

Abstract
Background
Best practice when performing culture-independent microbiological analysis of sputum samples involves their rapid freezing and storage at −80 °C. However, accessing biobanked collections can mean that material has been passed through repeated freeze–thaw cycles. The aim of this study was to determine the impact of these cycles on microbial community profiles.
Methods
Sputum was collected from eight adults with cystic fibrosis, and each sample was subjected to six freeze–thaw cycles. Following each cycle, an aliquot was removed and treated with propidium monoazide (PMA) prior to DNA extraction and 16S rRNA gene pyrosequencing.
Results
The impact of freeze–thaw cycles was greatest on rare members of the microbiota, with variation beyond that detected with within-sample repeat analysis observed after three cycles.
Conclusion
Four or more freeze thaw cycles result in a significant distortion of microbiota profiles from CF sputum.
Keywords: Microbiota, Microbiome, Biobank, Pyrosequencing, Propidium monoazide, Sputum
1. Introduction
The application of next-generation sequencing technologies for the investigation of lower respiratory tract infections in patients with cystic fibrosis has revealed complex and highly diverse microbial communities [1,2]. As technologies have improved and the associated costs have fallen, it is becoming possible to use these platforms, not only for research but also for diagnostic microbiology [3], making it more important than ever to identify and minimise the introduction of bias.
Spontaneously expectorated sputum is one of the most common specimen types used to investigate the microbial community responsible for lower respiratory infections in adults with CF. In order to perform culture-independent analysis on a representative airway sample, the methods used to collect and store specimens are hugely important. Current best practice involves the rapid stabilisation of sputum samples by freezing at −80 °C within 12 h of collection [4]. To allow profiling of the viable microbial community, samples can be treated with propidium monoazide (PMA) to remove the impact of extracellular DNA or DNA from dead and damaged cells, prior to DNA extraction and DNA sequencing [5]. Received wisdom suggests that once defrosted, respiratory samples may not be refrozen and sub-sampled again at a later date without incurring significant changes in the microbial community.
There is an increasing awareness of the importance of in-depth analysis for the investigation of the microbial communities responsible for infection. This has lead to many clinics collecting large detailed sample biobanks in ultralow temperature freezers, which can be accessed in order to address a wide range of clinical questions. However, biobanked samples may be accessed multiple times for culture independent analysis, thus passing though several freeze thaw cycles, a process that could result in changes to the microbial community. To date, no studies have used next-generation sequencing technologies to define how multiple freeze–thaw cycles affect the microbial community within collected sputum. We hypothesized that microbial community profiles would be significantly altered with each additional freeze–thaw cycle when analysed using 16S rRNA gene pyrosequencing.
2. Methods
2.1. Sample collection
Sputum samples were collected, under full ethical approval from the Southampton and South West Hampshire Research Ethics Committee (06/Q1704/26), from eight patients attending the regional Cystic Fibrosis Centre in Southampton General Hospital. All patients were chronically colonised with Pseudomonas aeruginosa. Patients were selected based on their ability to typically produce more than 2 ml of sputum. Sputum samples were collected and frozen at −80 °C within 1 h. Each sputum sample was subjected to six freeze–thaw cycles. Samples were removed from the −80 °C freezer, a 250 μl aliquot removed for DNA extraction, and the remaining sample allowed to completely thaw at room temperature for 30 min before being returned to −80 °C for 24 h.
2.2. DNA extraction and pyrosequencing
Sputum samples were washed three times with 1× phosphate buffered saline to remove saliva, as previously described [1]. Extracellular DNA and DNA from non-viable cells were excluded from analysis via crosslinking with PMA [6,7] prior to DNA extraction, as described previously [8]. Bacterial Golay barcode-encoded FLX amplicon pyrosequencing was performed using the primer 338F (3′- ACTCCTACGGGAGGCAGCAG) and 926R (3′- CCGTCAATTCMTTTRAGT). Initial generation of 16S rRNA gene amplicons involved a one step PCR of 25 cycles using AccuPrime™ Taq DNA Polymerase High Fidelity (Invitrogen, Carlsbad, CA). 454 pyrosequencing using the Lib-L kit was performed at the Wellcome Trust Sanger Institute, Hinxton, UK.
Resulting data were analysed using the Mothur sequencing analysis platform [9] as described previously [4]. The raw sequence data generated within the current study have been submitted to the NCBI Short Read Archive database under the study accession number SRP040968. The barcodes associated with each sample are shown in Table S1. Two aliquots were excluded due to insufficient number of sequence reads generated.
