binding to cyclic nucleotides [18].
Fig. 1.

hERG channel topology and sample current. A. Schematic of the hERG K+ channel with key functional regions as indicated. The PAS domain is shown in red and the Cap domain is shown in blue. Together these comprise the eag domain. The cyclic nucleotide-binding homology domain is shown in grey. B. Sample hERG channel current in response to the pulse protocol indicated. With a depolarizing step, the channels transition from a closed (C) state to an open (O) state and then rapidly enter an inactive state (I). Upon repolarization, the channels rapidly recover from inactivation (I to O), and re-enter the open (O) state before slowly entering a closed (C) state. The dotted line represents zero current.
The voltage-dependent opening and closing (activation and deactivation gating) properties of hERG are somewhat unique for K+ channels. In response to depolarization, hERG channels activate relatively slowly, but inactivate rapidly, and conduct a relatively small amount of outward current (Fig. 1B). However, in response to subsequent repolarization, hERG recovers rapidly from inactivation and then closes, or deactivates, very slowly, and consequently conducts a relatively large outward current (Fig. 1B). In the heart, ventricular action potentials have a rapid rise and long plateau phase, suggesting that the majority of hERG current is conducted during the repolarization phase of ventricular cardiac action potentials [4, 19–21]. Thus, the unique kinetics of hERG channels perfectly tune them for their role in the heart.
Similar to other voltage-activated K+ channels, voltage-dependent activation and deactivation gating in hERG channels is mediated by the charged voltage-sensor domain (VSD) located in transmembrane segments S1–S4 [22–25] (Fig. 1A). Movement of the VSD is thought to open a channel gate located in the lower S6 transmembrane region that controls access to the pore [26, 27]. Interactions of the intracellular S4–S5 linker with the lower S6 of hERG couple the movement of the VSD to the opening of the gate during channel activation and deactivation [28–31]. hERG channels also have a type of inactivation gating which is perturbed by mutations at the outer mouth of the pore region [21, 32], similar to the C-type inactivation mechanism described in Shaker K+ channels [33]. Voltage-dependent gating in hERG channels is regulated by the intracellular N- and C-terminal domains of the channel. In particular, deactivation gating is regulated by the N-terminal eag domain. The eag domain makes a direct interaction with another intracellular region (or regions) of the hERG channel (Fig. 2A). One intriguing property of the eag domain is that it does not require a peptide bond linking it to the rest of the channel in order to regulate gating [11, 34] (Fig. 2B). The key role of the eag domain in hERG channel physiology is underscored by mutations in the eag domain that were linked to LQT2 [35–47] and which disrupt regulation of gating [34, 35, 39, 41, 45, 48].
Fig. 2.
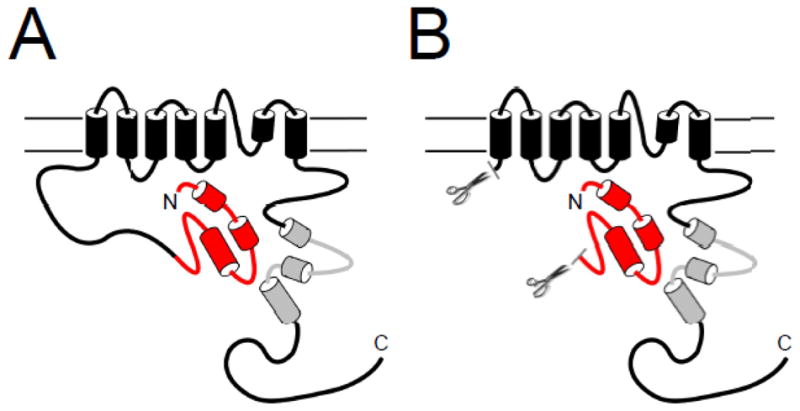
Model of eag Domain interaction with the channel. A. The eag domain forms a direct interaction with another region of the hERG channel. B. The eag domain interacts directly with the channel even in the absence of the proximal N-terminal linker region.
The precise mechanism for eag domain regulation of gating is not known. Here, we discuss recent advances regarding structural information about the eag domain, and new information regarding the identity of intracellular sites, including the C-linker/CNBHD in the C-terminal region, that interact with the eag domain to regulate deactivation gating.
2. Structural components of the eag domain
The structure of the hERG N-terminal region was solved by X-ray crystallography and revealed that amino acids 26–135 were a PAS homology domain [11]. The PAS domain was found to contain five antiparallel β-sheets flanked by 3 α-helices (Fig. 3A). The PAS domain appeared to be a monomer [11], although earlier work suggested that PAS could assemble into an oligomer [49]. The first 25 amino acids were present in the crystallized protein, but were presumed to be disordered. Recently, several groups have obtained NMR solution structures for the entire eag domain, including the first 25 amino acids [50–52] (Fig. 3B, C, D). The PAS domain part of the structures solved with NMR demonstrate good alignment and general agreement with the structure solved by X-ray crystallography, particularly in the proportion and orientation of α-helix and the orientation of β-sheet and loop regions (Fig. 4A, B). One notable difference was a dramatic reduction in the proportion of β-sheet in one of the structures, which was not seen in the other NMR structures [51] (Fig. 3C). Overall, the similarity of multiple independent structures provides strong validation of the PAS domain configuration. The NMR data also provided the first structural information about amino acids 1–25 of the eag domain, which “cap” the PAS domain (Figs. 3, 4). All three NMR structures identified the N-terminal Cap as an amphipathic helix between amino acids 13 and 23 and a disordered region between amino acids 1 and 12. The first twelve amino acids were highly mobile in relation to the amphipathic helix and the PAS domain [50–52]. Dynamic CAP regions that are located N-terminal and adjacent to PAS domains are found in several other PAS domain-containing proteins [12–15].
