

SUMMARY
We analyze de novo synonymous mutations identified in autism spectrum disorders (ASD) and schizophrenia (SCZ) with potential impact on regulatory elements using data from whole exome sequencing (WES) studies. Focusing on five types of genetic regulatory functions, we found that de novo near-splice site synonymous mutations changing exonic splicing regulators and those within frontal cortex-derived DNase I hypersensitivity sites are significantly enriched in ASD and SCZ, respectively. These results remained significant, albeit less so, after incorporating two additional ASD datasets. Among the genes identified, several are hit by multiple functional de novo mutations, with RAB2A and SETD1A showing the highest statistical significance in ASD and SCZ, respectively. The estimated contribution of these synonymous mutations to disease liability is comparable to de novo protein-truncating mutations. These findings expand the repertoire of functional de novo mutations to include “functional” synonymous ones and strengthen the role of rare variants in neuropsychiatric disease risk.
eTOC Blurb
The role of de novo non-synonymous mutations in neuropsychiatric disorders is well established. In this study, Takata et al. explore the role of a different class of genetic variants, synonymous or “silent” mutations, and discover that they too contribute to autism and schizophrenia by affecting regulatory elements.
INTRODUCTION
Recent whole-exome sequencing (WES) studies of family samples have pointed out the important role of de novo germline mutations in various genetically complex neuropsychiatric diseases, including autism spectrum disorders (ASD) (Iossifov et al., 2012; Neale et al., 2012; O’Roak et al., 2012; Sanders et al., 2012) and schizophrenia (SCZ) (Fromer et al., 2014; Girard et al., 2011; Gulsuner et al., 2013; McCarthy et al., 2014; Xu et al., 2011; Xu et al., 2012).
These studies mainly focused on the impact of mutations on the protein coding properties. Although there is variation in their estimated contribution, analysis of collective data from these studies confirms a prominent enrichment of gene-disruptive de novo loss-of-function (LOF) mutations (i.e. nonsense, frameshift and canonical splice site mutations) as well as a more moderate enrichment of de novo missense and inframe insertion/deletion (indel) mutations in the cases, while there was no significant global enrichment of de novo synonymous mutations (Figure S1). However, there is accumulating evidence that certain types of synonymous mutations have substantial contribution to complex human diseases, mediated by mechanisms other than changing protein sequences (reviewed in Sauna and Kimchi-Sarfaty, 2011)
To test whether specific subsets of synonymous mutations contribute to the genetic architecture of ASD and SCZ, we comprehensively analyzed those de novo synonymous mutations with potential impact on five different types of gene regulatory function: splicing regulation, transcription factor binding, microRNA (miRNA) binding, codon optimality and RNA secondary structure, by using publicly available WES datasets for ASD, SCZ and control subjects (Table S1) as well as various bioinformatics tools and data sources (Figure 1). Subtypes of synonymous mutations that showed significant enrichment after correcting for multiple testing and genes hit by these mutations were further analyzed for their properties and relative contribution to the disease etiology.
Figure 1. Schematic of the Methodology for the Analysis of De Novo Synonymous Mutations in ASD and SCZ.
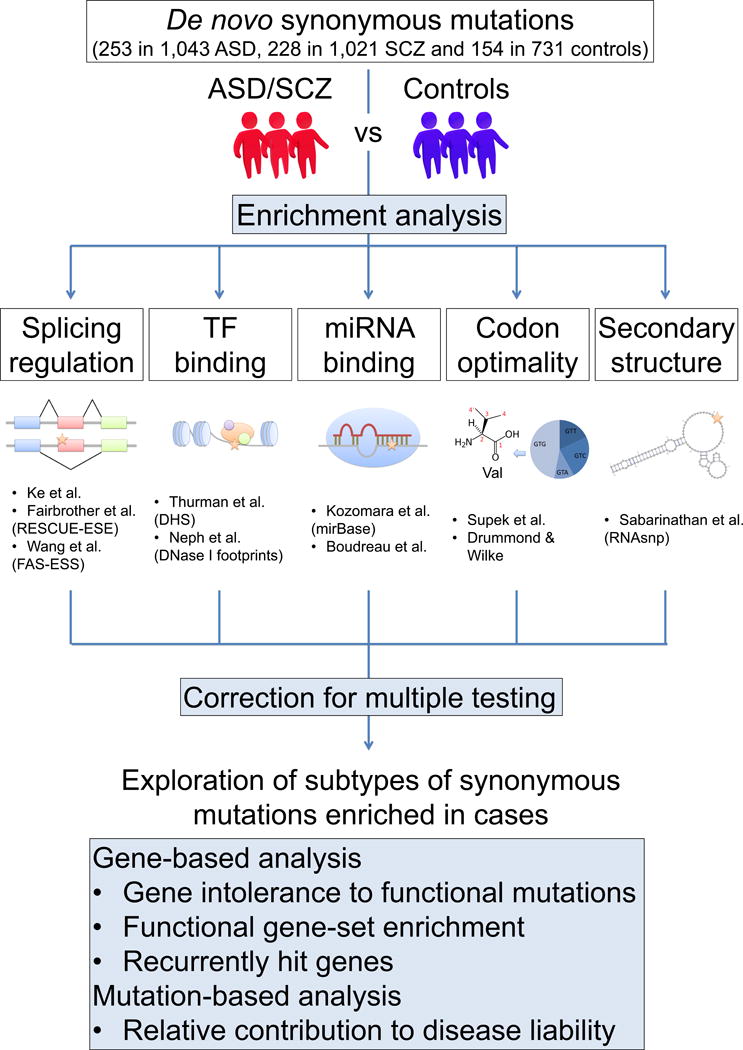
Published WES studies (ASD; (Fromer et al., 2014; Iossifov et al., 2012; Neale et al., 2012; O’Roak et al., 2012; Sanders et al., 2012), SCZ; (Fromer et al., 2014; Girard et al., 2011; Gulsuner et al., 2013; McCarthy et al., 2014; Xu et al., 2011; Xu et al., 2012) and control subjects; (Gulsuner et al., 2013; Iossifov et al., 2012; O’Roak et al., 2012; Rauch et al., 2012; Sanders et al., 2012; Xu et al., 2012)) were used to assess enrichment of synonymous mutations likely to affect genetic regulatory elements in the case groups using Fisher’s exact tests (1-tailed). The potential impact of each de novo synonymous mutation on five different types of genetic regulatory functions (splicing regulation, transcription factor (TF) binding, miRNA binding, codon optimality and mRNA secondary structure) was evaluated using various bioinformatics tools and data sources (see Supplemental Experimental Procedures). After performing exploratory enrichment analyses, we applied correction for multiple testing (Benjamini–Hochberg procedure) for the number of hypotheses tested to identify subtypes of potentially “functional” synonymous mutations robustly enriched in the case groups and were further analyzed for gene intolerance against functional mutations, functional gene-set enrichment, recurrence of damaging mutations (gene-based analysis) and relative contribution to the disease liability (mutation-based analysis). DHS; DNase I hypersensitive sites.
