

Abstract
Ferritin is an iron-sequestering protein that is generally cytoplasmic; however, our previous studies have shown that in avian corneal epithelial (CE) cells ferritin is nuclear. We have also observed that this nuclear localization involves a tissue-specific nuclear transporter that we have termed ferritoid, and that nuclear ferritin protects DNA from oxidative damage. Recently we have determined that ferritoid functions not only as a nuclear transporter, but also, within the nucleus, it remains associated with ferritin as a heteropolymeric complex. This ferritoid–ferritin complex has unique properties such as being half the size of a typical ferritin molecule and showing preferential binding to DNA. It is likely that the association between ferritoid and ferritin is involved both in the nuclear transport of ferritin and in determining certain of the properties of the complex; therefore, we have been examining the mechanisms involved in regulating the association of these two components. As the ferritoid sequence contains six putative phosphorylation sites, we have examined here whether phosphorylation is one such mechanism. We have determined that ferritoid in the nuclear ferritoid–ferritin complexes is phosphorylated, and that inhibition of this phosphorylation, using inhibitors of PKC, prevents its interaction with ferritin. Furthermore, in an experimental model system in which the nuclear transport of ferritin normally occurs (i.e., the co-transfection of COS-1 cells with full length constructs for ferritin and ferritoid), when phosphorylation sites in ferritoid are mutated, the interaction between ferritoid and ferritin is inhibited, as is the nuclear transport of ferritin
Keywords: FERRITOID, FERRITIN, PHOSPHORYLATION, CORNEA, NUCLEUS
Iron is vital for virtually all life, serving, for example, as a component of many enzymes and oxygen carrier proteins. However, free iron can also produce oxidative damage to cellular components, including DNA. This results from its ability to catalyze, through the Fenton Reaction, the conversion of hydrogen peroxide (H2O2) to the hydroxyl radical (·OH) which is the most energetic reactive oxygen species (ROS). Therefore, the intracellular concentration of iron must be tightly regulated [Stohs and Bagchi, 1995].
Ferritin is a multimeric iron sequestration molecule—composed of 24 ferritin subunits—that is capable of storing up to 4,500 atoms of iron as a core [Ford et al., 1984]. This action of ferritin keeps the level of free iron low, while maintaining it in a readily available form [Cazzola et al., 1990; Harrison and Arosio, 1996]. The importance of ferritin in iron homeostasis is emphasized by the observation that knock-outs of the ferritin gene in mice result in embryonic lethality that occurs prior to gastrulation [Ferreira et al., 2000].
In most cell types ferritin is a cytoplasmic molecule composed of two different types of ferritin subunits, termed heavy (H), and light (L) chains; however, previous studies in our laboratory have shown that in avian corneal epithelial (CE) cells, the ferritin molecule is largely nuclear and contains only H-ferritin [Cai et al., 1997; Beazley et al., 2008]. In this site ferritin protects DNA from damage by ROS, such as that induced by UV-radiation [for review see Linsenmayer et al., 2005], and by H2O2 [Cai et al., 2008].
In CE cells, the translocation of ferritin into the nucleus is mediated by a tissue-specific nuclear transporter that we have termed ferritoid for its similarities to ferritin. Structurally the ferritoid monomer has several domains, the largest being similar to the ferritin H chain (Fig. 1A), by both sequence analysis and molecular modeling [Millholland et al., 2003]. Sequence analyses also predict at least two additional domains. One is an N-terminal SV40-type nuclear localization sequence (NLS) that contains two consensus phosphorylation sites (one serine and one threonine), and which functional analyses (employing transfections with deletion constructs) showed to be necessary for nuclear transport. The other is a C-terminal tail, 78 amino acids long, that contains four consensus serine phosphorylation sites [Millholland et al., 2003].
Fig. 1.
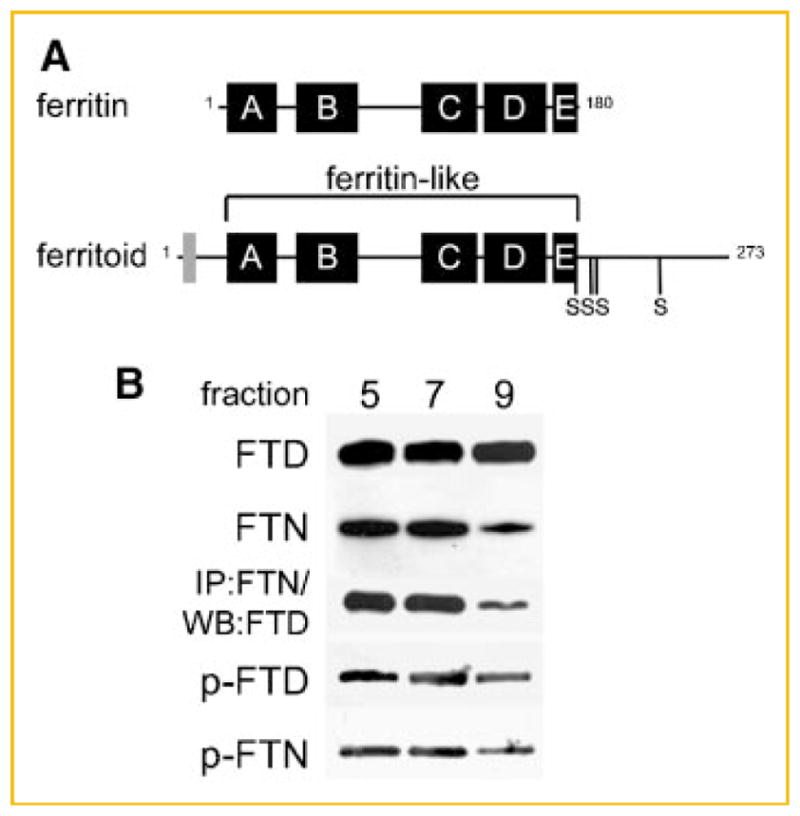
Analysis of phosphorylation in ferritoid–ferritin complexes. A: Schematic representation of ferritin and ferritoid sequences showing the 5 helical domains, A–E. In addition, the ferritoid sequence contains an SV40-type NLS (gray box) and a C-terminal tail containing four consensus phosphorylation sites at serines 186, 208, 212, and 240. B: Ferritinferritoid enriched protein lysate from E17 CE tissue, separated by gel-filtration chromatography on a Superdex-200 column, followed by SDS–PAGE and Western blot for ferritoid (FTD), ferritin (FTN), and phosphorylation of ferritoid and ferritin (p-FTD and p-FTN, respectively), or by immunoprecipitation with the anti-ferritin antibody followed by Western blot for ferritoid (IP:FTN/WB:FTD).
