

Abstract
Endosomes are emerging as specialized signaling compartments that endow receptors with distinct signaling properties. The diversity of endosomal signaling pathways and their contribution to various biological responses is still unclear. CD158d is an endosome-resident, killer cell Ig-like receptor (KIR2DL4) in natural killer cells that stimulates release of a unique set of pro-inflammatory and pro-angiogenic mediators in response to soluble HLA-G. We identify here the CD158d signaling cascade. In response to soluble agonist antibody or soluble HLA-G, signaling by CD158d was dependent on activation of NF-κB and Akt. CD158d associated with DNA-PKcs, promoted Akt recruitment to endosomes, and induced DNA-PKcs-dependent Akt phosphorylation. The sequential requirement for DNA-PKcs, Akt, and NF-κB in signaling by receptor CD158d delineates a new endosomal signaling pathway for a pro-inflammatory response.
INTRODUCTION
The classical view on the mechanism of transmembrane receptor signaling involves ligand binding and activation at the plasma membrane (PM), which initiates downstream signaling events. Most signals are terminated rapidly by negative feedback or by receptor internalization. This view is being challenged, however, with evidence indicating that signaling can not only continue after endocytosis, but can also be amplified in endosomal compartments (1, 2). Furthermore, some receptors may even initiate signaling in endosomes in the absence of signaling at the PM. Sorting to endosomes provides both spatial and temporal regulation, as well as platforms for the assembly of signaling complexes that can be distinct from those at the PM, thereby permitting unique signaling outcomes. Moreover, signaling in endosomes is generally more sustained (minutes to hours) than the typically transient signaling at the PM (seconds to minutes). Thus, the residence of receptors in endosomes can affect the duration, the strength, and the outcome of the signals they generate (3).
Examples of receptors that signal sequentially at the plasma membrane and in endosomes following internalization include growth factor receptors, such as epidermal growth factor receptor (EGFR) and nerve growth factor receptor (TrkA) (4). In these well-studied examples, unique endosomal scaffolds and adaptors mediate sustained signaling via PI3K and MAPK pathways. Compartmentalization of signal transduction by such receptors on the cell surface or in endosomes determines the choice of physiological fate, such as cell proliferation or differentiation (5). In addition, some cells of the immune system express members of the Toll-like receptor (TLR) family, such as TLR3, TLR7, and TLR9, which signal in endosomes. Other TLR family members signal from the PM. Regardless of the location of TLRs, recognition of pathogen-associated molecular patterns (PAMP) by these receptors leads to the activation of MyD88- and TRIF-dependent pathways for NF-κB and MAPK activation, resulting in pro-inflammatory cytokine responses and production of Type I interferon (6).
CD158d represents a new class of receptor that resides in, and signals primarily from endosomes (7). CD158d is the KIR2DL4 member of the killer cell Ig-like receptor (KIR) family and is expressed in all natural killer (NK) cells and in some T cells. In contrast to other KIR family members, CD158d has both a cytosolic immunoreceptor tyrosine-based inhibition motif (ITIM) and a charged residue in the transmembrane region via which it can associate with the FcRγ chain (8). Several members of the KIR family bind to major histocompatibility complex (MHC) class I molecules and inhibit NK and T cell effector functions such as cytokine secretion and cytotoxicity (9). CD158d binds to the non-classical class I molecule HLA-G (10). Activation of human, resting NK cells by CD158d induces cytokine secretion, but not cytotoxicity (10, 11). CD158d, unlike other KIRs, resides in Rab5-positive early endosomes and signals from this intracellular location (7). Soluble ligand (soluble HLA-G or an Fab to CD158d) was endocytosed by CD158d and induced a unique pro-inflammatory and pro-angiogenic response, distinct from the response of resting NK cells to a cell surface activation receptor such as CD16 (7). The transcriptional response to CD158d signals was up-regulation of cytokines such as IL-1β, IL-6, TNF-α, and IL-23, and chemokines such as IL-8, MIP-3α, MIP-1δ, and MIP-1α. This response is independent of the association of CD158d with the FcR γ chain (7).
How CD158d signals from endosomes is not known. The physiological relevance of endosomal signaling by CD158d is highlighted by the restricted expression of its ligand, soluble HLA-G, by fetal trophoblast cells that invade the maternal decidua during early pregnancy (12). Thus, endocytosis of soluble HLA-G by CD158d on NK cells (7) at the implantation site may lead to sustained expression of an array of pro-inflammatory and proangiogenic factors, which may promote vascular remodeling. Such remodeling of the maternal vasculature, which occurs over the first twelve weeks of pregnancy, is essential to establish sufficient blood supply for the fetus (13).
Here we identify a new endosomal signaling pathway used by the receptor CD158d. Unlike PI3K- and MAPK-dependent pathways used by activation receptors at the cell surface, and by growth factor receptors or TLRs in endosomes, CD158d engagement initiates a distinct serine-threonine kinase cascade that links to NF-κB activation and subsequent cytokine and chemokine secretion.
RESULTS
Determinants of endosomal localization and signaling for CD158d
Previous work has shown that expression of CD158d in the human embryonic kidney cell line 293T cells results in proper targeting of CD158d to endosomes and in constitutive secretion of the chemokine IL-8 (7). The cytoplasmic tail of CD158d is required for signaling but not for endosomal targeting (7). Secretion of IL-8 by transfected 293T cells was used as readout for CD158d signaling. Mutant and chimeric versions of CD158d (Ref. 7, and this study) were tested for their endosomal localization and their ability to induce IL-8 secretion. 293T cells were transfected with CD158d, gp49B (an irrelevant ITIM-containing cell surface receptor, LILRB4), or chimeras of CD158d and gp49B (Fig. 1A). The chimeric receptors consisted of either the extracellular portion of CD158d fused to the transmembrane and cytoplasmic tail of gp49B (Fig. 1A), denoted CD158d/gp49B, or the reverse chimera gp49B/CD158d (7). As determined by confocal microscopy, gp49B/CD158d was expressed at the cell surface (7 and Fig. 1B), implying a role for the extracellular domain of CD158d in targeting to endosomes. As shown with CD158d/gp49B, the extracellular domain of CD158d was sufficient for endosomal targeting (Fig. 1B). CD158d/gp49B was targeted to the same endosomal compartment as wild-type CD158d (Fig. S1), as shown by colocalization of CD158d-gfp and HA-tagged CD158d/gp49B. Neither one of the chimeric receptors was able to induce IL-8 secretion in 293T cells; crosslinking gp49B/CD158d at the PM does not elicit signaling (7) and CD158d/gp49B is unable to signal in endosomes, as it lacks the cytoplasmic tail of CD158d, which is required for signaling (Fig. 1C). Thus, the extracellular domain of CD158d controls endosomal localization and the receptor signals from endosomes.
Fig. 1.
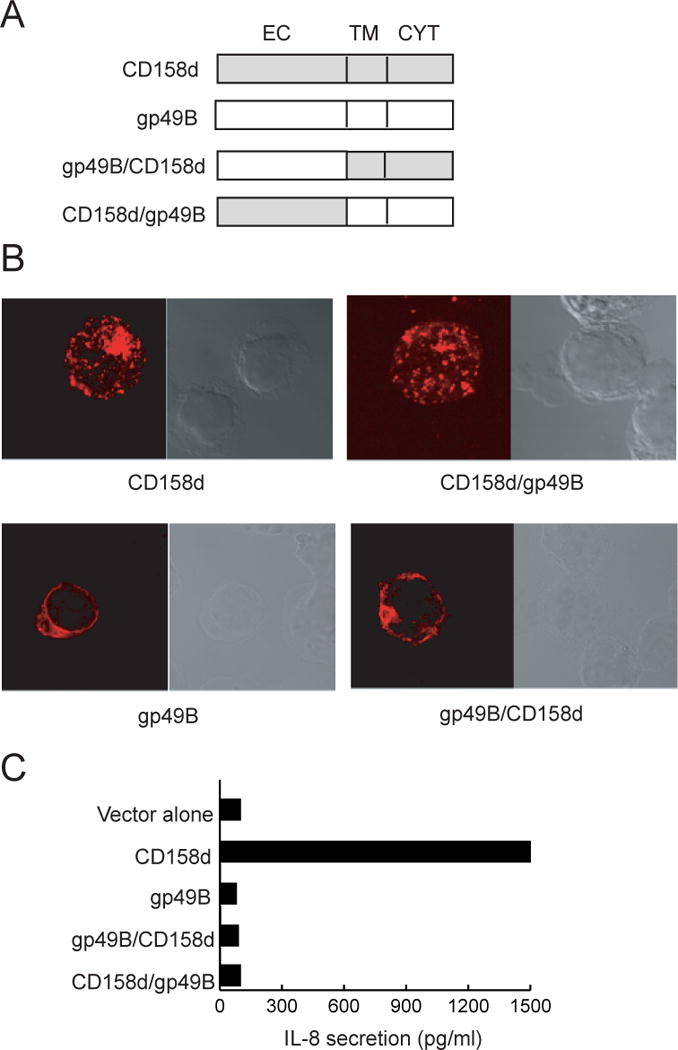
Determinants of endosomal localization and signaling by CD158d. (A) Schematic representation of CD158d, gp49B, and chimeric variants. Each construct carries an HA epitope tag at the N-terminus. EC: extracellular domain; TM: transmembrane region; CYT: cytoplasmic tail. (B) Receptor localization by confocal microscopy analysis. 293T cells transfected with the indicated constructs were stained with an anti-HA tag Ab after 48 h. (C) IL-8 secretion induced by the indicated plasmids in transfected 293T cells.