RESULTS
Enrichment of De Novo Synonymous Mutations in the Genetic Elements That Control Splicing Regulation in ASD and SCZ
A recent cancer study (Supek et al., 2014) specifically described global enrichment of synonymous mutations within 30 bp of the nearest splice site (referred to hereafter as “near-splice site” variants) and possible impact of these mutations on splicing regulation. Therefore, we first tested for enrichment of exonic near-splice site de novo synonymous mutations in ASD and SCZ, which has not been analyzed in the previous exome sequencing studies. We found that near-splice site de novo synonymous mutations are almost twice as frequent in ASD than controls (p = 0.00032, odds ratio (OR) = 1.96, 101 mutations in 1,043 ASD cases and 37 mutations in 731 controls, 1-tailed 2×2 Fisher’s exact test, Figure 2A). Significant enrichment was also observed in SCZ (p = 0.023, OR = 1.53, 78 mutations in 1,021 SCZ cases and 37 mutations in 731 controls) and the combined case group (ASD+SCZ: p = 0.0012, OR = 1.74, 179 mutations in 2,064 ASD+SCZ cases and 37 mutations in 731 controls). De novo synonymous mutations that are distant from the nearest splice site (i.e. distance to the nearest splice site > 30 bp) were not enriched (Figure 2A). These results suggest that, first, like the cancer study (Supek et al., 2014), only the near-splice site de novo synonymous mutations are enriched among cases. Second, this enrichment cannot be explained by the potential biases due to combination of the data from multiple studies (see Supplemental Information and Figure S2). When comparing the cumulative distributions of all de novo synonymous mutations according to their distance to the nearest splicing site between cases and controls, there is an overall significant differences for ASD (p = 0.0023, Two-sample Kolmogorov-Smirnov test), SCZ (p = 0.019) and ASD+SCZ (p = 0.0018) (Figure 2B). In agreement with Supek and colleagues, the difference in cumulative distributions between cases and controls was maximized for mutations within ~30 bp from the nearest splice site (Figure 2B, inset).
Figure 2. Enrichment Analyses of Near-Splice Site De Novo Synonymous Mutations in ASD and SCZ.
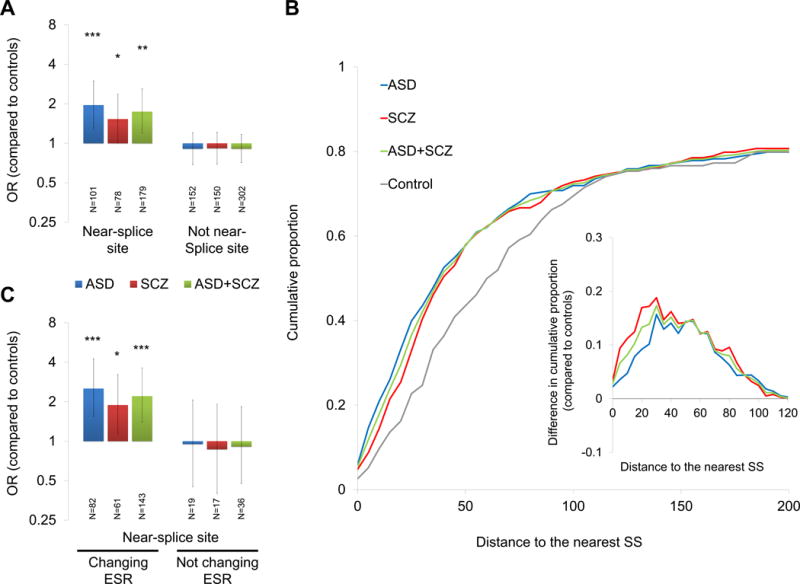
(A) Enrichment analyses of near-splice site de novo synonymous mutations (≤ 30 bp from the nearest splice site, left) and mutations distant from the nearest splice sites (> 30 bp, right) in the case groups as compared to controls. (B) Cumulative proportion curves for the distance to the nearest splice site. Proportion of the mutations with distance to the nearest splice site < or = to the number indicated in the X-axis was plotted in the Y-axis. Inset: Differences in cumulative proportion between cases and controls (i.e. [cumulative proportion in cases] − [cumulative proportion in controls]). (C) Enrichment analyses of near-splice site de novo synonymous mutations changing exonic splicing regulators (ESR) (left) and near-splice site mutations not changing ESR (right) compared to controls.
