

Abstract
Objective:
To image β-amyloid (Aβ) plaque burden in long-term survivors of traumatic brain injury (TBI), test whether traumatic axonal injury and Aβ are correlated, and compare the spatial distribution of Aβ to Alzheimer disease (AD).
Methods:
Patients 11 months to 17 years after moderate–severe TBI underwent 11C-Pittsburgh compound B (11C-PiB)-PET, structural and diffusion MRI, and neuropsychological examination. Healthy aged controls and patients with AD underwent PET and structural MRI. Binding potential (BPND) images of 11C-PiB, which index Aβ plaque density, were computed using an automatic reference region extraction procedure. Voxelwise and regional differences in BPND were assessed. In TBI, a measure of white matter integrity, fractional anisotropy, was estimated and correlated with 11C-PiB BPND.
Results:
Twenty-eight participants (9 with TBI, 9 controls, 10 with AD) were assessed. Increased 11C-PiB BPND was found in TBI vs controls in the posterior cingulate cortex and cerebellum. Binding in the posterior cingulate cortex increased with decreasing fractional anisotropy of associated white matter tracts and increased with time since injury. Compared to AD, binding after TBI was lower in neocortical regions but increased in the cerebellum.
Conclusions:
Increased Aβ burden was observed in TBI. The distribution overlaps with, but is distinct from, that of AD. This suggests a mechanistic link between TBI and the development of neuropathologic features of dementia, which may relate to axonal damage produced by the injury.
Traumatic brain injury (TBI) is the leading cause of disability in young adults.1 Survivors may deteriorate clinically many years after injury,2 and TBI is thought to be a major risk factor for dementia.3 However, the mechanisms relating acute injury to later neurodegeneration are unclear, and the prevalence of distinct types of dementia such as Alzheimer disease (AD) and chronic traumatic encephalopathy is uncertain.3
A mechanistic link between moderate to severe TBI and AD is suggested by the observation that β-amyloid (Aβ) aggregates are found in brains of up to a third of patients who die acutely after TBI,3 and in a similar proportion who survive for a year or more.4 Traumatic axonal injury (TAI), a pathology consistently observed after TBI,5 offers a potential mechanism for Aβ genesis.6 It is postulated that abundant amyloid precursor protein, which accumulates in damaged axons, is aberrantly cleaved to form Aβ, which subsequently aggregates as Aβ plaques.6 Immunohistochemical evidence also shows that the enzymes necessary for Aβ cleavage accumulate at sites of TAI.6
Localization of fibrillar Aβ pathology in vivo is possible using PET. The amyloid tracer 11C-Pittsburgh compound B (11C-PiB) shows robust retention in brains of patients with AD7 in a pattern that corresponds with neuropathologic studies of Aβ plaque distribution, with increases initially in the precuneus/posterior cingulate cortex (PCC), frontal cortex, and caudate nuclei, then lateral temporal and parietal cortex.8,9 Recently, a pilot 11C-PiB-PET study in patients with moderate to severe TBI less than 1 year after injury found increased uptake in cortical gray matter (GM) and striatum.10 These findings suggest that Aβ imaging in the chronic phase after TBI may inform our understanding of neurodegeneration in long-term survivors of TBI.
Diffusion tensor imaging (DTI) can be used to estimate in vivo the degree of axonal injury following TBI.11–14 In this study, we combined 11C-PiB-PET and DTI to test the following hypotheses: (1) Aβ pathology is present in long-term survivors of TBI without dementia; and (2) Aβ pathology after moderate to severe TBI is related to the amount and distribution of TAI.
METHODS
Study design and participants.
In this cross-sectional study, 9 patients with a history of a single moderate–severe TBI based on Mayo criteria15 were assessed with 11C-PiB-PET, structural T1 MRI, DTI, and neuropsychological examination. Patients were recruited at least 11 months after their injury (e-Methods on the Neurology® Web site at Neurology.org). For comparison of 11C-PiB binding, a group of patients with AD had 11C-PiB-PET and structural MRI (e-Methods). We used 3 healthy control groups: (1) for comparison of 11C-PiB binding, a group of healthy aged controls had PiB-PET and structural MRI; (2) for comparison of neuropsychological performance, a second group of healthy controls, age-matched to the patients with TBI, underwent neuropsychological assessment; and (3) for comparison of white matter (WM) integrity, a third group of healthy aged-matched controls underwent structural MRI and DTI.
Standard protocol approvals, registrations, and patient consents.
The project was approved by Hammersmith and Queen Charlotte's and Chelsea Research Ethics Committee. All participants gave written informed consent.
Procedures.
A neuropsychological test battery was performed on patients with TBI and age-matched controls (e-Methods). Patients with AD and healthy aged controls underwent the Mini-Mental State Examination.
