

Abstract
Objective:
To compare the efficacy, safety, and anti-inflammatory effects of cenicriviroc (CVC), an oral, once-daily C-C chemokine receptor types 5 and 2 antagonist, with those of efavirenz (EFV) in treatment-naive, HIV-1-infected adults.
Design:
A 48-week, randomized, double-blind, double-dummy phase 2b trial at 43 institutions (USA and Puerto Rico).
Methods:
Study participants (HIV-1 RNA ≥1000 copies/ml, CD4+ cell count ≥200 cells/μl, C-C chemokine receptor type 5-tropic virus) were randomized 2 : 2 : 1 to CVC 100 mg (CVC100), CVC 200 mg (CVC200), or EFV 600 mg, each administered with emtricitabine/tenofovir disoproxil fumarate. Key end points were virologic success (HIV-1 RNA <50 copies/ml) at week 24 (primary) and week 48 (secondary), safety/tolerability at weeks 24 and 48. Study sites and patients remained blinded until week 48.
Results:
A total of 143 patients were randomized (CVC100, n = 59; CVC200, n = 56; EFV, n = 28). Virologic success was obtained at week 24 in 76, 73, and 71% of study participants for CVC100, CVC200, and EFV, respectively (all P > 0.05 versus EFV), and at week 48 in 68, 64, and 50%, respectively (all P > 0.05 versus EFV). Resistance mutations emerged in five and zero CVC and EFV-treated study participants, respectively. Virologic nonresponse and nucleoside reverse transcriptase inhibitor resistance decreased when CVC minimum plasma concentration was at least 47.8 ng/ml. Treatment-related adverse events of at least grade 2 and discontinuations because of adverse events were less frequent in CVC-treated study participants. Total and low-density lipoprotein cholesterol decreased with CVC, but increased with EFV. C-C chemokine ligand type 2 (CCL2) (aka monocyte chemotactic protein-1) increased in a dose-dependent manner, whereas soluble CD14 levels decreased with CVC.
Conclusion:
CVC showed efficacy and favorable safety in treatment-naive HIV-1-infected study participants, supporting selection of CVC200 for phase 3 studies.
Trial registration:
Keywords: antiretroviral, C-C chemokine receptor type 2 antagonist, C-C chemokine receptor type 5 antagonist, cenicriviroc, HIV-1, randomized controlled trial
Introduction
C-C chemokine receptor type 5 (CCR5) or C-X-C chemokine receptor type 4 (CXCR4) is required for HIV entry into host cells, and antagonists of these receptors interfere with viral entry. CCR5-tropic virus is the most prevalent strain during the early stages of infection, whereas CXCR4 predominates during later stages [1–4]. CCR5 antagonists are active against viruses resistant to other drug classes.
Cenicriviroc (CVC), a novel, oral, once-daily, dual CCR5/C-C chemokine receptor type 2 (CCR2) antagonist, demonstrated potent antiviral activity and was generally well tolerated in HIV-1-infected, antiretroviral-experienced, CCR5 antagonist-naive study participants [5]. In addition, dose-dependent increases in the primary ligand of CCR2, monocyte chemotactic protein-1 (MCP-1), were observed during CVC treatment [5]. CCR2 and MCP-1 play critical roles in coordinating cell migration, including that of classical monocytes, and are intricately linked to processes that are associated with metabolic syndrome, insulin resistance, atherosclerosis, and hepatic fibrogenesis [6–9]. As morbidity and mortality in HIV-1-infected individuals are closely linked to HIV-associated chronic immune activation [10], potent CCR2 and CCR5 blockade suggests that CVC could also confer CCR2-mediated anti-inflammatory effects [5]. This is supported by nonclinical studies showing CVC antifibrotic activity in rodent models of thioacetamide-induced liver fibrosis and diet-induced nonalcoholic steatohepatitis [11,12]. Therefore, CVC is an attractive candidate for treatment of HIV infection, and may have benefit beyond its direct antiviral activity. The aim of this study was to compare the efficacy, safety, and anti-inflammatory properties of 100 and 200 mg of CVC with those of efavirenz (EFV) 600 mg, when each was administered in combination with emtricitabine/tenofovir disoproxil fumarate (FTC/TDF) in antiretroviral treatment-naive, HIV-1-infected adults with CCR5-tropic virus.
Methods
The study was conducted in accordance with the Declaration of Helsinki. Written informed consent was obtained, and the protocol, as well as any amendments, consent documents, and safety reports, were reviewed and study conduct was monitored by an Institutional Review Board at each site. CVC is an investigational drug that has not yet been approved by the Food and Drug Administration (FDA).
Study design
The phase 2b, 48-week, multicenter, randomized, double-blind, double-dummy study (652–2–202, NCT01338883) was conducted at 43 hospitals/institutions in Puerto Rico and the USA between May 2011 and 2013. Study participants were stratified by HIV-1 RNA of at least 100 000 or less than 100 000 copies/ml and randomized 2 : 2 : 1 in accordance with a computer-generated randomization schedule prepared by a nonstudy statistician to receive CVC 100 mg (CVC100), CVC 200 mg (CVC200), or EFV 600 mg (Sustiva; Bristol-Myers Squibb), in a double-blind manner, all given with open-label FTC/TDF (Truvada; Gilead Sciences). The drug kit number was obtained by study staff from an interactive voice response system (PharPoint Research, Inc.). The database was locked until the week 24 primary analysis, when the sponsor was unblinded. Study sites and patients remained blinded until the final week 48 analysis.
CVC was formulated as 50 mg tablets and each study participant received a total of six tablets daily, including placebos, on a twice-daily schedule: CVC/placebo with breakfast to enhance CVC absorption, EFV/placebo on an empty stomach at bedtime, and FTC/TDF at either time.