Next, we analyzed which of the individual mutations are more likely to affect splicing by referring to exonic splicing regulator (ESR) hexamer sequences in Ke et al. (2011), the RESCUE-ESE server (Fairbrother et al., 2002) and the FAS-ESS server (Wang et al., 2004). By mapping all de novo synonymous mutations to the list of exonic splicing enhancers (ESEs) and exonic splicing silencers (ESSs) found in any of the three datasets (in total 401 ESEs and 315 ESSs; see Supplemental Experimental Procedures), we found that near-splice site de novo mutations causing any type of ESR changes (i.e. ESE gain, ESE loss, ESS gain or ESS loss) showed further enrichment in the case groups (ASD: p = 3.4 × 10−5, OR = 2.52, 82 mutations in 1,043 ASD cases and 23 mutations in 731 controls; SCZ: p = 0.0056, OR = 1.89, 61 mutations in 1,021 SCZ cases and 23 mutations in 731 controls; ASD+SCZ: p = 0.00014, OR =2.21, 143 mutations in 2,064 ASD+SCZ cases and 23 mutations in 731 controls, 1-tailed 2×2 Fisher’s exact test, Figure 2C) and there was no enrichment of near-splice site mutations that do not change ESR (Figure 2C).
Enrichment of De Novo Synonymous Mutations within Brain-Derived DHS and DNase I Footprints in SCZ and ASD
Genetic variants also have the potential to affect transcription factor (TF) binding sites and are particularly enriched in DNase I hypersensitive sites (DHS) that represent open chromatin regions accessible to proteins such as TF. DHS are significantly enriched for signals from genome-wide association studies (GWAS) of various diseases (Maurano et al., 2012). Using the single integrated dataset generated by combining the data of 125 cell types (Thurman et al., 2012), we analyzed whether de novo synonymous mutations in ASD and SCZ are preferentially found in DHS and found no significant enrichment of de novo synonymous mutations within these DHS in any of the case groups as compared to the controls (Figure 3). We then refined our analysis based on the data derived from normal brain tissues in the ENCODE Experiment Matrix (https://genome.ucsc.edu/ENCODE/dataMatrix/encodeDataMatrixHuman.html). Three such available datasets were used originally: two derived from frontal cortex tissues (ENCODE Common Cell Types ID: “Cerebrum_frontal_OC” and “Frontal_cortex_OC”) and one derived from cerebellum tissues (ENCODE ID: “Cerebellum_OC”). With the DHS identified in the Cerebrum_frontal_OC data there was significant enrichment of de novo synonymous mutations within these DHS in SCZ (p = 0.00079, OR = 2.58, 50 mutations in 1,021 SCZ cases and 14 mutations in 731 controls, 1-tailed 2×2 Fisher’s exact test), ASD (p = 0.022, OR = 1.88, 38 mutations in 1,043 ASD cases and 14 mutations in 731 controls) and ASD+SCZ (p = 0.0018, OR = 2.23, 88 mutations in 2,064 ASD+SCZ cases and 14 mutations in 731 controls) (Figure 3). Interestingly, using the DHS identified in the Cerebellum_OC data, which are derived from the cerebellar tissues of the same individuals used for the Cerebrum_frontal_OC tissue, there was no significant enrichment of de novo synonymous mutations within DHS in any of the case groups (Figure 3, ASD: p = 1, OR = 0.60, 12 mutations in 1,043 ASD cases and 14 mutations in 731 controls; SCZ: p = 0.11, OR = 1.55, 30 mutations in 1,021 SCZ cases and 14 mutations in 731 controls; ASD+SCZ: p = 1, OR = 0.69, 42 mutations in 2,064 ASD+SCZ cases and 14 mutations in 731 controls, 1-tailed 2×2 Fisher’s exact test). Synonymous mutations hitting the frontal cortex-specific DHS in these datasets (DHS found in Cerebrum_frontal_OC and not found in Cerebellum_OC) were highly enriched in all of the case groups (ASD: p = 0.0046, OR = 2.96, 29 mutations in 1,043 ASD cases and 7 mutations in 731 controls; SCZ: p = 0.0044, OR = 2.91, 28 mutations in 1,021 SCZ cases and 7 mutations in 731 controls; ASD+SCZ: p = 0.0027, OR = 2.88, 57 mutations in 2,064 ASD+SCZ cases and 7 mutations in 731 controls). Using the data derived from an independent dataset with the frontal cortex tissues of two individuals (Frontal_cortex_OC), there was significant enrichment of de novo synonymous mutations within these DHS in SCZ (p = 0.012, OR = 1.8, 55 mutations in 1,021 SCZ cases and 22 mutations in 731 controls), but not in ASD or ASD+SCZ (Figure 3), suggesting a particularly robust enrichment of such mutations in SCZ. In addition, we observed significant enrichment of synonymous mutations within fetal brain-derived DNase I footprints (Neph et al., 2012), which indicate precise genomic locations bound by regulatory factors (Kavanagh et al., 2013), in SCZ and ASD+SCZ (p = 0.012 and 0.039 respectively, see Supplemental Information for details), further supporting a role of synonymous mutations affecting TF binding affinity in SCZ.
Figure 3. Enrichment Analyses of Synonymous Mutations within DNase I hypersensitive sites.