An additional property of ferritoid that we have recently observed is that its association with ferritin is not simply a transient one—existing only during the time that nuclear transport is occurring. Instead, the data suggest that following nuclear transport ferritoid remains associated with ferritin in a stable, high molecular weight ferritoid–ferritin complex. This nuclear ferritoid–ferritin complex, in addition, exhibits several unique properties, including: (1) a molecular weight of ~260 kDa—which is approximately half the size of a typical cytoplasmic ferritin—and (2) the ability to bind to DNA [Nurminskaya et al., 2009]. The molecular weight of the nuclear ferritoid–ferritin complex of CE cells suggests that it is comprised of 12 subunits, rather than the 24 subunits of all other vertebrate ferritins. For a vertebrate ferritin, this type of structure is unique; however, in bacteria there is precedence for such structures in the DNA binding proteins of starvation (Dps) [Zhao et al., 2002]. The bacterial Dps are a family of dodecomeric, ferritin-like proteins that protect DNA from damage, both through binding to DNA and sequestering iron. Thus they function in a manner analogous to that which we have suggested for the nuclear ferritoid–ferritin complex(es) of CE cells.
For the ferritoid–ferritin complex(es) to function in the protection of nuclear DNA, ferritoid and ferritin must associate with one another and undergo nuclear transport. Although all available evidence implicates the ferritoid component of this complex in the nuclear transport, a recent observation suggests that the presence of ferritoid, per se, is not sufficient for nuclear transport to occur. By immunofluorescence analysis of developing corneas, the youngest, least differentiated CE cells that contain both ferritoid and ferritin show both to be in a cytoplasmic location; but as differentiation progresses, both components become largely nuclear [Beazley et al., 2008].
As the nuclear localization of the 260 kDa ferritoid–ferritin complex(es) is critical for its proposed function in protecting the nuclear DNA from oxidative damage, we undertook the current study to determine further the mechanisms involved in regulating the association of ferritoid with ferritin, and their subsequent nuclear transport. We focused on phosphorylation as ferritoid contains consensus sites for phosphorylation (described above) and this modification has been shown to affect a wide variety of cellular processes, including protein–protein interactions and subcellular localization [Sprang et al., 1988; Hurley et al., 1989; Schmitz et al., 1991]. Here we show that in the nuclear ferritoid–ferritin complex(es) of avian CE cells both the ferritoid and ferritin are phosphorylated, and that inhibition of this phosphorylation by kinase inhibitors, or mutagenesis of ferritoid phosphorylation sites, prevents formation of the ferritoid–ferritin complex(es). Furthermore, when ferritoid phosphorylation sites are mutated, ferritin fails to undergo nuclear translocation. These results, when taken together, suggest that phosphorylation regulates the nuclear transport of ferritoid–ferritin complexes, and that it is the phosphorylation of the ferritoid component that is required for this to occur.
MATERIALS AND METHODS
WHOLE CORNEA ORGAN CULTURE
Chicken eggs (white leghorn) were obtained from Hyline (Elizabethtown, PA) and incubated at 38°C. Embryos were removed, rinsed in Hank’s balanced saline solution (Invitrogen), and staged both by chronological time of incubation and by the criteria of Hamburger and Hamilton [1951].
Whole cornea organ cultures were performed as described previously [Beazley et al., 2008]. For phosphorylation studies, the following kinase inhibitors (EMD Biosciences) were added to the culture medium either at their reported IC50 for cultured cells or, if IC50 values were nor reported, at their Ki (at a minimum of 1 μM): herbimycin A (10 μM), PD153035 (1 μM), PD98059 (10 μM), SB203580 (1 μM), TBB (1 μM), KT5720 (1 μM), H-89, dihydrochloride (30 μM), bisindolylmaleimide I (10 nM), H-89, or dihydrochloride (15 μM), and bisindolylmaleimide I (5 nM) together.
ISOLATION OF CE TISSUE
Whole corneas from E17 embryos or from organ cultures were rinsed in Hank’s balanced saline solution and the corneal epithelium was separated from the stroma by treatment with 0.5% Dispase in PBS at 4°C (2 h for E17 corneas or 30 min for organ culture corneas). Epithelia were rinsed with 50 mM HEPES buffer, pH 7.4 and pelleted by centrifugation (2,500 rpm for 5 min).
CELL CULTURE, PLASMID CONSTRUCTION, AND TRANSFECTION
The full-length ferritoid construct and the ferritoid construct in which the NLS was deleted (NLS−) were generated as previously described [Millholland et al., 2003]. An expression construct for full-length ferritoid in which four C-terminal serine residues were mutated to non-phosphorylatable glycine residues (FTD-Pmut) was generated from the full-length V5-tagged ferritoid construct [Millholland et al., 2003] by consecutive rounds of PCR using the QuikChange II site-directed mutagenesis kit (Stratagene). The primers used for mutagenesis were: 5′-GTTCCCAAGATGGCAGAAATGGTCTTGGGGATTACTTTATGG-3′ and 5′-CCATAAAGTAATCCCCAAGACCATTTCTGCCATCTTGGGAAC-3′ for S186 → G; 5′-GGCATAGAGCCAGAGTCTGGTCAGCAGTGCAGCCC-3′ and 5′-GGGCTGCACTGCTGACCAGACTCTGGCTCTATGCC-3′ for S208 → G; 5′-GTCTAGTCAGCAGTGCGGCCCTTGTCCACCTC-3′ and 5′-GAGGTGGACAAGGGCCGCACTGCTGACTAGAC-3′ for S212 → G; and 5′-CCCAGCACAGGAACGGCATAGGGCC-3′ and 5′-GGCCCTATGCCGTTCCTGTGCTGGG-3′ for S240 → G. The plasmid inserts were confirmed by sequencing, and plasmid preparations were made using the Plasmid Maxi Prep kit (Qiagen).
