

Abstract
Objective:
To assess the specificity of the dendritic protein neurogranin (Ng) in CSF from patients with a broad range of neurodegenerative diseases including a variety of dementias, tauopathies, and synucleinopathies.
Method:
An optimized immunoassay was used to analyze CSF Ng in a retrospective cohort of 331 participants with different neurodegenerative diseases, including healthy controls (n = 19), biomarker-proven Alzheimer disease (AD) (n = 100), genetic AD (n = 2), behavioral variant frontotemporal dementia (n = 20), speech variant frontotemporal dementia (n = 21), Lewy body dementia (n = 13), Parkinson disease (n = 31), progressive supranuclear palsy (n = 46), multiple system atrophy (n = 29), as well as a heterogeneous group with non-neurodegenerative cognitive impairment (n = 50). CSF Ng concentrations and correlations of CSF Ng with total tau, phosphorylated tau, and β-amyloid 42 concentrations, Mini-Mental State Examination score, and disease duration in the different groups were investigated.
Results:
Median CSF Ng concentration was higher in patients with AD compared to both controls (p < 0.001) and all other disease groups (all p < 0.001) except speech variant frontotemporal dementia. There were no significant differences in CSF Ng concentrations between any other neurodegenerative groups and controls. In addition, we found strong correlations between Ng and total tau (p < 0.001) and phosphorylated tau (p < 0.001).
Conclusions:
These results confirm an increase in CSF Ng concentration in patients with AD as previously reported and show that this is specific to AD and not seen in a range of other neurodegenerative diseases.
There is substantial evidence that synapse loss is an early event in Alzheimer disease (AD) preceding neuronal cell death and cognitive decline.1–3 Furthermore, synapse loss is a better correlate of cognitive decline than both plaque and tangle pathology.2,4,5 Neurogranin (Ng) is a neuron-specific postsynaptic protein that is mainly expressed in the cortex, hippocampus, and amygdala by excitatory neurons,6,7 i.e., the same brain regions that are affected in AD. It has a key role in synaptic plasticity, enhancing synaptic strength by regulating the availability of calmodulin.8–11 It has previously been shown that Ng levels are significantly lower in the cortex and hippocampus of patients with AD compared to controls.12,13
In a pilot study, CSF concentration of Ng was found to be increased in patients with AD, using a semiquantitative immunoprecipitation and Western blot method.14 The production of novel anti-Ng monoclonal antibodies has enabled quantification of low concentrations of Ng C-terminal peptides found in CSF.15 Consequently, Ng concentration was shown to be increased in CSF from patients with mild cognitive impairment due to AD, as well as from patients with AD compared to controls in several independent cohorts.15–17 Furthermore, Ng can predict conversion from mild cognitive impairment to AD, as well as predict faster cognitive decline and hippocampal atrophy rates in amyloid-positive patients with prodromal AD.15,18
Using a sensitive sandwich immunoassay with electrochemiluminescence detection to investigate the CSF from well-characterized patients with AD and a range of other neurodegenerative disorders associated with synapse dysfunction, we aimed to explore to what extent Ng increase is specific to AD.
METHODS
Standard protocol approvals, registrations, and patient consents.
The study was conducted in accordance with local clinical research regulations with appropriate approval from the London, Queen Square ethical committee.
Study participants.
This is an exploratory retrospective cross-sectional study of 331 participants seen at clinics at the National Hospital for Neurology and Neurosurgery, Queen Square, London, UK, with CSF available for analysis. Patients were classified using established diagnostic criteria into those with a primary dementia including AD and AD variants,19–21 Lewy body dementia (LBD),22 behavioral and speech variant frontotemporal dementia (bvFTD and svFTD, respectively),23 a parkinsonian condition including Parkinson disease (PD),24 multiple system atrophy (MSA),25 progressive supranuclear palsy (PSP),26 nonprogressive cognitive impairment, and healthy controls. Diagnosis was based on detailed clinical assessment including neuroimaging, neuropsychometry, and CSF biomarker data and had to fulfill clinical diagnosis criteria.
Furthermore, we only included patients with AD who satisfied the latest International Working Group 2 criteria19 with an AD-indicative CSF AD biomarker profile (β-amyloid 42 [Aβ42] <550 pg/mL, and total tau [t-tau]/Aβ42 >0.52) according to previously determined cutoffs.27 Two patients with AD had confirmed presenilin 1 (PSEN1) mutations (A426P and L424V). All patients with an unclear/mixed diagnosis were excluded from the analysis. Healthy controls were spouses or friends of the patients with no history or symptoms of neurodegenerative disease at the time of lumbar puncture. These individuals underwent a thorough neurologic examination as well as a standardized neuropsychological assessment using the Mattis Dementia Rating Scale.28,29 To ensure a “pure” group of healthy controls, we implemented stricter criteria and excluded those who had either Aβ42 <550 pg/mL or a t-tau/Aβ42 >0.52 or both.
Disease duration was recorded as the time in months from symptom onset to lumbar puncture. Most patients underwent Mini-Mental State Examination (MMSE) for grading of global cognitive ability30 as well as examinations of CSF biomarker profiles. At follow-up, one patient was diagnosed with prion disease and was excluded from the study.