Fig. 3.
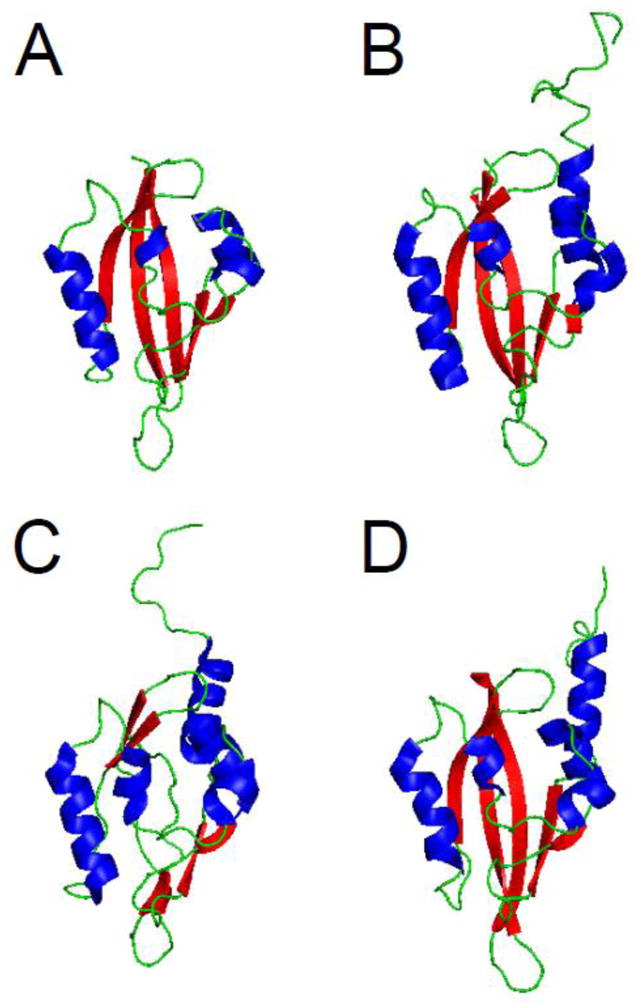
eag domain schematics. A. X- ray structure from Morais Cabral et al. 1998 (PDB ID: 1byw). Representative NMR structures, B. from Li et al. 2010 (PDB ID: 2l4r), C. from Muskett et al. 2010 (pdb: 2l1m), and D. from Ng et al. 2011 (pdb: 2l0w) [11, 50–52]. α-helix is shown in blue, β-sheet is shown in red, and loop regions are shown in green. Images were created using PyMOL (www.pymol.org).
Fig. 4.

A. Ribbon diagram and B. protein backbone overlays of the eag domain structures. The X-ray structure (from Morais Cabral et al. 1998) is shown in yellow and representative NMR structures are shown in green (Li et al. 2010), blue (Muskett et al. 2010) and red (Ng et al. 2011) [11, 50–52]. The N-terminal disordered region and α-helix identified in the NMR structures were found to be dynamic. Image was created using PyMOL.
3. Functional role of the eag domain in channel gating
The eag domain is necessary for regulation of deactivation gating in hERG channels. hERG channels with deletions of (approximately) just the eag domain (hERG Δ2–138) had deactivation kinetics that were accelerated 5 to 10-fold compared to deactivation in wild-type hERG channels [11]. Channels with deletions of just the eag domain had kinetics of deactivation that were indistinguishable from those measured in channels with deletions of most of the N-terminal region (hERG Δ2–354 or hERG Δ2–373) [11, 16, 53, 54], meaning that regulation of deactivation was localized to the eag domain.
Intriguingly, the eag domain does not require a peptide bond linking it to the rest of the channel protein in order to regulate gating. Purified eag domain polypeptides [11] or genetically-encoded eag domains [34] restored the slow kinetics of deactivation gating in N-terminal region-deleted hERG channels that had accelerated deactivation (see Fig. 2B). Genetically encoded eag domains changed deactivation gating in N-deleted channels such that the deactivation gating was indistinguishable from WT hERG channels, suggesting that the isolated eag domain and the S1 domain – C-terminus of hERG were sufficient for deactivation gating (and that the linker region between the eag domain and the S1 transmembrane domain was not a regulator of deactivation) (Fig. 2B). In optical experiments, isolated eag domains tagged with CFP (eag CFP) were in close physical proximity to N-terminal region-deleted hERG channels tagged with Citrine (hERG ΔN Citrine), as measured by Förster Resonance Energy Transfer (FRET) [34]. Thus, optical and functional experiments showed that eag domains make a direct interaction with the rest of the hERG channel and regulate deactivation gating.