NF-κB activation is required for signaling by CD158d
Inflammatory responses are often triggered through NF-κB signaling (14). We therefore tested the involvement of NF-κB in endosomal signaling by CD158d. 293T cells were transfected with CD158d together with plasmids carrying reporters for promoter activation by AP-1, CRE, NFAT, and NF-κB. Forty-eight hours after transfection, promoter activity was determined by a firefly luciferase assay with cell lysates. An internal control with renilla luciferase was used to normalize the responses to the transfection efficiency. CD158d induced an NF-κB-dependent reporter activity comparable to that induced by TNF-α, a potent stimulus for NF-κB activation (Fig. 2A). No stimulation of the other 3 reporters by CD158d was observed, although each responded to a known stimulus (data not shown). The cytoplasmic tail of CD158d was required for NF-κB-dependent reporter activity, as both a cytoplasmic tail-truncated receptor (CD158d-TR) and CD158d/gp49B failed to activate NF-κB reporter activity (Fig. 2B).
Fig. 2.
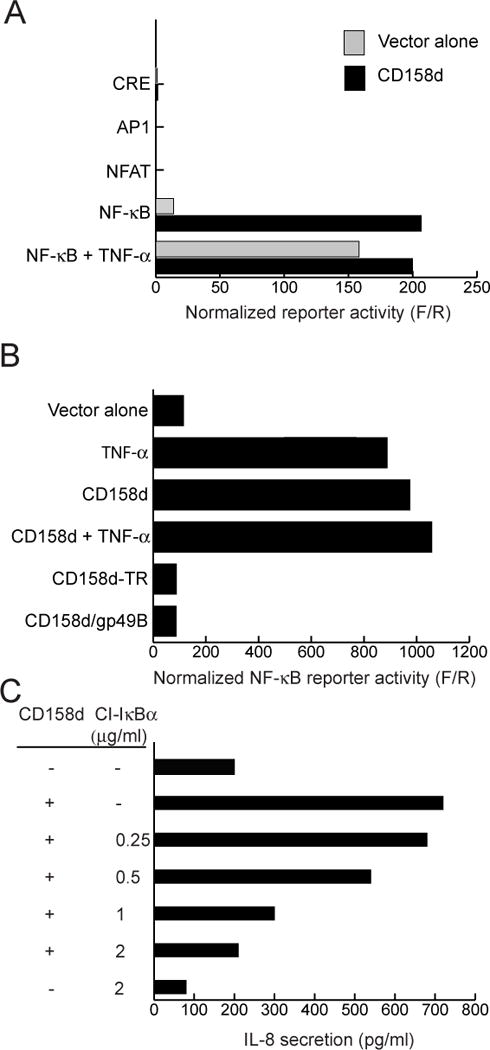
NF-κB activation is involved in signaling by CD158d. (A) Activity of the indicated reporter plasmids in 293T cells transfected with control vector (grey bars) or with a plasmid encoding CD158d (black bars). Stimulation with TNF-α (10 ng/ml) during the last 5 h of culture was used as a positive control for NF-κB activation. Results are presented as the ratio of Firefly luciferase activity to Renilla luciferase activity (F/R). The data are representative of three independent experiments. (B) NF-κB reporter activity in cells transfected with the indicated plasmids, as in (A). TNF-α (10 ng/ml) was present where indicated during the last 5 h of culture. The data are representative of three independent experiments. (C) IL-8 secretion by 293T cells transfected with plasmid encoding CD158d together with increasing amounts of a plasmid encoding the constitutive inhibitor CI-IκBα. Supernatants were tested for IL-8 48 h after transfection. The data are representative of three experiments. (D) Upper panels: IκBα phosphorylation in resting NK cells stimulated with 10 μg/ml of control antibody (cIg), CD158d mAb, or sHLA-G for 16 h. Cell lysates were immunoblotted for phospho-IκBα and tubulin (loading control). Lower panels: Translocation of p65 into the nucleus of resting NK cells stimulated with 10 μg/ml of cIg, CD158d mAb, or sHLA-G for 16 h. Cytoplasmic (cyt) and nuclear (nuc) fractions were lysed and analyzed by immunoblotting for p65, tubulin (a cytoplasmic marker), and histone deacetylase (HDAC, a nuclear marker). The data are representative of experiments done in resting NK cells from three different donors.
A constitutively inhibiting mutant of IκBα (CI-IκBα) (15) was used to test if the canonical NF-κB activation pathway was required for CD158d signaling. NF-κB is normally retained in the cytoplasm by the inhibitor IκBα. Inactivation of IκBα by phosphorylation and degradation allows NF-κB translocation into the nucleus. Mutations in serine 32 and serine 36 of IκBα prevent its phosphorylation and degradation, thereby blocking the canonical NF-κB activation pathway. 293T cells were transfected with CD158d together with CI-IκBα (S32G, S36A). Addition of increasing amounts of CI-IκBα plasmid blocked CD158d-dependent IL-8 secretion (Fig. 2C), providing strong evidence that NF-κB is required, and that the canonical pathway of NF-κB activation is utilized in this signaling pathway.
To assess the role of NF-κB in endosomal signaling by CD158d in resting NK cells, stimulation of resting NK cells with either control antibody, a mAb to CD158d, or the natural ligand, soluble HLA-G (sHLA-G), was carried out for 16 h. Cell lysates were then immuno-blotted for the phosphorylated form of IκBα. Phosphorylation of IκBα was detected after CD158d stimulation by both the agonist antibody and sHLA-G (Fig. 2D, upper panels). Since inactivation of IκBα by phosphorylation and its degradation leads to nuclear translocation of NF-κB, we next tested for the presence of p65 in the nucleus after CD158d signaling. Lysates of cytoplasmic and nuclear fractions from resting NK cells stimulated under the same conditions were immuno-blotted for p65. Nuclear translocation of p65 was detected upon activation via CD158d using both agonist Ab and sHLA-G (Fig. 2D, lower panels). Thus, endosomal signaling by CD158d in resting NK cells involves NF-κB activation.
CD158d signaling requires Akt
We sought to identify proximal signals, upstream of NF-κB, in the CD158d signaling pathway. Previous work had identified two unusual features of the NK cell response to CD158d: (i) soluble ligand, either agonist mAb or soluble HLA-G, is sufficient to induce signaling by CD158d in the absence of further crosslinking, and (ii) signaling occurs in endosomes, as a receptor engineered to traffic to the cell surface did not signal (7). Early events in signaling by cell surface immunoreceptors are primarily initiated by Src-family tyrosine kinases and by class I phosphatidylinositol 3-kinases (PI3K) (16, 17). The contribution of these types of kinases in CD158d signaling was tested in resting NK cells stimulated with soluble antibody to CD158d. For comparison, resting NK cells were stimulated with a combination of IL-12 and IL-18, which are potent stimulators of IFN-γ secretion by NK cells. CD158d engagement induced a comparable IFN-γ secretion (Fig. 3A). However, the IFN-γ secretion induced by CD158d was unaffected by the Src-family kinase inhibitor PP2 and the PI3K inhibitor wortmannin (Fig. 3A). As expected, these inhibitors blocked the NK cell response to IL-12 and IL-18. These results indicated that initiation of the CD158d signaling pathway does not involve Src-family kinases or PI3K. Several other inhibitors tested had no effect on CD158d-mediated secretion of IFN-γ (Table S1). These findings led us to undertake a screening strategy to identify early signaling components.
Fig. 3.
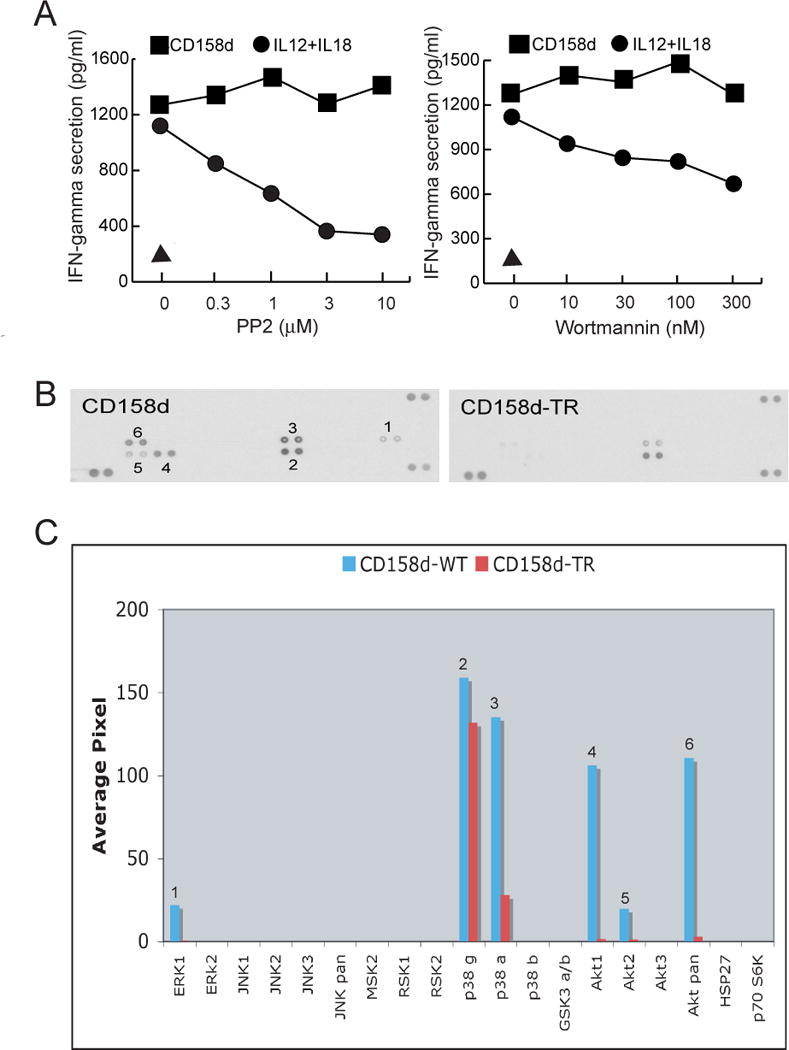
CD158d signaling induces Akt phosphorylation. (A) Inhibition of IFN-γ release by resting NK cells with PP2 or wortmannin at the indicated concentrations. Inhibitors were added for 1 h at 37°C before activation with CD158d mAb or with a combination of IL-12 (10 ng/ml) and IL-18 (25 ng/ml) for 16 h in the continuing presence of the inhibitor. The triangle represents the IFN-γ secretion induced by isotype-matched control antibody. (B) Kinase phosphorylation profile during CD158d signaling. Lysates of 293T cells transfected with either CD158d or tail-deleted receptor (CD158d-TR) were assayed with the Human Phospho-Kinase Profiler Array (RnD Systems #ARY002). (C) Signals from scanned X-ray film images in (B) were plotted as a function of average pixel density. Numbers represent corresponding data on the scan and the graph.