In separate analyses, we also grouped the 331 participants according to their CSF Aβ42 and tau profiles, disregarding their clinical diagnoses. In addition to typical AD (CSF Aβ42 <550 pg/mL and CSF t-tau/Aβ42 >0.52)27 and non-AD groups (CSF Aβ42 >550 pg/mL and CSF t-tau/Aβ42 <0.52), we identified a gray zone group with intermediate results. We excluded all healthy controls in this analysis.
CSF collection, storage, and analysis.
A standardized protocol for the collection and storage of CSF was followed.31 Briefly, CSF was collected in sterile polypropylene tubes, centrifuged at 4,000 rpm for 10 minutes at +4°C. The supernatant was divided into 0.5-mL aliquots that were stored at −80°C. Blood-contaminated samples were excluded by visual inspection.
CSF t-tau, phosphorylated tau at threonine 181 (p-tau181), and Aβ42 were analyzed using INNOTEST ELISAs (Fujirebio Europe NV, Gent, Belgium). All analyses were performed by board-certified laboratory technicians blinded to clinical information.
Sandwich immunoassay for Ng.
The monoclonal antibody Ng7, which recognizes the C-terminus of Ng (epitope Ng52–65), was found to be optimal for ELISA during method development and was used as a capturing antibody.15 Ng7 was coated on 96-well microtiter plates at a final concentration of 2.0 μg/mL (40 μL/well) in phosphate-buffered saline (PBS) and placed on a shaker for 10 minutes before incubating overnight at room temperature. After plates were washed 4 times with PBS containing 0.05% Tween (PBS-Tween), the remaining protein binding sites were blocked with blocking solution (150 μL/well) consisting of MSD blocker A (1.25 g), MQ H2O (20 mL), and PBS (1.25×) for 1 hour at room temperature. Plates were washed 4 times with PBS-Tween. The Ng calibrators (full-length recombinant Ng with concentrations ranging between 31.3 and 4,000 pg/mL), blanks, internal controls, and CSF samples were incubated in duplicate with the primary rabbit anti-Ng antibody (Upstate) diluted 1:20,000 in 0.1% BSA-PBS-Tween (all 50 μL/well) for 1 hour on the shaker and then overnight at room temperature. After washing 4 times with PBS-Tween, 0.5 μg/mL of detection antibody (goat anti-rabbit sulfo-tag) was added and incubated for 2 hours on a shaker. After washing 4 times with PBS-Tween, 150 μL of read buffer (2×) was added per well and the plate read using electrochemiluminescence (Meso Scale Discovery, Rockville, MD). All analyses were performed on one occasion with randomized samples using one batch of reagents by board-certified laboratory technicians blinded to clinical information to avoid bias. The lower and higher limits of quantification were 120 pg/mL and 4,000 pg/mL, respectively. Intra-assay coefficients of variation were 21.9% for the high-concentration Ng control and 29.8% for the low-concentration Ng control.
Statistical analysis.
We compared Ng levels between the different clinical groups and also according to the CSF AD biomarker profile. Because of small numbers, we excluded those with genetic AD from the statistical analyses, but no other data were missing or excluded. To compare demographic, CSF biomarker, and clinical data between groups, we used the nonparametric Kruskal–Wallis test with the Dunn test to correct for multiple comparisons and report p values both with and without correction for multiple comparisons. To further account for multiple testing, we set the threshold for statistical significance to p < 0.01. Fisher exact test was used to compare the distribution of categorical data across groups. Data are shown as medians and interquartile ranges for numerical data or as a percentage for categorical data. Correlations between biomarker data were assessed using Spearman rank correlation. The optimal cutoff point of CSF Ng to differentiate patients with AD from healthy controls was identified by selecting the CSF Ng concentration that produced the highest Youden32 index (J = sensitivity + specificity – 1). This cutoff point was used to calculate specificities against non-AD diagnoses as a whole group, as well as against each non-AD diagnosis. Statistical analysis was performed using commercial software (GraphPad Prism version 6.00 for Windows; GraphPad Software, San Diego CA).
RESULTS
Demographics.
We measured Ng concentrations in CSF from 331 participants including patients with sporadic AD (n = 100), genetic AD (n = 2), bvFTD (n = 20), svFTD (n = 21), LBD (n = 13), PD (n = 31), PSP (n = 46), MSA (n = 29), and those who had non-neurodegenerative cognitive problems (non-ND, n = 50). Clinical and biomarker characteristics are shown in table 1. We found an overall difference in sex, age, and disease duration across the groups (see table 1). MMSE scores were lower in the AD, bvFTD, LBD, PD, PSP, and non-ND groups compared with healthy controls. In patients with non-AD diagnoses, we found no overall difference in the proportion of those with Aβ42 <550 pg/mL. We found an overall difference in the number who satisfied t-tau/Aβ42 >0.52 with LBD having the highest proportion (69%) and those with PSP having the lowest proportion (28%). We found an overall difference in the proportion who satisfied both Aβ42 <550 pg/mL and t-tau/Aβ42 >0.52 with LBD having the highest proportion (54%) and PSP having the lowest proportion (13%).