The first 25 randomized study participants (CVC100, eight; CVC200, 10; and EFV, seven) underwent intensive 24-h pharmacokinetic sampling on day 14, and took CVC/placebo after breakfast (approximately 500–700 kilocalories, less than 15–20 g fat) and EFV/placebo with FTC/TDF on an empty stomach at bedtime. Following confirmation that both CVC doses provided plasma exposure levels within the target range [average plasma concentration (C avg) of 55.2–646 ng/ml], the remaining study participants were enrolled.
Treatment was discontinued for confirmed virologic failure/inadequate virologic response (Supplemental Digital Content 1). The original criterion for virologic failure was measurement of HIV-1 RNA more than 50 copies/ml at any time on or after week 24, confirmed within 7–14 days. This criterion resulted in premature discontinuation of study participants with transient low-level viremia who spontaneously resuppressed, and who, therefore, did not appear to constitute true treatment failures. Prior to unblinding for the week 24 primary analysis, the protocol was amended to redefine the confirmatory HIV-1 RNA criterion for virologic failure as more than 400 copies/ml within 14 days, instead of more than 50 copies/ml.
Study participants
Antiretroviral treatment-naive, HIV-1-infected adults (≥18 years of age) with CCR5-tropic virus, plasma HIV-1 RNA level of at least 1000 copies/ml, and CD4+ cell count of at least 200 cells/μl were enrolled. Tropism was determined by phenotype (Enhanced Sensitivity Trofile assay, Monogram Biosciences) and genotype [reflex strategy (triplicate population sequencing) followed by ultradeep sequencing, Quest Diagnostics]; study participants had to be CCR5-tropic by both assays. Exclusion criteria included the presence of primary resistance mutations or phenotypic resistance to TDF, FTC, or EFV and/or mutations associated with multidrug nucleoside/nucleotide resistance; serum alanine aminotransferase or aspartate aminotransferase (AST) at least 2.6 times the upper limit of normal (ULN); total bilirubin of at least the ULN; history of HIV-2, hepatitis B and/or C, cirrhosis, or any other known active or chronic liver disease.
Study objectives
The primary objectives were to determine the proportion of study participants with plasma HIV-1 RNA less than 50 copies/ml at week 24 (virologic success according to the US FDA Snapshot algorithm), and to compare the safety and tolerability of two different doses of CVC with those of EFV. Secondary objectives were to determine the proportion of study participants with HIV-1 RNA less than 50 copies/ml and less than 400 copies/ml at week 48, change from baseline in CD4+ and CD8+ cell counts at week 48, incidence of treatment-emergent drug resistance and tropism changes in study participants with virologic failure, effects of CVC-mediated CCR2 antagonism as measured by inflammatory and immune activation biomarkers and metabolic parameters, and the dose of CVC associated with optimal plasma safety and virologic response.
Study assessments
HIV-1 RNA levels, CD4+, and CD8+ cell counts were measured at every visit [two screening visits; baseline (day 1); weeks 1, 2, 4, and every 4 weeks until week 48]. Viral load was measured by the TaqMan assay (Applied Biosciences, Life Technologies, Carlsbad, California, USA). The impact of viral ‘blips’ was evaluated using a preplanned secondary analysis to determine the proportion of patients who achieved HIV-1 RNA less than 400 copies/ml at weeks 24 and 48. Virologic resistance and tropism changes were assessed in study participants meeting criteria for protocol-defined virologic failure (PDVF). Resistance-associated mutations were identified by GenoSureTM assay (Monogram Biosciences, South San Francisco, California, USA). Tropism testing was carried out as previously described and phenotypic CVC susceptibility was analyzed by PhenoSense Entry assay (Monogram Biosciences).
Safety and tolerability were evaluated by serum chemistries, complete blood counts, prothrombin time and partial thromboplastin time, urinalysis, adverse event monitoring from screening until 4 weeks after treatment completion, physical examinations, and ECGs. Severity of adverse events and laboratory abnormalities was assessed using the National Institute of Allergy and Infectious Diseases Division of AIDS toxicity grading scale. Fasting metabolic studies were performed at baseline and at weeks 4, 12, 24, and 48, including glucose control indicators (glucose and insulin for homeostatic model assessment-insulin resistance and hemoglobin A1c) and lipid profiles.
Inflammatory [high sensitivity C-reactive protein (hs-CRP), IL-6, MCP-1, D-dimer, fibrinogen, and soluble CD14 (sCD14)] and immune activation (CD4/CD38/CD3/human leukocyte antigen-DR and CD8/CD38/CD3/HLA-DR) biomarkers were measured at baseline and at weeks 4 (except for MCP-1 and sCD14), 12, 24, 32 (MCP-1 and sCD14 only), and 48. hs-CRP was measured by immunochemiluminometric assay using a quantitative C-reactive protein kit (Roche Diagnostics, Basel, Switzerland), IL-6 by ELISA using a Quantikine human IL-6 kit (R&D Systems, Minneapolis, Minnesota, USA), D-dimer by immunoturbidimetric assay using a D-dimer assay (Liatest) kit (Diagnostica Stago, Asnieres, France) and fibrinogen by polymerization function by the Clauss method using the Stago Fibrinogen kit (Diagnostica Stago); these biomarkers were measured by LabCorp Clinical Trials (Cranford, New Jersey, USA). sCD14 and MCP-1 were measured by R&D Systems using a solid phase sandwich ELISA with a human sCD14 and human C-C chemokine ligand type 2/MCP-1 Quantikine ELISA kit, respectively (R&D systems). Immune activation markers were measured by flow cytometry using cryopreserved peripheral blood mononuclear cells.
Samples for CVC trough levels were collected predose on day 1 and at weeks 4, 24, and 48, and additional random samples were collected at weeks 2, 8, 12, 16, 20, 32, and 40 in all study participants.