COS-1 cells were cultured as previously described [Millholland et al., 2003]. COS-1 cells were co-transfected [using FuGENE 6 (Roche Biochemicals)] with either the full-length wild-type ferritoid construct (FTD-wt) or the mutant ferritoid construct (FTD-Pmut), and with a c-Myc-tagged construct for full-length chicken H-ferritin [Cai et al., 1997]. For COS-1 cells cultured in four-chamber slides (BD Biosciences; for immunofluorescence), 1.0 μg of ferritoid plasmid plus 0.5 μg of ferritin plasmid were transfected per chamber; for COS-1 cells cultured in 60 mm plates (for Western blot), 4.0 μg of ferritoid plasmid plus 2.0 μg of ferritin plasmid were used. Forty-eight hours after transfection, the cells were fixed and processed for immunofluorescence or harvested for Western blot analysis.
IMMUNOFLUORESCENCE ANALYSIS
Cultured corneas were removed, fixed in 4% paraformaldehyde (20 min on ice), and rinsed with PBS. They were then cryoprotected in 8% sucrose (30 min at 4°C) and embedded in Tissue Tek OCT. Frozen sections (10 mm) were cut using a cryostat (Microm), and were mounted on 12-spot slides (ThermoShandon Scientific) coated with BIOBOND (Electron Microscopy Sciences).
Transfected COS-1 cells were fixed in 4% paraformaldehyde (10 min on ice), rinsed with PBS, and permeabilized (100% methanol for 10 min at −20°C).
Indirect immunofluorescence was performed as described previously [Cai et al., 1997]. The primary antibodies used were: a monoclonal antibody against chicken ferritin (6D11) [Zak and Linsenmayer, 1983], a monoclonal antibody against c-myc epitope tag (9E10) [Evan et al., 1985], and a monoclonal antibody against V5 epitope tag (Invitrogen). Samples were incubated in primary antibodies overnight at 4°C. The samples were then washed thoroughly with PBS and incubated with rhodamine-conjugated goat anti-mouse IgG1, alone or together with FITC-conjugated goat anti-mouse IgG2a (Pierce) secondary antibody for 45–90 min at room temperature. Samples were mounted in 95% glycerol in PBS containing Hoechst 33258 to stain nuclei (100 ng/mL; Sigma) and visualized by conventional immunofluorescence using a Nikon Fluophot microscope, equipped with a SPOT RT real time CCD camera (Diagnostic Instruments, Inc.).
PREPARATION OF PROTEIN LYSATES AND GEL FILTRATION CHROMATOGRAPHY
Total protein lysates and protein lysates enriched for ferritoid–ferritin complexes were prepared as described previously [Beazley et al., 2008]. For phosphorylation studies, HALT phosphatase inhibitor cocktail (Pierce) was included during lysate preparation. Protein lysates enriched for ferritoid and ferritin were fractionated by gel filtration chromatography on a Superdex 200 HR 10/30 column (Pharmacia) in 140 mM NaCl in phosphate buffer, pH 7.4, 0.02% Triton X-100 at 4°C. Absorbance of the eluate was monitored at 280 nm while 0.5 mL fractions were collected. The molecular weight of eluted proteins was determined from standard curves generated using Sigma Gel Filtration Calibration Standards.
IMMUNOPRECIPITATION AND WESTERN BLOT
Immunoprecipitations were performed as described previously [Beazley et al., 2008]. The antibodies used for immunoprecipitation were purified hybridoma 6D11, anti-phospho-serine (Qiagen), or anti-ferritoid IgY. Following immunoprecipitation, samples were heated (95°C, 5 min) to denature proteins, spun briefly, and electrophoresed (12% SDS–PAGE; BioRad).
Proteins separated by SDS–PAGE were transferred to polyvinylidene difluoride (PVDF) membranes (BioRad). For antibodies against ferritoid or ferritin, Western blots were performed as previously described [Beazley et al., 2008].
For the antibody directed against phospho-serine residues, Western blots were performed using the following protocol: PVDF membranes were treated with blocking solution [5% bovine serum albumin (Sigma) in TBST (TBS containing 0.1% Tween-20)] for 1 h at room temperature with shaking. The phospho-serine primary antibody (Qiagen) was diluted in blocking solution (1:200), and incubated overnight at 4°C with shaking. Membranes were then washed (4 × 10 min) with TBST to remove unbound antibody. Secondary antibody [HRP-conjugated rabbit anti-mouse IgG + IgM (Pierce) at] was diluted in TBST 1:10,000, placed onto membranes for 1 h at room temperature and membranes were washed again in TBST as above.
All Western blots were incubated in chemiluminescent substrate (Denville Scientific) for 5 min prior to exposure to photographic film. Western blots bands were quantified by densitometry using Scion Image software.
KINASE ACTIVITY ASSAY
PKC activity was analyzed using an ELISA-based kinase assay kit (EMD Biosciences). Total protein lysates were incubated with assay buffer and co-factors (25 mM Tris–HCl, 5 mM β-mercaptoethanol, 3 mM MgCl2, 2 mM CaCl2, 1 mM EGTA, 0.5 mM EDTA, 0.1 mM ATP, 50 μg/ml phosphatidylserine, pH 7.0) and added to individual wells of a 96-well plate pre-coated with a PKC peptide pseudosubstrate for 20 min at room temperature. Phosphorylation of the peptide was detected by colorimetric reaction. The absorbance was read at 492 nm in a microplate reader (BioRad). For samples cultured in the presence of specific kinase inhibitors, these inhibitors were included during the assay at the same concentrations used in culture. Assays were repeated in triplicate.