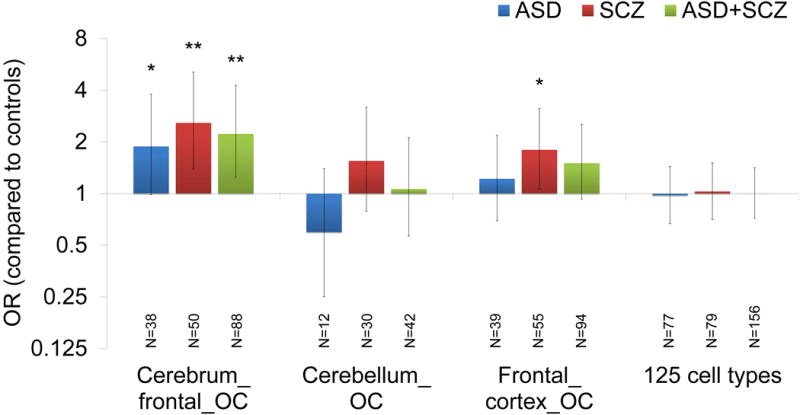
Enrichment analyses of de novo synonymous mutations within DNase I hypersensitive sites (DHS) using the data derived from brain tissues (Cerebrum_frontal_OC, Cerebellum_OC and Frontal_cortex_OC) and the integrated data of 125 cell types. The numbers below the bars indicate the number of each type of synonymous mutations. Error bars indicate 95% CI. P values were calculated by 1-tailed 2×2 Fisher’s exact tests. *p < 0.05, ** p < 0.005, *** p < 0.0005. ASD; autism spectrum disorders, OR; odds ratio, SCZ; schizophrenia, SS; splice site.
In contrast, when we evaluated whether synonymous mutations likely affecting miRNA binding sites in the coding regions (we could not include mutations in 3′ UTR, the main targets of miRNAs), codon optimality or RNA secondary structure are enriched in case subjects, we did not observe strong enrichment considering the number of hypotheses tested in our study (Figure S3, see Supplemental Information for details).
After these exploratory analyses, we applied multiple testing correction (Benjamini-Hochberg procedure) for the number of all hypotheses tested (87 in total, Table S2) and found that enrichment of near-splice site synonymous mutations, especially those changing ESR, in ASD (pcorrected = 0.003) and enrichment of synonymous mutations within the frontal cortex-derived DHS (Cerebrum_frontal_OC) in SCZ (pcorrected = 0.017) remain statistically significant (Table S2). Enrichment of these types of synonymous mutations was confirmed by a permutation-based analysis, and supported by analyses using independent datasets including the data of DHS from the Roadmap Epigenomics Project (Roadmap Epigenomics et al., 2015) (see Supplemental Information and Figure S4 for details).
Confirmation of Enrichment of De Novo Near-Splice Site Synonymous Mutations Changing ESR in ASD by a Joint Analysis Using New Large-Scale Datasets
In addition to the analyses described thus far, where we examined 253 mutations in 1,043 ASD probands, 228 mutations in 1,021 SCZ probands, and 154 mutations in 731 controls, we conducted a joint analysis by incorporating two additional large-scale WES datasets for ASD that were published later (De Rubeis et al., 2014; Iossifov et al., 2014) to test whether enrichment of de novo near-splice site synonymous mutations changing ESR persists in the combined dataset that comprises 1,562 synonymous mutations (1,046 in ASD subjects and 516 in controls). In SCZ, no new datasets were available for comparison. Near-splice site synonymous mutations changing ESR remained significantly enriched in the overall combined dataset that is three to four times larger than the initial dataset (p = 0.00049, OR = 1.55, 273 mutations in ASD and 96 mutations in controls, 1-tailed Fisher’s exact test, see also Supplemental Experimental Procedures). The observed OR in the joint analyses was lower than the one observed in our initial analyses (1.55 versus 2.52). This could be partly explained by phenotypic heterogeneity among the datasets used in the joint analyses (e.g. more non-sporadic probands (De Rubeis et al., 2014) and more moderately severe cases, such as ASD males without ID (Iossifov et al., 2014), in the newer datasets). Furthermore, this may also, in part, be a manifestation of the “winner’s curse”, the upward bias in the estimated effect of a newly identified risk variant, often observed in genetic association studies (Kraft, 2008). Regardless, confirmation of our findings warrants further large-scale studies in independent and well-phenotyped cohorts to further confirm enrichment of these mutations in ASD and to estimate their effect size. The same is true for our finding in SCZ, given the small number of synonymous mutations within frontal-cortex derived DHS analyzed in this study (50, 38 and 14 in SCZ, ASD and controls, respectively).
Genes Hit by Potentially “Functional” Synonymous Mutations in ASD Are Involved in Synaptic and Neuronal Functions
We tested whether genes hit by these potentially “functional” de novo synonymous mutations are more likely to be among those intolerant to functional genetic variation as defined by the Residual Variation Intolerance Score (RVIS) (Petrovski et al., 2013) and saw significant enrichment of the “intolerant” genes (see Supplemental Experimental Procedures for the definition of these genes) among both genes with near-splice site synonymous mutations changing ESR in ASD (p = 0.030, binominal exact test) and genes with synonymous mutations within the frontal cortex-derived DHS in SCZ (p = 0.012) (Figure S5). By contrast, there was no enrichment in the control gene sets, which are the genes with near-splice site synonymous mutations not changing ESR or mutations distant from the splice site in ASD and genes outside the frontal cortex-derived DHS in SCZ.
With functional gene-set enrichment analyses using the ToppGene Suite (Chen et al., 2009) there was significant enrichment of genes related to calcium channels and transporters (e.g. “GO: Molecular Function; voltage-gated calcium channel activity”, Benjamini–Hochberg corrected p value [pBH] = 0.0062 and “Gene Family; Calcium channels”, pBH = 2.1 × 10−7), neuronal function and localization (e.g. “GO: Cellular Component; neuron part”, pBH = 0.048 and “REACTOME; Depolarization of the Presynaptic Terminal Triggers the Opening of Calcium Channels”, pBH = 0.00077), and abnormal neuronal phenotypes in mice (e.g. “Mouse Phenotype; abnormal spike wave discharge”, pBH = 0.0046 and “Mouse Phenotype; absence seizures”, pBH = 0.039) (Table S3) among genes hit by near-splice site de novo synonymous mutations changing ESR in ASD (82 genes). Enrichment of genes related to synaptic and neuronal function was further confirmed by significant overrepresentation of synaptic genes defined by Synaptome Database (Pirooznia et al., 2012) among the same input genes (p = 0.046, OR = 1.71, 1-tailed 2×2 Fisher’s exact test). Interestingly, significant enrichment of synaptic genes was observed for those in the presynaptic region (p = 0.030, OR = 2.83), especially in the active zone (p = 0.007, OR = 4.59). It is also notable that there was enrichment of “Pathway Interaction Database; Regulation of Wnt-mediated beta-catenin signaling and target gene transcription” (pBH = 0.042), since this signaling pathway has implicated roles in the pathophysiology of ASD (Krumm et al., 2014).