3.1. Functional role of the PAS domain
What residues in the eag domain are critical for its function? Analysis of the eag domain structure identified a grouping of hydrophobic residues on the surface of the PAS domain [11] (Fig. 5). Point mutations at hydrophobic residues F29 and Y43 speed up deactivation kinetics, as do mutations at other sites that are nearby in the three-dimensional structure, such as K28E, N33T, R56Q, and M124R [11, 34, 48], suggesting that these point mutations might disrupt the interaction of the eag domain with the rest of the channel (Fig. 6A).
Fig. 5.

eag domain residues involved in LQT2 and gating. A. CPK model of the eag domain. B. eag domain model from A rotated 180° around a vertical axis. Green and red residues are part of the hydrophobic patch [11]. Green residues have not been characterized, while mutations in red residues have been shown to speed channel deactivation gating. Yellow residues are mutation sites which speed deactivation gating, and include LQT2 sites (K28, N33, and R56) adjacent to the hydrophobic patch. Blue residues are identified LQT2 mutation sites which do not form functional channels (T65, C66, L86) or do not affect deactivation gating (G53, H70, A78) in mammalian cells. Images were created using PyMOL (based on the structure from Ng et al. ; PDB ID: 2l0w) [52].
Fig. 6.
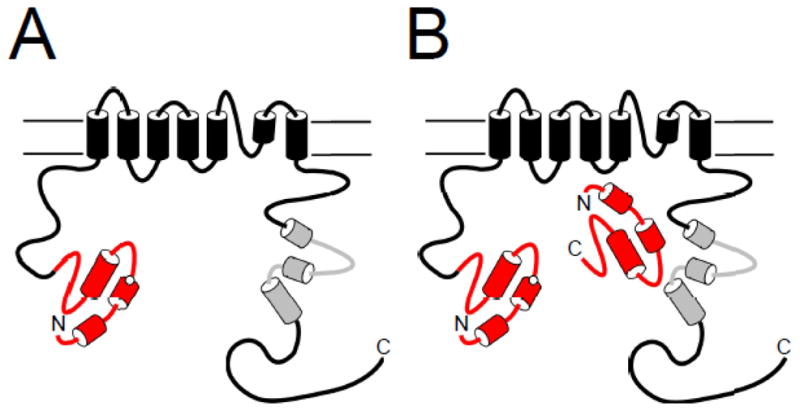
Model of disruption of eag domain interaction with the channel by LQT2 mutations. A. An eag domain mutation which disrupts slow deactivation gating also interferes with the interaction of the eag domain with the channel. B. A genetically encoded eag domain fragment is able to replace the mutated domain and modulate channel gating.
Experiments combining functional and optical recording showed that mutations in separate eag domains (eag [Y43A] CFP and eag [R56Q] CFP) showed reduced regulation of deactivation gating and reduced FRET, or no regulation of gating and no FRET [34], suggesting that the mutations disrupted an interaction between the isolated eag domains and the rest of the hERG channel [34]. In a correlative experiment, separate eag domains (eag CFP) regulated slow deactivation gating in full-length channels with K28E, N33T, Y43A, R56Q, and M124R mutations [34, 48] (Fig. 6B). FRET experiments showed that the eag domains (eag CFP) were in close proximity to the channels with Y43A and R56Q mutations (but not WT channels) [34]. These results suggest that the mutations weakened the interaction of the eag domain with the rest of the channel sufficiently to allow the mutant eag domains to be replaced by separate eag domains (Fig. 6).
Thus, mutations in the PAS domain at positions nearby each other in the three-dimensional structure disrupt the interaction between the PAS domain and the rest of the channel. One explanation for this result is that these residues form a direct interaction with the rest of the channel, however it is also possible that the mutations have an allosteric effect and that the PAS domain interacts with the channel at other sites or additional sites. Finally, in addition to hydrophobic sidechains (like Y43), charged sidechains (like R56) are important for the interaction of the PAS domain with the rest of the hERG channel.
3.2. Functional role of the eag Cap domain
The Cap domain region of the eag domain is necessary for regulation of deactivation gating, since channels with deletions of amino acids within the Cap domain (Δ2–9, Δ2–12, Δ2–23, Δ2–25, Δ2–26) had accelerated deactivation kinetics compared to wild-type channels [11, 52, 53]. Channels with point mutations within the first 25 amino acids also had accelerated deactivation [51, 52]. The amphipathic nature of the helix in the Cap region was important for slow deactivation gating, as channels with charge-reversal point mutations on the positive face of the helix had faster deactivation kinetics [51, 52] (Fig. 5). It was proposed that these mutations altered deactivation gating by disrupting the interaction of the Cap domain with another part of channel. It was also shown that a point mutation in the Cap domain disrupted FRET between an eag domain and the rest of the channel [55]. It is important to consider that, like PAS domain mutations, this result could be due to direct Cap domain interaction with the channel, or could be an allosteric effect. Indeed, deletions of the Cap domain disrupt the folding and three dimensional structure of the PAS domain in photoactive yellow protein (PYP) [56, 57]. There is evidence that the Cap domain can have an effect on channels in the absence of the PAS domain. Perhaps the clearest evidence for a distinct role of the Cap domain is that a purified peptide encoding part of the Cap domain (amino acids 1–16) partially recovered slow deactivation gating when applied to excised membrane patches that contained N-truncated hERG (Δ2–354) channels [16]. Structural (NMR) information showing that the Cap domain is dynamic is in agreement with idea that the Cap domain function can be distinct from PAS domain [51, 52].
