

Abstract
Glucokinase and hexokinase from pathogenic protozoa Trypanosoma cruzi are potential drug targets for antiparasitic chemotherapy of Chagas’ disease. These glucose kinases phosphorylate D-glucose with co-substrate ATP and yields glucose 6-phosphate and are involved in essential metabolic pathways, such as glycolysis and the pentose phosphate pathway. An inhibitor class was conceived that is selective for T. cruzi glucokinase (TcGlcK) using structure-based drug design involving glucosamine having a linker from the C2 amino that terminates with a hydrophobic group either being phenyl, p-hydroxyphenyl, or dioxobenzo[b]thiophenyl groups. The synthesis and characterization for two of the four compounds are presented while the other two compounds were commercially available. Four high-resolution X-ray crystal structures of TcGlcK inhibitor complexes are reported along with enzyme inhibition constants (Ki) for TcGlcK and Homo sapiens hexokinase IV (HsHxKIV). These glucosamine analogue inhibitors include three strongly selective TcGlcK inhibitors and a fourth inhibitor, benzoyl glucosamine (BENZ-GlcN), which is a similar variant exhibiting a shorter linker. Carboxybenzyl glucosamine (CBZ-GlcN) was found to be the strongest glucokinase inhibitor known to date, having a Ki of 0.71 ± 0.05 μM. Also reported are two biologically active inhibitors against in vitro Trypanosoma cruzi culture that were BENZ-GlcN and CBZ-GlcN, with intracellular amastigote growth inhibition IC50 values of 16.08 ± 0.16 μM and 48.73 ± 0.69 μM, respectively. These compounds revealed little to no toxicity against mammalian NIH-3T3 fibroblasts and provides a key starting point for further drug development with this class of compound.
Keywords: Chagas’ disease, Trypanosoma cruzi, Glucokinase, Hexokinase, Structure-based drug design
Graphical abstract
1. Introduction
Trypanosoma cruzi protozoa are human pathogenic Kinetoplastid parasites that cause Chagas’ disease. There are approximately 6–7 million people infected worldwide with T. cruzi parasites and a majority are from 21 Latin American countries (1). The Centers for Disease Control and Prevention estimates that there are over 300,000 people infected in the United States (2). The endemic regions span from Argentina to Mexico (3–5) and T. cruzi parasites are an increasing danger to the U.S. public health (6) since 10 states (primarily in the Southwest) have reported T. cruzi-infected specimens of triatomine insect vector species (7). First-line clinically available drugs used in Latin America, such as benznidazole and nifurtimox (Figure 1), which have still been in use since 1985 and earlier, are highly effective against T. cruzi parasites in the acute stage with parasitological cures upwards to 80% (8).
Figure 1.
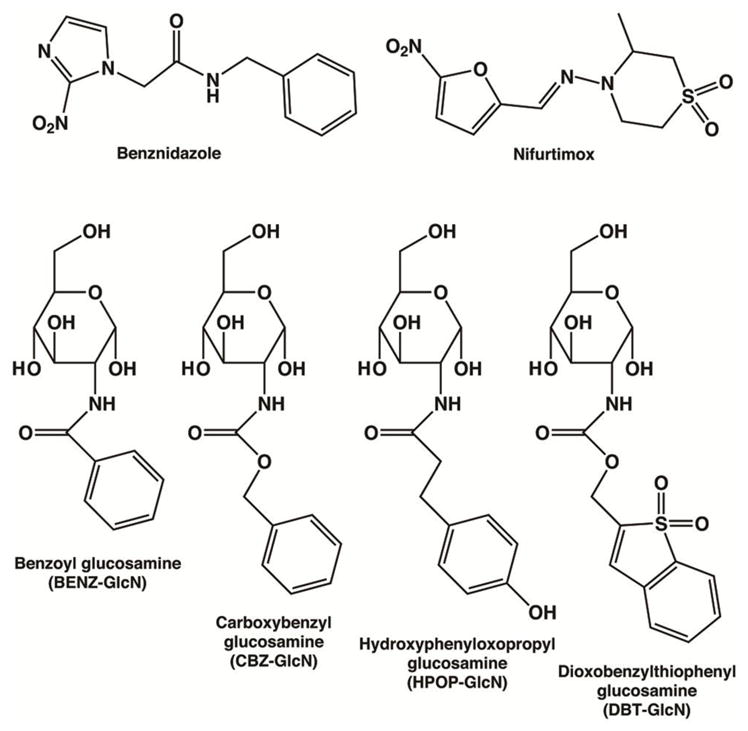
Chemical structures of known drugs and glucosamine analogue inhibitors for the antiparasitic treatment of Chagas’ disease. Benznidazole and nifurtimox are clinically available and extensively used drugs for Trypanosoma cruzi infections. Glucosamine analogues BENZ-GlcN, CBZ-GlcN, HPOP-GlcN, and DBT-GlcN were tested in this study against T. cruzi glucokinase as potentially potent and/or selective inhibitors.