Enrichment of these terms as well as synaptic genes was not observed among genes with de novo synonymous mutations distant from the nearest splice site (> 30 bp) or near-splice site mutations not changing ESR in ASD (171 genes) and genes with synonymous mutations changing ESR in controls (23 genes) (Table S3).
In contrast, when we used the genes with de novo synonymous mutations within the frontal cortex-derived DHS in SCZ (60 genes) as input, there was no term significantly enriched after performing Benjamini–Hochberg correction (pBH < 0.05, Table S3). This may suggest that the pathways affected by these genes in SCZ are more heterogeneous and the number of the input genes was too small to identify enrichment of particular biological pathways.
Significant Contribution of Potentially Functional De Novo Synonymous Mutations to Disease Liability
We examined the relative contribution of functional synonymous mutations to variability on the liability scale as compared to other mutation types and estimate that near-splice site synonymous mutations changing ESR in ASD and synonymous mutations within the frontal cortex-derived DHS in SCZ can explain 1.5% and 0.6% of the variability on the liability scale, respectively (Table S4, see Supplemental Experimental Procedures for details). These estimated contributions are comparable to that of de novo LOF mutations (1.3% for ASD and 0.74% for SCZ) and much higher than that of de novo missense mutations (0.1% for both ASD and SCZ). It is also notable that the per-individual mutation rates and relative risks compared to controls for potentially functional synonymous mutations are similar to or not greatly different from those for LOF mutations (Table S4).
Candidate ASD and SCZ Genes That Are Recurrently Hit by Damaging De Novo Mutations Including Potentially Functional Synonymous Mutations
Identification of these potentially functional de novo synonymous mutations can help discover new genes involved in disease, as genes with recurrent functional de novo mutations including LOF, damaging missense, and now “functional” synonymous mutations are more likely to be associated with disease. We evaluated the statistical significance of observing multiple functional de novo mutations, including potentially functional synonymous mutations in the same gene, by applying a framework for the interpretation of de novo mutations (Samocha et al., 2014) which takes into consideration gene length and local sequence context. Among the genes hit by near-splice site synonymous mutations changing ESR in ASD, eight are hit by other functional de novo mutations in ASD (Table 1). Of these, RAB2A, in which a LOF mutation was previously reported (Sanders et al., 2012), showed the highest significance (p = 4.48 × 10−6). The p value for this gene is close to the genome-wide significance threshold (p = 2.74 × 10−6) considering the number of genes tested (N = 18,271) (Samocha et al., 2014), and the corresponding p value conservatively corrected for the number of tested genes is 0.081. Among the genes identified in this study in SCZ, seven genes are hit by additional functional mutations in SCZ (Table 1). It is notable that one potentially functional synonymous mutation was identified in the SETD1A gene, in which two LOF mutations were previously reported in SCZ (Takata et al., 2014). This gene harbors significantly more functional mutations than expected after performing genome-wide correction (praw = 1.79 × 10−6, pcorrected = 0.033). In addition, another SCZ patient with a de novo LOF mutation in this gene was identified in a recent study (Guipponi et al., 2014). In total there are three de novo LOF and one de novo functional synonymous mutations in SETD1A among 1074 probands, and the corresponding p value is 1.2 × 10−8, further demonstrating that this gene is very likely to be a genuine disease susceptibility gene.
Table 1.
Genes with recurrent mutations including synonymous mutations potentially affecting regulatory elements and protein-altering mutations
| Gene | Gene size (bp) | Disease | Mutation types | # of functional synonymous mutations* | # of LOF mutations | # of missense or inframe indel mutations | Expected # of mutations | p values considering synonumous mutations | p values not considering synonumous mutations | Tests |
|---|---|---|---|---|---|---|---|---|---|---|
| RAB2A | 639 | ASD | Near-SS synonymous changing ESR, Nonsense | 1 | 1 | 0 | 0.003 | 4.4 8 × 10−6 | 2.32 × 10−3 | Near-SS synonymous changing ESR+LOF |
| EIF3G | 963 | ASD | Near-SS synonymous changing ESR, Missense | 1 | 0 | 1 | 0.032 | 5.02 × 10−4 | 2.96 × 10−2 | Near-SS synonymous changing ESR+LOF+Missense |
| SLC22A9 | 1662 | ASD | Near-SS synonymous changing ESR, Missense | 1 | 0 | 1 | 0.036 | 6.35 × 10−4 | 3.38 × 10−2 | Near-SS synonymous changing ESR+LOF+Missense |
| PPP2R1B | 1806 | ASD | Near-SS synonymous changing ESR, Missense | 1 | 0 | 1 | 0.044 | 9. 60 × 10−4 | 4.14 × 10−2 | Near-SS synonymous changing ESR+LOF+Missense |
| MCM4 | 2592 | ASD | Near-SS synonymous changing ESR, Missense | 1 | 0 | 1 | 0.068 | 2. 21 × 10−3 | 6.25 × 10−2 | Near-SS synonymous changing ESR+LOF+Missense |
| NUP133 | 3471 | ASD | Near-SS synonymous changing ESR, Missense | 1 | 0 | 1 | 0.080 | 3.01 × 10−3 | 7.30 × 10−2 | Near-SS synonymous changing ESR+LOF+Missense |