Table 1.
Demographics of controls and patients with different neurodegenerative diseases
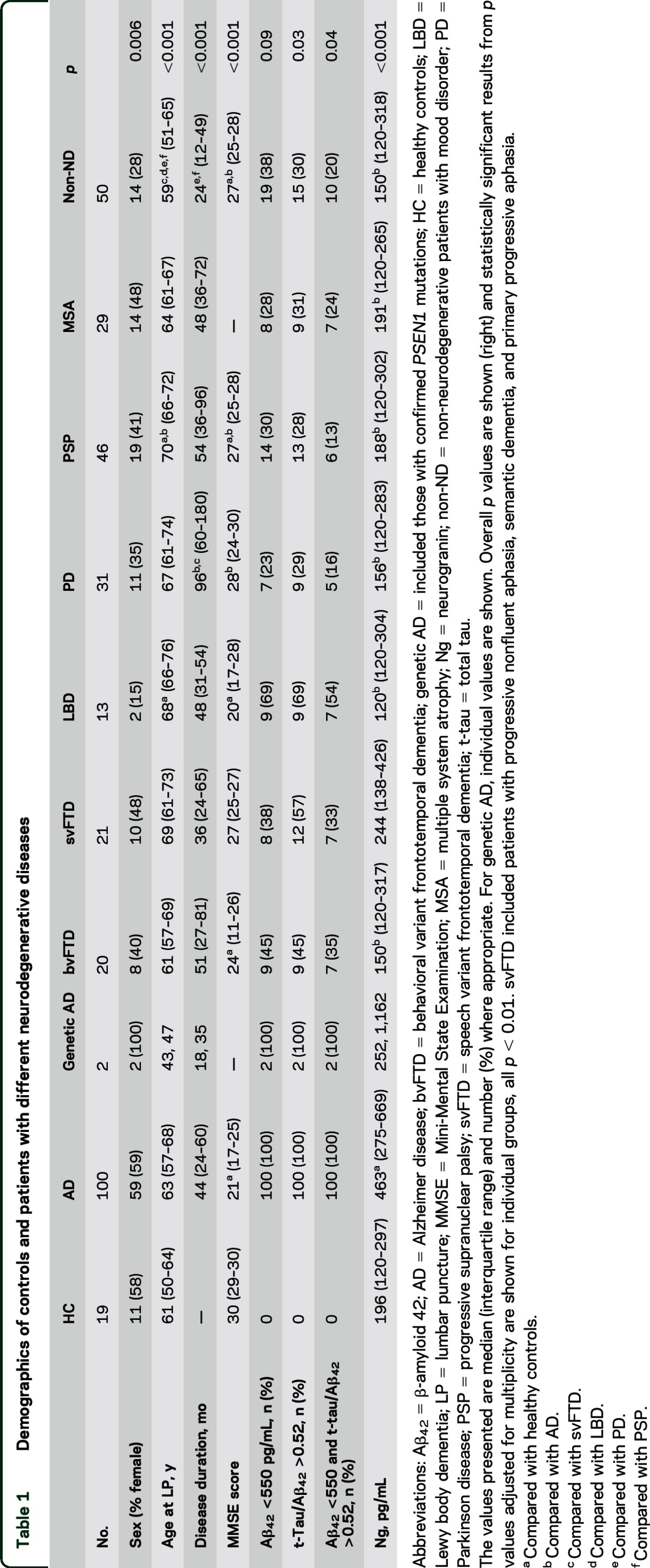
CSF Ng concentrations across different neurodegenerative disorders.
The AD group had higher CSF Ng concentrations (236% of control levels) compared with controls, bvFTD, LBD, PD, PSP, MSA, and non-ND groups (all p < 0.001; table 1, figure 1). CSF Ng concentrations for the 2 patients with genetic AD were quite different with one clearly increased CSF Ng concentration of 1,162 pg/mL in a PSEN1 A426P mutation carrier and another concentration in the upper normal range (252 pg/mL in a PSEN1 L424V mutation carrier). There was no evidence that any other disease group (61%–124% of control levels), apart from AD, had increased CSF Ng concentrations compared with controls.
Figure 1. Increased CSF Ng concentrations in patients with AD.