RESULTS
ANALYSIS OF FERRITOID AND FERRITIN PHOSPHORYLATION IN THE CE
To determine the role of phosphorylation in the ferritin–ferritoid interaction, we first examined whether the ferritoid and ferritin in the complex(es) from CE cells are phosphorylated, and, if so, ones of which molecular weight(s). For this we analyzed protein lysates of CE tissue from E17 embryos—a time when both ferritin and ferritoid are being maximally synthesized [Cai et al., 1997; Millholland et al., 2003] and are present together in supramolecular complexes [Nurminskaya et al., 2009]. Before analysis, the supernatants of the lysates were enriched for ferritoid–ferritin complexes—which are highly thermo-stable—by heating to 70°C for 10 min followed by rapid cooling and then centrifugation to remove the denatured proteins [Beazley et al., 2008]. The subsequent analyses of these ferritoid–ferritin enriched fractions employed a combination of gel filtration chromatography, immunoprecipitation, and Western blot.
For molecular weight separation, the heat-enriched lysates were fractionated by gel-filtration chromatography on a Superdex-200 column, which was capable of separating material with molecular weights ranging from 600 kDa to 29 kDa (collected as 18 fractions). Each fraction was then analyzed by Western blot for ferritoid (FTD) and ferritin (FTN) which showed most of both components eluting in fractions 5–9 (Fig. 1B) with a median molecular weight of 260 kDa, which is approximately half that of a typical ferritin. This corresponds to our previous results showing that the major ferritoid–ferritin complex present in CE cells is nuclear and has a molecular weight of 260 kDa [Nurminskaya et al., 2009].
To confirm that the ferritoid and ferritin in these fractions were co-assembled within the same supramolecular complex(es), we performed immunoprecipitations of these column fractions with the anti-ferritin antibody followed by Western blot of the precipitates for ferritoid (Fig. 1B, IP:FTN/WB:FTD). Consistent with our previous results using total CE cell lysates [Beazley et al., 2008], the ferritoid and ferritin in these fractions co-immunoprecipitate with one another.
To determine whether, in these complexes, the ferritoid and/or ferritin are phosphorylated, we performed Western blot on these fractions using an antibody against phosphorylated serine residues. The results show that both ferritoid, identified as the 30 kDa band (Fig. 1B, p-FTD), and ferritin, identified as the 19 kDa band (Fig. 1B, p-FTN), are phosphorylated. Furthermore, phosphorylation is not seen in the lower molecular weight fractions corresponding to ferritoid and ferritin monomers and homodimers (data not shown). Taken together, these results show a correlation between phosphorylation of ferritoid and the formation of the 260 kDa nuclear ferritoid–ferritin complex(es).
To confirm phosphorylation of ferritoid in vivo, we employed tandem mass spectrometry [Mann et al., 2002] on a band corresponding to that for ferritoid in Figure 1B (cut from a corresponding, coomassie-stained gel). The results show serine 208 is phosphorylated. That this residue lies within the 78 amino acid C-terminal tail of ferritoid is consistent with the possibility that it sterically influences the ability of ferritoid to participate in supramolecular assembly by altering its conformation (see Discussion Section).
INHIBITION OF FERRITIN NUCLEAR TRANSPORT AND THE FERRITOID–FERRITIN INTERACTION BY MUTATION OF FERRITOID PHOSPHORYLATION SITES
The ferritoid sequence contains six consensus phosphorylation sites—two located in the N-terminus and four in the C-terminus. Previous studies in our laboratory showed that when COS-1 cells are co-transfected with full-length constructs for ferritoid and H-ferritin, nuclear transport of both occurs. However, in co-transfections where the construct for ferritoid lacks the N-terminal NLS (NLS−), neither protein undergoes nuclear translocation, demonstrating that the NLS of ferritoid is necessary for nuclear transport to occur [Millholland et al., 2003].
As the ferritoid construct without the NLS also lacks the two N-terminal phosphorylation sites, we used this construct to determine whether these sites were necessary for the interaction of ferritoid with ferritin. For this we co-transfected COS-1 cells with constructs for full-length H-ferritin and for either wild-type ferritoid (Fig. 2A, NLS+) or the ferritoid construct lacking the N-terminal NLS (Fig. 2A, NLS−). Then, to determine whether the ferritoid and ferritin had interacted, lysates of the cells were immunoprecipitated with a ferritin antibody followed by Western blot for ferritoid. The results (Fig. 2A) showed that, even in the absence of the NLS region, ferritoid still interacts with ferritin, thus eliminating the involvement of this region and its putative phosphorylation sites in this interaction.
Fig. 2.

Analysis of the ferritoid–ferritin interaction and nuclear transport in COS-1 cells. A: COS-1 cells co-transfected with ferritin and either a wild-type ferritoid construct (NLS+) or a ferritoid construct lacking the NLS (NLS−), and analyzed by immunoprecipitation with ferritin antibody followed by Western blot for ferritoid (IP:FTN/WB:FTD). B: Schematic of a wild-type ferritoid construct (FTD-wt) and a full-length ferritoid construct containing point mutations in C-terminal serine residues 186, 208, 212, 240 (FTD-Pmut). C: Immunofluorescence micrographs of COS-1 cells co-transfected with a full-length myc-tagged ferritin construct and the V5-tagged construct for either wild-type ferritoid (FTD-wt) or ferritoid containing point mutations (FTD-Pmut) from (B). Ferritoid is shown in green, ferritin in red, and a merged image is shown to highlight subcellular distribution of the proteins. Scale bar in top right panel = 50 μm.
We then examined whether the four consensus phosphorylation sites in the C-terminal region of ferritoid (serine residues 186, 208, 212, and 240) are involved in ferritoid–ferritin complex formation and nuclear transport. For these analyses, we co-transfected COS-1 cells with a wild-type H-ferritin construct and a construct for ferritoid that was either wild-type (Fig. 2B, FTD-wt) or in which the four C-terminal serines were mutated to non-phosphorylatable glycines (Fig. 2B, FTD-Pmut).
First, to analyze whether the C-terminal sites in ferritoid are required for the nuclear transport of ferritin, we visualized the localization of both proteins by double-label immunofluorescence.