| CACNA1E | 6942 | ASD | Near-SS synonymous changing ESR, Missense | 1 | 0 | 1 | 0.185 | 1.51 × 10−2 | 1.61 × 10−1 | Near-SS synonymous changing ESR+LOF+Missense |
| NAV2 | 7467 | ASD | Near-SS synonymous changing ESR, Missense | 1 | 0 | 1 | 0.201 | 1. 78 × 10−2 | 1.73 × 10−1 | Near-SS synonymous changing ESR+LOF+Missense |
| SETD1A | 5124 | SCZ | Synonymous within FC DHS, Frameshift, Splice site | 1 | 2 | 0 | 0.022 | 1.79 × 10−6 | 8.65 × 10−5 | Synonymous within FC DHS+LOF |
| TNRC18 | 8907 | SCZ | Synonymous within FC DHS, Nonsense | 1 | 1 | 0 | 0.026 | 3.44 × 10−4 | 1.56 × 10−2 | Synonymous within FC DHS+LOF |
| ADCY7 | 3243 | SCZ | Synonymous within FC DHS, Missense | 1 | 0 | 1 | 0.093 | 4.03 × 10−3 | 8.31 × 10−2 | Synonymous within FC DHS+LOF+Missense |
| PDE4DIP | 7041 | SCZ | Synonymous within FC DHS, Missense | 1 | 0 | 1 | 0.155 | 1.08 × 10−2 | 1.37 × 10−1 | Synonymous within FC DHS+LOF+Missense |
| NCOR2 | 7545 | SCZ | Synonymous within FC DHS, Missense | 1 | 0 | 1 | 0.224 | 2.17 × 10−2 | 1.89 × 10−1 | Synonymous within FC DHS+LOF+Missense |
| CELSR2 | 8772 | SCZ | Synonymous within FC DHS, Missense | 1 | 0 | 1 | 0.262 | 2. 89 × 10−2 | 2.19 × 10−1 | Synonymous within FC DHS+LOF+Missense |
| SSPO | 15444 | SCZ | Synonymous within FC DHS, Missense | 1 | 0 | 1 | 0.354 | 4. 96 × 10−2 | 2.84 × 10−1 | Synonymous within FC DHS+LOF+Missense |
Near-splice site synonymous changing ESR for ASD and synonymous mutation within the frontal cortex-derived DHS for SCZ.
DHS; D N ase I hypersensitive site, ESR; exonic splicing regulator, FC; frontal cortex, LOF, loss-of-function, SS; splice site
In addition, both SETD1A and RAB2A were amongst the highly intolerant genes in the human genome in a recent variation analysis of over 60,000 humans in ExAC (http://biorxiv.org/content/early/2015/10/30/030338), (probability of being LOF intolerant, pLI = 0.999 for SETD1A and 0.971 for RAB2A), indicating that LOF variants within these genes are under natural selection in the human genome.
DISCUSSION
Our analyses found significant enrichment among cases of near-splice site mutations changing ESR in ASD and of synonymous mutations within frontal-cortex derived DHS in SCZ. The finding in ASD was confirmed in subsequent analyses that integrated the initial dataset with additional, newly published, large datasets (De Rubeis et al., 2014; Iossifov et al., 2014). Biologically interpretable results in our exploration of the properties of genes hit by these potentially “functional” synonymous mutations further support contribution of these mutations to disease etiology. Thru analyzing genes hit by potentially functional synonymous mutations using gene intolerance scores, we found a pattern similar to de novo LOF and missense mutations reported in earlier studies. We also saw enrichment of genes related to brain function and biological processes previously implicated in the disease (e.g. calcium channels and Wnt/beta-catenin signaling for ASD). Notably, near-splice site synonymous mutations, especially those changing ESR, have been shown to be significantly enriched in patients with epileptic encephalopathies (Epi4K_Consortium et al., 2013), indicating they may contribute to other neuropsychiatric disorders (Figure S6, see Supplemental Information for details).
By evaluating genes hit by multiple de novo damaging mutations, including potentially functional synonymous mutations, we identified RAB2A and SETD1A as promising candidate genes for ASD and SCZ respectively. These results are particularly important for development of valid animal and cellular disease models, an essential step towards clinical translation (Karayiorgou et al., 2012). RAB2A encodes a small guanosine triphosphatase (GTPase) protein that is required for protein transport from the endoplasmic reticulum (ER) to the Golgi complex (Tisdale et al., 1992). Association between common SNPs in this gene and the density of calbindin-positive GABAergic neurons in postmortem prefrontal cortex was reported in a GWAS (Kim and Webster, 2011), suggesting a role of RAB2A in prefrontal inhibitory neural circuits. In addition, a RAB2A polymorphism was recently shown to impact prefrontal cortical morphology, functional connectivity, and working memory (Li et al., 2015), all key deficits implicated in the etiology of neurodevelopmental disorders, including SCZ and ASD (Schubert et al., 2015). In addition, RAB2A is a target of CHD8, a well-established ASD gene involved in prenatal human neurodevelopment (Cotney et al., 2015).
SETD1A, in which three de novo LOF mutations were previously reported (Guipponi et al., 2014; Takata et al., 2014), encodes a catalytic unit of the histone H3 lysine 4 (H3K4) methyltransferase complex. Identification of an additional potentially functional de novo synonymous mutation in this gene and the accumulating evidence for involvement of the chromatin regulation pathway in neuropsychiatric diseases, including ASD and SCZ, further supports that SETD1A is a genuine susceptibility gene for SCZ. Notably, strong association of LOF mutations in SETD1A (also known as KMT2F) with SCZ was confirmed by a recent large-scale study examining several tens of thousands of WES data (Singh et al., 2016).