2.3. Statistical analysis
Statistical analysis was performed in R [10]. Changes in bacterial diversity were assessed using three complementary measures: species richness (S*, the total number of species), Shannon–Wiener (H′, a metric accounting for both number and relative abundance of species), and Simpson’s (1-D, a measure of the probability that two species randomly selected from a sample will differ) indices of diversity as described previously [4,11]. The Bray–Curtis (SBC, which accounts for the number and abundance of species present in each community and those that are shared), resulting in a value between 0 and 1 (higher values indicating greater similarity) measure of similarity was used to assess changes in community composition with each freeze–thaw cycle.
To avoid potential bias, all measures were calculated using randomised resampling to a uniform number of sequence reads per sample [5]. Mean diversity measures were calculated from the re-sampling of the reads from each specimen to the lowest number of sequence reads among all specimens (n = 261) for 1000 iterations. SBC was calculated by re-sampling to the minimum number of sequence reads per specimen within each patient and comparing community composition to the original sample for 1000 iterations. Bacterial species detected at the first point for each patient were partitioned into common and rare species using rank abundance curves [12]. The R package nlme [13] was used to fit mixed effect models to investigate the relationships between measures of diversity, similarity, and number of freeze thaw cycles. r2 values were calculated using the MuMIn package [14].
3. Results
To test the study hypothesis, sputum samples from eight CF patients were subjected to six freeze–thaw cycles. Aliquots of sputum were removed for DNA extraction and 16S rRNA gene pyrosequencing to assess the bacterial community after each cycle, and the remaining sample was allowed to defrost completely before being returned to −80 °C. All samples were treated with PMA prior to DNA extraction to focus the analysis on the viable bacterial community. A total of 106,065 sequences (mean ± standard error (SE) per sample 2306 ± 239) were generated from 46 samples, identifying 49 genera and 76 distinct operational taxonomic units (OTUs) classified to species level (Table S2).
3.1. Bacterial diversity
Species richness, S*, was found to be highly variable between patients (max = 49, min = 4), no pattern in raw richness values was observed over the freeze–thaw cycles either within patients (P = 0.6, r2 = 0.0004) or when using randomised resampling to calculate mean S* (P = 0.7, r2 = 0.0001). High levels of variability in bacterial diversity were also observed between patients as measured by H′ and 1-D. However, as with S*, no pattern in either of these diversity measures was observed over the 6 freeze–thaw cycles H′ (P = 0.4, r2 = 0.001), 1-D (P = 0.1, r2 = 0.003) (Fig. 1).
Fig. 1.
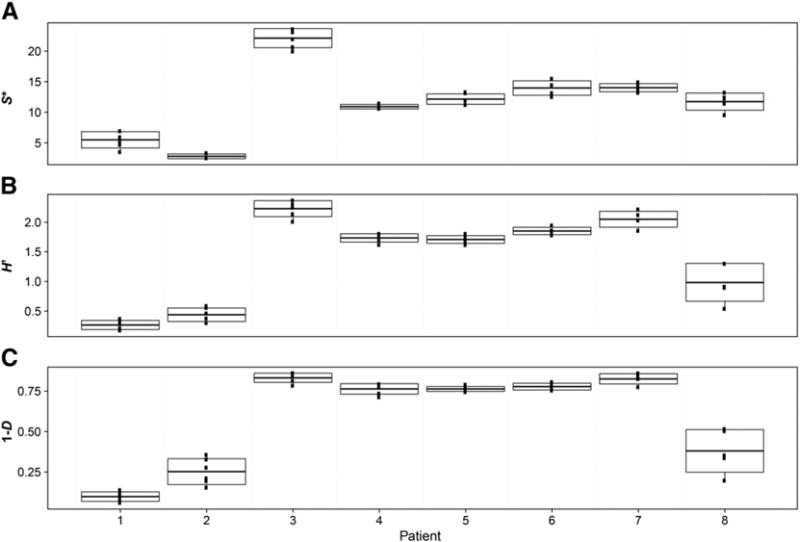
Boxplots from each patient showing variation in diversity over the 6 freeze–thaw cycles. Values of (A) species richness (S*), (B) Shannon–Wiener index of diversity (H′), and (C) Simpson’s index of diversity (1-D) were calculated with a uniform re-sample size following 1000 iterations in each instance. Lines in boxplot represent mean and standard deviation of the mean (n = 6); whiskers represent 25th and 75th percentiles.