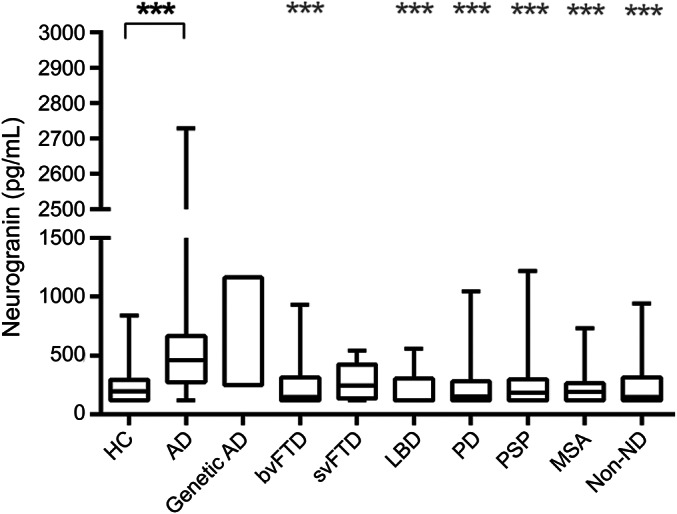
Boxplots showing CSF Ng concentrations across different diagnostic groups. Ng concentrations were significantly higher in the AD group compared to control participants. The lower, upper, and middle lines correspond to the 25th centile, 75th centile, and median, respectively. The whiskers extend to the minimum and maximum Ng data points. For genetic AD, the lower and upper lines of the box correspond to the individual CSF Ng concentrations. The differences between the groups were assessed using Kruskal–Wallis test followed by the Dunn multiple comparisons test. ***Compared to control, p < 0.001; ***compared to AD, p < 0.001. AD = Alzheimer disease (includes typical and atypical); bvFTD = behavioral variant frontotemporal dementia; genetic AD = those with confirmed PSEN1 mutations; HC = healthy controls; LBD = Lewy body dementia; MSA = multiple system atrophy; Ng = neurogranin; non-ND = non-neurodegenerative patients with mood disorder; PD = Parkinson disease; PSP = progressive supranuclear palsy; svFTD = speech variant frontotemporal dementia (includes patients with progressive nonfluent aphasia, semantic dementia, and primary progressive aphasia).
CSF Ng concentrations in patients grouped according to CSF AD biomarkers.
Of the 312 patients, independent of clinical diagnosis, we determined that 109 patients had a non-AD-indicative CSF biomarker profile, 151 an AD-indicative CSF profile, with the remaining 52 having intermediate gray zone results. CSF Ng levels were higher in the AD biomarker-positive group (median, 361 pg/mL) compared with controls (median, 180 pg/mL) as well as the gray group (median, 152 pg/mL) (both p < 0.001) (figure 2).
Figure 2. Elevated CSF Ng concentrations in patients with AD-indicative CSF profiles.
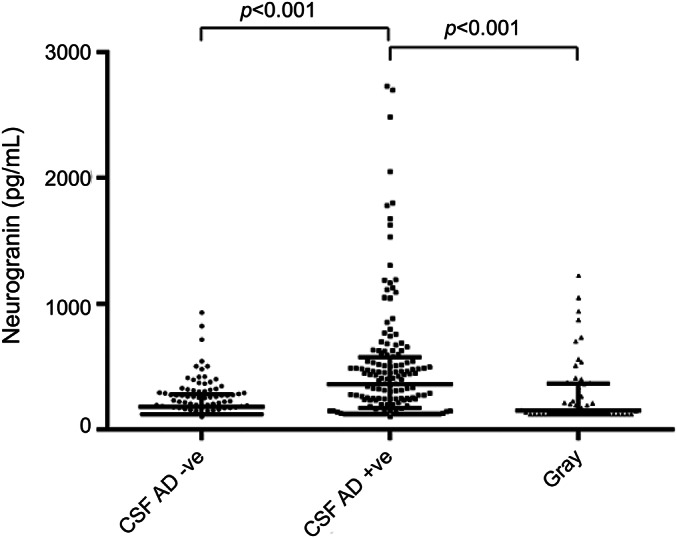
Scatter plots displaying CSF Ng concentrations in patients with non-AD-indicative (n = 109) and AD-indicative (n = 151) CSF profiles, as well as with gray zone values (n = 52). The middle line shows the median. The lower and upper lines correspond to interquartile range. The differences between the groups were assessed using Kruskal–Wallis test followed by the Dunn multiple comparisons test. A p value <0.01 was considered statistically significant. AD = Alzheimer disease; Ng = neurogranin.
Diagnostic values for Ng.
