

Abstract
This Letter describes the natural product guided synthesis of unnatural analogs of the marine bromopyrrole alkaloid dispyrin, and the resulting SAR of H3 antagonism. Multiple rounds of iterative parallel synthesis improved human H3 IC50 ~33-fold, and afforded a new class of H3 antagonists based on the novel bromotyramine core of dispyrin.
Keywords: H3 antagonist, Dispyrin, Marine natural product, Alkaloid
The neurotransmitter histamine exerts its action through four distinct Class A GPCRs (H1–H4).1–7 The histamine H3 receptor, a Gi/o-coupled receptor in the CNS, is a pre-synaptic auto- and heteroreceptor that not only controls the release of histamine, but also other neurotransmitters (acetylcholine, noradrenaline, dopamine, GABA and serotonin).1–7 Preclinically, H3 antagonists/inverse agonists have demonstrated efficacy in a number of CNS pathologies including schizophrenia, epilepsy, depression, pain, decreasing food intake, drug abuse and addiction, sleep disorders/narcolepsy and cognitive enhancement.1–7 Early reference H3 antagonists contained imidazole moieties, such as thioperimide 1 and Perceptin (GT-2331) 2 (Fig. 1). Effort from multiple companies then focused on non-imidazole H3 antagonists and include compounds such as UCL 1972 3, ABT-239 4, JNJ’s 5, Novo Nordisk’s 6, Eli Lilly’s 7 and GSK189254 8 to exemplify a few (Fig. 1). This intense effort from the pharmaceutical industry led to the evolution of a refined H3 antagonist pharmacophore model 9.1–8
Figure 1.

Imidiazole and non-imidazole H3 antagonists 1–8 leading to a refined H3 pharmacophore model 9.
We recently completed the first total synthesis of dispyrin 10,9 a bromopyrrole alkaloid with a novel bromotyramine core, isolated by Crews in 200710 from the marine sponge Agelas dispar (Fig. 2). Upon recognition that dispyrin 10 possessed the basic features of the refined H3 pharmacophore model 9, we evaluated our synthetic dispyrin against the human H3 receptor. Gratifyingly, dispyrin was found to have modest activity as an H3 antagonist (IC50 = 2.35 μM, Ki = 1.04 μM).9 Based on these data, we initiated a natural product guided synthesis effort, employing iterative parallel synthesis11 for molecular editing, aimed at improving H3 inhibition and binding; moreover, we wanted to validate the marine natural product dispyrin 10 as a viable lead molecule due to the novel scaffold providing intellectual property in extremely crowded chemical space.
Figure 2.
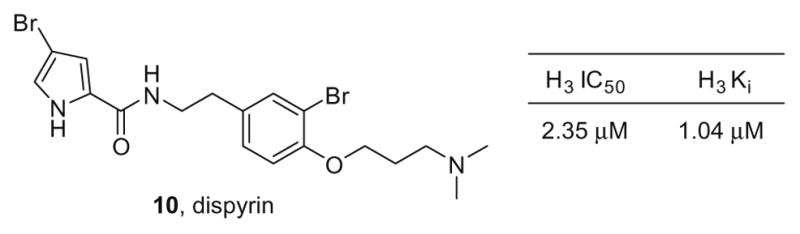
Dispyrin 10, a novel bromopyrrole alkaloid from Agelas dispar with a bromotyramine core unprecedented in marine natural products.
The first generation 25-member library was based on a 5 × 5 two-dimensional design wherein the core was held constant and the amide R1 and aminoalkyl moieties R2 varied (Scheme 1). The library synthesis began with a simple DIC amide coupling employing commercially available 3-bromo-4-methoxyphenylethylamine 11 with one of five heterocyclic carboxylic acids R1. These five scaffolds were then treated with BBr3 to remove the methyl ether liberating the free phenols 13. Each of the five phenols 13 was then alkylated with one of five aminoalkyl chlorides to install R2 under microwave-assisted conditions to afford unnatural dispyrin analogs 14 (Table 1).
Scheme 1.