However, in the chronic stage, the same antiparasitic therapy has substantially lower efficacy for reasons that are not well understood (9). These drugs have tolerability issues because they produce harsh side effects, including allergic dermopathy, anorexia, peripheral polyneuropathy, and vomiting (10, 11), and as a result, there is weak patient compliance. Furthermore, different T. cruzi strains were observed to respond much differently to certain inhibitors from in vitro biological inhibition assays, such as posaconazole, indicating that a drug that gives rise to good biological activity from one strain may not have the same effectiveness towards another (12). In order to develop new and improved drugs, the major challenges such as drug efficacy of chronic stage Chagas’ disease, drug tolerability, and universal compound effectiveness simply need to be overcome.
Glycolysis and the pentose phosphate pathway (PPP) are implicated as targets in T. cruzi for antiparasitic drug development (13, 14). Glycolytic enzymes have been studied as potential drug targets for the treatment of trypanosomatid diseases (13–15), especially the hexokinases from T. cruzi (16–18) and T. brucei (19–22). Metabolic studies have demonstrated that T. cruzi trypomastigotes and amastigotes use glucose as their preferred carbon source in glucose-rich media (23). Additionally, a metabolic control analysis study involving RNA interference (RNAi) against five important T. brucei glycolytic enzymes was assessed for the role of glycolytic flux (i.e., as glucose consumption and pyruvate production) on the growth trend of the parasite’s bloodstream life-stage and good evidence was revealed for T. brucei hexokinase (TbHxK) as a validated drug-target (24). T. cruzi parasites are highly dependent on glucose for cell growth and differentiation (25–27) and inhibition of mostly any of the glycolytic enzymes in T. cruzi will likely lead to prompt cell death (14).
Two enzymes responsible for the catalysis of D-glucose to glucose 6-phosphate (G6P), in the presence of ATP and Mg2+, include hexokinase and glucokinase. The inhibition of these enzymes appears to be a good strategy for targeting since glycolysis and the PPP should encounter a direct impact by functioning with lower metabolic flux. T. cruzi hexokinase (TcHxK) has higher activity and is found at a higher percentage than T. cruzi glucokinase (TcGlcK) in the parasite (28), it is currently proposed that TcHxK is a potential drug target (16), but the status of TcGlcK is unclear. Genetic validation by RNAi cannot be carried out in T. cruzi parasites due to a nonfunctional RNAi pathway (29, 30). However, the structure of TcGlcK was solved in recent years through X-ray crystallography by Cordeiro and colleagues (31) and there is still no solved crystal structure for any trypanosomal (T. cruzi or T. brucei) hexokinase.
Hexokinases have a broad substrate range where various monosaccharides can be phosphorylated at appreciable rates, including D-glucose, D-mannose, D-galactose, and D-fructose; in addition, hexokinases are inhibited by G6P. Hexokinases also have a higher affinity for D-glucose than glucokinases. Glucokinases exhibit high specificity for D-glucose catalysis, they are not inhibited by G6P, and they have lower affinity for D-glucose (compared to hexokinases) (32). Glucose kinases (hexokinases or glucokinases) are categorized into two nonhomologous families, the hexokinase family and the ribokinase family. Within the hexokinase family (the subject of the work herein), there are three groups where glucose kinases are further subdivided on the basis of conserved amino acid 1° structure, which include (1) the hexokinase group, (2) group A, and (3) group B. Between these groups, conservation is lost when 1° structure sequence alignments are performed (e.g., protein BLAST). Additionally, the molecular mass of a hexokinase group enzyme (50–54 kDa per subunit) is generally higher on a subunit basis compared to group A (35–42 kDa per subunit) or group B (30–36 kDa per subunit) enzymes (32). T. cruzi has a hexokinase (from the hexokinase group; subunit MW of ~52 kDa) and a glucokinase (from group A; subunit MW of ~42 kDa), which is especially unusual because trypanosomatids are the only organisms known to have both enzymes present; various Leishmania spp. also have both a hexokinase and a group A glucokinase. Moreover, the functionality of both TcGlcK and TcHxK are quite similar because they perform as true glucokinases (as mentioned above) (16, 28). On the basis of percent sequence identity from a BLAST against 1° structure of hexokinases found in the GenBank database, TcHxK aligns with various existing conserved motifs, but this is not the case when aligning against glucokinases, such as TcGlcK. TcHxK and TcGlcK also have an approximate 17-fold difference in their KM values for D-glucose catalysis of 0.060 mM (16) and 1.00 mM (28), respectively.