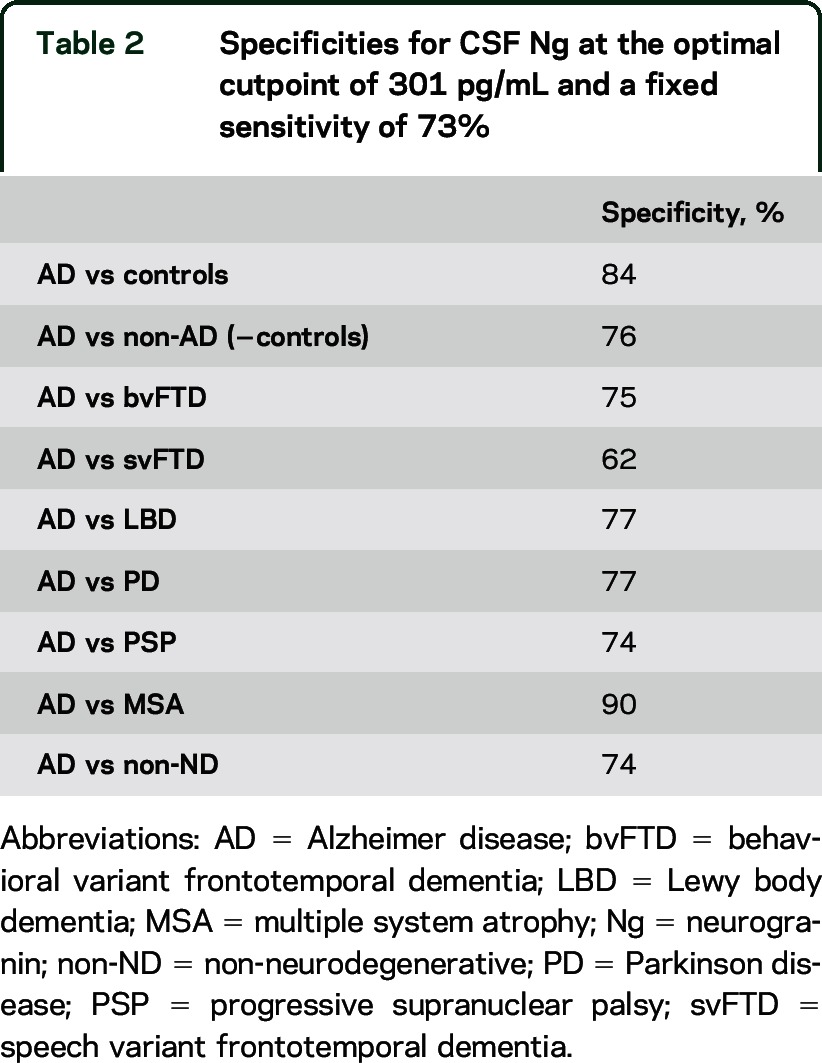
Using the Youden index, we found that the optimal cutoff point for CSF Ng to differentiate patients with AD from healthy controls was 301 pg/mL, which gave a sensitivity of 73% and a specificity of 84% (table 2). At this cutoff point, the specificities for differentiating AD from non-AD patients, as well as from patients with specific non-AD diagnoses, were above 70% for all comparisons, except for svFTD (62%) (table 2).
Table 2.
Specificities for CSF Ng at the optimal cutpoint of 301 pg/mL and a fixed sensitivity of 73%
Correlation between AD biomarkers and Ng.
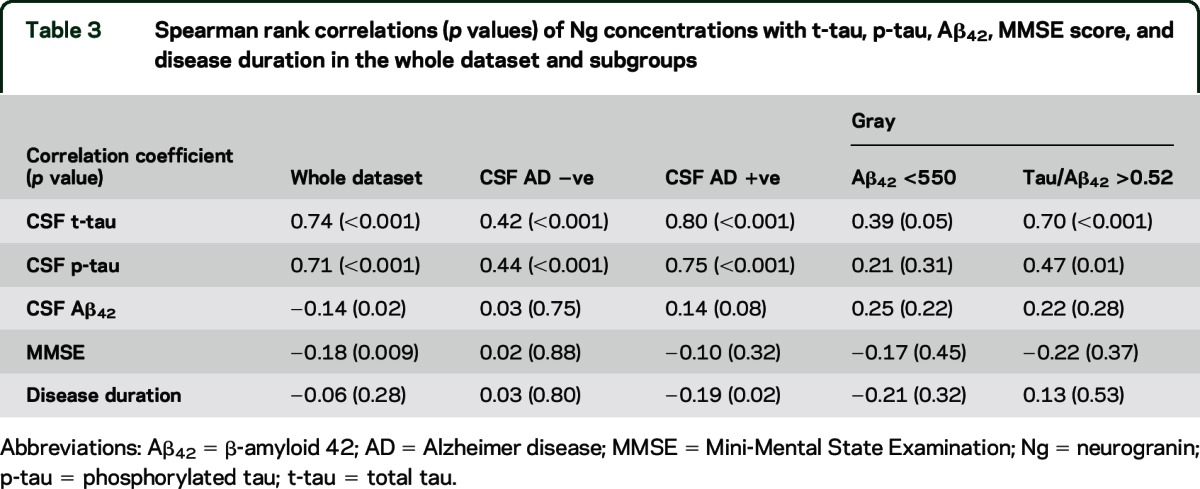
We investigated correlations between CSF Ng concentrations and AD biomarkers, including t-tau, p-tau, Aβ42, MMSE scores, and disease duration in patients with non-AD, AD, and gray CSF profiles (table 3). As Ng levels increased, t-tau and p-tau concentrations increased (rs = 0.74 and 0.71, respectively, both p < 0.001) in the whole dataset, as well as in the CSF AD biomarker-negative and -positive groups (table 3). A weaker association of CSF Ng with tau was found in the subgroup of patients who satisfied the Aβ42 <550 pg/mL criterion only, with a stronger correlation in those who satisfied the t-tau/Aβ42 >0.52 criterion. We found that high CSF Ng concentrations were associated with low Aβ42 concentrations in the whole dataset; however, the correlation was weak (rs = −0.14, p = 0.02). Similarly, high CSF Ng concentrations were associated with low MMSE scores in the whole dataset. Again, this was a weak correlation (rs = −0.18, p = 0.009), which was absent in the subgroups. No overall correlation was found between CSF Ng and disease duration in the whole dataset, but in the CSF AD biomarker-positive group, we found that high CSF Ng concentration correlated weakly with short disease duration (rs = −0.19, p = 0.02).