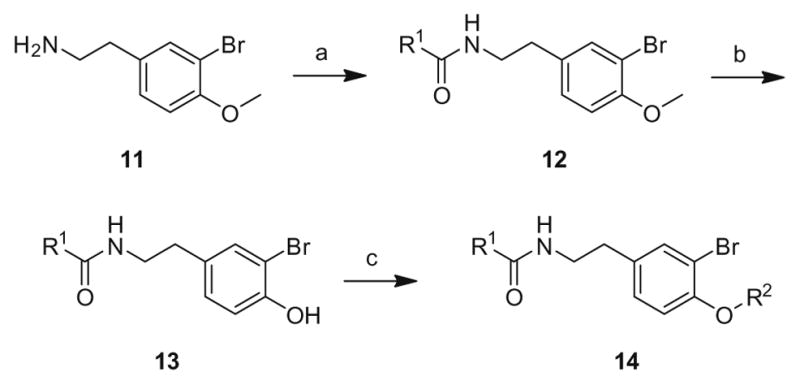
First generation library synthesis. Reagents and conditions: (a) R1COOH, DIC, HOBt, DIEA, DCM, rt, 12 h (69–99%); (b) BBr3, DCM, −78 °C–rt, 1.5 h (50–95%); (c) 1.3 equiv ClR2, CsCO3, KI, DMF, mw, 160 °C, 20 min (72–93%). All analogs purified to >98% by mass-directed preparative HPLC.12
Table 1.
Structures and activities of dispyrin analogs 14
| ||||
|---|---|---|---|---|
| Compd | R1 | R2 | H3Kia (μm) | H3IC50a (μm) |
| 14a |
|
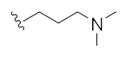
|
1.04 | 2.35 |
| 14b |
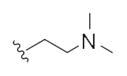
|
1.61 | 3.72 | |
| 14c |
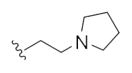
|
0.19 | 0.43 | |
| 14d |
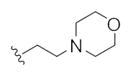
|
12.9 | 29.1 | |
| 14e |
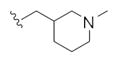
|
1.33 | 2.94 | |
| 14f |
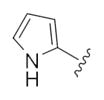
|
|
1.36 | 2.92 |
| 14g |
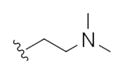
|
1.55 | 3.43 | |
| 14h |
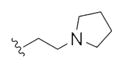
|
0.15 | 0.38 | |
| 14i |
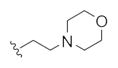
|
26.4 | 59.2 | |
| 14j |
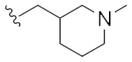
|
1.52 | 3.63 | |
| 14k |
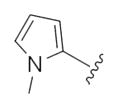
|
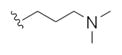
|
1.76 | 3.99 |
| 14l |
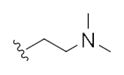
|
2.13 | 4.54 | |
| 14m |
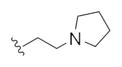
|
0.13 | 0.31 | |
| 14n |
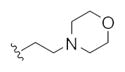
|
17.6 | 39.6 | |
| 14o |
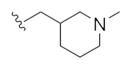
|
1.65 | 3.62 | |
| 14p |
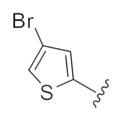
|
|
1.61 | 3.61 |
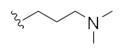
| 14q |
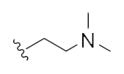
|
0.67 | 1.52 | |
| 14r |
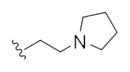
|
0.08 | 0.18 | |
| 14s |
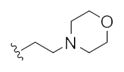
|
13.5 | 30.4 | |
| 14t |
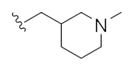
|
1.25 | 3.73 | |
| 14u |
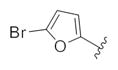
|
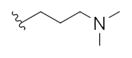
|
2.29 | 4.81 |
| 14v |
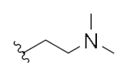
|
2.72 | 5.93 | |
| 14w |
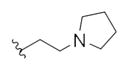
|
0.17 | 0.39 | |
| 14x |
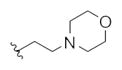
|
32.5 | 73.2 | |
| 14y |
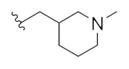
|
2.11 | 4.56 | |
Average of three independent determinations with human H3.