In the COS-1 cells co-transfected with ferritin and wild-type ferritoid, both proteins become co-localized to the nucleus (Fig. 2C, FTD-wt), as verified in the merged image. However, when ferritin is co-transfected along with the ferritoid construct with the mutated C-terminal phosphorylation sites (Fig. 2C, FTD-Pmut), only ferritoid is preferentially located within the nucleus, while ferritin is uniformly distributed throughout the cell (i.e. in both the nucleus and cytoplasm) as we have described previously [see Discussion Section and Millholland et al., 2003]. These results show that the mutated ferritoid is still capable of undergoing nuclear transport; however it is no longer able to transport ferritin along with it—as can be seen in the merged image—suggesting interference in its ability to associate with ferritin.
Then, to examine directly whether these C-terminal serine point mutations do block phosphorylation, and as such inhibit ferritoid–ferritin interactions, we analyzed, by co-immunoprecipitation, protein lysates of the co-transfected COS-1 cells (Fig. 3). First, we verified that both the wild-type ferritoid construct (wt) and the construct containing the point mutations (Pmut) had equal transfection efficiencies (as shown in Fig. 3A). Next, to examine the phosphorylation state of the wild-type ferritoid versus the mutated form, the cell lysates were immunoprecipitated with the anti-ferritoid antibody followed by Western blot for phospho-serine (Fig. 3B). Wild-type ferritoid (wt) is phosphorylated (demarcated with an asterisk), but the mutated form (Pmut) shows little if any phosphorylation. (In the lanes the higher molecular weight bands represent the IgY used for the immunoprecipitation.)
Fig. 3.

Analysis of the ferritoid–ferritin interaction in COS-1 cells following mutation of ferritoid phosphorylation sites. COS-1 cells co-transfected with ferritin and either the wild-type ferritoid (wt) or mutant ferritoid constructs (see Fig. 2B) analyzed for: (A) the presence of ferritin (FTN) and ferritoid (FTD) by Western blot; (B) the phosphorylation of ferritoid by immunoprecipitation with the anti-ferritoid antibody followed by Western blot for phospho-serine; or (C) ferritoid–ferritin binding by immunoprecipitation with the anti-ferritin antibody (IP:FTN) followed by Western blot for ferritoid (FTD) or ferritin (FTN). In (B) the ferritoid bands is demarcated by an asterisk. Higher molecular weight bands in (B) are due to reactivity of the phospho-serine antibody with the IgY antibody. In (C) the ferritin bands are demarcated by a double asterisk; higher molecular weight bands are due to reactivity of the secondary antibody with the IgG in the sample. A molecular weight marker is shown on the left of the figure with molecular weights for ferritoid (FTD) and ferritin (FTN) marked with labeled arrows.
To determine whether the mutated ferritoid is able to bind to ferritin, we performed immunoprecipitations with the anti-ferritin antibody on protein lysates of COS-1 cells co-transfected with ferritin, and either wild-type or mutated ferritoid, and then assayed the immunoprecipitates for ferritoid (FTD) or ferritin (FTN) by Western blot (Fig. 3C). The results show that the ferritoid and ferritin co-precipitated only when the ferritoid was wild-type (wt), and not when it had been mutated (Pmut). For these studies, as a positive control, the ferritin immunoprecipates were analyzed for ferritin itself (Fig. 3C, FTN); the ferritin (demarcated with a double asterisk) was detected equally whether the co-transfected ferritoid was wild-type or mutant. (In the FTN lanes, the additional heavy bands of reactivity represent the IgG from the immunoprecipitation and the secondary anti-IgG antibody used for the Western blot.) Taken together these data support the hypothesis that ferritin interacts only with phosphorylated ferritoid.
INHIBITION OF FERRITOID PHOSPHORYLATION AND ITS INTERACTION WITH FERRITIN BY INHIBITORS OF PKC ACTIVITY
The results from the mutational analyses above suggest that in transfected COS-1 cells the phosphorylation of ferritoid is necessary for its interaction with ferritin—which is required for its subsequent nuclear transport. To determine whether this reflects what actually occurs in CE cells, we examined the nuclear localization of ferritin in primary cultures of CE cells treated with a variety of specific kinase inhibitors (Table I). In initial studies we surveyed a panel of different inhibitors, which eliminated, as potential candidates, tyrosine kinases p60 and EGFR, p38 and p42 MAP kinases, casein kinase 2, and PKA—as inhibitors of these kinases had no effect on the nuclear localization of ferritin.
TABLE I.
Effect of Kinase Inhibitors on the Nuclear Localization of Ferritin in CE Cells
| Kinase inhibitor | Conc. | Kinase specificity | Ki or IC50 | Nuclear ferritin |
|---|---|---|---|---|
| Herbimycin A | 10 μM | p60 tyrosine kinase | 12 μM (IC50) | + |
| PD153035 | 1 μM | EGFR tyrosine kinase | 6 pM (Ki) | + |
| PD98059 | 10 μM | MEK (p42 MAPKK) | 2 μM (IC50) | + |
| SB203580 | 1 μM | p38 MAP kinase | 600 nM (IC50) | + |
| TBB | 1 μM | Casein kinase 2 | 900 nM (IC50) | + |
| KT5720 | 1 μM | PKA | 56 nM (Ki) | + |
| H-89 | 30 μM | PKA/PKC | 50 nM/32 μM (Ki) | − |
| Bisindolylmaleimide-I | 10 nM | PKC | 10 nM (IC50) | − |
However, the nuclear localization of ferritin was prevented by two other kinase inhibitors, bisindolylmaleimide-I and H-89. Bisindolylmaleimide-1 is a potent, selective inhibitor for PKC, with a median inhibition concentration (IC50) of approximately 10 nM [Toullec et al., 1991]. H-89, although predominantly an inhibitor of PKA [with a dissociation constant (Ki) of 50 nM], also inhibits PKC at a higher concentration (Ki for PKC = 32 μM) [Chijiwa et al., 1990; Hidaka and Kobayashi, 1992]. As the common activity of both of these inhibitors is against PKC, this suggests that PKC is the naturally occurring effector of ferritoid phosphorylation. That the other inhibitory activity of H-89—against PKA—is not involved in the phosphorylation of ferritoid is supported by the additional observation that another highly selective inhibitor of PKA (KT5720) does not prevent the nuclear localization of ferritin.