An overview of the imaging methods is shown in figure e-1. All patients and healthy aged controls had 11C-PiB-PET using a Siemens ECAT EXACT HR+ scanner (Siemens Medical Systems, Erlangen, Germany). 11C-PiB was manufactured and supplied by Hammersmith Imanet (London, UK). All participants had an IV bolus injection of 11C-PiB, mean dose 370 MBq, and dynamic PET emission scans were acquired over 90 minutes.
To generate nondisplaceable binding potential (BPND) images of 11C-PiB, we used a supervised clustering procedure for automatic reference region extraction.16 T1 images were automatically segmented into GM and WM. The tissue segmentations were warped to an average group template image using a diffeomorphic nonlinear image registration procedure (DARTEL).17 The group template image was then registered to Montreal Neurological Institute (MNI) space. Each individual's 11C-PiB BPND image was coregistered to their T1 image, then the individual flow-fields and template registration obtained from the DARTEL procedure were used to warp the BPND images to MNI space. The normalized BPND images were masked using the thresholded GM template and smoothed (8 mm full width at half maximum) (e-Methods).
11C-PiB binding potentials were also sampled from anatomically defined regions of interest (ROIs). The MAPER (multi-atlas propagation with enhanced registration) procedure was used to generate native-space ROIs.18 To improve sampling accuracy, ROI masks were intersected with thresholded tissue probability maps (e-Methods). To confirm that the hippocampal ROI results were not an effect of mislabeling due to atrophy, sampling was repeated on hippocampal masks that were manually segmented using a harmonized protocol.19
In patients with focal injuries, lesions apparent on T1 imaging were manually segmented and excluded from ROI and voxelwise analyses. We also investigated 11C-PiB binding within a lesion, the lesion penumbra, and normal-appearing GM in the same hemisphere (e-Methods).
Patients with TBI and a group of healthy aged-matched controls underwent DTI (e-Methods). Voxelwise maps of fractional anisotropy (FA), a measure of WM tract integrity after TBI, were calculated using the FSL Diffusion Toolkit.20 The FA maps were skeletonized using tract-based spatial statistics (TBSS).21 We calculated the mean FA of the TBSS skeleton and also of selected tracts from the Johns Hopkins University WM Tractography Atlas.22 We chose tracts connected to GM regions that had shown increased 11C-PiB binding in TBI. We also sampled the corticospinal tract as a control, since this was not connected to these regions.
Statistical analysis.
Group differences in neuropsychological measures were examined using independent sample t tests and Mann–Whitney U tests in SPSS version 21 (IBM Corp., Armonk, NY). Voxelwise differences in BPND between groups were assessed using nonparametric permutation tests in FSL with 10,000 permutations. This approach incorporated a tool that uses voxelwise regressors to exclude individual lesions from the analysis.23 Results were cluster-corrected using threshold-free cluster enhancement and a family-wise error rate of <0.05. For presentation, images were thresholded at p < 0.001 uncorrected. For ROI analysis, regional BPND was compared using repeated-measures analysis of variance (ANOVA), in SPSS. Mean FA values of WM tracts were compared between patients with TBI and controls using unpaired 2-sample t tests. Regional 11C-PiB was correlated with mean FA values, age, time since injury, and neuropsychological test scores (e-Methods). Mean FA values were correlated with age and time since injury. To correct for multiple comparisons, a false discovery rate threshold was calculated using q = 0.05.
RESULTS
Nine patients with TBI (mean age 44.1 ± 4.9 years, range 38–54) were recruited 11 months to 17 years after injury (table 1). Ten patients with AD (mean age 67.3 ± 4.5, range 58–76) and 9 healthy aged controls (62.3 ± 4.3, range 55–66) were also assessed. In addition, a group of 15 age-matched controls (37.3 ± 11.3, range 19–60) underwent neuropsychological assessment and a separate group of 11 age-matched controls (40.9 ± 5.4, range 35–51) underwent MRI and DTI. None of the patients had a clinical diagnosis of posttraumatic stress disorder or anxiety disorder. One patient had a diagnosis of depression following the TBI. Structural T1 scans were reviewed by a senior neuroradiologist. Four patients with TBI had no abnormalities. The remaining 5 had focal lesions, with damage in the frontal (n = 3) or temporal (n = 3) lobes (figure e-2). One patient had undergone a parietotemporal lobectomy following TBI.
Table 1.
Demographics and clinical data of all patients with traumatic brain injury
Neuropsychological impairment after TBI.
The patients with TBI showed impairments in neuropsychological performance compared to age-matched healthy controls. Significantly poorer responses were seen across a range of tasks, including tests of attention, information processing speed, and cognitive flexibility (table e-1). In other tests, the patients were well matched with controls. As expected, the AD group had a lower Mini-Mental State Examination score (mean 21.1/30 ± 4.1) than healthy aged controls (all 30/30, t = −6.54, df = 9, p < 0.001).
Amyloid pathology after TBI is detected by 11C-PiB binding.