Sample size
A sample size of 60 study participants in each CVC arm and 30 study participants in the EFV arm was proposed to provide a preliminary evaluation of the safety and efficacy of CVC, and to sufficiently detect an 18% difference in the proportion of patients with HIV-1 RNA less than 50 copies/ml at week 24 (primary end point) between groups (P = 0.05; t-test for two independent proportions; 80% power; 80% EFV rate) or −0.92 log10 copies/ml difference between each CVC group and the corresponding active control group in HIV-1 RNA (SD = 1.4, P = 0.05; 80% power; t-test with nonparametric adjustment to the normal distribution). Sample size statistics are based on Power Analysis and Sample Size 2008 software.
Statistical analyses
Study participants with HIV-1 RNA less than 50 and less than 400 copies/ml at weeks 24 and 48 were summarized by treatment group. Change from baseline was assessed by treatment arm for CD4+ and CD8+ cell counts, and inflammatory and immune activation biomarkers. Descriptive analyses of viral resistance and tropism changes were summarized by treatment arm in study participants meeting PDVF criteria. Preplanned statistical analyses were conducted to assess treatment differences for: proportion with HIV-1 RNA less than 50 and less than 400 copies/ml, changes from baseline in HIV-1 RNA levels, and in CD4+ and CD8+ cell counts; other analyses were conducted posthoc. Unless specified otherwise, all statistical testing was two sided, and used a Cochran–Mantel–Haenszel test, controlling for HIV-1 RNA at baseline; statistical significance level was set as P < 0.05. An analysis of variance model with treatment as the fixed effect was also used to assess differences in demographics and baseline characteristics among arms. A Breslow–Day test was used to assess the homogeneity of virologic response across the baseline HIV-1 RNA stratification variable (HIV-1 RNA ≥100 000 and <100 000 copies/ml). A van Elteren test, controlling for baseline HIV-1 RNA, was used to determine differences in cholesterol levels between arms and for MCP-1 mean changes from baseline. Least squares means were calculated for sCD14 from a linear mixed model including treatment, baseline sCD14 value, baseline HIV-1 RNA, visit, and treatment-by-visit interaction as fixed effects. All statistical analyses were conducted with the SAS System, version 9.1.3 or higher (SAS Institute Inc., Cary, North Carolina, USA).
Noncompartmental CVC pharmacokinetic parameters were calculated using descriptive statistics. A population analysis was performed on all samples, using a two-compartment model to predict CVC plasma exposure [13]. Pharmacokinetic/pharmacodynamic modeling evaluated the relationship between CVC parameters and virologic outcomes. A Classification and Regression Tree analysis was used to further investigate the association between week 24 CVC minimum plasma concentration (C min) and virologic outcomes.
Results
Study population and disposition
Of the 392 study participants screened, 143 were randomized to CVC or EFV, in combination with FTC/TDF (CVC100, 59; CVC200, 56; and EFV, 28) (Supplemental Digital Content 2). Demographic and baseline characteristics are described in Table 1.
Table 1. Demographics and baselinea characteristics of randomized study participants per treatment group.
| CVC 100 mg (n = 59) | CVC 200 mg (n = 56) | EFV 600 mg (n = 28) | All (n = 143) | P value | |
| Men, n (%) | 54 (92) | 56 (100) | 25 (89) | 135 (94) | 0.061 b |
| Mean age (years) (minimum–maximum) | 36 (19–63) | 36 (21–57) | 32 (19–49) | 35 (19–63) | 0.251 c |
| Race, n (%) | 0.117 b | ||||
| White | 34 (58) | 36 (64) | 18 (64) | 88 (62) | |
| Black or African-American | 24 (41) | 13 (23) | 9 (32) | 46 (32) | |
| Other | 1 (2) | 7 (13) | 1 (4) | 9 (6) | |
| Hispanic ethnicity, n (%) | 7 (12) | 18 (32) | 10 (36) | 35 (24) | 0.013 b |
| Mean BMI (kg/m2) (minimum–maximum) | 26.6 (18.3–41.7) | 26.1 (19.8–37.5) | 25.5 (18.1–34.3) | 26.2 (18.1–41.7) | 0.582 c |
| Median HIV-1 RNA (log10 copies/ml) (minimum–maximum) | 4.50 (3.42–5.55) | 4.66 (3.03–5.65) | 4.56 (3.35–5.86) | 4.57 (3.03–5.86) | 0.301 c |
| HIV-1 RNA by stratification factor, n (%) | 0.409 b | ||||
| ≥100 000 copies/ml | 10 (17) | 14 (25) | 4 (14) | 28 (20) | |
| <100 000 copies/ml | 49 (83) | 42 (75) | 24 (86) | 115 (80) | |
| Median CD4+ cell count (cells/μl) (minimum–maximum) | 396 (188–749) | 388 (77–1090) | 310 (191–641) | 385 (77–1090) | 0.232 c |
ANOVA, analysis of variance; CVC, cenicriviroc; EFV, efavirenz.
aDefined as the mean of the screening visit 2 and baseline visit values.
bA Cochran–Mantel–Haenszel test compared all three treatments.
cThe overall treatment effect was assessed from an ANOVA model with treatment as the fixed effect.
Premature discontinuation rates were not significantly different between CVC and EFV arms (P = 0.237; Table 2). The most common reasons for discontinuation were PDVF in the CVC arms, and adverse events in the EFV arm (Table 2). Of the 15 study participants meeting the original PDVF criteria, 11 met the amended criteria; the remaining four study participants (CVC100, three; and EFV, one) were withdrawn prior to the protocol amendment, with viral load values consistent with ‘blips’ (transient HIV-1 RNA 50–400 copies/ml). Three of four study participants had HIV-1 RNA less than 50 copies/ml at a visit occurring 48 h or less after withdrawal; the fourth had an HIV-1 RNA value of 91 copies/ml 1 day after the last CVC dose. Virologic data at week 48 were missing for three and two study participants in the CVC and EFV arms, respectively.