We next examined whether inhibiting the activity of PKC would inhibit the phosphorylation of ferritoid, and, if so, whether this inhibition would also alter the binding of ferritoid to ferritin. To examine these possibilities we employed an organ culture system using whole corneas from E8 embryos. E8 is 3 days before ferritoid and ferritin synthesis would be initiated in ovo; however, when this stage cornea is cultured in vitro the synthesis of both proteins is initiated precociously (i.e., within 24 h) [Beazley et al., 2008]. To inhibit phosphorylation by PKC, the E8 corneas were cultured in the presence of a combination of the two PKC inhibitors (bisindolylmaleimide-1 and H-89) that had demonstrated an effect on the nuclear localization of ferritin (Table I). In combination, we empirically found that each of these inhibitors could be used at a lower concentration, which improved cell viability (data not shown). After 48 h in culture CE tissue was harvested and examined both for ferritoid–ferritin binding and for the phosphorylation of ferritoid.
The relationships between the phosphorylation of ferritoid and its association with ferritin were determined by co-immunoprecipitations (Fig. 4A). Control corneas, cultured in the absence of the PKC inhibitors [Fig. 4A, PKCi (−)], demonstrated the presence of phospho-serine in ferritoid, as immunoprecipitation with an antibody against phospho-serine, followed by Western blot for ferritoid, showed a strong band for ferritoid (WB:FTD; IP:pSer).
Fig. 4.

Analysis of the ferritoid–ferritin interaction in CE protein lysates from E8 whole corneas cultured in the presence (+) or absence (−) of PKC inhibitors. A: Western blot for ferritoid (WB:FTD) following immunoprecipitation of protein lysates with the anti-phospho-serine (IP:pSer) or the anti-ferritin (IP:FTN) antibodies. B: Quantitation of the bands from (A). Density of ferritoid bands in the presence of kinase inhibitors are expressed as a percentage of the density of ferritoid bands in the absence of inhibitors. All band densities are normalized to ferritoid bands in the corresponding total lysates. C: Western blot for ferritoid following immunoprecipitation with the anti-ferritoid antibody (WB:FTD/IP:FTD) or Western blot for ferritin following immunoprecipitation with the anti-ferritin antibody (WB:FTN/IP:FTN). The ferritin bands in B are marked by an asterisk. Higher molecular weight bands are due to reactivity of the secondary antibody with the IgG in the sample. D: Western blots of total lysates for tubulin, ferritoid (FTD), and ferritin (FTN). In (A,C,D), molecular weight markers are shown on the left. E: Kinase activity assay for PKC in the presence of PKC inhibitors normalized to PKC activity in the absence of inhibitors. Colorimetric reaction assayed at 492 nm and run in triplicate.
Also, these control cultures [PKCi (−)] demonstrated that when phosphorylation proceeds normally, ferritoid–ferritin binding occurs—as determined by co-immunoprecipitation. In such cultures, when immunoprecipitation was performed with the anti-ferritin antibody, followed by Western blot for ferritoid, the ferritin immunoprecipates showed a strong band for ferritoid (Fig. 4A, WB:FTD; IP:FTN).
For the experimental corneas cultured in the presence of the inhibitors [Fig. 4A, PKCi (+)], the amount of ferritoid immunoprecipitated with the phospho-serine antibody (IP:pSer; WB:FTD) was reduced by an average of 57% compared to control (Fig. 4B, pSer). Consistent with an involvement of phosphorylation in the binding of ferritoid to ferritin, in these experimental cultures the amount of ferritoid that co-immunoprecipitated with ferritin (IP:FTN; WB:FTD) was also reduced—by an average of 86% compared to controls (Fig. 4B, FTN).
To eliminate the possibility that these observed decreases might be artifactual—resulting, for example, from the presence of the PKC inhibitors in the extracts interfering with the antibody reactions themselves—immunoprecipitations and Western were also performed in which the inhibitors were added directly to each antibody reaction mixture. Figure 4C shows that for both ferritoid (WB:FTD, IP:FTD) and ferritin (WB:FTN, IP:FTN) the immunoprecipitations are equally efficient whether performed in the presence (+) or absence (−) of the inhibitors. (In this figure, the ferritin band is demarcated by the asterisk, with the additional heavy bands of reactivity reflecting the presence of IgG from the immunoprecipitation reacting with the secondary, anti-IgG antibody used for the Western blot. These bands are not seen in the ferritoid immunoprecipitates as these employed streptavidin rather than a secondary antibody.)
That the overall production of ferritoid and ferritin is not appreciably affected by the kinase inhibition is evidenced by the observation that they were present at similar levels in the presence or absence of the inhibitors (Fig. 4D, FTD and FTN). Also, that equal amounts of protein were loaded is shown by Western blots of the lysates assayed for tubulin (Fig. 4D, tubulin).
Lastly, the relative inhibition of the PKC activity effected by the inhibitors was determined using an ELISA-based kinase activity assay (Fig. 4E). In the presence of the PKC inhibitors, the overall kinase activity is reduced by 30–40%, as compared to the controls. This level of inhibition is consistent with—and likely to be responsible for—the reduction of ferritoid phosphorylation being only partial (see Fig. 4A,B and Discussion Section).
DISCUSSION
In the present study we have examined the regulation of the nuclear translocation of ferritin in CE cells. Overall, the results show that phosphorylation of ferritoid plays a major role in the association between this nuclear transporter and its ferritin cargo, and if this phosphorylation event is interrupted, the requisite complex fails to form and the subsequent nuclear transport of ferritin fails to occur.
Our previous studies showed that ferritoid is the nuclear transporter for ferritin in the CE [Millholland et al., 2003]. The data presented here confirm this observation. In addition they demonstrate that the nuclear transport of ferritin requires an interaction with ferritoid and that ferritoid needs to be phosphorylated for this interaction to occur. Mutations of consensus serine phosphorylation sites in the C-terminal domain of ferritoid abrogate its interaction with ferritin and also inhibit the nuclear transport of ferritin.
Studies by others suggest that glycosylation of ferritin is important for regulating its nuclear localization [Surguladze et al., 2005]. However, we have observed that ferritin is more glycosylated in the liver—where it is cytoplasmic—than it is in the CE, indicating that glycosylation is unlikely to affect the nuclear localization of ferritin in these cells (unpublished observations).