3.3. Differences in PAS and CAP domain functions
There are some important differences between the functional role of the Cap domain and the functional role of the PAS domain. Mutations and deletions in both the Cap domain and the PAS domain alter channel deactivation; in contrast, mutations in these regions have different effects on inactivation. Channels with deletions that include the PAS domain or point mutations in the PAS domain (such as R56Q) had inactivation gating that was slowed by approximately 2-fold and a steady-state inactivation curve that was shifted in a positive direction [19, 35, 48, 53, 54, 58, 59]. In contrast, channels with deletions of the first 25 amino acids did not show a change in channel inactivation kinetics [19, 53, 54, 58]. These results suggest that the PAS domain (amino acids 26–135) plays a role in inactivation gating, but that the CAP domain (amino acids 1–25) is not involved in inactivation gating. Perhaps the first 25 amino acids are the functional determinants of slow deactivation gating, whereas the core of the eag domain serves as a positional regulator of slow deactivation gating and modulates inactivation.
4. eag domain binding site
Since separate eag domains regulate gating and showed FRET with hERG channels bearing N-terminal region deletions, the eag domain must bind to some other region of the hERG channel. Current studies have focused on two regions as potential eag domain binding sites: the S4–S5 linker and the C-terminal C-linker/CNBHD.
4.1. A possible role for the S4–S5 linker region
Previously, it was suggested that the eag domain formed hydrophobic interactions with another region of the hERG channel, and the S4–S5 linker was suggested as a possible interacting site [11]. Point mutations within the S4–S5 linker have been shown to speed channel deactivation gating [28], consistent with the involvement of this region in regulation of deactivation. Experiments using NEM modification of a introduced cysteine at a site in the S4–S5 linker (G546C) also showed a disruption of slow deactivation gating [53]. Mutation at the same residue (G546C) also disrupted FRET between the channel and separate eag domain [55]. Recently, NMR experiments have further suggested an interaction between the eag domain and the S4–S5 linker [50]. An S4–S5 linker peptide was purified and combined in solution with purified eag domain protein. A shift was noted in the position of several amino acids in the eag domain NMR structure when the peptide was present [50], suggesting that the S4–S5 linker peptide interacted with some region of the eag domain. This experiment, however, lacked confirmation of an intact structure for the S4–S5 linker peptide and also lacked a peptide control to show specificity of the effect with the S4–S5 linker peptide [50]. In another study, a putative eag domain interaction with the S4–S5 linker was investigated by substituting cysteines in several positions within the first amino acids in the Cap region of the eag domain and positions within the S4–S5 linker. Evidence for disulfide cross-linking between cysteines in the Cap region and those in the S4–S5 linker was detected, suggesting close proximity (< 7Å) between those regions [55].
While these experiments suggest that the S4–S5 linker was involved in deactivation gating, the S4–S5 linker and eag domain are not sufficient for regulation of deactivation. Channels with intact eag domains and S4–S5 linkers, but with deletions in the C-terminal regions, including deletions of the CNBHD, had fast deactivation gating, indicating that the CNBHD was also necessary for eag domain modulation of deactivation (as discussed below) [60]. The region of the eag domain which may interact with the S4–S5 linker has not been conclusively determined.
4.2. Role of the C-terminal regions
The hERG channel C-terminal region C-linker/CNBHD is also a site of interaction for the eag domain. FRET experiments demonstrated that the N- and C- termini of hERG were in proximity, in support of an HCN channel homology model, which would position the CNBHD below the channel pore [34, 61]. Channels with mutations in or deletion of the CNBHD were found to have abnormal gating kinetics [51, 60, 62]. A series of lysine mutations in the CNBHD had faster deactivation gating, and the affected residues were found to cluster in a hydrophobic band on the surface on a homology model of the hERG CNBHD, which is based on the HCN2 CNBD structure [62] (Fig. 7). Another report showed that channels with point mutations in the CNBHD had faster deactivation kinetics, and proposed a model in which the CAP region of the eag domain bound in a cleft formed by CNBHDs of adjacent subunits [51] (Fig. 7). This interaction model was developed based on the hydrophobic band identified in the CNBHD and the hydrophobic patch identified in the eag domain (Figs. 5, 7). We found that channels with deletions of the CNBHD also had fast deactivation gating, suggesting a role for the CNBHD in deactivation gating [60]. We detected a biochemical interaction between an eag domain fusion protein and a C-linker/CNBHD fusion protein [60]. We also found evidence that the mechanism for regulation of deactivation was an intersubunit interaction between the eag domain and C-linker/CNBHD [60] (Fig. 8). Overall, these results indicated that the eag domain directly interacted with the C-linker/CNBHD, and that this interaction was necessary for eag domain regulation of hERG.
Fig. 7.