E. coli glucokinase (EcGlcK) has a representative X-ray crystal structure for group A glucokinases (32). A structure-based sequence alignment between TcGlcK (PDB entry 2Q2R) (31) and EcGlcK (PDB entry 1SZ2) (33) was found to be consistent with secondary structural elements (31) even though the 1° sequence alignment was less than 16% identical. The quaternary structure of TcGlcK (dimeric form) is also very similar to that of EcGlcK. Additionally, there are four Homo sapiens hexokinase (HsHxK) isoenzymes I–IV and HsHxKIV has a representative structure among them. The crystal structures were solved for all four isoenzymes and they are among the hexokinase group even though HsHxKIV has been termed glucokinase based on its in vivo physiological activity (31). The structural comparison provided by Lunin et al. (33) for EcGlcK and the C-terminal half of HsHxKI (residues 475–917) showed that the tertiary structures and active sites were similar. The 3° structure of EcGlcK (the group A structural prototype) compares very well to both TcGlcK and HsHxKI (C-terminal half).
The basis of the presented work was to use structure-based drug design (SBDD) and a surrogate X-ray crystallography approach, in which we conceived of competitive inhibitors by using SBDD on TcGlcK (PDB entry 2Q2R) with the intention of inhibiting TcGlcK (only) or both TcGlcK and TcHxK. In order to achieve selectivity from an inhibitor design (e.g., the inhibitor can bind stronger to TcGlcK and avoid binding with any of the four human hexokinase isoenzymes), visual assessments were made after the crystal structure of TcGlcK was superimposed onto the crystal structure of HsHxKIV. Another goal of the study was to determine if the TcGlcK inhibitors would also inhibit TcHxK because both enzymes phosphorylate D-glucose through catalysis. The active site regions in TcGlcK and TcHxK are assumed to compare well structurally, albeit a 19% global pairwise 1° structural alignment between the two enzymes. Furthermore, we sought to determine if TcGlcK was a chemically-validated drug-target by using glucosamine analogue inhibitors.
Here, we report the X-ray crystal structures of TcGlcK in its complexes with inhibitors BENZ-GlcN, CBZ-GlcN, HPOP-GlcN, and DBT-GlcN (Figure 1). These inhibitors present the molecular details for affinity in the active site and may disclose more inhibitor design possibilities that can be used for optimized selectivity and affinity. We present inhibitor kinetic parameters of the aforementioned inhibitors with enzymes TcGlcK and HsHxKIV. The synthesis and characterization are shown for HPOP-GlcN and DBT-GlcN whereas inhibitors BENZ-GlcN and CBZ-GlcN were commercially available. Finally, we show effective in vitro inhibition assays against T. cruzi amastigote parasites for BENZ-GlcN and CBZ-GlcN.
2. Materials and methods
2.1. Materials
N-Cbz-D-glucosamine (CBZ-GlcN), 2-benzamido-2-deoxy-D-glucopyranose (BENZ-GlcN), and isopropyl β-D-thiogalactopyranoside (IPTG) were purchased from Carbosynth. Dulbecco’s modified Eagle’s medium (DMEM) was purchased from CellGro. N-Benzyl-2-nitro-1H-imidazole-1-acetamide (benznidazole, 97%), hexadeuterodimethyl sulfoxide (D6-DMSO, 99.96 atom%), tetrahydrofuran (anhydrous grade), 3-(4-hydroxyphenyl)propionic acid N-hydroxysuccinimide ester (≥97.0%), ammonium formate (HPLC grade, ≥99.0%), acetonitrile (HPLC grade), chlorophenol red-β-D-galactoside (CPRG), ethylenediaminetetraacetic acid tetrasodium salt hydrate (>99.0%), imidazole (99+%), bovine pancreas deoxyribonuclease I (DNase I), bovine pancreas ribonuclease A (RNase A), D-(+)-glucosamine hydrochloride (≥99%), triethanolamine (≥99.0%), sodium citrate tribasic dihydrate (≥99.0%), carboxymethyl (CM) cellulose, Saccharomyces cerevisiae glucose 6-phosphate dehydrogenase (type XV), β-nicotinamide adenine dinucleotide phosphate hydrate (NADP+, ≥95%), adenosine 5′-triphosphate disodium salt hydrate (ATP, 99%), D-glucose 6-phosphate sodium salt (≥98%), and all buffer salts (≥98%) were purchased from Sigma. Cobalt-nitrilotriacetic acid (Co-NTA) resin, methanol, ethyl acetate, lysozyme (type VI), triethylamine, DL-dithiothreitol, 2xYT broth, lysogeny broth (LB), kanamycin sulfate, protease inhibitor tablets (EDTA-free), 1,1-dioxobenzo[b]thiophen-2-ylmethyl N-succimidyl carbonate (95%), and all other chemicals were purchased from Fisher Scientific.