This first generation library was highly informative. In general, all R1s and R2s afforded modestly potent (Kis and IC50s in the low micromolar range) H3 antagonists. Potent H3 antagonists (Kis < 200 nM, IC50s < 430 nM) resulted for all of the heterocyclic amides R1 in combination with the ethyl pyrrolidinyl R2 (14c, 14h, 14m, 14r and 14w). In contrast, the ethyl morpholino congeners (14d, 14i, 14n, 14s and 14x) were uniformly weak (Kis > 12 μM, IC50s > 29 μM). The most potent H3 antagonist from the first generation library was 14r (R1 = 4-bromo-thiophene, R2 = ethyl pyrrolidine) with a Ki of 80 nM and an IC50 of 180 nM—a 13-fold improvement over the parent natural product dispyrin 10 (IC50 = 2.35 μM, Ki = 1.04 μM). Based on these data, the next library maintained R1 = 4-bromo-thiophene and surveyed functionalized pyrrolidines at R2 (Scheme 2).
Scheme 2.

Second generation library synthesis. Reagents and conditions: (a) BrCH2CH(OMe)2, Cs2CO32−, KI, DMF, reflux (70%); (b) TosOH, mw, 160 °C, 10 min (60%); (c) functionalized pyrrolidine, MP-(OAc)3H, DCM, rt (40–85%). All analogs purified to >98% by mass-directed preparative HPLC.12
Following Scheme 1, a large quantity of 15 was prepared. Then, the phenol was alkylated with 2-bromo-1,1-dimethoxy ethane to provide 16, which was then converted to the corresponding aldehyde 17 by treatment with tosylic acid. Finally, reductive amination employing a functionalized pyrrolidine and MP-B(OAc)3H provided analogs 18. As shown in Table 2, analogs 18 were weaker H3 antagonists than 14r, and there was no evidence of enantioselective inhibition (18a vs 18b). In agreement with the H3 pharmacophore model, incorporation of β-fluorine atoms such as in 18c and 18d, which lowers the pKa on the pyrrolidine nitrogen from 11 to 9, afforded diminished H3 inhibition.13
Table 2.
Structures and activities of dispyrin analogs 18
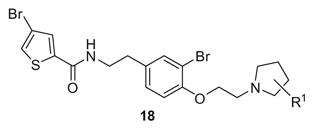
| |||
|---|---|---|---|
| Compd | R1 | H3Kia (μm) | H3IC50a (μm) |
| 18a | (S)-2-Me | 0.31 | 0.71 |
| 18b | (S)-2-Me | 0.33 | 0.75 |
| 18c | (S)-3-F | 1.10 | 2.11 |
| 18d | (S)-3-F | 1.15 | 2.52 |
Average of three independent determinations.
We then prepared two singleton compounds following the synthetic route depicted in Scheme 1 with the appropriate substitutions, wherein the bromine in 14r was replaced with a chlorine 19 and a truncated version 20 (Fig. 3). A twofold diminution in potency was noted for 19, relative to 14r, and the truncated benzyl version lost over 13-fold compared to 14r; however, this highlighted that the heavy bromine atom was not required for H3 inhibition.
Figure 3.

Chlorinated version and chlorinated truncated version of 14r.
The final library iteration was directed at surveying a wider range of alternative amides (heterocycles and functionalized aromatic moieties) while holding the preferred ethyl pyrrolidine ether and bromotyramine core constant. The synthesis began with 11 and conversion to the phthalimide congener 21. Standard BBr3 deprotection provided 22 which was alkylated with chloroethyl pyrrolidine to deliver 23. Hydrolysis of the phthalimide with hydrazine, followed by amide coupling with a diverse collection of aryl and heteroaryl carboxylic acids generated library 24 (Scheme 3).
Scheme 3.