The estimated proportion of disease liability explained by these mutations is comparable to de novo LOF mutations and suggests that potentially functional synonymous mutations can contribute to the disease risk as much as LOF mutations. Although the overall contribution of potentially functional synonymous mutations to disease liability may not be very large (~1%) because these mutations are very rare, it should be noted that each mutation could greatly increase the disease risk. Indeed, ORs observed for potentially functional synonymous mutations are comparable to those observed for de novo LOF mutations (see Figure 2 and S1). Furthermore, genetic regulatory elements are spread throughout the genome. While the datasets used in our analyses cover only the coding regions that comprise ~1% of the human genome, it is likely that mutations affecting regulatory elements outside the coding regions can explain a substantial part of disease liability. Detailed investigation of the impact of mutations on regulatory elements both inside and outside the coding regions, using whole-genome or “regulome” sequencing along with the rapidly increasing knowledge on various types of functional DNA elements in the human genome (Encode_Project_Consortium et al., 2012), will help us better understand the genetic architecture of complex neuropsychiatric diseases.
EXPERIMENTAL PROCEDURES
Experimental procedures are available in Supplemental Information.
Supplementary Material
Highlights.
De novo synonymous mutations likely affecting splicing regulation are enriched in ASD
De novo synonymous mutations within frontal cortex-derived DHS are enriched in SCZ
“Functional” synonymous mutations significantly contribute to disease liability
Functional synonymous mutations support role of SETD1A and RAB2A in neuropsychiatry
Acknowledgments
This work was partially supported by National Institute of Mental Health (NIMH) grants MH061399 (to M.K.), MH097879 (to J.A.G.) and MH095797 (to I.I.L.) and by a National Alliance for Research in Schizophrenia and Depression (NARSAD) Young Investigator Award (to B.X.). A.T. was supported by the JSPS Postdoctoral Fellowship for Research Abroad.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
AUTHOR CONTRIBUTIONS
A.T. and B.X. designed research; A.T., B.X., I.I.-L., J.A.G. and M.K. performed research; A.T., B.X. and I.I.-L. analyzed data; and A.T., B.X., I.I.-L., J.A.G., and M.K. wrote the paper.
References
- Chen J, Bardes EE, Aronow BJ, Jegga AG. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic acids research. 2009;37:W305–311. doi: 10.1093/nar/gkp427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotney J, Muhle RA, Sanders SJ, Liu L, Willsey AJ, Niu W, Liu W, Klei L, Lei J, Yin J, et al. The autism-associated chromatin modifier CHD8 regulates other autism risk genes during human neurodevelopment. Nature communications. 2015;6:6404. doi: 10.1038/ncomms7404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Ercument Cicek A, Kou Y, Liu L, Fromer M, Walker S, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014 doi: 10.1038/nature13772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Encode_Project_Consortium. Bernstein BE, Birney E, Dunham I, Green ED, Gunter C, Snyder M. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epi4K_Consortium, Epilepsy Phenome/Genome, P. Allen AS, Berkovic SF, Cossette P, Delanty N, Dlugos D, Eichler EE, Epstein MP, Glauser T, et al. De novo mutations in epileptic encephalopathies. Nature. 2013;501:217–221. doi: 10.1038/nature12439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairbrother WG, Yeh RF, Sharp PA, Burge CB. Predictive identification of exonic splicing enhancers in human genes. Science. 2002;297:1007–1013. doi: 10.1126/science.1073774. [DOI] [PubMed] [Google Scholar]
- Fromer M, Pocklington AJ, Kavanagh DH, Williams HJ, Dwyer S, Gormley P, Georgieva L, Rees E, Palta P, Ruderfer DM, et al. De novo mutations in schizophrenia implicate synaptic networks. Nature. 2014;506:179–184. doi: 10.1038/nature12929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girard SL, Gauthier J, Noreau A, Xiong L, Zhou S, Jouan L, Dionne-Laporte A, Spiegelman D, Henrion E, Diallo O, et al. Increased exonic de novo mutation rate in individuals with schizophrenia. Nature genetics. 2011;43:860–863. doi: 10.1038/ng.886. [DOI] [PubMed] [Google Scholar]
- Guipponi M, Santoni FA, Setola V, Gehrig C, Rotharmel M, Cuenca M, Guillin O, Dikeos D, Georgantopoulos G, Papadimitriou G, et al. Exome sequencing in 53 sporadic cases of schizophrenia identifies 18 putative candidate genes. PloS one. 2014;9:e112745. doi: 10.1371/journal.pone.0112745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulsuner S, Walsh T, Watts AC, Lee MK, Thornton AM, Casadei S, Rippey C, Shahin H, Consortium on the Genetics of, S., Group, P.S. et al. Spatial and temporal mapping of de novo mutations in schizophrenia to a fetal prefrontal cortical network. Cell. 2013;154:518–529. doi: 10.1016/j.cell.2013.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, Yamrom B, Lee YH, Narzisi G, Leotta A, et al. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74:285–299. doi: 10.1016/j.neuron.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iossifov I, O’Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, Stessman HA, Witherspoon KT, Vives L, Patterson KE, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014 doi: 10.1038/nature13908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karayiorgou M, Flint J, Gogos JA, Malenka RC, Genetic and Neural Complexity in Psychiatry Working, G. The best of times, the worst of times for psychiatric disease. Nature neuroscience. 2012;15:811–812. doi: 10.1038/nn.3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavanagh DH, Dwyer S, O’Donovan MC, Owen MJ. The ENCODE project: implications for psychiatric genetics. Molecular psychiatry. 2013;18:540–542. doi: 10.1038/mp.2013.13. [DOI] [PubMed] [Google Scholar]