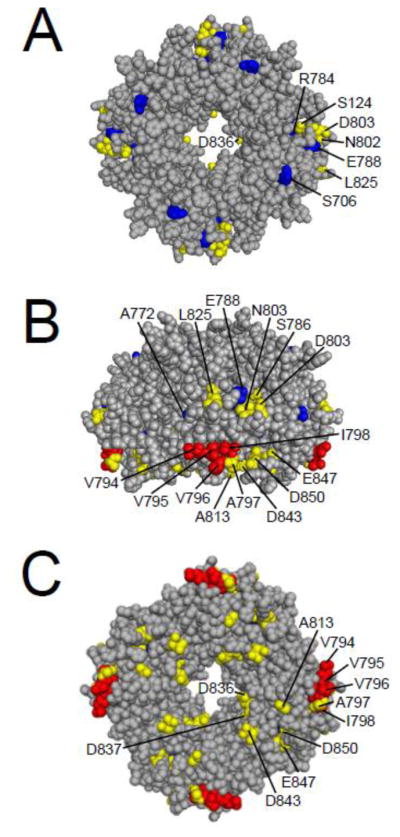
Cyclic nucleotide-binding homology domain residues involved in LQT2 and gating. A. CPK model of the CNBHD viewed looking down from the plasma membrane. B. CNBHD model from A rotated 90° to show a side view. C. CNBHD model from A rotated 180° to show the cytoplasmic face. Red residues are hydrophobic residues which have been shown to speed channel deactivation gating, blue residues are identified LQT2 mutation sites that have been shown to speed deactivation gating, and yellow residues are all others which have been shown to speed deactivation gating. Images were created using PyMOL (based on Zagotta et al.; PDB ID:1Q5O) [17].
Fig. 8.
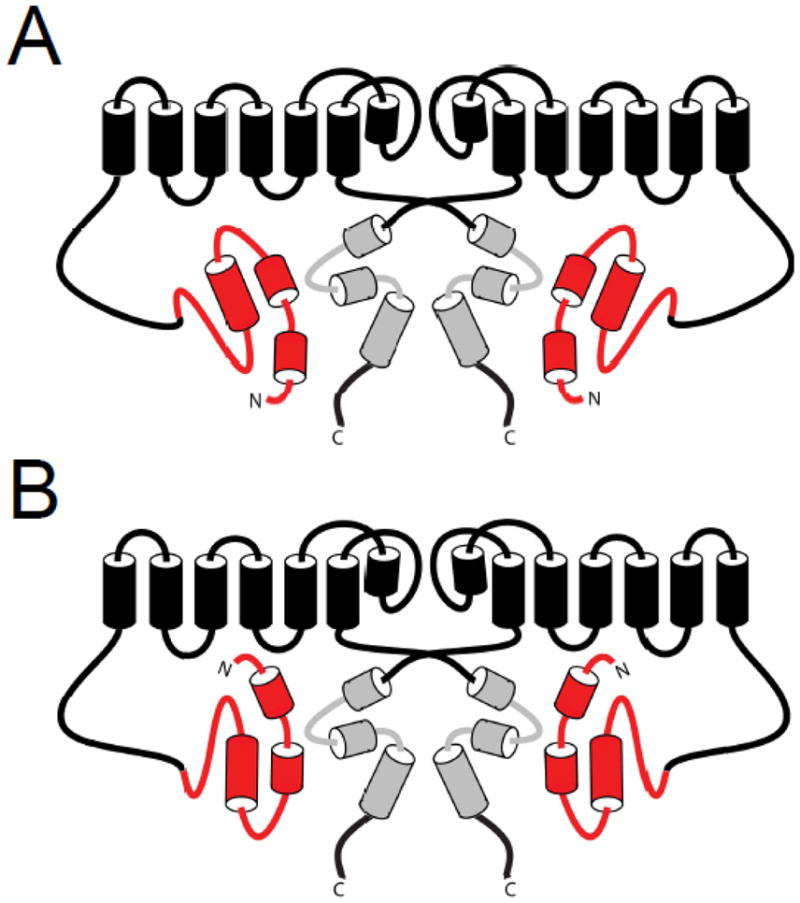
A. Model of eag domain interaction showing the Cap domain and the PAS domain interacting with the C-terminal CNBHD. B. Model of eag domain interaction showing the Cap domain interacting with the S4–S5 linker and the PAS domain interacting with the C-terminal CNBHD.
5. The eag domain and Long QT Syndrome
Forty-five eag domain mutations have been linked to LQT2 and account for approximately 15% of all known LQT2 mutations [35–47]. Twelve of these were characterized with electrophysiological recordings (K28E, F29L, I31S, N33T, G53R, R56Q, T65P, C66G, H70R, A78P, L86R, and M124R), and were found to have changes in deactivation gating compared to wild-type channels, as well as some defects in protein biogenesis [34, 35, 39, 41, 45, 48]. The altered kinetics of one hERG eag domain mutant channel, hERG R56Q, were shown to prolong the ventricular action potential and were pro-arrhythmogenic in a computational model of cardiac excitability [63]. When expressed in mammalian cells, five LQT2 mutations with the largest speeding of deactivation gating (K28E, F29L, N33T, R56Q, and M124R) occur in or near a hydrophobic patch on one face of the eag domain which was proposed to form a binding interface with another part of the channel [11, 48] (Fig. 5). These five LQTS mutants disrupted the interaction of the eag domain with the rest of the hERG channel (Fig. 6A), based in part on the ability of a separate eag domain to interact with mutant channels and restore gating [34, 48] (Fig. 6B).