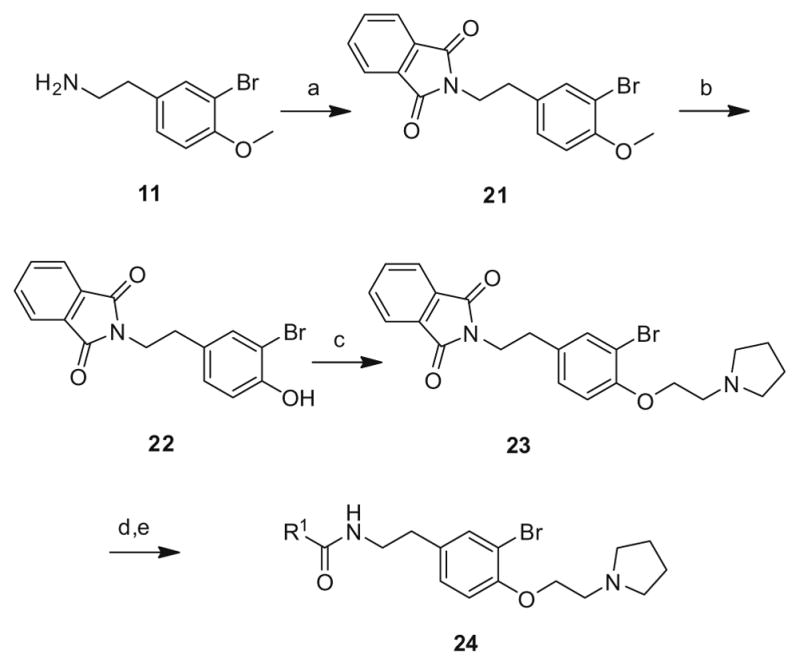
Third generation library synthesis. Reagents and conditions: (a) 1,2-dicarboxybenzene, DIC, HOBt, DIEA, DCM, rt, 12 h (99%); (b) BBr3, DCM, −78 °C–rt, 1.5 h (95%); (c) 1.3 equiv Chloroethyl pyrrolidine, CsCO3, KI, DMF, mw, 160 °C, 20 min (93%); (d) (i) N2H4, mw, 160 °C; (ii) SCX; (e) R1COOH, DIC, HOBt, DIEA, DCM, rt, 12 h (64–98%). All analogs purified to >98% by mass-directed preparative HPLC.12
This third generation library was uniformly active, providing H3 antagonists in the sub-micromolar range. Six-member heterocycles, such as pyridine 24a, were active, as were aryl amides with halogens (Cl and Br) or trifluoromethyl groups in the 3-position (24f–h). Five-member heterocycles (24b–e) proved optimal, with a 5-oxazole 24b (Ki = 32 nM, IC50 = 83 nM) and 2-thiazole 24d (Ki = 32 nM, IC50 = 72 nM) affording the most potent H3 antagonists of the unnatural dispyrin analogs. For example, 24d improved the H3 Ki and IC50 ~33-fold over the natural product dispyrin, and required only three iterations of molecular editing and 40 analogs. Moreover, as dispyrin represented a novel chemotype, we were able to obtain composition of matter patents for the dispyrin analogs as H3 antagonists within an incredibly crowded intellectual property landscape.14 This effort highlights the value of employing natural products as leads for therapeutically relevant targets (Table 3).
Table 3.
Structures and activities of dispyrin analogs 24
| |||
|---|---|---|---|
| Compd | R1 | H3Kia (μm) | H3IC50a (μm) |
| 24a |
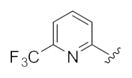
|
0.27 | 0.56 |
| 24b |
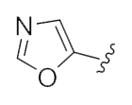
|
0.03 | 0.08 |
| 24c |
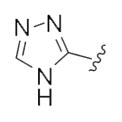
|
0.43 | 0.97 |
| 24d |
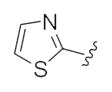
|
0.03 | 0.07 |
| 24e |
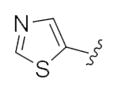
|
0.12 | 0.26 |
| 24f |
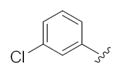
|
0.25 | 0.55 |
| 24g |
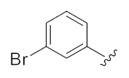
|
0.21 | 0.45 |
| 24h |
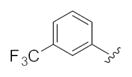
|
0.44 | 0.98 |
Average of three independent determinations.