- Ke S, Shang S, Kalachikov SM, Morozova I, Yu L, Russo JJ, Ju J, Chasin LA. Quantitative evaluation of all hexamers as exonic splicing elements. Genome research. 2011;21:1360–1374. doi: 10.1101/gr.119628.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Webster MJ. Integrative genome-wide association analysis of cytoarchitectural abnormalities in the prefrontal cortex of psychiatric disorders. Molecular psychiatry. 2011;16:452–461. doi: 10.1038/mp.2010.23. [DOI] [PubMed] [Google Scholar]
- Kraft P. Curses–winner’s and otherwise–in genetic epidemiology. Epidemiology. 2008;19:649–651. doi: 10.1097/EDE.0b013e318181b865. discussion 657–648. [DOI] [PubMed] [Google Scholar]
- Krumm N, O’Roak BJ, Shendure J, Eichler EE. A de novo convergence of autism genetics and molecular neuroscience. Trends in neurosciences. 2014;37:95–105. doi: 10.1016/j.tins.2013.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Liu B, Chen C, Cui Y, Shang L, Zhang Y, Wang C, Zhang X, He Q, Zhang W, et al. RAB2A Polymorphism impacts prefrontal morphology, functional connectivity, and working memory. Hum Brain Mapp. 2015;36:4372–4382. doi: 10.1002/hbm.22924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, Reynolds AP, Sandstrom R, Qu H, Brody J, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337:1190–1195. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy SE, Gillis J, Kramer M, Lihm J, Yoon S, Berstein Y, Mistry M, Pavlidis P, Solomon R, Ghiban E, et al. De novo mutations in schizophrenia implicate chromatin remodeling and support a genetic overlap with autism and intellectual disability. Molecular psychiatry. 2014 doi: 10.1038/mp.2014.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale BM, Kou Y, Liu L, Ma’ayan A, Samocha KE, Sabo A, Lin CF, Stevens C, Wang LS, Makarov V, et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485:242–245. doi: 10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neph S, Vierstra J, Stergachis AB, Reynolds AP, Haugen E, Vernot B, Thurman RE, John S, Sandstrom R, Johnson AK, et al. An expansive human regulatory lexicon encoded in transcription factor footprints. Nature. 2012;489:83–90. doi: 10.1038/nature11212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, Levy R, Ko A, Lee C, Smith JD, et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485:246–250. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrovski S, Wang Q, Heinzen EL, Allen AS, Goldstein DB. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS genetics. 2013;9:e1003709. doi: 10.1371/journal.pgen.1003709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirooznia M, Wang T, Avramopoulos D, Valle D, Thomas G, Huganir RL, Goes FS, Potash JB, Zandi PP. SynaptomeDB: an ontology-based knowledgebase for synaptic genes. Bioinformatics. 2012;28:897–899. doi: 10.1093/bioinformatics/bts040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch A, Wieczorek D, Graf E, Wieland T, Endele S, Schwarzmayr T, Albrecht B, Bartholdi D, Beygo J, Di Donato N, et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet. 2012;380:1674–1682. doi: 10.1016/S0140-6736(12)61480-9. [DOI] [PubMed] [Google Scholar]
- Roadmap Epigenomics, C. Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–330. doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samocha KE, Robinson EB, Sanders SJ, Stevens C, Sabo A, McGrath LM, Kosmicki JA, Rehnstrom K, Mallick S, Kirby A, et al. A framework for the interpretation of de novo mutation in human disease. Nature genetics. 2014 doi: 10.1038/ng.3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, Ercan-Sencicek AG, DiLullo NM, Parikshak NN, Stein JL, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–241. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauna ZE, Kimchi-Sarfaty C. Understanding the contribution of synonymous mutations to human disease. Nature reviews Genetics. 2011;12:683–691. doi: 10.1038/nrg3051. [DOI] [PubMed] [Google Scholar]
- Schubert D, Martens GJ, Kolk SM. Molecular underpinnings of prefrontal cortex development in rodents provide insights into the etiology of neurodevelopmental disorders. Mol Psychiatry. 2015;20:795–809. doi: 10.1038/mp.2014.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh T, Kurki MI, Curtis D, Purcell SM, Crooks L, McRae J, Suvisaari J, Chheda H, Blackwood D, Breen G, et al. Rare loss-of-function variants in KMT2F are associated with schizophrenia and developmental disorders. bioRxiv. 2016 doi: 10.1038/nn.4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supek F, Minana B, Valcarcel J, Gabaldon T, Lehner B. Synonymous mutations frequently act as driver mutations in human cancers. Cell. 2014;156:1324–1335. doi: 10.1016/j.cell.2014.01.051. [DOI] [PubMed] [Google Scholar]
- Takata A, Xu B, Ionita-Laza I, Roos JL, Gogos JA, Karayiorgou M. Loss-of-Function Variants in Schizophrenia Risk and SETD1A as a Candidate Susceptibility Gene. Neuron. 2014;82:773–780. doi: 10.1016/j.neuron.2014.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB, Wang H, Vernot B, et al. The accessible chromatin landscape of the human genome. Nature. 2012;489:75–82. doi: 10.1038/nature11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tisdale EJ, Bourne JR, Khosravi-Far R, Der CJ, Balch WE. GTP-binding mutants of rab1 and rab2 are potent inhibitors of vesicular transport from the endoplasmic reticulum to the Golgi complex. The Journal of cell biology. 1992;119:749–761. doi: 10.1083/jcb.119.4.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Rolish ME, Yeo G, Tung V, Mawson M, Burge CB. Systematic identification and analysis of exonic splicing silencers. Cell. 2004;119:831–845. doi: 10.1016/j.cell.2004.11.010. [DOI] [PubMed] [Google Scholar]
- Xu B, Roos JL, Dexheimer P, Boone B, Plummer B, Levy S, Gogos JA, Karayiorgou M. Exome sequencing supports a de novo mutational paradigm for schizophrenia. Nature genetics. 2011;43:864–868. doi: 10.1038/ng.902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B, Ionita-Laza I, Roos JL, Boone B, Woodrick S, Sun Y, Levy S, Gogos JA, Karayiorgou M. De novo gene mutations highlight patterns of genetic and neural complexity in schizophrenia. Nature genetics. 2012;44:1365–1369. doi: 10.1038/ng.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.