To identify kinase(s) that may be involved in CD158d signaling, kinase phosphorylation during CD158d activation was profiled in 293T cells transiently transfected for 48 h with CD158d or with the cytoplasmic tail-deleted receptor (CD158d-TR). Assays of the lysates on an antibody array revealed that Akt1 (henceforth referred to as Akt) and the MAPK p38α were selectively phosphorylated in response to CD158d but not CD158-TR (Fig. 3B, C). Weak phosphorylation of Erk was also detected (Fig. 3B, C). However, activation of resting NK cells by CD158d, as determined by IFN-γ secretion, was insensitive to the MEK-1 inhibitor PD98059, thereby ruling out a role for Erk (11). In contrast, CD158d signaling was sensitive to the p38 MAPK inhibitor SB203580 (11). However, further experiments with SB203580 have shown that p38 MAPK was not required for IFN-γ gene transcription but for post-transcriptional control of IFN-γ secretion (Y. Bryceson, personal communication). We conclude that Akt is likely to be involved in CD158d signaling.
Phosphorylation of Akt upon activation by CD158d was then validated in primary, resting NK cells. Resting NK cells were stimulated with soluble control antibody, soluble CD158d mAb, and the natural ligand, sHLA-G, for 16 h, followed by immuno-blotting of cell lysates for Akt phosphorylated at Serine 473 (Fig. 4A). CD158d-mediated activation of resting NK cells resulted in Akt phosphorylation at S473, a site located in the hydrophobic motif in the C-terminus of Akt. There was no detectable phosphorylation at Threonine 308 (Fig. S2), a site in the activation loop that is usually phosphorylated by PDK-1 (18). Known kinases that phosphorylate Akt at S473 include the related mTOR-Rictor complex, ATM, and DNA-PK.
Fig. 4.
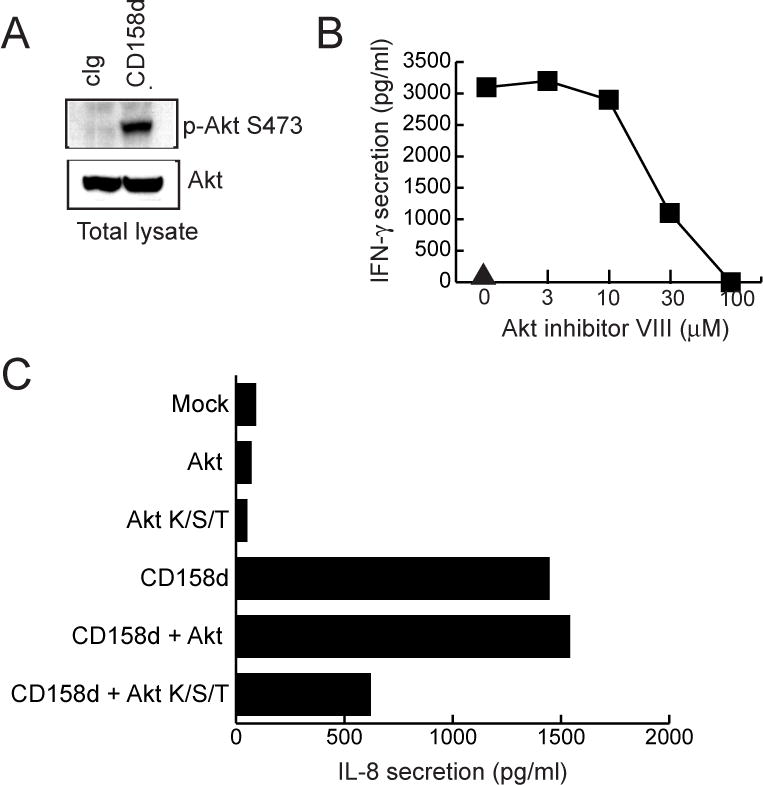
Akt activation is required for CD158d signaling. (A) Akt phosphorylation in resting NK cells stimulated with 10 μg/ml of control antibody (cIg), CD158d mAb, and sHLA-G for 16 h. Cell lysates were immunoblotted for Akt S473 phosphorylation and for tubulin (loading control). (B) IFN-γ and IL-8 secretion by resting NK cells treated with Akt inhibitor VIII at 37°C for 1 h before stimulation and for 16 h during stimulation with IgG CD158d mAb. The triangle denotes IFN-γ and IL-8 secretion in the presence of isotype-matched control mAb. (C) Inhibition of Akt phosphorylation by resting NK cells with Bafilomycin A1 (1 μM) or Dynasore (50 μM). Inhibitors were added for 1 h at 37°C before activation with CD158d mAb for 16 h in the continued presence of inhibitor. Cells lysates were immunoblotted for Akt phosphorylation at S473 and for tubulin (loading control). Data shown in A-C are representative of experiments done using resting NK cells from three different donors. (D) Dominant-negative Akt (Akt K/S/T) impairs CD158d function. IL-8 secretion by 293T cells transfected with the indicated plasmids. The average inhibition of IFN-γ secretion by Akt K/S/T, relative to wild type Akt, in 4 independent experiments was 62.15% ± 3.65.
A role for Akt in CD158d-mediated signaling was tested using an inhibitor (Akt inhibitor VIII) that blocks through binding Akt in its inactive conformation, thereby preventing S473 phosphorylation by steric hindrance (19). Akt inhibitor VIII is far more specific than other inhibitors of Akt, which interfere with binding of the Akt PH domain to PIP3. Akt inhibitor VIII blocked CD158d-induced secretion of IFN-γ and IL-8 in resting NK cells (Fig. 4B). Furthermore, to test whether Akt phosphorylation was dependent on endocytosis, two inhibitors of endosomal function were used. NK cells were pre-treated with Dynasore, a non-competitive, cell-permeable inhibitor of the GTPase activity of dynamin (20). Dynasore blocks dynamin-dependent endocytosis, and earlier work showed that the internalization of CD158d into endosomes is dynamin-dependent (7). Second, resting NK cells were treated with Bafilomycin A1, a vacuolar proton pump inhibitor that induces fragmentation of early endosomes, with little effect on late endosomes or lysosomes (21). Both inhibitors blocked the IFN-γ and IL-8 secretion induced by CD158d in response to soluble CD158d antibody (Fig. S3). Next, the effect of these inhibitors on the Akt phosphorylation induced by CD158d was tested. As shown by immuno-blotting for Akt phosphorylated at S473, both inhibitors blocked Akt phosphorylation in resting NK cells stimulated by CD158d (Fig. 4C)
More direct evidence for a role of Akt in CD158d signaling was obtained using a dominant-negative Akt, which carries a point mutation in the catalytic site and two mutations in phosphorylation sites (Akt K179M/T308A/S473A; denoted here as Akt K/S/T). 293T cells transfected with CD158d were co-transfected with either Akt or the dominant-negative Akt K/S/T. While transfection of Akt did not affect the IL-8 secretion induced by CD158d, transfection of Akt K/S/T impaired the IL-8 secretion (Fig. 4D). These results identify Akt as a key molecule during endosomal signaling by CD158d.
Akt activation typically requires its recruitment to the plasma membrane through binding of its pleckstrin homology (PH) domain to PtdIns(3,4)P2 and PtdIns(3,4,5)P3 and phosphorylation by PDK-1, which is recruited to PtdIns(3,4,5)P3. Akt is distributed primarily in the cytosol and low levels have been detected in the nucleus (18). The role of Akt in CD158d signaling suggests that Akt may be recruited to endosomes containing CD158d. Previous work showed that CD158d localizes to a Rab5-positive compartment (7). When over-expressed, the constitutively active form of Rab5, Rab5Q79L, promotes homotypic fusion of Rab5+ endosomes, which results in enlargement of early endosomes (22). Expression of Rab5Q79L together with CD158d in 293T cells showed that CD158d colocalized with Rab5Q79L in large early endosomes (Fig. S4). To examine the effect of Rab5Q79L on CD158d signaling, IL-8 secretion by 293T cells expressing CD158d in the presence of Rab5 or Rab5Q79L was tested. There was a marked increase in IL-8 secretion induced by CD158d in the presence of Rab5Q79L (Fig. 5A), consistent with signaling by CD158d in endosomes. To facilitate imaging of signaling molecules in endosomal compartments, 293T cells were transiently transfected with CD158d and Rab5Q79L. Furthermore, since there is abundant Akt in the cytosol, a “cytosol leak” was performed for 1 min prior to fixation and permeabilization to improve the visualization of intracellular membranes such as endosomal membranes (22). To visualize Akt and CD158d in endosomes enlarged in the presence of Rab5Q79L, the Rab5 mutant was GFP-tagged, Akt was HA-tagged, and CD158d was detected by the uptake of soluble Ab for 2 h at 37°C. Co-localization of Akt with CD158d and Rab5Q79L was obvious in cells that expressed all three molecules (Fig. 5C, D; Fig. S5, Fig. S6). In contrast, Akt was not detected in Rab5Q79L-positive endosomes in cells that did not co-express CD158d (Fig. 5B). We conclude that Akt is recruited to endosomes carrying CD158d.