Table 2. Study participant disposition at week 48 (final analysis) and reasons for early discontinuation per treatment group.
| n (%) | CVC 100 mg (n = 59) | CVC 200 mg (n = 56) | All CVC (n = 115) | EFV 600 mg (n = 28) |
| Completed | 42 (71) | 41 (73) | 83 (72) | 17 (61) |
| Discontinued early | 17 (29) | 15 (27) | 32 (28) | 11 (39) |
| P value versus EFV a | 0.332 | 0.246 | 0.237 | |
| Reasons for early discontinuation: | ||||
| Confirmed virologic failure according to original withdrawal criteria | 7 (12) | 6 (11) | 13 (11) | 2 (7) |
| Lost to follow-up | 5 (8) | 2 (4) | 7 (6) | 2 (7) |
| Adverse event | 0 | 1 (2) | 1 (1) | 6 (21) |
| Consent withdrawn | 2 (3) | 2 (4) | 4 (3) | 0 |
| Noncompliance | 1 (2) | 3 (5) | 4 (3) | 0 |
| Termination by sponsor/IRB/IEC b | 1 (2) | 0 | 1 (1) | 0 |
| Study participant incarcerated | 0 | 1 (2) | 1 (1) | 1 (4) |
| Study participant enrolled in error (prohibited medication) | 1 (2) | 0 | 1 (1) | 0 |
CVC, cenicriviroc; EFV, efavirenz; IEC, Independent Ethics Committee; IRB, Institutional Review Board.
aComparison between each CVC arm and the EFV treatment group based on Cochran–Mantel–Haenszel test controlling for HIV-1 RNA at baseline.
bStudy participant took CVC 200 mg because of dispensing of incorrect treatment kit. The study participant discontinued study medication on day 9 as instructed by the sponsor, because of an exclusionary entry criterion (history of an abnormal electrocardiogram).
Efficacy
The proportion of study participants with virologic success (HIV-1 RNA <50 copies/ml) was similar in all treatment arms at weeks 24 (primary end point) and 48 (secondary end point) (all P > 0.05; Table 3; Supplemental Digital Content 3). Also, rates of virologic nonresponse were not significantly different between all treatment groups at week 48 (all P > 0.05; Table 3).
Table 3. Virologic response at week 24 (primary end point) and week 48 (secondary end point) according to Food and Drug Administration Snapshot algorithma per treatment group.
| Week 24 | Week 48 | |||||
| n (%) | CVC 100 mg (n = 59) | CVC 200 mg (n = 56) | EFV 600 mg (n = 28) | CVC 100 mg (n = 59) | CVC 200 mg (n = 56) | EFV 600 mg (n = 28) |
| Virologic success (HIV-1 RNA <50 copies/ml) | 45 (76) | 41 (73) | 20 (71) | 40 (68) | 36 (64) | 14 (50) |
| P value versus EFV b | 0.606 | 0.683 | 0.110 | 0.169 | ||
| Treatment difference from EFV arm c , % (95% CI) | 5 (–16, 26) | 4 (–17, 25) | 18 (–5, 41) | 16 (–7, 39) | ||
| Virologic nonresponse d | 7 (12) | 8 (14) | 1 (4) | 9 (15) | 11 (20) | 3 (11) |
| P value e | 0.335 | 0.564 | ||||
| Reasons for no virologic data at time point | ||||||
| Discontinued study because of adverse event or death | 0 (0) | 1 (2) | 5 (18) | 0 (0) | 1 (2) | 6 (21) |
| Discontinued study for other reasons f | 6 (10) | 6 (11) | 2 (7) | 8 (14) | 7 (13) | 3 (11) |
| Missing data during window, but on study | 1 (2) | 0 (0) | 0 (0) | 2 (3) | 1 (2) | 2 (7) |
CI, confidence interval; CVC, cenicriviroc; EFV, efavirenz.
aStudy participants considered to have HIV-1 RNA less than 50 copies/ml, if the last on-treatment HIV-1 RNA value in the week 24 or 48 window was less than 50 copies/ml and the study participant did not have a protocol-excluded change in antiviral therapy prior to that value.
bComparison between each CVC arm and the EFV treatment group based on Cochran–Mantel–Haenszel test controlling for HIV-1 RNA at baseline.
cTreatment differences were estimated using stratum-adjusted Mantel–Haenszel proportions controlling for HIV-1 RNA at baseline; 95% CIs were provided based on this method.
dIncludes study participants who changed therapy in a manner not permitted per protocol prior to week 24 or 48, study participants who discontinued prior to week 24 or 48 for lack or loss of efficacy, and study participants who had at least 50 copies/ml in the week 24 or 48 window.
eComparison between treatment groups using an unadjusted Cochran–Mantel–Haenszel test.
fIncluding withdrawal of consent and lost to follow-up.
The proportion of study participants with HIV-1 RNA less than 400 copies/ml at week 48 was 71% for CVC100 and 50% for EFV (P = 0.057) and was significantly higher for CVC200 compared with EFV (73 versus 50%, P = 0.020).
Virologic success at week 48 was compared according to baseline viral load stratum. For CVC100, CVC200, and EFV, virologic success was 69, 69, and 50%, respectively, for HIV-1 RNA less than 100 000 copies/ml; and 60, 50, and 50%, respectively, for HIV-1 RNA of at least 100 000 copies/ml. No significant differences were observed in virologic success between study participants with low or high baseline viral load (P = 0.582).
Mean increases in CD4+ cell counts were robust and similar across all arms at week 48, with gains of 205 and 211 cells/μl for CVC100 and CVC200, respectively, and 147 cells/μl for EFV. Mean CD4+/CD8+ cell ratio increased from baseline in all groups (Supplemental Digital Content 4).