3.2. Bacterial community membership
Changes in community composition were compared using the SBC measure of similarity. A significant decrease in similarity was observed over the 6 cycles (P < 0.0001, r2 = 0.40) (Fig. 2). Variation in intra-sample composition could contribute to the effect associated with freeze–thaw cycles [4,15]. Therefore, to account for within-sample variation, a cut off value for similarity was calculated by comparing the within-sample similarities of triplicate sequence datasets of eight samples that were published previously [4]. Based on this data, Bray–Curtis similarity values below 0.682 were judged to differ significantly from the original sample.
Fig. 2.
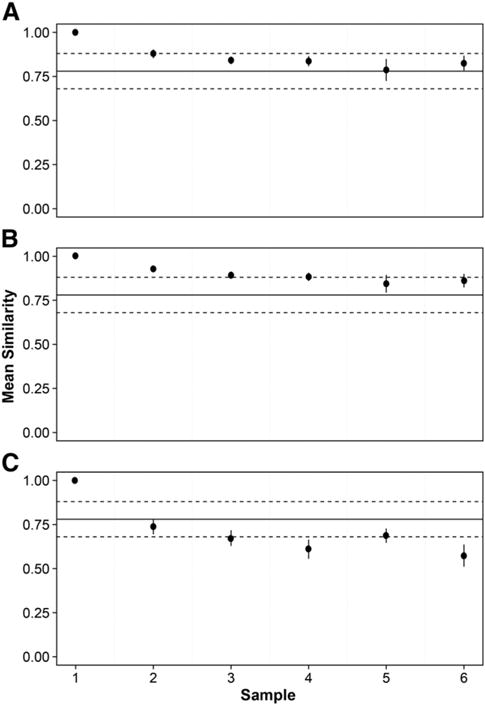
Mean changes in bacterial community composition over six freeze–thaw cycles, using Bray–Curtis index of similarity for (A) whole communities, (B) common and (C) rare species groups. Error bars represent the standard error of the mean (n = 6). Whole community similarity was calculated with a uniform re-sample size following 1000 iterations. Solid lines represent the overall mean similarity for within sample replicates and dashed lines represent the standard error of the mean (n = 24).
To minimise bias, changes in similarity were measured using randomised resampling to a uniform subsample size. Analysis showed that, despite a significant trend of decreasing similarity between aliquots within each sample over the six freeze–thaw cycles, none of these changes in within-sample similarity fell below the cut-off value of 0.682 for significant within-sample variation.
To investigate the drivers of this trend in community composition, we partitioned the community into common and rare species using rank abundance curves, indicating the breakpoint between the common and rare using the inflection point (Fig. S1) [12]. Once partitioned, the Bray–Curtis similarity was calculated for the common and rare species. The common and abundant species were most important in determining the overall trend in community similarity, while the rare species were shown to be more variable (Fig. 2). Over the six cycles, the common species were not found to fall below the cut-off value for within-sample variation. In contrast, however, substantial variation was observed in the detection of rare species, for which the mean change in similarity among aliquots from each sample dropped below the expected level for within-sample variation after four freeze–thaw cycles.
4. Discussion
The handling of respiratory samples for culture-independent analysis is vital if an unbiased picture of the microbial community is to be obtained. Storage of samples at −80 °C is the standard recognised method for maintenance of sample integrity during biobanking. However, whether repeated sub-sampling of these specimens, which will require repeated thawing and freezing, leads to sample degradation and changes in community composition detected has not been reported.
In searching for changes in bacterial diversity due to freeze–thaw cycles, no significant overall trend was observed. A significant negative trend in community similarity was observed using the Bray–Curtis measure of similarity. Despite this observation, over the 6 cycles the change in similarity never fell below the expected level for within-sample variation, indicating that repeat freeze–thaw cycles will not affect the overall bacterial community composition. However, the rare community fell below the level of within sample variation from 4 freeze–thaw cycles.
Our results challenge the long-held view that the microbial community within respiratory samples will significantly change once samples have been defrosted during subsampling.
More than three freeze–thaw cycles will result in significant divergence of the rare community from the original sample. If the sample is subjected to four or more cycles community analysis may be carried out on the common community but the rare should be interpreted with care. In practical terms, these findings support the aliquoting of samples to avoid unnecessary freeze–thaw cycles.
Supplementary Material
Acknowledgments
This study was supported by the UK Natural Environment Research Council (NE/H019456/1) to CJvdG, by the Wellcome Trust (WT 098051) to AWW and JP, and by the US National Institute of Health (K02HL105543 and P30 DK089507) to LRH.