The conclusion that the phosphorylation of ferritoid is required for nuclear transport is supported by two lines of evidence. One is that for CE tissue the 260 kDa ferritoid–ferritin complexes are phosphorylated, while the ferritoid and ferritin monomers are not, and it is these complexes that are located within the nuclear compartment [Nurminskaya et al., 2009]. The other is the demonstration that inhibition of ferritoid phosphorylation by two independent experimental methods (mutational analysis and chemical inhibition) prevents ferritoid–ferritin complex formation and nuclear localization.
Using a spectrum of specific kinase inhibitors on CE cells, we observed that only those that affect PKC activity prevented the nuclear translocation of ferritin. Furthermore, in corneas cultured in the presence of PKC inhibitors, both the phosphorylation of ferritoid and its interaction with ferritin are abrogated as compared to control cultures. Even a 30% decrease in PKC activity was sufficient to reduce the phosphorylation of ferritoid by half, and its interaction with ferritin by almost 90%. This suggests that PKC is the kinase responsible for regulating the interaction of these proteins. However, we cannot rule out the possibility that another kinase(s) contribute to this regulation in vivo.
One caveat in the experiments employing PKC inhibitors is that PKC may affect the phosphorylation of many proteins. Therefore, phosphorylated proteins other than ferritoid may be involved in regulating the nuclear transport of ferritin—or even in formation of the ferritoid–ferritin complex itself. For example, we observed that in the nuclear ferritoid–ferritin complex ferritin is also phosphorylated, and previous studies suggest that phosphorylation of ferritin can modulate its ability to interact with the chemokine receptor CXCR4—which promotes its nuclear translocation in HEK293 cells [Li et al., 2006]. However, in the COS-1 cells co-transfected with ferritin and the ferritoid construct in which the C-terminal serines were mutated, we demonstrate directly that phosphorylation of ferritoid is a critical regulator of binding to ferritin and its subsequent nuclear transport.
Here we observed that in COS-1 cells co-transfected with ferritin and the mutant ferritoid construct, ferritin is uniformly distributed throughout the cells while ferritoid is localized to the nucleus. We have previously observed that mutant ferritin constructs that cannot undergo supramolecular assembly can diffuse into the nucleus resulting in a uniform cellular distribution [Cai and Linsenmayer, 2001]. Therefore, it is likely that the uniform distribution seen here reflects the transfected ferritin being present in both a monomeric form, which can move into the nucleus, as well as in high-molecular weight complexes, which are restricted to the cytoplasm.
In the co-transfected COS-1 cells, the ferritin protein is unable to bind to the mutant, non-phosphorylated ferritoid. The most direct explanation for this is that it is caused by changes in ferritoid phosphorylation, but it is also possible that substituting serine residues with glycines in itself results in conformational changes in ferritoid that inhibit binding. We consider this unlikely, however, as similar effects on ferritoid phosphorylation and its interaction with ferritin are observed when phosphorylation is prevented by kinase inhibitors. Thus, these two different methods reinforce the proposed role for phosphorylation in regulating the interaction of ferritoid with ferritin.
The typical ferritin molecule is a multimeric protein complex formed by interactions between ferritin subunits [Ford et al., 1984; Lawson et al., 1991], and mutations that affect the conformation of the individual subunits prevent the assembly of this complex [Luzzago and Cesareni, 1989]. As phosphorylation has been shown to affect protein conformation [Sprang et al., 1988], it is likely that the phosphorylation of the ferritoid C-terminal tail domain induces a conformational change in ferritoid that is necessary for its interaction with ferritin.
Overall, the studies presented here, when taken together, suggest that in the CE the phosphorylation of ferritoid is involved in regulating the association between ferritoid and ferritin, thus affecting subsequent nuclear translocation of the complex.
Acknowledgments
Grant sponsor: National Institutes of Health; Grant number: EY13127.
This study was supported by the grant National Institutes of Health (EY13127 to T.F.L.).
References
- Beazley KE, Nurminskaya M, Talbot CJ, Linsenmayer TF. Corneal epithelial nuclear ferritin: Developmental regulation of ferritin and its nuclear transporter ferritoid. Dev Dyn. 2008;237:2529–2541. doi: 10.1002/dvdy.21691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai CX, Linsenmayer TF. Nuclear translocation of ferritin in corneal epithelial cells. J Cell Sci. 2001;114:2327–2334. doi: 10.1242/jcs.114.12.2327. [DOI] [PubMed] [Google Scholar]
- Cai CX, Birk DE, Linsenmayer TF. Ferritin is a developmentally-regulated nuclear protein of avian corneal epithelial cells. J Biol Chem. 1997;272:12831–12839. doi: 10.1074/jbc.272.19.12831. [DOI] [PubMed] [Google Scholar]
- Cai CX, Ching A, Lagace C, Linsenmayer T. Nuclear ferritin-mediated protection of corneal epithelial cells from oxidative damage to DNA. Dev Dyn. 2008;237:2676–2683. doi: 10.1002/dvdy.21494. [DOI] [PubMed] [Google Scholar]
- Cazzola M, Bergamaschi G, Dezza L, Arosio P. Manipulations of cellular iron metabolism for modulating normal and malignant cell proliferation: Achievements and prospects. Blood. 1990;75:1903–1919. [PubMed] [Google Scholar]