6. Conclusions
Several structures have now been identified which encompass the entire eag domain, and evidence was presented for interactions of this domain with both the hERG S4–S5 linker and the C-terminal C-linker/CNBHD. The evidence for S4–S5 linker and C-linker/CNBHD interaction is largely complementary, suggesting that both of these regions may be involved in interaction of the eag domain with the channel. In one model of eag domain interaction, the N-terminal cap of the eag domain are oriented towards the intracellular facing surface of the CNBHD [51] (Fig. 8A). This orientation would likely prohibit interaction of the first 25 amino acids of the eag domain with the S4–S5 linker, although another region of the PAS domain could possibly interact with the S4–S5 linker instead. In an alternate model, the Cap domain of the amphipathic helix could interact with the S4–S5 linker, and the PAS domain could form a stable interaction with the C-terminal region CNBHD, possibly mediated by the hydrophobic and charged residues which have been identified on the surfaces of the eag domain and the CNBHD [55, 60, 62] (Fig. 8B). Further research is needed to determine the precise orientation and the specific residues involved in the interaction of the eag domain with the S4–S5 linker region and/or the C-linker/CNBHD.
Highlights.
The N-terminal eag domain of hERG channels regulates the unusually slow kinetics of channel closing.
Like other PAS domain-containing proteins, the hERG eag domain comprises a PAS domain and an adjacent Cap domain
The eag domain makes a direct, regulatory interaction with the rest of the channel.
Long QT syndrome-linked mutations in the eag domain weaken its interaction with the rest of the channel
The N-terminal eag domain interacts with the C-terminal Cyclic Nucleotide-Binding Homology Domain (CNBHD) to regulate channel closing
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Warmke JW, Ganetzky B. Proc Natl Acad Sci U S A. 1994;91(8):3438–3442. doi: 10.1073/pnas.91.8.3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT. Cell. 1995;80(5):795–803. doi: 10.1016/0092-8674(95)90358-5. [DOI] [PubMed] [Google Scholar]
- 3.Sanguinetti MC, Jiang C, Curran ME, Keating MT. Cell. 1995;81(2):299–307. doi: 10.1016/0092-8674(95)90340-2. [DOI] [PubMed] [Google Scholar]
- 4.Trudeau MC, Warmke JW, Ganetzky B, Robertson GA. Science. 1995;269(5220):92–95. doi: 10.1126/science.7604285. [DOI] [PubMed] [Google Scholar]
- 5.Sanguinetti MC, Jurkiewicz NK. J Gen Physiol. 1990;96(1):195–215. doi: 10.1085/jgp.96.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Romano C. Lancet. 1965;1:658–659. doi: 10.1016/s0140-6736(65)91761-7. [DOI] [PubMed] [Google Scholar]
- 7.Schwartz PJ, Periti M, Malliani A. Am Heart J. 1975;109:378–390. doi: 10.1016/0002-8703(75)90089-7. [DOI] [PubMed] [Google Scholar]
- 8.Vincent GM, Timothy KW, Leppert M, Keating M. N Engl J Med. 1992;327(12):846–852. doi: 10.1056/NEJM199209173271204. [DOI] [PubMed] [Google Scholar]
- 9.Roden DM. J Intern Med. 2006;259(1):59–69. doi: 10.1111/j.1365-2796.2005.01589.x. [DOI] [PubMed] [Google Scholar]
- 10.Mitcheson JS, Chen J, Lin M, Culberson C, Sanguinetti MC. Proc Natl Acad Sci U S A. 2000;97(22):12329–12333. doi: 10.1073/pnas.210244497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morais Cabral JH, Lee A, Cohen SL, Chait BT, Li M, Mackinnon R. Cell. 1998;95(5):649–655. doi: 10.1016/s0092-8674(00)81635-9. [DOI] [PubMed] [Google Scholar]
- 12.Pellequer JL, Wager-Smith KA, Kay SA, Getzoff ED. Proc Natl Acad Sci U S A. 1998;95(11):5884–5890. doi: 10.1073/pnas.95.11.5884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Taylor BL, Zhulin IB. Microbiol Mol Biol Rev. 1999;63(2):479–506. doi: 10.1128/mmbr.63.2.479-506.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rajagopal S, Anderson S, Srajer V, Schmidt M, Pahl R, Moffat K. Structure. 2005;13(1):55–63. doi: 10.1016/j.str.2004.10.016. [DOI] [PubMed] [Google Scholar]