Virologic resistance and tropism in study participants with virologic failure
Fifteen study participants met the PDVF criteria [CVC, 13 (11%); EFV, two (7%); P = 0.797]. No mutations or tropism changes were detected in the four study participants withdrawn according to the original PDVF criteria (CVC100, three and EFV, one). Eleven study participants met the amended PDVF criteria (CVC100, four; CVC200, six; and EFV, one). Among the 10 CVC-treated study participants, two failed resistance testing for technical reasons, five had treatment-emergent nucleoside reverse transcriptase inhibitor (NRTI) resistance-associated mutations (M184I and/or V), and one had an NRTI resistance-associated mutation (K70R) at screening. At the time of virologic failure, two CVC-treated study participants had non-NRTI resistance-associated mutations that were found by retrospective deep sequencing to be preexisting at screening or baseline. Of the five study participants with paired tropism data, one (CVC200 arm) had a treatment-emergent tropism change to dual/mixed (R5X4) phenotype and had reduced phenotypic susceptibility to CVC; this study participant also developed an M184V mutation. No mutations or tropism changes were detected in the EFV-treated study participant who met the amended PDVF criteria.
Safety and tolerability
Overall, 88, 84, and 96% of study participants had at least one adverse event in the CVC100, CVC200, and EFV arms, respectively. Most adverse events were considered mild or moderate. The most frequent treatment-related adverse events, reported in at least 5% of CVC-treated study participants, regardless of severity, were nausea (12%), headache (10%), diarrhea (7%), and abnormal dreams (7%). No apparent dose relationship for adverse events was observed. Events in the EFV arm were typical of its known safety profile [14]. Adverse events of at least grade 2 reported in at least 5% of study participants and considered possibly related to study medication were less frequent among CVC recipients versus those receiving EFV (P = 0.002; Table 4). No significant differences were observed between treatment groups in the number of adverse events of at least grade 3 reported (P = 0.142). One grade 4 adverse event (suicidal ideation) was reported in the EFV arm only. There was a significant difference favoring CVC in adverse events leading to discontinuation between treatment groups (P < 0.001; Table 4).
Table 4. Incidence of adverse events a per treatment group.
| n (%) | CVC 100 mg (n = 58) | CVC 200 mg (n = 57) | EFV 600 mg (n = 28) | P value b |
| Treatment-emergent grade ≥3 adverse events | 2 (3) | 3 (5) | 4 (14) | 0.142 |
| Grade 3 | 2 (3) | 3 (5) | 3 (11) | |
| Grade 4 | 0 (0) | 0 (0) | 1 (4) | |
| Treatment-emergent grade ≥2 adverse events c | 5 (9) d | 5 (9) d | 10 (36) | 0.001 |
| Psychiatric disorders | 1 (2) | 2 (4) | 6 (21) | |
| Abnormal dreams | 1 (2) | 0 (0) | 3 (11) | |
| Insomnia | 0 (0) | 0 (0) | 3 (11) | |
| Gastrointestinal disorders | 2 (3) | 2 (4) | 2 (7) | |
| Nausea | 0 (0) | 2 (4) | 2 (7) | |
| Nervous system disorders | 1 (2) | 1 (2) | 3 (11) | |
| Skin and subcutaneous tissue disorders | 2 (3) | 0 (0) | 3 (11) | |
| Rash events | 1 (2) | 0 (0) | 2 (7) | |
| Adverse events leading to discontinuation | 0 (0) e | 1 (2) e | 6 (21) | <0.001 |
| Serious adverse events | 1 (2) | 1 (2) | 1 (4) | 0.833 |
| Deaths | 0 (0) | 0 (0) | 0 (0) |
CVC, cenicriviroc; EFV, efavirenz; ITT, intention-to-treat.
aIn study participants who had received at least one dose of study drug. One study participant was randomized to receive CVC 100 mg, but took CVC 200 mg (incorrect treatment kit); the study participant was included in the 100 mg arm in the ITT population, but in the 200 mg arm in the safety population.
bThe P values were assessed using a Cochran–Mantel–Haenszel test for differences between treatment groups in number of study participants with the adverse event type.
cAt least possibly related (as determined by study investigator) in at least 5% of study participants.
dPairwise comparisons with the EFV arm, using a Cochran–Mantel–Haenszel test, showed statistical significance (P = 0.002 for each CVC dose versus EFV).
ePairwise comparisons with the EFV arm, using a Cochran–Mantel–Haenszel test, showed statistical significance (P < 0.001 CVC100 versus EFV; P = 0.002 for CVC200 versus EFV).
Most laboratory abnormalities were grade 1 or 2 (Supplemental Digital Content 5). The proportion with grade 3 or 4 laboratory abnormalities was similar among all treatment arms (P = 0.409). All creatine phosphokinase elevations were transient, asymptomatic, and resolved upon continued treatment. All alanine aminotransferase and AST elevations were grade 1 or 2, except for one transient, asymptomatic grade 3 AST elevation (5.2 × ULN) observed in a CVC100-treated study participant. All bilirubin abnormalities were grade 1 or 2 and not different among treatment arms.
Fasting total cholesterol levels decreased significantly with CVC treatment compared with EFV up to week 48 (P < 0.001; Fig. 1). This was primarily associated with decreases in low-density lipoprotein cholesterol, with no meaningful change in high-density lipoprotein cholesterol in the CVC arms. Both low-density lipoprotein and high-density lipoprotein cholesterol increased with EFV. Fasting triglycerides remained below baseline with CVC treatment but tended to fluctuate with EFV. There were no notable differences in glucose, insulin, homeostatic model assessment-insulin resistance, or hemoglobin A1c (Supplemental Digital Content 6), and no clinically relevant ECG changes.
Fig. 1.
Mean changes from baselinea in fasting metabolic parameters in each treatment group over the 48-week duration of the study.