Fig. 5.
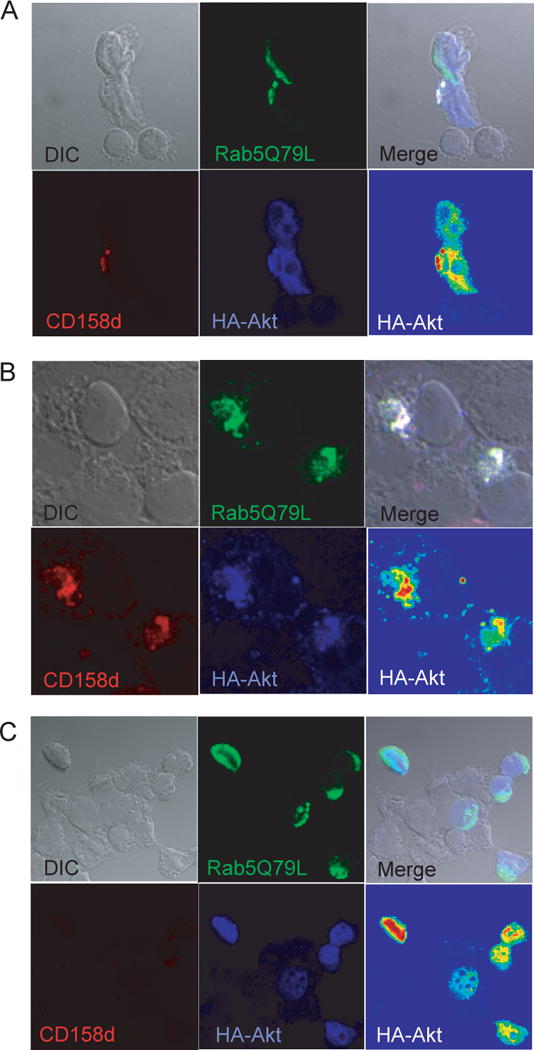
Akt is recruited to endosomes containing CD158d. (A) Rab5Q79L enhances CD158d function. IL-8 secretion by 293T cells transfected with the indicated plasmids for 48 h. IL-8 secretion collected during the last 16 h was determined. (B) A single confocal section of CD158d-negative 293T cells co-transfected with Rab5Q79L-GFP (green) and HA-Akt (blue). In 21 CD158d-negative cells analyzed, no co-localization of Akt with Rab5Q79L-positive endosomes was observed. The average colocalization coefficient for Akt and Rab5Q79L in the transfected cells in the field shown was 0.128 ± 0.142. (C, D) A single confocal section of 293T cells co-transfected with Rab5Q79L-GFP (green), CD158d (red), and HA-Akt (blue). An intensity scale (red=high, blue=low) is also displayed for HA-Akt in the bottom right panel. Cells were loaded with Cy3-labeled CD158d mAb for 2 h. Akt was labeled post-fixation and permeabilization with anti-HA mAb followed by Alexa-647 secondary Ab. For comparison, note that in the CD158d-negative cell at the top in (C), Akt did not co-localize with Rab5Q79L-positive endosomes. Additional images are shown in Figure S4. The colocalization coefficient for Akt and Rab5Q79L in the cells in Fig. 5C is as follows: CD158d-negative cell, top: 0.115; CD158d-positive cell, bottom: 0.799. Fig. 5D: 0.704 and 0.711 for the cells on the left and right, respectively. See Fig. S4 and S5 for additional colocalization image displays and profiles.
DNA-PKcs associates with CD158d
Akt is upstream of NF-κB activation in several growth factor receptor signaling pathways, such as the pro-inflammatory response induced by TNF-α and pro-survival signaling through PDGF (23, 24). Akt activates IKKα by phosphorylation of Threonine 23, which then phosphorylates IκBα, leading to IκBα degradation and NF-κB activation (23). In addition to this function, Akt is a ubiquitous signaling node that links to a staggering array of signaling components and key processes, such as survival, proliferation and metabolism (18). Akt signaling specificity hinges on its location, its phosphorylation by upstream regulators and the substrates that it acts upon. To identify potential links between CD158d and upstream regulators of Akt, a tandem-affinity purification (TAP) strategy combined with sequencing by mass spectrometry was undertaken. Recombinant TAP-tagged CD158d was expressed in 293T cells and isolated from an enriched endosomal fraction. Proteins associated with CD158d were subjected to sequencing by tandem-mass spectrometry (μLC/MS/MS) in bulk. Among the top eight proteins by rank were CD158d, calmodulin, and the 469 kDa catalytic subunit of the DNA-dependent protein kinase (DNA-PKcs) (Fig. 6). The other five, including actin, keratin, and a ribonucleoprotein, were likely contaminants. Calmodulin was most likely pulled down through the calmodulin binding protein (CBP) that is part of the tandem affinity tag. DNA-PKcs is a member of the PI3K-like kinase (PIKK) family, which includes ataxia telangiectasia mutated (ATM), ATM-Rad3 related (ATR), and mammalian target of rapamycin mTOR. PIKKs regulate diverse processes such as genome surveillance and responses to cellular stress such as DNA damage (25). Unlike PI3K, the kinase domain of ATM, ATR, mTOR, and DNA-PKcs has serine and threonine kinase activity rather than the lipid phosphorylation activity of PI3K. DNA-PKcs is a known upstream Akt regulator, in addition to the mTOR-Rictor complex, that phosphorylates Akt at the Serine 473 (26). Recent work has shown that Akt acts downstream of DNA-PK in the DNA repair signaling pathway (27).
Fig. 6.
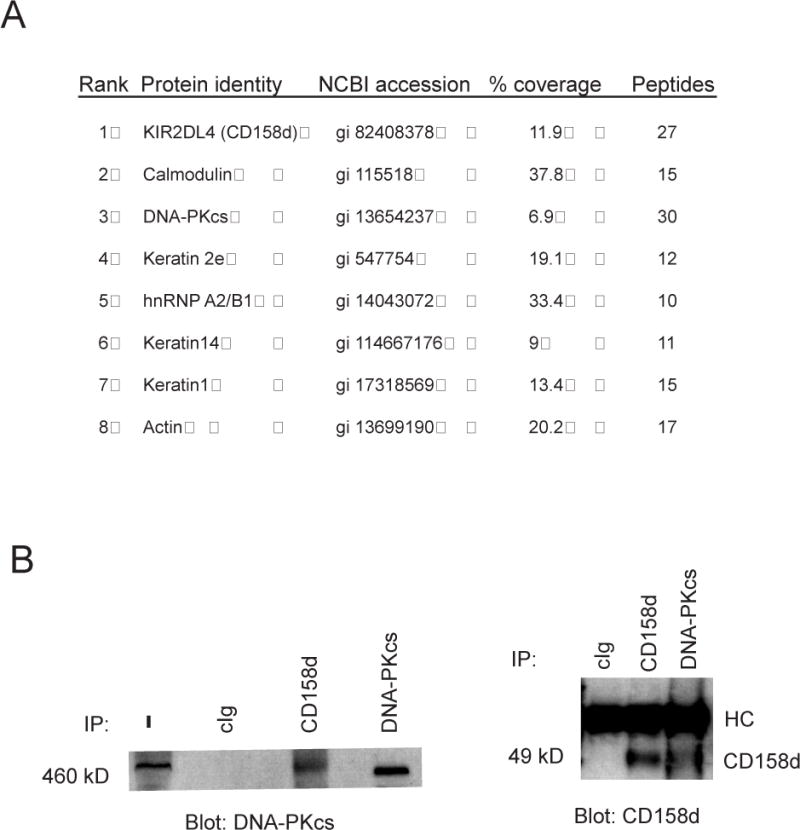
DNA-PKcs is associated with CD158d. (A) Proteins associated with TAP-tagged CD158d identified by μLC/MS/MS. Rank is based on the intensity of the MS/MS spectra. The number of unique peptides in the MS/MS spectra is shown. (B) Association of DNA-PKcs with CD158d in NK cells. NKL cells were stimulated with IgM CD158d mAbs for 2 h. Lysates without immunoprecipitation (−) or immunoprecipitated with control Ab (cIg) or Abs to CD158d and DNA-PKcs, as indicated, were immunoblotted for DNA-PKcs (left panel) and CD158d (right panel).
Association of DNA-PKcs with CD158d was examined in the NK cell line NKL. Cell lysates were immunoprecipitated with a CD158d mAb and analyzed by immuno-blotting for associated DNA-PKcs. Conversely, cell lysates were immunoprecipitated with anti-DNA-PKcs mAb and immuno-blotted for CD158d. In both sets of experiments association of DNA-PKcs with CD158d was detected (Fig. 6B).
Akt acts downstream of DNA-PKcs in the CD158d signaling pathway
To test whether DNA-PKcs is involved in endosomal signaling by CD158d, resting NK cells were stimulated with soluble CD158d mAb in the presence of NU7026, an inhibitor with a sixty-fold higher potency against DNA-PKcs than against PI3K, and with no activity towards other PIKK kinases (28). Pre-incubation of resting NK cells with different doses of NU7026 prior to activation by soluble CD158d mAb blocked IFN-γ secretion (Fig. 7A). In contrast, NU7026 had little effect on the IFN-γ secretion induced by IL-12 and IL-18 (Fig. 7A). Similar blocking of IL-8 secretion by resting NK cells in the presence of NU7026 was also seen (Fig. 7A). Next, we tested if the phosphorylation of Akt at S473 induced by CD158d signaling was dependent on DNA-PKcs. After stimulation of resting NK cells with soluble mAb to CD158d for 16 h, phosphorylation of Akt at S473 detected by immuno-blotting was abrogated in the presence of NU7026 (Fig. 7B). We conclude that Akt is downstream of DNA-PKcs in the CD158d signaling pathway.
Fig. 7.