Table 3.
Spearman rank correlations (p values) of Ng concentrations with t-tau, p-tau, Aβ42, MMSE score, and disease duration in the whole dataset and subgroups
DISCUSSION
We confirm that CSF concentrations of the synaptic protein Ng are higher in patients with AD who satisfy the latest International Working Group 2 criteria compared to controls. We extend this result by showing that this increase is specific to AD and not seen in other neurodegenerative diseases, including a variety of dementias. Finally, we demonstrate a positive correlation of CSF Ng with markers of neurodegeneration.
It is well established that in the AD-affected brain, there is substantial synapse loss in the cortex, which correlates better with cognitive decline than plaque and tangle pathology.1,12 Furthermore, Ng levels are reduced in the hippocampus and frontal lobes in patients with AD.12,13 Previous studies have confirmed that by contrast, CSF concentrations of this protein are elevated in AD,14–16,18 a result that we confirm. One potential explanation for the apparent dissociation between brain and CSF Ng concentrations in AD is that Ng brain levels relate to the total synapse density, which is considerably reduced in AD, while the CSF concentrations reflect ongoing further synapse loss. This causes a continuous leakage of Ng into the brain interstitial fluid, which is cleared into the CSF resulting in higher CSF Ng concentrations. An alternate explanation is that pathologic processes in AD somehow stimulate Ng secretion from dendrites into the CSF in a manner similar to how Aβ has been proposed to increase tau expression and release from neurons, also in the absence of frank neuronal death.33,34 Further studies are required to elucidate these mechanisms in detail.
Given that synapse degeneration occurs as part of any neurodegenerative process, it may be considered surprising that other canonical non-AD neurodegenerative disorders, e.g., FTD, did not have significantly elevated CSF Ng concentrations compared to controls. However, the main brain regions affected in AD, namely, the parietal and temporal cortices, amygdala, and hippocampus, are also the regions with the highest Ng protein expression,6,7 which may be the reason for the apparent AD specificity of CSF Ng. Of note, CSF Ng concentrations were slightly higher in svFTD compared with bvFTD, which might be due to greater synapse loss in the temporal lobes compared to the frontal cortex in this condition. This likely reflects that a proportion of these cases have AD pathology. The apparently AD-specific Ng elevation may thus not only be useful in differential diagnosis but could also provide important insights into selective neuronal vulnerability in neurodegenerative diseases.35
We confirm that Ng concentration positively correlates with both t-tau and p-tau, suggesting that Ng is linked to tau-related neurodegeneration.15 While there was a weak correlation between Ng and Aβ42 levels across the whole dataset, likely reflecting the proportion of individuals having underlying AD pathology, there were no correlations in individual subgroups, suggesting little or no relationship between plaque load and synapse loss. This supports the idea that Aβ may be necessary for initiation of disease pathology but that it is not directly related to synapse loss, which is more closely associated with neurodegeneration, itself relating to subsequent cognitive decline.36 While there was no relationship between disease duration and Ng across the cohort, there was a significant negative correlation between Ng and disease duration in those with an AD-biomarker picture, suggesting that Ng elevation in AD may be highest either in those with more rapid disease, or very early in the disease process.
One limitation of the study is the small sample size in some groups, most notably in those with genetic AD. With this caveat, it is of interest to note that one of the highest Ng values was observed in a PSEN1 mutation carrier (A426P, Ng 1,162 pg/mL)—whether this reflects familial AD in general or specific mutations/mechanisms needs to be assessed in larger cohorts. Another limitation of our study is the relatively high coefficients of variation of the Ng assay (20%–30%). This makes it harder to interpret individual CSF Ng concentrations in relation to fixed cutoff points, as well as to detect treatment-induced changes over time in CSF Ng concentrations in clinical trials. Developing more precise assays for CSF Ng is an important area for further study. A third limitation is that while the patients with AD had their clinical diagnosis supported by biomarkers, the other diagnoses were made on clinical grounds only, as diagnostically useful biomarkers for these disorders are presently lacking. Furthermore, all controls were AD biomarker-negative. This enrichment approach allows for a very pure control group to be defined, with the advantage that our results may reflect “real” AD vs “true” control differences; in less well characterized individuals and in particular given that a proportion of apparently healthy controls may have presymptomatic AD changes, results may be less discriminatory. However, the similar CSF Ng concentrations in the control group compared with the other non-AD groups suggest that this may not be a major issue in practice.
We report that CSF concentrations of Ng are significantly increased in patients with AD but not in patients with other neurodegenerative diseases. We propose that high CSF Ng concentrations reflect synapse degeneration in AD-affected brain regions and that CSF Ng has potential as a diagnostic marker for AD in combination with existing CSF biomarkers. Further studies are required to test the hypothesis that CSF Ng may have utility as a very early and potentially presymptomatic biomarker for AD, as a prognostic marker in the clinic, and as an outcome measure in clinical trials.
ACKNOWLEDGMENT
The authors thank Dr. Nicholas Counsell and Mr. Andre Lopes at the CRUK clinical trials center, UCL, for their general statistical advice.