- 15.Moglich A, Ayers RA, Moffat K. Structure. 2009;17(10):1282–1294. doi: 10.1016/j.str.2009.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang J, Myers CD, Robertson GA. J Gen Physiol. 2000;115(6):749–758. doi: 10.1085/jgp.115.6.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zagotta WN, Olivier NB, Black KD, Young EC, Olson R, Gouaux E. Nature. 2003;425(6954):200–205. doi: 10.1038/nature01922. [DOI] [PubMed] [Google Scholar]
- 18.Brelidze TI, Carlson AE, Zagotta WN. J Biol Chem. 2009;284(41):27989–27997. doi: 10.1074/jbc.M109.016337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spector PS, Curran ME, Zou A, Keating MT, Sanguinetti MC. J Gen Physiol. 1996;107(5):611–619. doi: 10.1085/jgp.107.5.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang S, Liu S, Morales MJ, Strauss HC, Rasmusson RL. J Physiol. 1997;502(Pt 1):45–60. doi: 10.1111/j.1469-7793.1997.045bl.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith PL, Baukrowitz T, Yellen G. Nature. 1996;379(6568):833–836. doi: 10.1038/379833a0. [DOI] [PubMed] [Google Scholar]
- 22.Hille B. Ion channels of excitable membranes. 3. Sunderland: Sinauer Associates; 2001. [Google Scholar]
- 23.Piper DR, Hinz WA, Tallurri CK, Sanguinetti MC, Tristani-Firouzi M. J Biol Chem. 2005;280(8):7206–7217. doi: 10.1074/jbc.M411042200. [DOI] [PubMed] [Google Scholar]
- 24.Smith PL, Yellen G. J Gen Physiol. 2002;119(3):275–293. doi: 10.1085/jgp.20028534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang M, Liu J, Tseng GN. J Gen Physiol. 2004;124(6):703–718. doi: 10.1085/jgp.200409119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Doyle DA, Morais Cabral J, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. Science. 1998;280(5360):69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 27.Liu Y, Holmgren M, Jurman ME, Yellen G. Neuron. 1997;19(1):175–184. doi: 10.1016/s0896-6273(00)80357-8. [DOI] [PubMed] [Google Scholar]
- 28.Sanguinetti MC, Xu QP. J Physiol. 1999;514(Pt 3):667–675. doi: 10.1111/j.1469-7793.1999.667ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tristani-Firouzi M, Chen J, Sanguinetti MC. J Biol Chem. 2002;277(21):18994–19000. doi: 10.1074/jbc.M200410200. [DOI] [PubMed] [Google Scholar]
- 30.Ferrer T, Rupp J, Piper D, Tristani-Firouzi M. J Biol Chem. 2006;281(18):12858–12864. doi: 10.1074/jbc.M513518200. [DOI] [PubMed] [Google Scholar]
- 31.Van Slyke AC, Rezazadeh S, Snopkowski M, Shi P, Allard CR, Claydon TW. Biophys J. 2010;99(9):2841–2852. doi: 10.1016/j.bpj.2010.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Herzberg IM, Trudeau MC, Robertson GA. J Physiol. 1998;511(Pt 1):3–14. doi: 10.1111/j.1469-7793.1998.003bi.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hoshi T, Zagotta WN, Aldrich RW. Neuron. 1991;7(4):547–556. doi: 10.1016/0896-6273(91)90367-9. [DOI] [PubMed] [Google Scholar]
- 34.Gustina AS, Trudeau MC. Proc Natl Acad Sci U S A. 2009;106(31):13082–13087. doi: 10.1073/pnas.0900180106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen J, Zou A, Splawski I, Keating MT, Sanguinetti MC. J Biol Chem. 1999;274(15):10113–10118. doi: 10.1074/jbc.274.15.10113. [DOI] [PubMed] [Google Scholar]
- 36.Napolitano C, Priori SG, Schwartz PJ, Bloise R, Ronchetti E, Nastoli J, Bottelli G, Cerrone M, Leonardi S. JAMA. 2005;294(23):2975–2980. doi: 10.1001/jama.294.23.2975. [DOI] [PubMed] [Google Scholar]
- 37.Splawski I, Shen J, Timothy KW, Lehmann MH, Priori S, Robinson JL, Moss AJ, Schwartz PJ, Towbin JA, Vincent GM, Keating MT. Circulation. 2000;102(10):1178–1185. doi: 10.1161/01.cir.102.10.1178. [DOI] [PubMed] [Google Scholar]
- 38.Tester DJ, Will ML, Haglund CM, Ackerman MJ. Heart Rhythm. 2005;2(5):507–517. doi: 10.1016/j.hrthm.2005.01.020. [DOI] [PubMed] [Google Scholar]
- 39.Paulussen A, Raes A, Matthijs G, Snyders DJ, Cohen N, Aerssens J. J Biol Chem. 2002;277(50):48610–48616. doi: 10.1074/jbc.M206569200. [DOI] [PubMed] [Google Scholar]
- 40.Lupoglazoff JM, Denjoy I, Berthet M, Neyroud N, Demay L, Richard P, Hainque B, Vaksmann G, Klug D, Leenhardt A, Maillard G, Coumel P, Guicheney P. Circulation. 2001;103(8):1095–1101. doi: 10.1161/01.cir.103.8.1095. [DOI] [PubMed] [Google Scholar]