In summary, a natural products guided synthesis effort in molecular editing, employing iterative parallel synthesis, quickly optimized the weak H3 antagonism of the marine natural product dispyrin 10 over 30-fold to afford unnatural analog 24d with low nanomolar potency and binding. By employing a novel natural product scaffold for lead optimization, we were able to establish an intellectual property position in an incredibly crowded intellectual property landscape. Although the role of natural products drug discovery efforts within the pharmaceutical industry is being significantly reduced, despite overwhelming success, the biological activity of dispyrin and its analogs argue further that natural products are viable drug leads and offer patenting advantages.
Acknowledgments
The authors thank the Vanderbilt Department of Pharmacology and the Vanderbilt Institute of Chemical Biology for support of this research. J.P.K. acknowledges a VICB predoctoral training fellowship.
References and notes
- 1.Celanire S, Wijtmans M, Talaga P, Leurs R, de Esch JP. Drug Discovery Today. 2005;10:1613. doi: 10.1016/S1359-6446(05)03625-1. [DOI] [PubMed] [Google Scholar]
- 2.Esbenshade TA, Brownman KE, Bitner RS, Strakhova M, Cowart MD, Brioni JD. Br J Pharmacol. 2008;154:1166. doi: 10.1038/bjp.2008.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berlin M, Boyce CM. Exp Opin Ther Patents. 2007;17:675. doi: 10.1517/13543776.17.6.675. [DOI] [PubMed] [Google Scholar]
- 4.Stocking EM, Letavic MA. Curr Top Med Chem. 2008;8:988. doi: 10.2174/156802608784936728. [DOI] [PubMed] [Google Scholar]
- 5.Jesudason CD, Beavers LS, Cramer JW, Dill J, Finley DR, Lindsley CW, Stevens FC, Gadski RA, Oldham SW, Pickard RT, Siedem CS, Sindelar DK, Singh A, Watson BM, Hipskind PA. Bioorg Med Chem Lett. 2006;16:3415. doi: 10.1016/j.bmcl.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 6.Wijtmans M, Leurs R, de Esch I. Exp Opin Invest Drugs. 2007;16:967. doi: 10.1517/13543784.16.7.967. [DOI] [PubMed] [Google Scholar]
- 7.Hudkins RL, Raddatz R. Ann Reports Med Chem. 2007;42:49. [Google Scholar]
- 8.Santora VJ, Covel JA, Hayashi R, Hofilena BJ, Ibarra JB, Pulley MD, Weinhouse MI, Sengupta D, Duffield JJ, Semple G, Webb RR, Sage C, Ren A, Pereira G, Knudsen J, Edwards JEL, Suarez M, Frazer J, Thomsen W, Hauser E, Whelan K, Grottick AJ. Bioorg Med Chem Lett. 2008;18:1490. doi: 10.1016/j.bmcl.2007.12.059. [DOI] [PubMed] [Google Scholar]
- 9.Kennedy JP, Brogan JT, Lindsley CW. J Nat Prod. 2008;71:1783. doi: 10.1021/np800339e. [DOI] [PubMed] [Google Scholar]
- 10.Pina IC, White KN, Cabera G, Rivero E, Crews P. J Nat Prod. 2007;70:613. doi: 10.1021/np0606042. [DOI] [PubMed] [Google Scholar]
- 11.Kennedy JP, Williams L, Bridges TM, Daniels RN, Weaver D, Lindsley CW. J Comb Chem. 2008;10:345. doi: 10.1021/cc700187t. [DOI] [PubMed] [Google Scholar]
- 12.Leister WH, Strauss KA, Wisnoski DD, Zhao Z, Lindsley CW. J Comb Chem. 2003;5:322. doi: 10.1021/cc0201041. [DOI] [PubMed] [Google Scholar]
- 13.Fadeyi O, Lindsley CW. Org Lett. 2009;11:943. doi: 10.1021/ol802930q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Provisional patent (61/059,975) filed. Jun 9, 2008. p. 3208. [Google Scholar]