GLOSSARY
- Aβ42
β-amyloid 42
- AD
Alzheimer disease
- bvFTD
behavioral variant frontotemporal dementia
- LBD
Lewy body dementia
- MMSE
Mini-Mental State Examination
- MSA
multiple system atrophy
- Ng
neurogranin
- non-ND
non-neurodegenerative
- PBS
phosphate-buffered saline
- PD
Parkinson disease
- PSEN1
presenilin 1
- PSP
progressive supranuclear palsy
- p-tau
phosphorylated tau
- svFTD
speech variant frontotemporal dementia
- t-tau
total tau
AUTHOR CONTRIBUTIONS
Study concept and design: Blennow, Schott, Zetterberg. Acquisition of data: Wellington, Törnqvist. Analysis and interpretation of data: Wellington, Paterson, Portelius, Törnqvist, Magdalinou, Fox, Blennow, Schott, Zetterberg. Drafting of the manuscript: Wellington, Paterson, Schott, Zetterberg. Critical revision of the manuscript for important intellectual content: Fox, Blennow, Portelius, Törnqvist, Magdalinou. Statistical analysis: Wellington, Schott, Zetterberg. Obtained funding: Fox, Blennow, Zetterberg. Administrative, technical, and material support: Törnqvist, Magdalinou, Paterson. Study supervision: Fox, Blennow, Schott, Zetterberg.
STUDY FUNDING
This study was supported by grants from the Wolfson Foundation, the Swedish Research Council, the Knut and Alice Wallenberg Foundation, Frimurarstiftelsen, Alzheimerfonden, Hjärnfonden, the Agneta Prytz-Folke and Gösta Folke Foundation, and the Torsten Söderberg Foundation. The DRC is an Alzheimer's Research UK coordinating center. This work was supported by the NIHR UCL/UCLH Biomedical Research Centre and the NIHR Queen Square Dementia Biomedical Research Unit. N.C.F. is an NIHR Senior Investigator. J.M.S. acknowledges the support of the NIHR Queen Square Dementia BRU, the NIHR UCL/H Biomedical Research Centre, Wolfson Foundation, EPSRC (EP/J020990/1), MRC (CSUB19166), ARUK (ARUK-Network 2012-6-ICE; ARUK-PG2014-1946), and European Commission (H2020-PHC-2014-2015-666992). No sponsors were included in the study design, data collection, analysis and interpretation, or writing of the manuscript.
DISCLOSURE
H. Wellington, R. Paterson, E. Portelius, U. Törnqvist, N. Magdalinou, and N. Fox report no disclosures relevant to the manuscript. K. Blennow is cofounder of Brain Biomarker Solutions, a GU Holding-based platform company at the University of Gothenburg, has served at advisory boards for IBL International and Roche Diagnostics, and has given lectures for Fujirebio Europe. J. Schott reports no disclosures relevant to the manuscript. H. Zetterberg is cofounder of Brain Biomarker Solutions, a GU Holding-based platform company at the University of Gothenburg. Apart from this, he reports no disclosures. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Dekosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alzheimer's disease: correlation with cognitive severity. Ann Neurol 1990;27:457–464. [DOI] [PubMed] [Google Scholar]
- 2.Terry RD, Masliah E, Salmon DP, et al. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol 1991;30:572–580. [DOI] [PubMed] [Google Scholar]
- 3.Selkoe DJ. Alzheimer's disease is a synaptic failure. Science 2002;298:789–791. [DOI] [PubMed] [Google Scholar]
- 4.Scheff SW, Price DA, Dekosky ST, Mufson EJ. Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology 2007;68:1501–1508. [DOI] [PubMed] [Google Scholar]
- 5.Blennow K, Bogdanovic N, Alafuzoff I, Ekman R, Davidsson P. Synaptic pathology in Alzheimer's disease: relation to severity of dementia, but not to senile plaques, neurofibrillary tangles, or the ApoE4 allele. J Neural Transm 1996;103:603–618. [DOI] [PubMed] [Google Scholar]
- 6.Represa A, Deloulme JC, Sensenbrenner M, Ben-Ari Y, Baudier J. Neurogranin: immunocytochemical localization of a brain-specific protein kinase C substrate. J Neurosci 1990;10:3782–3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guadaño-Ferraz A, Viñuela A, Oeding G, Bernal J, Rausell E. RC3/neurogranin is expressed in pyramidal neurons of motor and somatosensory cortex in normal and denervated monkeys. J Comp Neurol 2005;493:554–570. [DOI] [PubMed] [Google Scholar]
- 8.Zhong L, Gerges NZ. Neurogranin targets calmodulin and lowers the threshold for the induction of long-term potentiation. PLoS One 2012;7:e41275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhong L, Cherry T, Bies CE, Florence MA, Gerges NZ. Neurogranin enhances synaptic strength through its interaction with calmodulin. EMBO J 2009;28:3027–3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Watson JB, Sutcliffe JG, Fisher RS. Localization of the protein kinase C phosphorylation/calmodulin-binding substrate RC3 in dendritic spines of neostriatal neurons. Proc Natl Acad Sci USA 1992;89:8581–8585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martzen MR, Slemmon JR. The dendritic peptide neurogranin can regulate a calmodulin-dependent target. J Neurochem 1995;64:92–100. [DOI] [PubMed] [Google Scholar]