Abbreviations
- PMA
Propidium monoazide
- OTU
Operational taxonomic units
- S*
Species richness
- H′
Shannon–Wiener
- 1-D
Simpson’s
- SBC
Bray–Curtis
Appendix A. Supplementary data
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.jcf.2014.10.004.
Footnotes
Data deposition: The sequence data reported in this paper have been deposited in the NCBI Short Read Archive database (Accession number SRP040968).
Conflict of interest statement
The authors declare no conflict of interest.
References
- 1.Rogers GB, Carroll MP, Serisier DJ, Hockey PM, Jones G, Kehagia V, et al. Use of 16S rRNA gene profiling by terminal restriction fragment length polymorphism analysis to compare bacterial communities in sputum and mouthwash samples from patients with cystic fibrosis. J Clin Microbiol. 2006;44:2601–4. doi: 10.1128/JCM.02282-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Loens K, Van Heirstraeten L, Malhotra-Kumar S, Goossens H, Ieven M. Optimal sampling sites and methods for detection of pathogens possibly causing community-acquired lower respiratory tract infections. J Clin Microbiol. 2009;47:21–31. doi: 10.1128/JCM.02037-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pattison SH, Rogers GB, Crockard M, Elborn JS, Tunney MM. Molecular detection of CF lung pathogens: current status and future potential. J Cyst Fibros. 2013;12:194–205. doi: 10.1016/j.jcf.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cuthbertson L, Rogers GB, Walker AW, Oliver A, Hafiz T, Hoffman LR, et al. Time between sputum sample collection and storage significantly influences bacterial sequence composition from Cystic Fibrosis respiratory infections. J Clin Microbiol. 2014 doi: 10.1128/JCM.00764-14. http://dx.doi.org/10.1128/JCM.00764-14. [DOI] [PMC free article] [PubMed]
- 5.Rogers GB, Cuthbertson L, Hoffman LR, Wing PA, Pope C, Hooftman DA, et al. Reducing bias in bacterial community analysis of lower respiratory infections. ISME J. 2013;7:697–706. doi: 10.1038/ismej.2012.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nocker A, Sossa-Fernandez P, Burr MD, Camper AK. Use of propidium monoazide for live/dead distinction in microbial ecology. Appl Environ Microbiol. 2007;73:5111–7. doi: 10.1128/AEM.02987-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rogers GB, Stressmann FA, Koller G, Daniels T, Carroll MP, Bruce KD. Assessing the diagnostic importance of nonviable bacterial cells in respiratory infections. Diagn Microbiol Infect Dis. 2008;62:133–41. doi: 10.1016/j.diagmicrobio.2008.06.011. [DOI] [PubMed] [Google Scholar]
- 8.Rogers GB, Marsh P, Stressmann AF, Allen CE, Daniels TV, Carroll MP, et al. The exclusion of dead bacterial cells is essential for accurate molecular analysis of clinical samples. Clin Microbiol Infect. 2010;16:1656–8. doi: 10.1111/j.1469-0691.2010.03189.x. [DOI] [PubMed] [Google Scholar]
- 9.Schloss PD, Gevers D, Westcott SL. Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS One. 2011;6 doi: 10.1371/journal.pone.0027310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.R, Core, Team. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2013. [Google Scholar]
- 11.van der Gast C, Walker A, Stressmann F, Rogers G, Scott P, Daniels T, et al. Partitioning core and satellite taxa from within cystic fibrosis lung bacterial communities. ISME J. 2011;5:780–91. doi: 10.1038/ismej.2010.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Siqueira T, Bini LM, Roque FO, Couceiro SRM, Trivinho-Strixino S, Cottenie K. Common and rare species respond to similar niche processes in macroinvertebrate metacommunities. Ecography. 2012;35:183–92. [Google Scholar]
- 13.Pinheiro J, Bates D, DebRoy S, Sarkar D, The R Development Core Team nlme: Linear and Nonlinear Mixed Effects Models. R package version 31-110. 2013 [Google Scholar]
- 14.Barton K. MuMIn: Multi-Model Inference. R package version195. 2013 [Google Scholar]
- 15.Zhao JC, Li J, Schloss PD, Kalikin LM, Raymond TA, Petrosino JF, et al. Effect of sample storage conditions on culture-independent bacterial community measures in cystic fibrosis sputum specimens. J Clin Microbiol. 2011;49:3717–8. doi: 10.1128/JCM.01189-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.