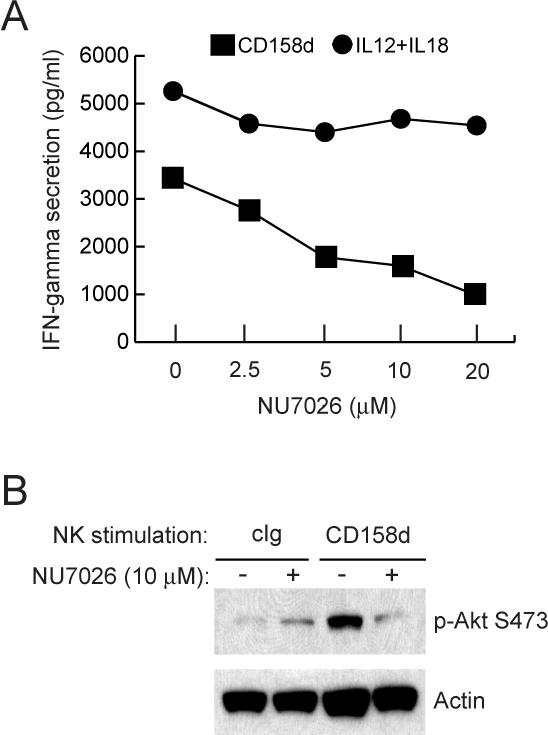
Inhibition of DNA-PKcs blocks CD158d signaling. (A) IFN-γ secretion by resting NK cells stimulated with IgG CD158b mAb or with IL-12 (10 ng/ml) and IL-18 (25 ng/ml) for 16 h. IL-8 secretion by resting NK cells stimulated with CD158d mAb is also shown. NK cells were treated with the indicated concentration of NU7026 for 1 h at 37°C before stimulation and during the 16 h of stimulation. The triangle denotes secretion in the presence of isotype-matched control antibody. (B) Akt S473 phosphorylation detected by immunoblotting in lysates of resting NK cells stimulated with cIg or CD158d mAb for 16 h. Where indicated (+), cells were pre-incubated with 10 μM NU7026 for 1 h prior to stimulation and during the 16 h of stimulation. Blots were re-probed for actin.
Inhibition of DNA-PKcs impairs CD158d signaling
To test the role of DNA-PKcs on CD158d signaling, 293T cells stably expressing CD158d were transfected with siRNA specific for DNA-PKcs. After 72 h, a partial reduction in the amount of DNA-PKcs was detected (Fig. 8A). IL-8 secretion was reduced in the cells transfected with specific DNA-PKcs siRNA, as compared with those transfected with control siRNA (Fig. 8B).
Fig. 8.
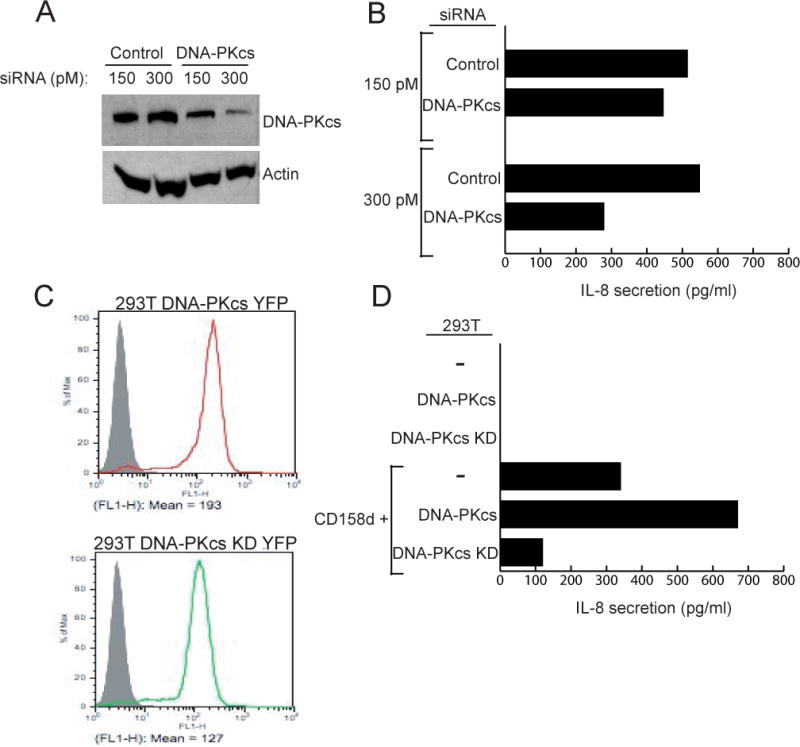
DNA-PKcs activity is required for CD158d signaling. (A) siRNA knockdown of DNA-PKcs in 293T-CD158d-GFP cells. 48 h after transfection with siRNA cell lysates were immunoblotted for DNA-PKcs and re-probed for actin. (B) IL-8 secretion by 293T-CD158d-GFP cells 48 h after transfection with the indicated siRNA. The average inhibition of IL-8 secretion by DNA-PKcs siRNA at 300 pM in 3 independent experiments was 49.47% ± 8.06%. (C) Stable expression of DNA-PKcs-YFP and a kinase-dead (KD) mutant of DNA-PKcs-YFP in 293T cells. The grey profile represents untransfected 293T cells. (D) IL-8 secretion by 293T cells, 293T-DNA-PKcs-YFP and 293T-DNA-PKcs-KD-YFP cells. As indicated, these cells were either not transfected or transiently transfected with a CD158d plasmid for 48 h. All cells were transfected with a Renilla luciferase reporter construct to normalize for transfection efficiency in the different cell lines. The data is representative of three independent experiments.
The requirement for the kinase activity of DNA-PKcs in the CD158d signaling pathway was tested by comparing the effect of DNA-PKcs and a kinase-dead mutant of DNA-PKcs (DNA-PKcs-KD) in 293T cells. The D3921N mutation in the kinase domain of DNA-PKcs abolishes its kinase activity (29). YFP-tagged versions of DNA-PKcs and DNA-PKcs-KD were transfected in 293T cells and clones with comparable expression were selected (Fig. 8C). These transfected 293T cells were then transiently transfected with CD158d and tested for IL-8 secretion. IL-8 secretion was increased in cells expressing wild-type DNA-PKcs, and greatly decreased in cells expressing DNA-PKcs-KD, as compared to wild type 293T cells (Fig. 8D). Thus, the kinase activity of DNA-PKcs is required for endosomal signaling by CD158d.
DISCUSSION
We describe here a new endosomal signaling pathway, which is utilized by immunoreceptor CD158d for the generation of an NF-κB-dependent, pro-inflammatory response. This unique pathway is different from other known examples of endosomal signaling such as those studied with a number of receptor tyrosine kinases (e.g. TrkA, EGFR), or with endosomal TLR (e.g. TLR3, TLR7, TLR9) in innate immune responses to viruses and bacteria (4, 6). The CD158d endosomal pathway described here does not require early signals typically associated with immunoreceptor signaling, such as Src-family kinases or PI3K. Rather, it generates signaling for a pro-inflammatory response through the serine-threonine kinases DNA-PKcs and Akt, and through NF-κB activation.
A strong illustration of the raison d’être of endosomal signaling is found in the nervous system where survival and differentiation signals must be transmitted over long distances from the axon tip of an innervating neuron to the cell body (5). Studies on the TrkA receptor show that signaling endosomes containing activated TrkA complexes are involved in the retrograde transport of key survival signals to the nucleus (30). In other situations, where receptors can signal sequentially at the cell surface and in endosomes, the physiological necessity for sustained endosomal signaling is less well understood. With respect to the signaling pathway described in this study, all the unique facets of endosomal signaling (and the inability to elicit signaling by crosslinking CD158d at the PM) are highly relevant to the potential physiological role of CD158d during early pregnancy. The CD158d ligand is soluble HLA-G, which has a restricted expression mostly limited to fetal trophoblast cells at the maternal-fetal interface (31). The decidua, which is the maternal tissue in contact with the endometrium of the pregnant uterus, is unusual in that NK cells constitute the major lymphocyte population at this site. (32). Production of pro-inflammatory cytokines in response to HLA-G in vitro has been detected in primary, resting NK cells (7), uterine NK cells (33), and decidual NK cells (34). The transcriptional response to engagement of CD158d includes up-regulation of a limited set of pro-inflammatory and pro-angiogenic genes (7). Therefore, NK cells in the uterus may be sensors of soluble HLA-G expressed by invading trophoblast cells from the fetus. Through the use of a soluble ligand and an endosomal receptor, this mode of fetal-maternal communication is greatly simplified compared to the complex array of receptor–ligand interactions that would occur during NK–trophoblast cell contacts. Trophoblast invasion is important for the conversion of maternal blood vessels into low-resistance vessels, in order to provide an adequate supply of blood to the fetus (13). Sustained endosomal signaling by CD158d in response to soluble HLA-G would lead to the prolonged secretion of cytokines and chemokines for the duration of trophoblast invasion, thereby contributing to the major vascular remodeling that occurs during the first trimester of pregnancy.