11C-PiB BPND images of the TBI group are shown for individual patients (figure 1). Slices from a representative patient with AD and a healthy aged control are shown. Direct comparison of patients with TBI and healthy aged controls showed areas of increased 11C-PiB BPND following TBI (figure 2A). Peaks of increased 11C-PiB BPND corrected for multiple comparisons were observed in the precuneus/PCC and cerebellum. There were no areas of decreased binding in patients with TBI compared to controls. We performed a confirmatory ROI analysis using anatomically defined regions (figure 3). ANOVA of BPND sampled from 10 ROIs in the TBI and healthy control groups showed a significant group-by-region interaction (F3.127, 50.036 = 2.984, p = 0.038, Greenhouse-Geisser correction applied). The partial η2 effect size estimate was 0.157. The interaction was driven by increased binding in the putamen of patients with TBI (t = 2.573, df = 16, p = 0.020) and a decrease in the superior frontal gyrus (t = −2.312, df = 16, p = 0.034), but nonsignificant differences elsewhere.
Figure 1. 11C-PiB binding following TBI.

Images of axial T1 MRI are superimposed with 11C-PiB BPND maps for all patients with TBI and a representative healthy aged control and a participant with AD. For patients with TBI, the interval in months from the time of TBI to PET scanning and the age in years of each participant at scanning is also shown. AD = Alzheimer disease; BPND = nondisplaceable binding potential; 11C-PiB = 11C-Pittsburgh compound B; TBI = traumatic brain injury.
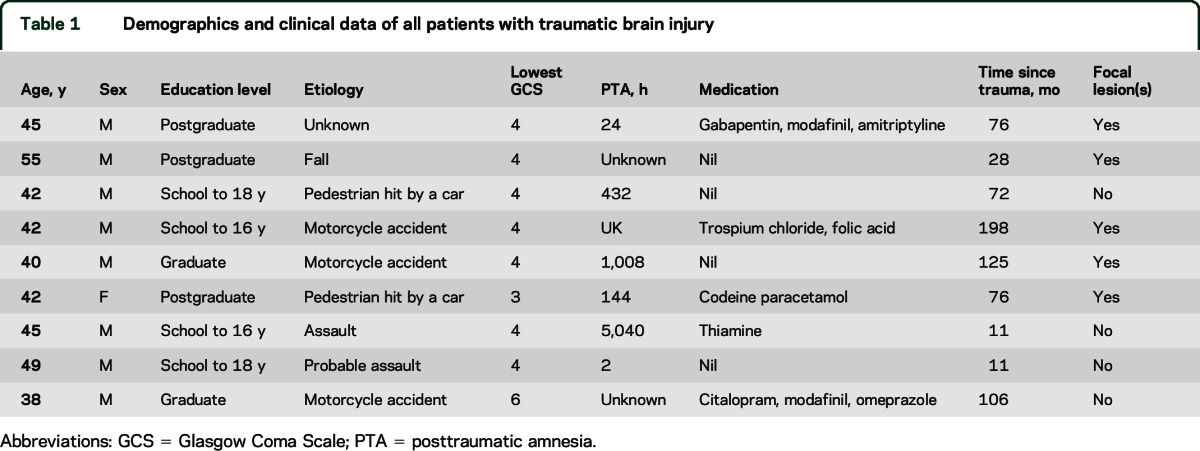
Figure 2. Increased 11C-PiB binding in TBI and AD.
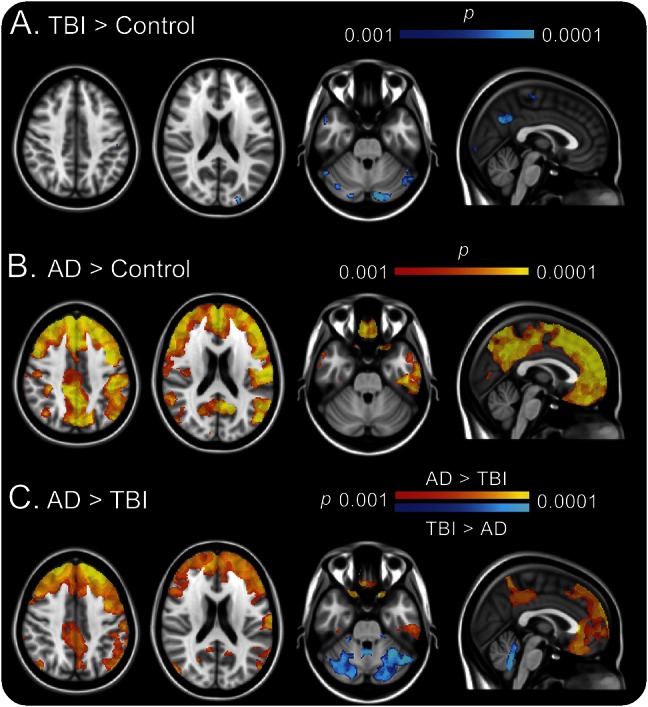
(A) Blue–light blue areas showed significantly increased 11C-PiB BPND in TBI compared to healthy aged controls. (B) Red–yellow areas showed significantly increased binding in AD compared to controls. (C) Blue–light blue areas showed significantly increased 11C-PiB BPND in TBI compared to AD. Red–yellow areas showed significantly increased binding in AD compared to TBI. Images are shown thresholded at p < 0.001 uncorrected. AD = Alzheimer disease; BPND = nondisplaceable binding potential; 11C-PiB = 11C-Pittsburgh compound B; TBI = traumatic brain injury.