- 41.Shushi L, Kerem B, Goldmit M, Peretz A, Attali B, Medina A, Towbin JA, Kurokawa J, Kass RS, Benhorin J. Ann Noninvasive Electrocardiol. 2005;10(3):334–341. doi: 10.1111/j.1542-474X.2005.00643.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hayashi K, Shimizu M, Ino H, Yamaguchi M, Terai H, Hoshi N, Higashida H, Terashima N, Uno Y, Kanaya H, Mabuchi H. Clin Sci (Lond) 2004;107(2):175–182. doi: 10.1042/CS20030351. [DOI] [PubMed] [Google Scholar]
- 43.Van Langen IM, Birnie E, Alders M, Jongbloed RJ, Le Marec H, Wilde AA. J Med Genet. 2003;40(2):141–145. doi: 10.1136/jmg.40.2.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Larsen LA, Andersen PS, Kanters J, Svendsen IH, Jacobsen JR, Vuust J, Wettrell G, Tranebjaerg L, Bathen J, Christiansen M. Clin Chem. 2001;47(8):1390–1395. [PubMed] [Google Scholar]
- 45.Rossenbacker T, Mubagwa K, Jongbloed RJ, Vereecke J, Devriendt K, Gewillig M, Carmeliet E, Collen D, Heidbuchel H, Carmeliet P. Circulation. 2005;111(8):961–968. doi: 10.1161/01.CIR.0000156327.35255.D8. [DOI] [PubMed] [Google Scholar]
- 46.Millat G, Chevalier P, Restier-Miron L, Da Costa A, Bouvagnet P, Kugener B, Fayol L, Gonzalez Armengod C, Oddou B, Chanavat V, Froidefond E, Perraudin R, Rousson R, Rodriguez-Lafrasse C. Clin Genet. 2006;70(3):214–227. doi: 10.1111/j.1399-0004.2006.00671.x. [DOI] [PubMed] [Google Scholar]
- 47.Jongbloed R, Marcelis C, Velter C, Doevendans P, Geraedts J, Smeets H. Hum Mutat. 2002;20(5):382–391. doi: 10.1002/humu.10131. [DOI] [PubMed] [Google Scholar]
- 48.Gianulis EC, Trudeau MC. J Biol Chem. 2011;286(25):22160–22169. doi: 10.1074/jbc.M110.205948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li X, Xu J, Li M. J Biol Chem. 1997;272(2):705–708. doi: 10.1074/jbc.272.2.705. [DOI] [PubMed] [Google Scholar]
- 50.Li Q, Gayen S, Chen AS, Huang Q, Raida M, Kang C. Biochem Biophys Res Commun. 2010;403(1):126–132. doi: 10.1016/j.bbrc.2010.10.132. [DOI] [PubMed] [Google Scholar]
- 51.Muskett FW, Thouta S, Thomson SJ, Bowen A, Stansfeld PJ, Mitcheson JS. J Biol Chem. 2010 doi: 10.1074/jbc.M110.199364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ng CA, Hunter MJ, Perry MD, Mobli M, Ke Y, Kuchel PW, King GF, Stock D, Vandenberg JI. PLoS One. 2011;6(1):e16191. doi: 10.1371/journal.pone.0016191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang J, Trudeau MC, Zappia AM, Robertson GA. J Gen Physiol. 1998;112(5):637–647. doi: 10.1085/jgp.112.5.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schonherr R, Heinemann SH. J Physiol. 1996;493(Pt 3):635–642. doi: 10.1113/jphysiol.1996.sp021410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.de la Pena P, Alonso-Ron C, Machin A, Fernandez-Trillo J, Carretero L, Dominguez P, Barros F. J Biol Chem. 2011;286(21):19065–19075. doi: 10.1074/jbc.M111.238899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.van der Horst MA, van Stokkum IH, Crielaard W, Hellingwerf KJ. FEBS Lett. 2001;497(1):26–30. doi: 10.1016/s0014-5793(01)02427-9. [DOI] [PubMed] [Google Scholar]
- 57.Vreede J, van der Horst MA, Hellingwerf KJ, Crielaard W, van Aalten DM. J Biol Chem. 2003;278(20):18434–18439. doi: 10.1074/jbc.M301701200. [DOI] [PubMed] [Google Scholar]
- 58.Viloria CG, Barros F, Giraldez T, Gomez-Varela D, de la Pena P. Biophys J. 2000;79(1):231–246. doi: 10.1016/S0006-3495(00)76286-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Berecki G, Zegers JG, Verkerk AO, Bhuiyan ZA, de Jonge B, Veldkamp MW, Wilders R, van Ginneken AC. Biophys J. 2005;88(1):566–578. doi: 10.1529/biophysj.104.047290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gustina AS, Trudeau MC. J Gen Physiol. 2011;137(3):315–325. doi: 10.1085/jgp.201010582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Miranda P, Manso DG, Barros F, Carretero L, Hughes TE, Alonso-Ron C, Dominguez P, de la Pena P. Biochim Biophys Acta. 2008;1783(10):1681–1699. doi: 10.1016/j.bbamcr.2008.06.009. [DOI] [PubMed] [Google Scholar]
- 62.Al-Owais M, Bracey K, Wray D. Eur Biophys J. 2009;38(5):569–576. doi: 10.1007/s00249-009-0408-2. [DOI] [PubMed] [Google Scholar]
- 63.Clancy CE, Rudy Y. Nature. 1999;400(6744):566–569. doi: 10.1038/23034. [DOI] [PubMed] [Google Scholar]