- 12.Reddy PH, Mani G, Park BS, et al. Differential loss of synaptic proteins in Alzheimer's disease: implications for synaptic dysfunction. J Alzheimers Dis 2005;7:103–117. [DOI] [PubMed] [Google Scholar]
- 13.Davidsson P, Blennow K. Neurochemical dissection of synaptic pathology in Alzheimer's disease. Int Psychogeriatr 1998;10:11–23. [DOI] [PubMed] [Google Scholar]
- 14.Thorsell A, Bjerke M, Gobom J, et al. Neurogranin in cerebrospinal fluid as a marker of synaptic degeneration in Alzheimer's disease. Brain Res 2010;1362:13–22. [DOI] [PubMed] [Google Scholar]
- 15.Kvartsberg H, Duits FH, Ingelsson M, et al. Cerebrospinal fluid levels of the synaptic protein neurogranin correlates with cognitive decline in prodromal Alzheimer's disease. Alzheimers Dement 2015;11:1180–1190. [DOI] [PubMed] [Google Scholar]
- 16.De Vos A, Jacobs D, Struyfs H, et al. C-terminal neurogranin is increased in cerebrospinal fluid but unchanged in plasma in Alzheimer's disease. Alzheimers Dement 2015;11:1461–1469. [DOI] [PubMed] [Google Scholar]
- 17.Kvartsberg H, Portelius E, Andreasson U, et al. Characterization of the postsynaptic protein neurogranin in paired cerebrospinal fluid and plasma samples from Alzheimer's disease patients and healthy controls. Alzheimers Res Ther 2015;7:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Portelius E, Zetterberg H, Skillbäck T, et al. Cerebrospinal fluid neurogranin: relation to cognition and neurodegeneration in Alzheimer's disease. Brain 2015;138:3373–3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dubois B, Feldman HH, Jacova C, et al. Advancing research diagnostic criteria for Alzheimer's disease: the IWG-2 criteria. Lancet Neurol 2014;13:614–629. [DOI] [PubMed] [Google Scholar]
- 20.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 1991;82:239–259. [DOI] [PubMed] [Google Scholar]
- 21.Gorno-Tempini ML, Hillis AE, Weintraub S, et al. Classification of primary progressive aphasia and its variants. Neurology 2011;76:1006–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McKeith IG, Galasko D, Kosaka K, et al. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the Consortium on DLB International Workshop. Neurology 1996;47:1113–1124. [DOI] [PubMed] [Google Scholar]
- 23.The Lund and Manchester Groups. Clinical and neuropathological criteria for frontotemporal dementia. J Neurol Neurosurg Psychiatry 1994;57:416–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson's disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry 1992;55:181–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gilman S, Wenning G, Low P, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology 2008;71:670–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Litvan I, Agid Y, Jankovic J, et al. Accuracy of clinical criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome). Neurology 1996;46:922–930. [DOI] [PubMed] [Google Scholar]
- 27.Duits FH, Teunissen CE, Bouwman FH, et al. The cerebrospinal fluid “Alzheimer profile”: easily said, but what does it mean? Alzheimers Dement 2014;10:713–723. [DOI] [PubMed] [Google Scholar]
- 28.Magdalinou NK, Paterson RW, Schott JM, et al. A panel of nine cerebrospinal fluid biomarkers may identify patients with atypical parkinsonian syndromes. J Neurol Neurosurg Psychiatry 2015;86:1240–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schmidt K, Mattis P, Adams J, Nestor P. Alternate-form reliability of the Dementia Rating Scale–2. Arch Clin Neuropsychol 2005;20:435–441. [DOI] [PubMed] [Google Scholar]
- 30.Folstein MF, Folstein SE, McHugh PR. “Mini-Mental State”: a practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975;12:189–198. [DOI] [PubMed] [Google Scholar]
- 31.Blennow K, Hampel H, Weiner M, Zetterberg H. Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat Rev Neurol 2010;6:131–144. [DOI] [PubMed] [Google Scholar]
- 32.Youden WJ. Index for rating diagnostic tests. Cancer 1950;3:32–35. [DOI] [PubMed] [Google Scholar]
- 33.Moore S, Evans LDB, Andersson T, et al. APP metabolism regulates tau proteostasis in human cerebral cortex neurons. Cell Rep 2015;11:689–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maia LF, Kaeser SA, Reichwald J, Hruscha M, Martus P. Changes in amyloid-β and tau in the cerebrospinal fluid of transgenic mice overexpressing amyloid precursor protein. Sci Transl Med 2013;5:194re2. [DOI] [PubMed] [Google Scholar]
- 35.Warren JD, Rohrer JD, Schott JM, Fox NC, Hardy J, Rossor MN. Molecular nexopathies: a new paradigm of neurodegenerative disease. Trends Neurosci 2013;36:561–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Spires-Jones TL, Hyman B. The intersection of amyloid beta and tau at synapses in Alzheimer's disease. Neuron 2014;82:756–771. [DOI] [PMC free article] [PubMed] [Google Scholar]