Figure 3. 11C-PiB BPND region of interest analysis.

Mean group 11C-PiB BPND ± SEM is shown for patients with TBI (green), patients with AD (red), and healthy aged controls (yellow). ACC = anterior cingulate cortex; AD = Alzheimer disease; BPND = nondisplaceable binding potential; 11C-PiB = 11C-Pittsburgh compound B; Caud = caudate; Cereb = cerebellum; Hipp = hippocampus; IFG = inferior frontal gyrus; OL = occipital lobe; PCC = posterior cingulate cortex; Put = putamen; SFG = superior frontal gyrus; Skel = skeleton; TBI = traumatic brain injury; Thal = thalamus.
11C-PiB binding is decreased around focal lesions.
Visual inspection of individual TBI BPND images showed no binding in the vicinity of focal cortical lesions evident on structural MRI. To confirm this, we sampled binding in ROIs placed in and around the most prominent lesion in each brain. As expected, there was no specific binding in the focal lesion. In addition, binding in the penumbra was reduced compared to normal-appearing GM in the same hemisphere (t = −11.54, df = 4, p < 0.001).
11C-PiB binding after TBI is correlated with WM damage and time since injury.
We next examined whether Aβ plaque pathology in the PCC was associated with the degree of TAI in the patients with TBI. We tested the hypothesis that regional GM 11C-PiB binding increases with lower FA (indicative of axonal injury) in the cingulum bundles that were directly connected to the PCC (figure 4A). Mean FA in all tracts examined was reduced as expected (figure 4B). PCC BPND was negatively correlated in both the left cingulum (R = −0.733, p = 0.031) and right cingulum (R = −0.750, p = 0.025, figure 4C), a relationship that survived correction for the age of the patient (R = −0.758, p = 0.029; R = −0.787, p = 0.020). The mean FA of the WM skeleton also showed a correlation with PCC binding (R = −0.733, p = 0.031), although this was only of borderline significance when correcting for age (R = −0.694, p = 0.056). There was no significant correlation found with the corticospinal tract FA. 11C-PiB binding in the PCC also increased with time since injury duration (R = 0.767, p = 0.021), although this was not significant after correcting for age (R = 0.625, p = 0.097). Of the 4 FA measures, the mean FA of the left cingulum also correlated with time since injury (R = −0.717, p = 0.037). There was no independent relationship between 11C-PiB binding and FA after correction for time since injury. There was also no correlation between patient age and 11C-PiB binding or FA.
Figure 4. Relationship between white matter damage and regional 11C-PiB BPND in patients with TBI.
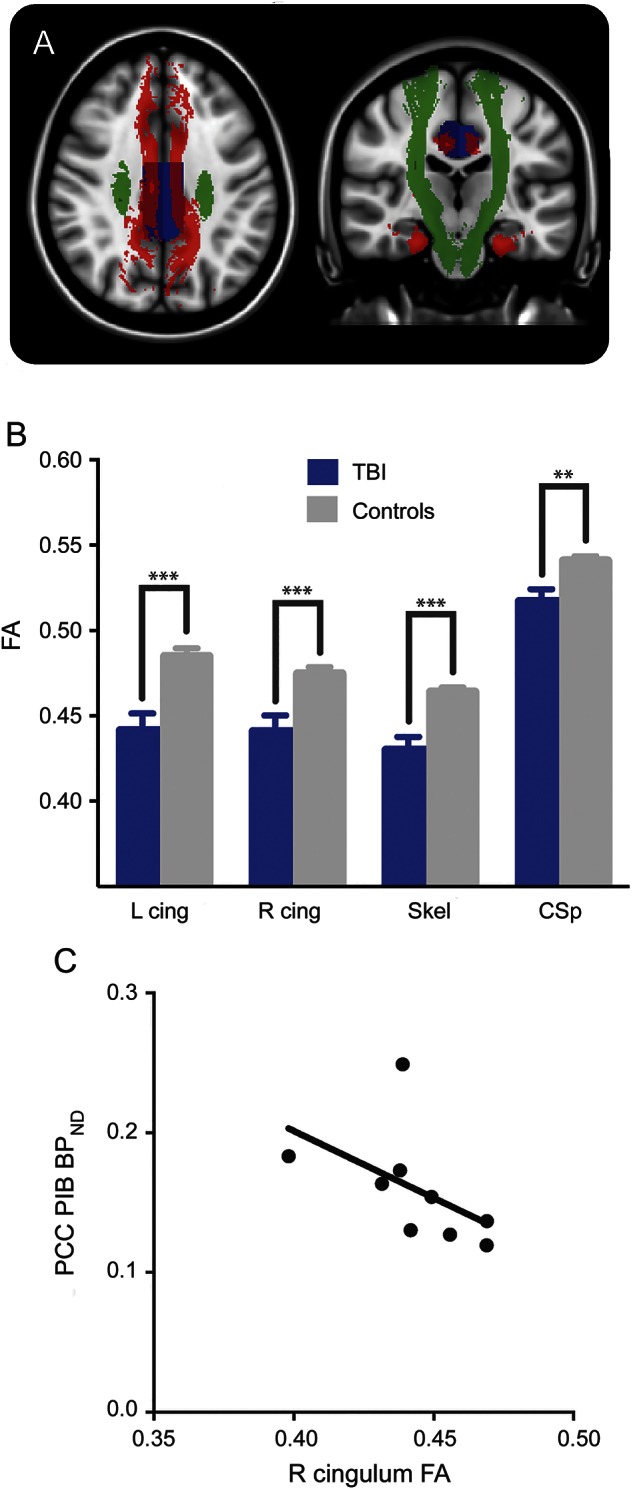
(A) Selected white matter tracts from the Johns Hopkins University tractography atlas and region of interest from the MAPER (multi-atlas propagation with enhanced registration) segmentation are shown on an MNI152 standard image. The tracts in red are the left and right cingulum–cingulate bundle combined with left and right cingulum–hippocampus tract. The regional segmentation of the PCC is shown (blue), which receives connections from these tracts. The corticospinal tract (green) is not connected to the PCC. FA, a measure of white matter integrity, was sampled from the tracts in patients with TBI using diffusion tensor imaging and related to regional 11C-PiB BPND sampled in the PCC. (B) The mean FA of all tracts tested was reduced in TBI (blue) compared to controls (gray) (**p < 0.01, ***p < 0.001). (C) 11C-PiB BPND in the PCC increased with decreasing FA in the right cingulum. BPND = nondisplaceable binding potential; 11C-PiB = 11C-Pittsburgh compound B; cing = cingulum; CSp = corticospinal tract; FA = fractional anisotropy; PCC = posterior cingulate cortex; TBI = traumatic brain injury.