- Chijiwa T, Mishima A, Hagiwara M, Sano M, Hayashi K, Inoue T, Naito K, Toshioka T, Hidaka H. Inhibition of forskolin-induced neurite outgrowth and protein phosphorylation by a newly synthesized selective inhibitor of cyclic AMP-dependent protein kinase, N-[2-(p-bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide (H-89), of PC12D pheochromocytoma cells. J Biol Chem. 1990;265:5267–5272. [PubMed] [Google Scholar]
- Evan GI, Lewis GK, Ramsay G, Bishop JM. Isolation of a monoclonal antibody specific for human c-myc proto-oncogen product. Mol Cell Biol. 1985;5:3610–3616. doi: 10.1128/mcb.5.12.3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira C, Bucchini D, Martin ME, Levi S, Arosio P, Grandchamp B, Beaumont C. Early embryonic lethality of H ferritin gene deletion in mice. J Biol Chem. 2000;275:3021–3024. doi: 10.1074/jbc.275.5.3021. [DOI] [PubMed] [Google Scholar]
- Ford GC, Harrison PM, Rice DW, Smith JM, Treffry A, White JL, Yariv J. Ferritin: Design and formation of an iron-storage molecule. Philos Trans Roy Soc Lond Ser B Biol Sci. 1984;304:551–565. doi: 10.1098/rstb.1984.0046. [DOI] [PubMed] [Google Scholar]
- Hamburger V, Hamilton HL. A series of normal stages in the development of the chick embryo. J Morphol. 1951;88:49–92. [PubMed] [Google Scholar]
- Harrison PM, Arosio P. The ferritins: Molecular properties, iron storage function and cellular regulation. Biochim Biophys Acta. 1996;1275:161–203. doi: 10.1016/0005-2728(96)00022-9. [DOI] [PubMed] [Google Scholar]
- Hidaka H, Kobayashi R. Pharmacology of protein kinase inhibitors. Annu Rev Pharmacol Toxicol. 1992;32:377–397. doi: 10.1146/annurev.pa.32.040192.002113. [DOI] [PubMed] [Google Scholar]
- Hurley TR, Luo K, Sefton BM. Activators of protein kinase C induce dissociation of CD4, but not CD8, from p56lck. Science. 1989;245:407–409. doi: 10.1126/science.2787934. [DOI] [PubMed] [Google Scholar]
- Lawson DM, Artymiuk PJ, Yewdall SJ, Smith JMA, Livingstone JC, Treffry A, Luzzago A, Levi S, Arosio P, Cesareni G, Thomas CD, Shaw WV, Harrison PM. Solving the structure of human H ferritin by genetically engineering intermolecular crystal contacts. Nature. 1991;349:541–544. doi: 10.1038/349541a0. [DOI] [PubMed] [Google Scholar]
- Li R, Luo C, Mines M, Zhang J, Fan GH. Chemokine CXCL12 induces binding of ferritin heavy chain to the chemokine receptor CXCR4, alters CXCR4 signaling, and induces phosphorylation and nuclear translocation of ferritin heavy chain. J Biol Chem. 2006;281:37616–37627. doi: 10.1074/jbc.M607266200. [DOI] [PubMed] [Google Scholar]
- Linsenmayer TF, Cai CX, Millholland JM, Beazley KE, Fitch JM. Nuclear ferritin in corneal epithelial cells: Tissue-specific nuclear transport and protection from UV-damage. Prog Retin Eye Res. 2005;24:139–159. doi: 10.1016/j.preteyeres.2004.08.004. [DOI] [PubMed] [Google Scholar]
- Luzzago A, Cesareni G. Isolation of point mutations that affect the folding of the H chain of human ferritin in E. coli. EMBO J. 1989;8:569–576. doi: 10.1002/j.1460-2075.1989.tb03411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann M, Ong SE, Gronborg M, Steen H, Jensen ON, Pandey A. Analysis of protein phosphorylation using mass spectrometry: Deciphering the phosphoproteome. Trends Biotechnol. 2002;20:261–268. doi: 10.1016/s0167-7799(02)01944-3. [DOI] [PubMed] [Google Scholar]
- Millholland JM, Fitch JM, Cai CX, Gibney EP, Beazley KE, Linsenmayer TF. Ferritoid, a tissue-specific nuclear transport protein for ferritin in corneal epithelial cells. J Biol Chem. 2003;278:23963–23970. doi: 10.1074/jbc.M210050200. [DOI] [PubMed] [Google Scholar]
- Nurminskaya M, Talbot CJ, Nurminsky DI, Beazley KE, Linsenmayer TF. Nuclear ferritin: A ferritoidferritin complex in corneal epithelial cells. Invest Ophthalmol Vis Sci. 2009 doi: 10.1167/iovs.08-3170. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz ML, Henkel T, Baeuerle PA. Proteins controlling the nuclear uptake of NF-kappa B, Rel and dorsal. Trends Cell Biol. 1991;1:130–137. doi: 10.1016/0962-8924(91)90118-s. [DOI] [PubMed] [Google Scholar]
- Sprang SR, Acharya KR, Goldsmith EJ, Stuart DI, Varvill K, Fletterick RJ, Madsen NB, Johnson LN. Structural changes in glycogen phosphorylase induced by phosphorylation. Nature. 1988;336:215–221. doi: 10.1038/336215a0. [DOI] [PubMed] [Google Scholar]
- Stohs SJ, Bagchi D. Oxidative mechanisms in the toxicity of metal ions. Free Radical Biol Med. 1995;18:321–336. doi: 10.1016/0891-5849(94)00159-h. [DOI] [PubMed] [Google Scholar]
- Surguladze N, Patton S, Cozzi A, Fried MG, Connor JR. Characterization of nuclear ferritin and mechanism of translocation. Biochem J. 2005;388:731–740. doi: 10.1042/BJ20041853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toullec D, Pianetti P, Coste H, Bellevergue P, Grand-Perret T, Ajakane M, Baudet V, Boissin P, Boursier E, Loriolle F. The bisindolylmaleimide GF 109203X is a potent and selective inhibitor of protein kinase C. J Biol Chem. 1991;266:15771–15781. [PubMed] [Google Scholar]
- Zak NB, Linsenmayer TF. Monoclonal antibodies against developmentally regulated corneal antigens. Dev Biol. 1983;99:373–381. doi: 10.1016/0012-1606(83)90287-7. [DOI] [PubMed] [Google Scholar]
- Zhao GH, Ceci P, Ilari A, Giangiacomo L, Laue TM, Chiancone E, Chasteen ND. Iron and hydrogen peroxide detoxification properties of DNA-binding protein from starved cells—A ferritin-like DNA-binding protein of Escherichia coli. J Biol Chem. 2002;277:27689–27696. doi: 10.1074/jbc.M202094200. [DOI] [PubMed] [Google Scholar]