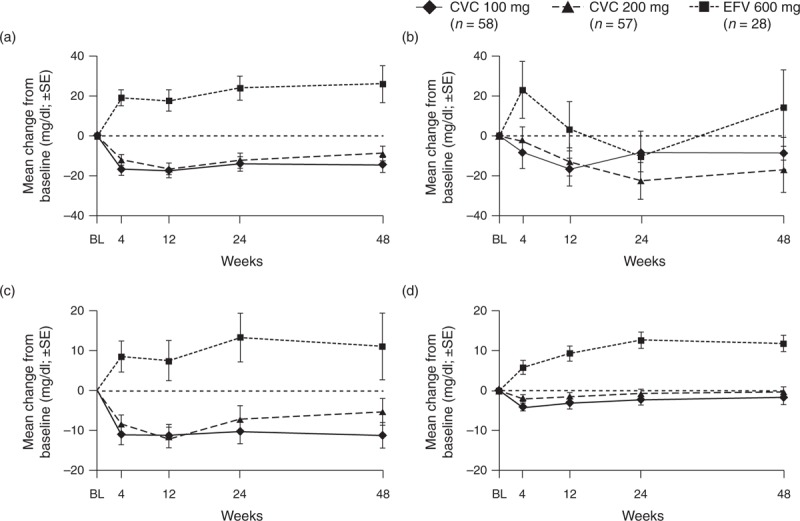
(a) Total cholesterol (b) triglycerides (c) LDL cholesterol (d) HDL cholesterol (safety population). BL, baseline; CVC, cenicriviroc; EFV, efavirenz; HDL, high-density lipoprotein; LDL, low-density lipoprotein; SE, standard error. aBaseline defined as the last nonmissing assessment prior to initiation of study treatment.
Biomarkers of inflammation and immune activation
CVC led to a dose-dependent, compensatory increase in MCP-1 because of CCR2 blockade, whereas no change was observed with EFV (Supplemental Digital Content 7). At week 48, mean change from baseline was significantly different between the CVC and EFV arms (P < 0.001). During the first 12 weeks, sCD14 (a marker of monocyte activation) levels decreased and remained below baseline in the CVC arms throughout the study; whereas in the EFV arm, sCD14 levels increased and remained above baseline values (Supplemental Digital Content 7). CVC effects on sCD14 were independent of changes in HIV-1 RNA, in contrast to EFV, where greater viral load decline was associated with smaller changes in sCD14 levels (Supplemental Digital Content 8). Least squares mean changes from baseline in sCD14 were significantly different between the CVC and EFV arms at most time points throughout the study (P < 0.05; except for CVC200 at week 32). No significant differences across the arms were observed for other inflammatory (hs-CRP, fibrinogen, IL-6, and D-dimer) or immune activation (total CD38+ expression and total HLA DR+ expression on CD4+ or CD8+ T cells) biomarkers.
Cenicriviroc pharmacokinetic analysis
The week 24 and 48 population pharmacokinetic analyses included 110 and 109 study participants, respectively, 18 with day 14 24-h sampling, and 92 and 91, respectively, with trough and/or random sampling. At week 48, both C avg and C min were associated with virologic failure (Supplemental Digital Content 9). At week 24, the median C min in virologic nonresponders was 43% lower than in virologic responders (42.9 versus 74.8 ng/ml; P = 0.029). Increasing C min values by quartile were associated with a decreasing proportion of virologic nonresponders. Classification and Regression Tree analysis identified a C min breakpoint concentration of 47.8 ng/ml, with study participants reaching or exceeding this concentration less likely to experience virologic nonresponse than those with lower concentrations (7.5 versus 29.4%, respectively). In four of five CVC-treated study participants with NRTI mutations at codon 184, predicted C min values were less than 47.8 ng/ml. The pharmacokinetic model predicted that 42% (CVC100) and 84% (CVC200) of study participants would achieve C min of at least 50 ng/ml; this was confirmed in the trial.
Discussion
CVC100 and CVC200 were efficacious and well tolerated in antiretroviral treatment-naive, HIV-1-infected adults with CCR5-tropic virus. At weeks 24 (primary end point) and 48 (secondary end point), virologic success was similar among treatment arms. Overall, virologic failure was 11% in the CVC arms and 7% in the EFV arm. Notably, three CVC-treated study participants were withdrawn because of virologic ‘blips’ prior to a protocol amendment designed to prevent premature discontinuation for transient low-level viremia.
Treatment-emergent, NRTI-associated mutations were observed in five CVC-treated study participants with virologic failure and none in the EFV arm; importantly, four of the five mutations emerging at codon 184 occurred in study participants with a suboptimal CVC plasma concentration (C min < 47.8 ng/ml). Pharmacokinetic analyses demonstrated a trend toward improved virologic outcomes with increasing CVC plasma concentrations; study participants with C min more than 47.8 ng/ml were more likely to achieve virologic success. A tropism change to R5X4 was observed in one study participant receiving CVC200.
CVC was better tolerated than EFV, with fewer treatment-related adverse events of at least grade 2 and adverse events leading to discontinuation. Higher rates of adverse events-related discontinuation among EFV recipients led to a greater proportion of study participants with no virologic data at weeks 24 and 48.
Notable study limitations include a complex dosing regimen and considerable pill burden that may have reduced adherence across all arms and contributed to high rates of missing data, in spite of retention measures. These measures included missed-visit alerts for sites, transportation assistance, and patient-assistance programs to enable access to poststudy HIV regimens following 48-week study completion.
The complex dosing regimen also made enrollment challenging at a time when once-daily regimens with lower pill burdens were the standard of care. This was further exacerbated by a high screen-failure rate (primarily because of having X4 or dual/mixed-tropic virus, CD4 less than 200 cells/μl, primary resistance to antiretroviral regimens in the study, and HIV-1 RNA <1000 copies/ml). As a result, the number of sites was needed to complete study enrollment was very high. To ensure that compliance and quality assurance were maintained between the large number of study sites, standardized audits of data collection and analysis functions were conducted throughout the study period, as well as targeted audits of eight study sites with high enrollment. Underrepresentation of women precluded any sex-specific conclusions. The low rate of virologic success in the EFV arm at week 48 (50% of study participants with HIV-1 RNA <50 copies/ml) contrasts sharply with results from other phase 3 studies where EFV-based therapy was used as a comparator (80–84% of study participants with HIV-1 RNA levels <50 copies/ml; intention-to-treat, FDA Snapshot Algorithm) [15–17]. Of note, a lower than expected rate of virologic response was also reported in two contemporary studies (71% at week 48) [18,19]. Our results may underestimate EFV efficacy because of the small sample size (n = 28), high proportion of week 48 missing data (39%), and potential nonadherence associated with a complex dosing regimen. Discontinuation because of adverse events associated with EFV accounted for a high proportion of missing data (21%).