11C-PiB binding is not correlated with neuropsychological impairment in TBI.
There were no significant correlations between PCC binding and behavioral performance in the patients with TBI.
Distinct distributions of 11C-PiB binding in TBI and AD.
The direct contrast of AD and controls showed increased 11C-PiB binding in AD association cortex and cingulate (figure 2B). Conjunction analysis showed that 11C-PiB binding was increased in a cluster within the precuneus/PCC in both AD and TBI compared to controls. In general, 11C-PiB binding was higher in AD than TBI across regions, but the TBI cases had relatively increased binding in the cerebellum (figure 2C). Interrogating ROI data with ANOVA confirmed the voxel-level findings. Increased 11C-PiB binding was seen in cortical association and cingulate regions in AD whereas increased cerebellar binding was seen in TBI (e-Results). There was no correlation between patient age and regional 11C-PiB binding within any of the 3 participant groups.
DISCUSSION
TBI can predispose patients to various types of dementia, but there is no consensus about how post-TBI dementia syndromes should be classified or diagnosed. Patients often clinically deteriorate years after TBI,2 but it is difficult to determine whether this is related to the prior head injury. Improved methods of characterizing neurodegenerative processes triggered by TBI are needed. We investigated amyloid pathology using 11C-PiB-PET. For the first time, we show in vivo that increases in 11C-PiB binding are present in long-term survivors of TBI in a distribution overlapping with AD but also involving the cerebellum.24 A mechanistic link between axonal injury and amyloid pathology is suggested by the relationship between cortical 11C-PiB binding and WM damage in connected tracts.
In AD, Aβ deposition usually begins in inferior frontal and cingulate association cortex, extending into other association cortical regions. Early deposition is seen in the PCC,25 and we observed increased 11C-PiB uptake in both patients with TBI and those with AD. While the ventromedial frontal cortex is affected early in AD, the hippocampus and cerebellum are not usually involved until much later in the disease.24 In keeping with this pattern, we observed strong 11C-PiB binding in the prefrontal cortex in our patients with AD, but relatively low levels in the hippocampus and cerebellum. However, a different pattern was observed in our patients with TBI, who had increased cerebellar 11C-PiB binding relative to both AD and controls. The distinct distribution of 11C-PiB binding in the 2 contexts suggests that amyloid pathology is triggered by a different mechanism after TBI, which is likely to relate to biomechanical forces underlying the distinctive pattern of Aβ plaque pathology seen in cases of chronic traumatic encephalopathy.26 TBI might also accelerate an aging process27 and our results may reflect this change in aging trajectory, particularly considering that the increased 11C-PiB binding after TBI was observed in comparison to a much older aged control group. However, in keeping with studies of AD,8 11C-PiB binding did not correlate with cognitive impairment.