In view of the potential physiological relevance of endosomal signaling by CD158d, we set out to delineate the signaling pathway that underlies this unique pro-inflammatory and pro-angiogenic response. A proteomics approach identified DNA-PKcs as a protein associated with CD158d. Using siRNA targeting, a selective DNA-PKcs kinase inhibitor, and cells expressing a kinase-dead mutant of DNA-PKcs, we show that DNA-PKcs and its kinase activity are required for the phosphorylation and activation of Akt in response to CD158d engagement, and for the production of cytokines and chemokines by primary NK cells and transfected 293T cells. DNA-PKcs was immunoprecipitated together with CD158d, and vice versa, in NKL cells. It is still unknown if the interaction of DNA-PKcs with CD158d is direct or indirect, as part of a multi-protein signaling complex. DNA-PKcs is a phosphoinositide-3-kinase-related protein kinase (PIKK) family member with a prominent role in the DNA damage response required to maintain genomic integrity. It promotes DNA repair via non-homologous end joining after binding double-stranded breaks (28). In DNA repair, DNA-PKcs forms a complex with the Ku heterodimer of 70 and 86 kD (28). However, DNA-PKcs functions also in other contexts in the absence of the Ku heterodimer. In vitro studies have shown that the Ku subunits are not essential for the kinase activity of DNA-PKcs (28). These two Ku subunits were not present in the CD158d-associated proteins identified here by mass spectrometry. During DNA damage due to ionizing radiation, DNA-PKcs activates Akt via phosphorylation of the hydrophobic motif at S473 (27, 35, 36). In addition to a role in the DNA damage response, DNA-PKcs has also been implicated in feeding-dependent metabolic gene activation during insulin signaling (37) and in class switch recombination in lymphocytes (38). A wider role for DNA-PKcs, beyond double-stranded DNA break, is beginning to be appreciated. It is interesting that although humans express much higher levels of DNA-PKcs than rodents, this excess does not correlate with increased ability to repair DNA, suggesting other important functions for this molecule in humans (39, 40).
We have identified Akt phosphorylation as a response to CD158d signaling by kinase phosphorylation profiling, and confirmed it directly in primary NK cells. Using IFN-γ and IL-8 secretion as functional readouts, we show that CD158d signaling was impaired in the presence of a selective Akt inhibitor, which binds to the hydrophobic motif in Akt. Akt phosphorylation in resting NK cells was abrogated in the presence of inhibitors of endocytosis and early endosomal acidification, indicating that the signaling is occurring in endosomes. CD158d signaling was also impaired by expression of a dominant-negative mutant of Akt. Akt is an important regulator of a vast array of cellular processes, including growth, survival, and proliferation. Consequently, Akt activation is tightly regulated and its specificity is dictated in part by its subcellular location and its substrate selection. The traditional view of Akt signaling places it at the PM downstream of PI3K-dependent PtdIns(3,4,5)P3 synthesis. Akt and its kinase PDK1 are recruited to PtdIns(3,4,5)P3 at the PM, resulting in Akt phosphorylation at Threonine 308. A number of kinases, including mTOR, ATM, and DNA-PK, can potentially phosphorylate Akt at S473 (26). Here we have shown that, as a result of CD158d signaling, Akt is phosphorylated primarily at S473 by DNA-PKcs. This result is consistent with a report showing exchange protein activated by cAMP (EPAC)-triggered phosphorylation of Akt by DNA-PKcs at S473, but not T308, during repair of etoposide-induced double stranded breaks (41).
In addition to the PM and nuclei, other locations of Akt signaling have been described, including mitochondria (42, 43). Akt signals in the nucleus to initiate pro-survival signaling after DNA damage. After γ-irradiation, foci of Akt phosphorylated at S473 were detected in nuclei at the site of double-stranded breaks (27). Transient partitioning of Akt into APPL-containing endosomes after growth factor stimulation has also been reported (44). In such signaling endosomes, Akt phosphorylates GSK3 to promote survival during zebrafish development. In our study, detection of Akt on endosomes occurred only in cells that co-expressed CD158d. Such endosomal segregation of Akt would give it access to qualitatively different signaling scaffolds, including DNA-PKcs in association with CD158d.
We found that NF-κB activation is required for the pro-inflammatory response generated by CD158d signaling. In resting NK cells, evidence for NF-κB involvement included the phosphorylation of IκBα and the nuclear translocation of p65 during CD158d signaling induced by both agonist antibody and the physiological ligand, sHLA-G. This result is consistent with many studies showing a close link between NF-κB activation and the control of inflammatory responses (14). A previous study showed that IKKβ and IκBα are phosphorylated after CD158d crosslinking with Abs at the surface of an activated NK cell line (45). At endosomes, the canonical NF-κB activation pathway is used downstream of CD158d, as evidenced by the profound effect of a constitutively inhibiting IκBα mutant on CD158d signaling. Activated Akt stimulates IKKα activity by phosphorylating it at Thr23 (23). Phosphorylation of IκBα by IKK leads to NF-κB activation. Thus, we provide evidence for the sequential requirement of DNA-PKcs, Akt, and NF-κB in the pro-inflammatory signaling pathway triggered by CD158d from endosomes.
There is growing evidence for the role of endosomes as signaling organelles that provide outputs distinct from those generated at the PM (46). Those receptors that signal in endosomes are capable of delivering prolonged signals, which can last beyond the duration of ligand exposure at the cell surface. This feature is illustrated by growth factor receptors that initiate signaling at the PM, and continue to signal in endosomes after ligand-induced internalization to generate distinct functional outcomes (46). Residence time of growth factor receptors in endosomes has been correlated with signal strength (47). Most transmembrane receptors in the immune system, including other KIR family members besides CD158d, signal from the PM. In contrast, CD158d signals from endosomes in primary NK cells for a unique proinflammatory/proangiogenic response (7). It is important to note that the signaling pathway described here occurs in primary, resting NK cells, which do not express detectable CD158d on the cell surface, and that stimulation occurs only after CD158d engagement with soluble ligand or soluble agonist Ab (11) (7). CD158d crosslinking at the surface of an IL-2-activated NK cell line induced cytotoxicity and IFN-γ secretion, probably through association with the FcR γ chain, and MAPK activation independently of the FcR γ chain (45). Thus, in IL-2-activated NK cells, a low level of cell surface CD158d can signal for MAPK activation upon antibody stimulation. In contrast, in primary, resting NK cells, most of CD158d resides in endosomes from where it signals after binding to soluble ligand. Furthermore, the endosomal targeting of CD158d and IL-8 secretion in a human embryonic kidney cell line transfected with CD158d suggest that this pathway may represent a broad mode of endosomal signaling in different cell types.
Accumulation of soluble HLA-G in an endocytic compartment containing CD158d may serve to provide more sustained signaling, as has been observed with endocytosed growth factor receptors. In Rab5-positive endosomes, CD158d couples with the serine-threonine kinase DNA-PKcs, which phosphorylates Akt. In turn, Akt activates NF-κB to trigger a pro-inflammatory response. Our study expands the role of DNA-PKcs beyond its well-established function in DNA repair. It also reveals a novel functional outcome of endosomal Akt signaling, which is independent of the PI3K pathway. In conclusion, our study has revealed a new pathway of endosomal signaling, which utilizes the serine-threonine kinases DNA-PKcs and Akt, to generate NF-κB-dependent activation of a pro-inflammatory and pro-angiogenic response.
MATERIALS AND METHODS
Cells and cell culture
Polyclonal NK cells were isolated from peripheral blood lymphocytes (PBL) using the MACS NK cell negative isolation kit (Miltenyi Biotech, Auburn, CA). NK cells were greater than 95% CD3 negative and CD56 positive. These resting NK cells were cultured in Iscoves medium containing 10% human serum without added IL-2 or feeder cells. They were used within 24 h. Resting NK cells were stimulated with either isotype matched control mAb or anti-CD158d mAb #33 at 10 μg/ml or with IL-12 (10 ng/ml) and IL-18 (25 ng/ml) for 16 h. NKL cells were a gift from M. Robertson (Indiana School of Medicine, Indianapolis, IN). They were cultured in RPMI 1640 medium containing 10% fetal calf serum, 1% glutamine, 1% sodium pyruvate, and 200 U/ml of recombinant IL-2 (National Cancer Institute-FCRDC, Frederick, MD). 293T cells were obtained from ATCC (American Type Culture Collection, Manassas, VA) and cultured in Iscoves medium containing 10% fetal calf serum and 1% glutamine. 293T cells stably transfected with gfp-tagged CD158d have been described previously (7). The 293T cells were transiently transfected with LipofectAMINE 2000 (Invitrogen, Frederick, MD) according to the supplier’s instructions. Cells were tested 48 h after transfection for IL-8 secretion. Supernatants were collected from the final 16 h of culture. siRNAs targeted against DNA-PKcs were purchased from Thermo Scientific Dharmacon Products (Lafayette, CO). Control scrambled oligos were purchased from IDT. 293T cells stably transfected with gfp-tagged CD158d were transfected with either 150 or 300 pM of siRNA using LipofectAMINE 2000. After 16 h, medium was replaced and after 72 h, culture supernatants were harvested to check for IL-8 secretion. Cell lysates were also prepared from siRNA treated cells to check for knockdown by immuno- blotting for DNA-PKcs.
Antibodies and Reagents
mAbs to CD158d (#33, IgG1; #36 and #64, IgM) and an affinity purified rabbit polyclonal antibody to the extracellular portion of CD158d were generated in our laboratory (11). Anti-HA antibodies were purchased from Covance (Princeton, NJ). Rabbit antibodies to actin, HDAC, p65, DNA-PKcs, Akt, phospho-S473 Akt and phospho-IκBα were purchased from Cell Signaling (Danvers, MA). Mouse mAb to DNA-PKcs was purchased from Thermo Scientific (Rockford, IL). Antibody to tubulin was purchased from Invitrogen (Frederick, MD). All isotype control antibodies were obtained from Sigma (St. Louis, MO). All secondary goat anti-mouse antibodies conjugated to Alexa Fluor 488, 564 or 647 were purchased from Molecular Probes (Eugene, OR). The following inhibitors: NU7026; Akt inhibitor VIII; isozyme selective, Akt 1/2, PP2; wortmannin and Bafilomycin A1 were obtained from Calbiochem (San Diego, CA). Dynasore was purchased from Sigma (St. Louis, MO). Proteome Profiler Human Phospho MAPK Array Kit (CAT#ARY002) was obtained from RnD Systems (Minneapolis, MN). Recombinant, single-chain soluble HLA-G (48) was obtained from Organon NV (Oss, the Netherlands).