Data on virologic response, pharmacokinetic/pharmacodynamic, and safety and tolerability support the selection of CVC 200 mg once daily for further clinical evaluation. Phase 3 studies in antiretroviral treatment-naive, HIV-1-infected study participants are planned to evaluate fixed-dose combination of CVC/lamivudine as a novel backbone versus TDF/FTC, coadministered with guideline-recommended third agents.
Our findings demonstrated a dose-dependent increase in MCP-1, suggesting potent blockade of CCR2. The monocyte activation biomarker sCD14, an independent predictor of mortality in people with HIV [20], decreased and remained below baseline with CVC treatment, but increased with EFV. CVC has demonstrated CCR2-mediated antifibrotic activity in animal models of liver and kidney fibrosis [11,12,21]; a phase 2b study in non-HIV-infected patients with nonalcoholic steatohepatitis and liver fibrosis is fully enrolled (CENTAUR Study 652-2-203; NCT02217475). These intriguing findings support further exploration of the potential anti-inflammatory effects of CVC in HIV infection and other inflammatory syndromes.
Acknowledgements
Editorial support was provided by Camille Bonomelli of Alpharmaxim Healthcare Communications. The investigators thank the study volunteers for their participation.
This work was supported by Tobira Therapeutics, Inc.
The authors are grateful to Will Chang and Helen Jenkins (Tobira Therapeutics, Inc.) and to Amy Flynt (biometrics at PharPoint Research).
Study 202 investigators: Anthony Scarsella, Chiu-Bin Hsiao, Javier Morales-Ramirez, Laveeza Bhatti, Frederick Cruickshank, Debbie Hagins, Louis Sloan, Lawrence Waldman, Robert Bolan, Craig Dietz, Anthony LaMarca, Ronald Nahass, Olayemi Osiyemi, Gary Richmond, Douglas Ward, Bisher Akil, Dushyantha Jayaweera, Jason Leider, Moti Ramgopal, Jorge Rodriguez, Michael Wohlfeiler, Cynthia Brinson, Shannon Schrader, Stephen Brown, Jerome Ernst, Annie Luetkemeyer, Martin Markowitz, Norman Markowitz, Anthony Mills, Mahmoud Mustafa, Jorge Santana, Steven Santiago, Pablo Tebas, Carmen Zorrilla-Maldonado, Marc Tribble, David Prelutsky.
Authors’ contributions: M.T., M.S., E.D., J.G., J.L., A.L.L., J.C., J.E., E.L., and J.F. made substantial contributions to the conception or design of the work, or the acquisition, analysis, or interpretation of data for the work; made substantial contributions to drafting the work or revising it critically for important intellectual content; provided final approval of the version to be published; and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Conflicts of interest
M.T. is affiliated with an institution that received clinical trial funding from Tobira Therapeutics, Inc., BMS, GeoVax Inc., Gilead, GSK, Merck, ViiV, Kowa Research Institute; and is on the Data Safety Monitoring Boards of GSK/ViiV, and Janssen. M.S. is affiliated with an institution that received research grants from Tobira Therapeutics, Inc., Merck, BMS, Gilead, ViiV, and GSK. E.D. has been a consultant for Gilead, Janssen, and Vertex and has been a speaker in conferences supported by Gilead and Janssen. J.G. has been a consultant or speaker in conferences supported by AbbVie, BMS, GSK, ViiV, Janssen, Merck, and Gilead; is affiliated with an institution that received research grants from AbbVie, BMS, GSK, ViiV, Boehringer Ingelheim, Pfizer, Janssen, Merck Frost, Gilead, and Tibotec (Janssen) Therapeutics; has served an investigator for Abbott, Avexa, Boehringer Ingelheim, Gilead, GSK, Merck, Pfizer, Roche Laboratories, Parexel, Hiesped, and Tibotec Therapeutics; and his institution has received honoraria for speaking or chairing engagements from Abbvie, BMS, GSK, Gilead, Merck, Pfizer, and Tibotec Therapeutics. J.L. reported no potential conflicts of interest. A.L.L. is a board member of SAB Merck. J.C. is affiliated with an institution that received a research grant from Tobira Therapeutics, Inc. and consultancy fees from Gilead Pharmaceuticals; he has been a speaker in conferences supported by Gilead, ViiV, Abbvie, Janssen, and Merck. J.E. is a former employee of Tobira Therapeutics, Inc. E.L. is an employee of Tobira Therapeutics, Inc. J.F. has received travel support from Tobira Therapeutics, Inc. and is is affiliated with an institution that received a research grant from Tobira Therapeutics, Inc.; she has been a consultant for Merck, ViiV, Janssen, Gilead, and BMS; has received payments for lectures or speaker bureaus from Merck, Janssen, ViiV, and Gilead; and her institution has received research grants from the CDC, OH, Department of Health, NIH, HRSA, NIAID, Durr Foundation, MAC Foundation, and Internet for Health.