Axonal damage produced at the time of injury may act as an initial trigger for Aβ production and accumulation of amyloid pathology.6 In keeping with this possibility, we observed an association between the extent of WM damage and 11C-PiB binding in the PCC following TBI. The biomechanical effects of torsional and shear stress on WM tracts produce TAI, and this is thought to be an important factor driving overproduction of Aβ, leading to its aggregation in the acute phase.3 Axons and their surrounding myelin are damaged, and the pathologic effects of injury remain visible for many years, particularly in long-distance WM tracts.28 Animal models and human autopsy studies provide evidence that Aβ is produced at the site of axonal injury shortly after TBI.6
The relationship between 11C-PiB binding and WM damage was seen in the cingulum bundles, which connect to the PCC. The relationship was not observed in the corticospinal tract, which is not directly connected to the PCC, suggesting a more specific link between the 2 observations. Misfolded proteins, including Aβ, have the capacity to move from neuron to neuron via prion-like transsynaptic spread,29,30 and computational simulations show that a simple diffusion mechanism can produce the complex patterns of brain atrophy observed in AD if large-scale WM structure is factored into the model.31 The implication for TBI is that the WM may be both a source of Aβ and a conduit for Aβ diffusion. The correlation between measures of TAI and Aβ pathology in the PCC may reflect its role as a highly connected cortical hub,32 which integrates damage that spreads from damaged WM tracts. The time elapsed since a patient's injury also correlated with 11C-PiB binding, suggesting there is a progressive neurodegenerative process. Our results suggest that 11C-PiB binding, WM structure, age, and time since injury are interrelated, and longitudinal studies with larger numbers will be needed to clarify the causal relationships. Such studies should also examine 11C-PiB binding in the context of host genotype, particularly APOE,33 which was not addressed here.
Our findings are broadly consistent with a previous 11C-PiB study in patients with TBI scanned less than 1 year after injury (median 11 days). Hong et al.10 showed increased cortical and striatal 11C-PiB binding early after TBI. Of note, the validity of in vivo neuroimaging was supported by [3H]PiB autoradiography and Aβ immunohistochemistry. In contrast to our study, this earlier work used the cerebellum as a reference region for quantification of 11C-PiB binding, assuming that there was minimal Aβ plaque density in the cerebellum and that the ratio of cortical to cerebellar binding provided a measure of cortical Aβ burden.34 Hong et al. provide evidence to support this assumption early after TBI. However, our results demonstrate that this is not the case in the chronic phase after TBI. Our initial analyses in TBI using the cerebellum as a reference region suggested decreased cortical 11C-PiB binding. Therefore, we used a procedure for automatic reference region extraction that has been validated in familial AD and does not require a single anatomically defined reference region.16
Our study has a number of potential limitations. First, given the small sample size, our findings should be regarded as preliminary. Second, the 11C-PiB healthy controls were age-matched to the AD group, and so were older than the TBI group. Although 2 separate age-matched control groups would have been preferable, Aβ pathology increases with age35 and so a comparison with older healthy controls is likely to have reduced our sensitivity to detect a relative increase in the younger TBI group. Therefore, the presence of abnormalities in a relatively young TBI group is even more striking. Third, it is possible that GM tissue differences such as atrophy, associated with AD or aging, could have biased our group contrast results. A number of analysis steps were used to minimize this possibility: an advanced algorithm for optimized registration of brain images into standard space (DARTEL)36; 11C-PiB binding was only assessed in regions where the GM probability was high; and ROI analyses, based on both automated segmentations, were used to provide confirmatory results. To control for the possible effects of focal injury after TBI, we also excluded lesioned areas from the analysis. It is possible that the extent of focal lesions was underestimated as we used T1 imaging to segment the lesions. However, since 11C-PiB binding was reduced in visible lesions, this possibility would have biased the analysis against detecting increases in 11C-PiB.
We provide 11C-PiB-PET evidence for the presence of amyloid pathology many years after injury in patients with TBI without dementia. The distribution of 11C-PiB binding partially overlapped with that seen in typical AD but also affected the cerebellum, unlike in AD. This suggests a different mechanism for amyloid plaque genesis. Our findings support the hypothesis that amyloid plaque pathology is related to the presence of axonal damage produced subsequent to the TBI.
Supplementary Material
ACKNOWLEDGMENT
The authors thank all the patients and controls who participated in this work.