DNA plasmids
Expression constructs for CD158d have been described (7). The CD158d/gp49B chimera was engineered using PCR to fuse the extracellular domain of CD158d ending at S223 with the transmembrane and tail of gp49B beginning at position T227. The amino acids at the boundary are NPSS/TPTE. The constitutively inhibiting IκBα was a gift from U. Siebenlist (NIAID, NIH). HA-Akt, Akt-WT, and Akt K179M/S473A/T308A constructs have been described (49) and were obtained from Addgene (www.addgene.org). The Rab5-GFP and Rab5Q79L-GFP constructs were a gift from J. Bonifacino (NICHD, NIH). Luciferase reporter constructs for AP1, CRE, NFAT, and NF-κB were obtained from Howard Young (NCI-FCRDC, NIH). AP1-luc, NF-κB-luc, and CRE-luc were in pLuc-MCS vector (Stratagene). The NFAT-luc was in pGL3 Basic vector (50). Reporter activity was read out using Dual-Luciferase Reporter Assay System Promega, (Madison, WI). YFP-tagged DNA-PKcs and YFP-tagged DNA-PKcs-KD constructs were obtained from B.P.C. Chen and D.J. Chen (University of Texas Southwestern Medical Center, Dallas, TX). The DNA-PKcs cDNA is 14 kb in size. Due to its large insert size, the DNA-PKcs plasmid is unstable and had to be amplified from bacteria grown on large LB agar plates rather than in liquid culture.
Protein Isolation for M/S Analysis
293T cells were transfected with CD158d expressed in the CTAP expression vector (Stratagene, La Jolla, CA) for 72 h. 1 × 109 cells were harvested and cell pellets were resuspended in homogenization buffer (0.25 M sucrose, 1 mM EDTA, pH 7.4). To isolate CD158d-containing endosomes, after homogenization, cells were overlaid on a 2.5 M sucrose-27% percoll gradient and centrifuged at 34,000 × g for 1 h. One ml fractions were collected and tested for the presence of CD158d by immuno-blotting for the CBP tag. After overnight dialysis of positive fractions in PBS, organelles were pelleted and lysed in 0.5% Triton X-100, 150 mM NaCl, 20 mM Tris pH 7.4. CD158d and interacting partners were then isolated using the Interplay TAP Purification Kit (Stratagene, La Jolla, CA). Eluted samples were concentrated by trichloro-acetic acid precipitation and loaded on a SDS-PAGE. The proteins were allowed to completely enter the gel matrix and a 1 cm × 0.5 cm gel slice containing the entire protein sample was isolated. Sequence analysis was performed at the Harvard Microchemistry and Proteomics Analysis Facility by microcapillary reverse-phase HPLC nano-electrospray tandem mass spectrometry (μLC/MS/MS) on a Thermo LTQ-Orbitrap mass spectrometer.
Fractionation of lysates and Immunoprecipitations
The preparation and fractionation of cytoplasmic and nuclear extracts from resting NK cells were done using the NE-PER Nuclear and Cytoplasmic Extraction Kit from Pierce Biotechnology (Rockford, IL). NKL cells were lysed in 0.5% NP40, 20 mM Tris (pH7.4), 150 mM NaCl, 1 mM PMSF, and 8 mM iodoacetamide. Samples were either used as total lysates or immunoprecipitated with mAbs for 3 h, followed by protein G agarose beads for 1 h. Immuno-blots were developed using either polyclonal antibodies to CD158d or mAb to DNA-PKcs, followed by anti-mouse or anti-rabbit IgG peroxidase (Amersham, Piscataway, NJ) and Super Signal substrate (Pierce).
Immunostaining and Confocal Microscopy
48 h after transfection, 293T cells were trypsinized and allowed to settle on poly-L-lysine coated two-well culture slides (BioCoat, BD, Bedford, MA) for 30 min prior to fixation in PBS and 4% paraformaldehyde. Cells were permeabilized and blocked for 30 min with PBS, 10% normal donkey serum, and 0.5% Triton-X-100. Cells were stained with the relevant primary antibody for 1 h and revealed with Alexa Fluor conjugated secondary antibodies in PBS, 3% normal donkey serum, and 0.5% Triton X-100. Cells were then washed and mounted in slides using ProLong Anti-fade kit (Molecular Probes, Eugene OR). For experiments involving the triple transfection of CD158d, Akt, and Rab5Q79L, a cytosol leak was performed for 1 min. after the permeabilization of cells in 0.01% saponin, 80 mM PIPES, 5 mM EGTA, and 1 mM MgCl2, prior to fixation in 4% paraformaldehyde. Images were processed using a confocal laser-scanning microscope (Axiovert 200M LSM 510 META; Zeiss Jena, Germany). Images were acquired as previously described (7). The “colocalization” function was used to analyze images processed on the LSM 510 META. Colocalization coefficients were calculated from the relative number of colocalizing pixels in channel 1 or 2, respectively, as compared to the total number of pixels above threshold. A value range of 0 to 1 was used, where 0 is no colocalization and 1 refers to the colocalization of all pixels.
Cellular Assays
For cytokine and chemokine assays, resting NK cells were incubated with soluble mAbs at 10 μg/ml or with a combination of IL-12 (10 ng/ml) and IL-18 (25 ng/ml). After 16 h, supernatants were removed and tested for the presence of cytokines by ELISA. ELISA kits for IFN-γ (Pierce) and IL-8 (Biosource, Camarillo, CA) were used. For inhibitor studies, resting NK cells were incubated with inhibitor for 1 h at 37°C prior to activation by mAbs or cytokines in the continuing presence of inhibitor for 16 h. Cell supernatants were tested for IFN-γ secretion and whole cell lysates were analyzed biochemically by immuno-blotting. To assess reporter gene expression, 293T cells were transfected with reporter constructs using LipofectAMINE. 48 h after transfection, cell lysates were analyzed using the Dual Luciferase Reporter Assay System (Promega) to measure firefly and renilla luciferase activity. Renilla luciferase activity was also tested from lysates of DNA-PKcs transfectants of 293T cells that were transiently transfected with CD158d to normalize for differences in transfection efficiency between different cell lines.
Supplementary Material
Acknowledgments
This work was supported by the Intramural Research Program of the National Institutes of Health, National Institute of Allergy and Infectious Diseases. We thank W. Lane (Harvard Microchemistry) for mass spectrometry analysis; H. Young, U. Siebenlist, J. Bonifacino, Y. Bryceson, B.P. Chen and D.J. Chen for plasmids.
Footnotes
REFERENCES AND NOTES
- 1.Miaczynska M, Pelkmans L, Zerial M. Not just a sink: endosomes in control of signal transduction. Curr Opin Cell Biol. 2004;16:400–406. doi: 10.1016/j.ceb.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 2.Polo S, Di Fiore PP. Endocytosis conducts the cell signaling orchestra. Cell. 2006;124:897–900. doi: 10.1016/j.cell.2006.02.025. [DOI] [PubMed] [Google Scholar]
- 3.von Zastrow M, Sorkin A. Signaling on the endocytic pathway. Curr Opin Cell Biol. 2007;19:436–445. doi: 10.1016/j.ceb.2007.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hoeller D, Volarevic S, Dikic I. Compartmentalization of growth factor receptor signalling. Curr Opin Cell Biol. 2005;17:107–111. doi: 10.1016/j.ceb.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 5.Sorkin A, Von Zastrow M. Signal transduction and endocytosis: close encounters of many kinds. Nat Rev Mol Cell Biol. 2002;3:600–614. doi: 10.1038/nrm883. [DOI] [PubMed] [Google Scholar]
- 6.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 7.Rajagopalan S, Bryceson YT, Kuppusamy SP, Geraghty DE, van der Meer A, Joosten I, Long EO. Activation of NK cells by an endocytosed receptor for soluble HLA-G. PLoS Biol. 2006;4:e9. doi: 10.1371/journal.pbio.0040009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kikuchi-Maki A, Catina TL, Campbell KS. Cutting edge: KIR2DL4 transduces signals into human NK cells through association with the Fc receptor gamma protein. J Immunol. 2005;174:3859–3863. doi: 10.4049/jimmunol.174.7.3859. [DOI] [PubMed] [Google Scholar]
- 9.Long EO. Negative signaling by inhibitory receptors: the NK cell paradigm. Immunol Rev. 2008;224:70–84. doi: 10.1111/j.1600-065X.2008.00660.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rajagopalan S, Long EO. A human histocompatibility leukocyte antigen (HLA)-G-specific receptor expressed on all natural killer cells. J Exp Med. 1999;189:1093–1100. doi: 10.1084/jem.189.7.1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rajagopalan S, Fu J, Long EO. Cutting edge: induction of IFN-gamma production but not cytotoxicity by the killer cell Ig-like receptor KIR2DL4 (CD158d) in resting NK cells. J Immunol. 2001;167:1877–1881. doi: 10.4049/jimmunol.167.4.1877. [DOI] [PubMed] [Google Scholar]
- 12.Moffett-King A. Natural killer cells and pregnancy. Nat Rev Immunol. 2002;2:656–663. doi: 10.1038/nri886. [DOI] [PubMed] [Google Scholar]
- 13.Moffett A, Loke C. Immunology of placentation in eutherian mammals. Nat Rev Immunol. 2006;6:584–594. doi: 10.1038/nri1897. [DOI] [PubMed] [Google Scholar]