Supplementary Material
References
- 1. Brumme ZL, Goodrich J, Mayer HB, Brumme CJ, Henrick BM, Wynhoven B, et al. Molecular and clinical epidemiology of CXCR4-using HIV-1 in a large population of antiretroviral-naive individuals . J Infect Dis 2005; 192:466–474. [DOI] [PubMed] [Google Scholar]
- 2. Moyle GJ, Wildfire A, Mandalia S, Mayer H, Goodrich J, Whitcomb J, et al. Epidemiology and predictive factors for chemokine receptor use in HIV-1 infection . J Infect Dis 2005; 191:866–872. [DOI] [PubMed] [Google Scholar]
- 3. Shepherd JC, Jacobson LP, Qiao W, Jamieson BD, Phair JP, Piazza P, et al. Emergence and persistence of CXCR4-tropic HIV-1 in a population of men from the multicenter AIDS cohort study . J Infect Dis 2008; 198:1104–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Demarest J, Bonny T, Vavro C, Labranche C, Kitrinos K, McDanal C, et al. HIV-1 co–receptor tropism in treatment naive and experienced subjects . 44th Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC); 31 October 2004; Washington, District of Columbia, USA. [Google Scholar]
- 5. Lalezari J, Gathe J, Brinson C, Thompson M, Cohen C, Dejesus E, et al. Safety, efficacy, and pharmacokinetics of TBR-652, a CCR5/CCR2 antagonist, in HIV-1-infected, treatment-experienced, CCR5 antagonist-naive subjects . J Acquir Immune Defic Syndr 2011; 57:118–125. [DOI] [PubMed] [Google Scholar]
- 6. Miura K, Yang L, van Rooijen N, Ohnishi H, Seki E. Hepatic recruitment of macrophages promotes nonalcoholic steatohepatitis through CCR2 . Am J Physiol Gastrointest Liver Physiol 2012; 302:G1310–G1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Saiman Y, Friedman SL. The role of chemokines in acute liver injury . Front Physiol 2012; 3:213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Seki E, De Minicis S, Gwak GY, Kluwe J, Inokuchi S, Bursill CA, et al. CCR1 and CCR5 promote hepatic fibrosis in mice . J Clin Invest 2009; 119:1858–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Seki E, De Minicis S, Inokuchi S, Taura K, Miyai K, van Rooijen N, et al. CCR2 promotes hepatic fibrosis in mice . Hepatology 2009; 50:185–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Aberg JA. Aging, inflammation, and HIV infection . Top Antivir Med 2012; 20:101–105. [PMC free article] [PubMed] [Google Scholar]
- 11. Lefebvre E, Hashiguchi T, Jenkins H, Nabhan A, Yoneyama H, Friedman SL, et al. Anti-fibrotic and anti-inflammatory activity of the dual CCR2 and CCR5 antagonist cenicriviroc in a mouse model of NASH . Hepatology 2013; 58 Suppl 1:221A–222A. [Google Scholar]
- 12. Hong F, Chou H, Friedman SL. Significant antifibrotic activity of cenicriviroc, a dual CCR2/CCR5 antagonist, in a rat model of thioacetamide-induced liver fibrosis and cirrhosis . 64th Annual Meeting of the American Association of the Study of Liver Diseases (AASLD); 1 November 2013; Washington, District of Columbia, USA. [Google Scholar]
- 13. Marier JF, Trinh M, Pheng LH, Palleja SM, Martin DE. Pharmacokinetics and pharmacodynamics of TBR-652, a novel CCR5 antagonist, in HIV-1-infected, antiretroviral treatment-experienced, CCR5 antagonist-naive patients . Antimicrob Agents Chemother 2011; 55:2768–2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bristol-Myers Squibb Pharma EEIG. Sustiva Summary of Product Characteristics. 2014. [Google Scholar]
- 15. Cohen CJ, Molina JM, Cahn P, Clotet B, Fourie J, Grinsztejn B, et al. Efficacy and safety of rilpivirine (TMC278) versus efavirenz at 48 weeks in treatment-naive HIV-1-infected patients: pooled results from the phase 3 double-blind randomized ECHO and THRIVE Trials . J Acquir Immune Defic Syndr 2012; 60:33–42. [DOI] [PubMed] [Google Scholar]
- 16. Sax PE, DeJesus E, Mills A, Zolopa A, Cohen C, Wohl D, et al. Co-formulated elvitegravir, cobicistat, emtricitabine, and tenofovir versus co-formulated efavirenz, emtricitabine, and tenofovir for initial treatment of HIV-1 infection: a randomised, double-blind, phase 3 trial, analysis of results after 48 weeks . Lancet 2012; 379:2439–2448. [DOI] [PubMed] [Google Scholar]
- 17. Walmsley SL, Antela A, Clumeck N, Duiculescu D, Eberhard A, Gutiérrez F, et al. Dolutegravir plus abacavir-lamivudine for the treatment of HIV-1 infection . N Engl J Med 2013; 369:1807–1818. [DOI] [PubMed] [Google Scholar]
- 18. Gatell JM, Morales-Ramirez JO, Hagins DP, Thompson M, Keikawus A, Hoffmann C, et al. Forty-eight-week efficacy and safety and early CNS tolerability of doravirine (MK-1439), a novel NNRTI, with TDF/FTC in ART-naive HIV-positive patients . J Int AIDS Soc 2014; 17:19532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Margolis DA, Brinson CC, Smith GH, de Vente J, Hagins DP, Eron JJ, et al. Cabotegravir plus rilpivirine, once a day, after induction with cabotegravir plus nucleoside reverse transcriptase inhibitors in antiretroviral-naive adults with HIV-1 infection (LATTE): a randomised, phase 2b, dose-ranging trial . Lancet Infect Dis 2015; 15:1145–1155. [DOI] [PubMed] [Google Scholar]
- 20. Sandler NG, Wand H, Roque A, Law M, Nason MC, Nixon DE, et al. Plasma levels of soluble CD14 independently predict mortality in HIV infection . J Infect Dis 2011; 203:780–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Moyle G, Richards TL, Plato CF, Jenkins H, Wolfgang GHI, Lefebvre E. Antifibrotic activity of dual CCR5/CCR2 antagonist cenicriviroc in a mouse model of renal fibrosis . 20th International AIDS Conference (AIDS 2014); 22 July 2014; Melbourne, Australia. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.