GLOSSARY
- Aβ
β-amyloid
- AD
Alzheimer disease
- ANOVA
analysis of variance
- BPND
nondisplaceable binding potential
- 11C-PiB
11C-Pittsburgh compound B
- DTI
diffusion tensor imaging
- FA
fractional anisotropy
- GM
gray matter
- MNI
Montreal Neurological Institute
- PCC
posterior cingulate cortex
- ROI
region of interest
- TAI
traumatic axonal injury
- TBI
traumatic brain injury
- TBSS
tract-based spatial statistics
- WM
white matter
Footnotes
Supplemental data at Neurology.org
Editorial, page 798
AUTHOR CONTRIBUTIONS
Dr. Scott performed the analysis and interpretation of the data and wrote the manuscript. Dr. Ramlackhansingh contributed to the study design and coordination, and acquisition of data. Dr. Edison revised the manuscript and contributed to the acquisition of data. Dr. Hellyer contributed to the analysis of the data and revised the manuscript. Dr. Cole contributed to the analysis of the data and revised the manuscript. Dr. Veronese contributed to the analysis of the data. Dr. Leech contributed to the analysis of the data. Dr. Greenwood revised the manuscript and contributed to the acquisition of data. Prof. Turkheimer contributed to the analysis of the data and revised the manuscript. Prof. Gentleman revised the manuscript. Prof. Heckemann contributed to the analysis of the data and revised the manuscript. Prof. Matthews revised the manuscript. Prof. Brooks revised the manuscript. Prof. Sharp contributed to the study concept, design and coordination, acquisition of data, supervision of the study, and wrote the manuscript.
STUDY FUNDING
No targeted funding reported.
DISCLOSURE
G. Scott was supported by a clinical research fellowship awarded in the Wellcome Trust–GlaxoSmithKline Translational Medicine Training Programme. This work was supported by the Imperial College Healthcare Trust Biomedical Research Centre. A. Ramlackhansingh, P. Edison, P. Hellyer, and J. Cole report no disclosures relevant to the manuscript. M. Veronese is supported by an MRC PET programme grant (G1100809/1). R. Leech and R. Greenwood report no disclosures relevant to the manuscript. F. Turkheimer is supported by an MRC PET programme grant (G1100809/1). S. Gentleman and R. Heckemann report no disclosures relevant to the manuscript. P. Matthews has consulted or received honoraria for lectures from GlaxoSmithKline, Biogen, IDEC, IXICO, and Novartis, and has research support from the MS Society of Great Britain, the Progressive MS Alliance, the MRC, and GlaxoSmithKline, and personal support from the Edmond J. Safra Foundation and from Lily Safra. Matthews is an NIHR Senior Investigator. D. Brooks has been a consultant and part time employee for GE Healthcare in the past. D. Sharp receives personal and research support from the National Institute for Health Research and the Medical Research Council (UK). Go to Neurology.org for full disclosures.
REFERENCES
- 1.Fleminger S, Ponsford J. Long term outcome after traumatic brain injury. BMJ 2005;331:1419–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Whitnall L, McMillan TM, Murray GD, Teasdale GM. Disability in young people and adults after head injury: 5–7 year follow up of a prospective cohort study. J Neurol Neurosurg Psychiatry 2006;77:640–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smith DH, Johnson VE, Stewart W. Chronic neuropathologies of single and repetitive TBI: substrates of dementia? Nat Rev Neurol 2013;9:211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johnson VE, Stewart W, Smith DH. Widespread tau and amyloid-beta pathology many years after a single traumatic brain injury in humans. Brain Pathol 2012;22:142–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johnson VE, Stewart W, Smith DH. Axonal pathology in traumatic brain injury. Exp Neurol 2013;246:35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johnson VE, Stewart W, Smith DH. Traumatic brain injury and amyloid-beta pathology: a link to Alzheimer's disease? Nat Rev Neurosci 2010;11:361–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Quigley H, Colloby SJ, O'Brien JT. PET imaging of brain amyloid in dementia: a review. Int J Geriatr Psychiatry 2011;26:991–999. [DOI] [PubMed] [Google Scholar]
- 8.Rowe CC, Ng S, Ackermann U, et al. Imaging beta-amyloid burden in aging and dementia. Neurology 2007;68:1718–1725. [DOI] [PubMed] [Google Scholar]
- 9.Ikonomovic MD, Klunk WE, Abrahamson EE, et al. Post-mortem correlates of in vivo PiB-PET amyloid imaging in a typical case of Alzheimer's disease. Brain 2008;131:1630–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hong YT, Veenith T, Dewar D, et al. Amyloid imaging with carbon 11–labeled Pittsburgh compound B for traumatic brain injury. JAMA Neurol 2014;71:23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mac Donald CL, Dikranian K, Bayly P, Holtzman D, Brody D. Diffusion tensor imaging reliably detects experimental traumatic axonal injury and indicates approximate time of injury. J Neurosci 2007;27:11869–11876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sharp DJ, Ham TE. Investigating white matter injury after mild traumatic brain injury. Curr Opin Neurol 2011;24:558–563. [DOI] [PubMed] [Google Scholar]