- 14.Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol. 2009;27:693–733. doi: 10.1146/annurev.immunol.021908.132641. [DOI] [PubMed] [Google Scholar]
- 15.Brown K, Gerstberger S, Carlson L, Franzoso G, Siebenlist U. Control of I kappa B-alpha proteolysis by site-specific, signal-induced phosphorylation. Science. 1995;267:1485–1488. doi: 10.1126/science.7878466. [DOI] [PubMed] [Google Scholar]
- 16.Salmond RJ, Filby A, Qureshi I, Caserta S, Zamoyska R. T-cell receptor proximal signaling via the Src-family kinases, Lck and Fyn, influences T-cell activation, differentiation, and tolerance. Immunol Rev. 2009;228:9–22. doi: 10.1111/j.1600-065X.2008.00745.x. [DOI] [PubMed] [Google Scholar]
- 17.Fruman DA, Bismuth G. Fine tuning the immune response with PI3K. Immunol Rev. 2009;228:253–272. doi: 10.1111/j.1600-065X.2008.00750.x. [DOI] [PubMed] [Google Scholar]
- 18.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Calleja V, Laguerre M, Parker PJ, Larijani B. Role of a novel PH-kinase domain interface in PKB/Akt regulation: structural mechanism for allosteric inhibition. PLoS Biol. 2009;7:e17. doi: 10.1371/journal.pbio.1000017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Macia E, Ehrlich M, Massol R, Boucrot E, Brunner C, Kirchhausen T. Dynasore, a cell-permeable inhibitor of dynamin. Dev Cell. 2006;10:839–850. doi: 10.1016/j.devcel.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 21.Duclos S, Corsini R, Desjardins M. Remodeling of endosomes during lysosome biogenesis involves ‘kiss and run’ fusion events regulated by rab5. J Cell Sci. 2003;116:907–918. doi: 10.1242/jcs.00259. [DOI] [PubMed] [Google Scholar]
- 22.Stenmark H, Parton RG, Steele-Mortimer O, Lutcke A, Gruenberg J, Zerial M. Inhibition of rab5 GTPase activity stimulates membrane fusion in endocytosis. EMBO J. 1994;13:1287–1296. doi: 10.1002/j.1460-2075.1994.tb06381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ozes ON, Mayo LD, Gustin JA, Pfeffer SR, Pfeffer LM, Donner DB. NF-kappaB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature. 1999;401:82–85. doi: 10.1038/43466. [DOI] [PubMed] [Google Scholar]
- 24.Romashkova JA, Makarov SS. NF-kappaB is a target of AKT in anti-apoptotic PDGF signalling. Nature. 1999;401:86–90. doi: 10.1038/43474. [DOI] [PubMed] [Google Scholar]
- 25.Abraham RT. PI 3-kinase related kinases: ‘big’ players in stress-induced signaling pathways. DNA Repair (Amst) 2004;3:883–887. doi: 10.1016/j.dnarep.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 26.Bozulic L, Hemmings BA. PIKKing on PKB: regulation of PKB activity by phosphorylation. Curr Opin Cell Biol. 2009;21:256–261. doi: 10.1016/j.ceb.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 27.Bozulic L, Surucu B, Hynx D, Hemmings BA. PKBalpha/Akt1 acts downstream of DNA-PK in the DNA double-strand break response and promotes survival. Mol Cell. 2008;30:203–213. doi: 10.1016/j.molcel.2008.02.024. [DOI] [PubMed] [Google Scholar]
- 28.Collis SJ, DeWeese TL, Jeggo PA, Parker AR. The life and death of DNA-PK. Oncogene. 2005;24:949–961. doi: 10.1038/sj.onc.1208332. [DOI] [PubMed] [Google Scholar]
- 29.Kurimasa A, Kumano S, Boubnov NV, Story MD, Tung CS, Peterson SR, Chen DJ. Requirement for the kinase activity of human DNA-dependent protein kinase catalytic subunit in DNA strand break rejoining. Mol Cell Biol. 1999;19:3877–3884. doi: 10.1128/mcb.19.5.3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zweifel LS, Kuruvilla R, Ginty DD. Functions and mechanisms of retrograde neurotrophin signalling. Nat Rev Neurosci. 2005;6:615–625. doi: 10.1038/nrn1727. [DOI] [PubMed] [Google Scholar]
- 31.Kovats S, Main EK, Librach C, Stubblebine M, Fisher SJ, DeMars R. A class I antigen, HLA-G, expressed in human trophoblasts. Science. 1990;248:220–223. doi: 10.1126/science.2326636. [DOI] [PubMed] [Google Scholar]
- 32.Trowsdale J, Moffett A. NK receptor interactions with MHC class I molecules in pregnancy. Semin Immunol. 2008;20:317–320. doi: 10.1016/j.smim.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 33.van der Meer A, Lukassen HG, van Cranenbroek B, Weiss EH, Braat DD, van Lierop MJ, Joosten I. Soluble HLA-G promotes Th1-type cytokine production by cytokine-activated uterine and peripheral natural killer cells. Mol Hum Reprod. 2007;13:123–133. doi: 10.1093/molehr/gal100. [DOI] [PubMed] [Google Scholar]
- 34.Li C, Houser BL, Nicotra ML, Strominger JL. HLA-G homodimer-induced cytokine secretion through HLA-G receptors on human decidual macrophages and natural killer cells. Proc Natl Acad Sci U S A. 2009;106:5767–5772. doi: 10.1073/pnas.0901173106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Park J, Feng J, Li Y, Hammarsten O, Brazil DP, Hemmings BA. DNA-dependent protein kinase-mediated phosphorylation of protein kinase B requires a specific recognition sequence in the C-terminal hydrophobic motif. J Biol Chem. 2009;284:6169–6174. doi: 10.1074/jbc.C800210200. [DOI] [PubMed] [Google Scholar]
- 36.Feng J, Park J, Cron P, Hess D, Hemmings BA. Identification of a PKB/Akt hydrophobic motif Ser-473 kinase as DNA-dependent protein kinase. J Biol Chem. 2004;279:41189–41196. doi: 10.1074/jbc.M406731200. [DOI] [PubMed] [Google Scholar]
- 37.Wong RH, Chang I, Hudak CS, Hyun S, Kwan HY, Sul HS. A role of DNA-PK for the metabolic gene regulation in response to insulin. Cell. 2009;136:1056–1072. doi: 10.1016/j.cell.2008.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Callen E, Jankovic M, Wong N, Zha S, Chen HT, Difilippantonio S, Di Virgilio M, Heidkamp G, Alt FW, Nussenzweig A, Nussenzweig M. Essential role for DNA-PKcs in DNA double-strand break repair and apoptosis in ATM-deficient lymphocytes. Mol Cell. 2009;34:285–297. doi: 10.1016/j.molcel.2009.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Allen C, Halbrook J, Nickoloff JA. Interactive competition between homologous recombination and non-homologous end joining. Mol Cancer Res. 2003;1:913–920. [PubMed] [Google Scholar]
- 40.Meek K, Dang V, Lees-Miller SP. DNA-PK: the means to justify the ends? Adv Immunol. 2008;99:33–58. doi: 10.1016/S0065-2776(08)00602-0. [DOI] [PubMed] [Google Scholar]
- 41.Huston E, Lynch MJ, Mohamed A, Collins DM, Hill EV, MacLeod R, Krause E, Baillie GS, Houslay MD. EPAC and PKA allow cAMP dual control over DNA-PK nuclear translocation. Proc Natl Acad Sci U S A. 2008;105:12791–12796. doi: 10.1073/pnas.0805167105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kunkel MT, Ni Q, Tsien RY, Zhang J, Newton AC. Spatio-temporal dynamics of protein kinase B/Akt signaling revealed by a genetically encoded fluorescent reporter. J Biol Chem. 2005;280:5581–5587. doi: 10.1074/jbc.M411534200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sasaki K, Sato M, Umezawa Y. Fluorescent indicators for Akt/protein kinase B and dynamics of Akt activity visualized in living cells. J Biol Chem. 2003;278:30945–30951. doi: 10.1074/jbc.M212167200. [DOI] [PubMed] [Google Scholar]
- 44.Schenck A, Goto-Silva L, Collinet C, Rhinn M, Giner A, Habermann B, Brand M, Zerial M. The endosomal protein Appl1 mediates Akt substrate specificity and cell survival in vertebrate development. Cell. 2008;133:486–497. doi: 10.1016/j.cell.2008.02.044. [DOI] [PubMed] [Google Scholar]
- 45.Miah SM, Hughes TL, Campbell KS. KIR2DL4 differentially signals downstream functions in human NK cells through distinct structural modules. J Immunol. 2008;180:2922–2932. doi: 10.4049/jimmunol.180.5.2922. [DOI] [PubMed] [Google Scholar]
- 46.Sadowski L, Pilecka I, Miaczynska M. Signaling from endosomes: location makes a difference. Exp Cell Res. 2009;315:1601–1609. doi: 10.1016/j.yexcr.2008.09.021. [DOI] [PubMed] [Google Scholar]
- 47.Zoncu R, Perera RM, Balkin DM, Pirruccello M, Toomre D, De Camilli P. A phosphoinositide switch controls the maturation and signaling properties of APPL endosomes. Cell. 2009;136:1110–1121. doi: 10.1016/j.cell.2009.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Van Lierop MJ, Wijnands F, Loke YW, Emmer PM, Lukassen HG, Braat DD, van der Meer A, Mosselman S, Joosten I. Detection of HLA-G by a specific sandwich ELISA using monoclonal antibodies G233 and 56B. Mol Hum Reprod. 2002;8:776–784. doi: 10.1093/molehr/8.8.776. [DOI] [PubMed] [Google Scholar]
- 49.Ramaswamy S, Nakamura N, Vazquez F, Batt DB, Perera S, Roberts TM, Sellers WR. Regulation of G1 progression by the PTEN tumor suppressor protein is linked to inhibition of the phosphatidylinositol 3-kinase/Akt pathway. Proc Natl Acad Sci U S A. 1999;96:2110–2115. doi: 10.1073/pnas.96.5.2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim LJ, Seto AG, Nguyen TN, Goodrich JA. Human Taf(II)130 is a coactivator for NFATp. Mol Cell Biol. 2001;21:3503–3513. doi: 10.1128/MCB.21.10.3503-3513.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.