- 13.Sharp DJ, Scott G, Leech R. Network dysfunction after traumatic brain injury. Nat Rev Neurol 2014;10:156–166. [DOI] [PubMed] [Google Scholar]
- 14.Magnoni S, Mac Donald CL, Esparza TJ, et al. Quantitative assessments of traumatic axonal injury in human brain: concordance of microdialysis and advanced MRI. Brain 2015;138:2263–2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Malec JF, Brown AW, Leibson CL, et al. The Mayo classification system for traumatic brain injury severity. J Neurotrauma 2007;24:1417–1424. [DOI] [PubMed] [Google Scholar]
- 16.Ikoma Y, Edison P, Ramlackhansingh A, Brooks DJ, Turkheimer FE. Reference region automatic extraction in dynamic [11C]-PIB. J Cereb Blood Flow Metab 2013;33:1725–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ashburner J. A fast diffeomorphic image registration algorithm. Neuroimage 2007;38:95–113. [DOI] [PubMed] [Google Scholar]
- 18.Heckemann RA, Keihaninejad S, Aljabar P, Rueckert D, Hajnal JV, Hammers A. Improving intersubject image registration using tissue-class information benefits robustness and accuracy of multi-atlas based anatomical segmentation. Neuroimage 2010;51:221–227. [DOI] [PubMed] [Google Scholar]
- 19.Frisoni GB, Jack CR. Harmonization of magnetic resonance-based manual hippocampal segmentation: a mandatory step for wide clinical use. Alzheimers Dement 2011;7:171–174. [DOI] [PubMed] [Google Scholar]
- 20.Smith SM, Jenkinson M, Woolrich MW, et al. Advances in functional and structural MR image analysis and implementation as FSL. Neuroimage 2004;23(suppl 1):S208–S219. [DOI] [PubMed] [Google Scholar]
- 21.Smith SM, Jenkinson M, Johansen-Berg H, et al. Tract-based spatial statistics: voxelwise analysis of multi-subject diffusion data. Neuroimage 2006;31:1487–1505. [DOI] [PubMed] [Google Scholar]
- 22.Hua K, Zhang J, Wakana S, et al. Tract probability maps in stereotaxic spaces: analyses of white matter anatomy and tract-specific quantification. Neuroimage 2008;39:336–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blumbergs PC, Jones NR, North JB. Diffuse axonal injury in head trauma. J Neurol Neurosurg Psychiatry 1989;52:838–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thal DR, Rub U, Orantes M, Braak H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 2002;58:1791–1800. [DOI] [PubMed] [Google Scholar]
- 25.Tosun D, Schuff N, Mathis CA, Jagust W, Weiner MW. Spatial patterns of brain amyloid-beta burden and atrophy rate associations in mild cognitive impairment. Brain 2011;134:1077–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stein T, Montenigro P, Alvarez V, et al. Beta-amyloid deposition in chronic traumatic encephalopathy. Acta Neuropathol 2015;130:21–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cole JH, Leech R, Sharp DJ; Alzheimer's Disease Neuroimaging Initiative. Prediction of brain age suggests accelerated atrophy after traumatic brain injury. Ann Neurol 2015;77:571–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johnson VE, Stewart JE, Begbie FD, Trojanowski JQ, Smith DH, Stewart W. Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain 2013;136:28–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harris JA, Devidze N, Verret L, et al. Transsynaptic progression of amyloid-beta-induced neuronal dysfunction within the entorhinal-hippocampal network. Neuron 2010;68:428–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Polymenidou M, Cleveland DW. Prion-like spread of protein aggregates in neurodegeneration. J Exp Med 2012;209:889–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Raj A, Kuceyeski A, Weiner M. A network diffusion model of disease progression in dementia. Neuron 2012;73:1204–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Crossley NA, Mechelli A, Scott J, et al. The hubs of the human connectome are generally implicated in the anatomy of brain disorders. Brain 2014;137:2382–2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ponsford J, McLaren A, Schonberger M, et al. The association between apolipoprotein E and traumatic brain injury severity and functional outcome in a rehabilitation sample. J Neurotrauma 2011;28:1683–1692. [DOI] [PubMed] [Google Scholar]
- 34.Rowe CC, Villemagne VL. Brain amyloid imaging. J Nucl Med 2011;52:1733–1740. [DOI] [PubMed] [Google Scholar]
- 35.Rowe CC, Ellis KA, Rimajova M, et al. Amyloid imaging results from the Australian Imaging, Biomarkers and Lifestyle (AIBL) Study of Aging. Neurobiol Aging 2010;31:1275–1283. [DOI] [PubMed] [Google Scholar]
- 36.Klein A, Andersson J, Ardekani BA, et al. Evaluation of 14 nonlinear deformation algorithms applied to human brain MRI registration. Neuroimage 2009;46:786–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.