2.2. Cloning
The following genes: Trypanosoma cruzi glucokinase, strain CL Brener (GenBank accession number XP_821474) and Homo sapiens hexokinase IV (UniProtKB accession code P35557) were cloned into separate kanamycin–resistant pET-28a(+) Escherichia coli expression vectors at restriction sites 5′ NcoI and 3′ HindIII at Genewiz, Inc. (South Plainfield, NJ). Codons in all plasmids were optimized for protein expression. These plasmid constructs were designated as pET-TcGlcK-xtal and pET-HsHxKIV-xtal, respectively. Each construct encodes for an N-terminal hexahistidine tag. The pET-TcGlcK-xtal plasmid encodes the segment MGRGSHHHHHHGMA that precedes the start methionine and the 9-residue segment VGKKQKAQL at the C-terminal region was not included. The CL Brener strain of T. cruzi glucokinase differs at eight positions compared to the previously reported construct used by Cordeiro and colleagues in the first X-ray crystal structure determination of TcGlcK; see PDB entry 2Q2R. These positions in the CL Brener strain of TcGlcK include the following changes: A22V, I65L, M81I, H125R, L213I, F232L, H327R, and S344T. The pET-HsHxKIV-xtal plasmid encodes the segment MGHHHHHHENLYFQGM that precedes residue K12 (N-terminal segment M1 – A11 of HsHxKIV was not inlcuded) and the 8-residue segment KKACMLGQ at the C-terminal region was also not included. This pET-HsHxKIV-xtal construct was based off of one used in X-ray crystallography experiments (34).
2.3. Standard Methods
Standard methods are presented in the Supplementary Information section for the following: (1) the expression and purification of TcGlcK and HsHxKIV, (2) activity assays of TcGlcK and HsHxKIV, (3) crystallization of TcGlcK, (4) X-ray crystal structure determinations, (5) general procedures for the synthesis of glucosamine analogues, (6) the synthesis of HPOP-GlcN, (7) the synthesis of DBT-GlcN, and (8) biological assays. X-ray diffraction data collection and refinement statistics for the TcGlcK inhibitor complexes are shown in Table 1.
Table 1.
Data Collection and Refinement Statistics.
| TcGlcK-BENZ-GlcN | TcGlcK-CBZ-GlcN | TcGlcK-HPOP-GlcN | TcGlcK-DBT-GlcN | |
|---|---|---|---|---|
| Data Collection | ||||
| resolution limits (Å) | 50.0 - 2.40 | 50.0 - 2.50 | 50.0 - 2.10 | 50.0 - 1.90 |
| total/unique reflections | 136171/31494 | 105321/28549 | 160176/46434 | 251662/62066 |
| measured | ||||
| space group symmetry | P21 | P21 | P21 | P21 |
| unit cell dimensions | ||||
| a, b, c (Å) | 68.6, 78.8, 76.6 | 68.7, 79.0, 76.4 | 68.1, 80.0, 75.8 | 68.2, 78.7, 75.4 |
| α, β, γ (deg) | 90.0, 94.5, 90.0 | 90.0, 94.7, 90.0 | 90.0, 95.0, 90.0 | 90.0, 94.8, 90.0 |
| Rmergea,b | 0.079 (0.822) | 0.101 (0.694) | 0.067 (0.389) | 0.040 (0.440) |
| <I/σ(I)>a | 12.9 (1.4) | 13.26 (2.17) | 18.96 (2.86) | 26.73 (3.00) |
| completeness (%)a | 98.5 (92.0) | 99.8 (100) | 98.5 (97.4) | 99.1 (98.6) |
| Refinement | ||||
| reflections used in | 59783/3023 | 28332/1434 | 46407/2342 | 62037/3148 |
| refinement/test set | ||||
| Rworkc | 0.1929 | 0.1902 | 0.1905 | 0.1972 |
| Rfreed | 0.2564 | 0.2304 | 0.2368 | 0.2373 |
| protein chainse | 2 | 2 | 2 | 2 |
| protein residuese | 734 | 734 | 728 | 731 |
| protein atomse | 5696 | 5696 | 5654 | 5675 |
| solvent moleculese | 87 | 4 | 244 | 201 |
| ligand moleculese | 2 | 2 | 2 | 2 |
| Root Mean Square Deviationf | ||||
| bonds (Å) | 0.009 | 0.010 | 0.009 | 0.009 |
| angles (deg) | 1.30 | 1.20 | 1.20 | 1.20 |
| Wilson B-factorg (Å2) | 51 | 53 | 44 | 41 |
| Average B-factorsh (Å2) | ||||
| main chain | 43 | 79 | 42 | 53 |
| solvent | 32 | 60 | 40 | 48 |
| ligand | 31 | 73 | 38 | 48 |
| Ramachandran Plotf (%) | ||||
| allowed | 89.7 | 89.1 | 91.4 | 90.5 |
| additionally allowed | 10.0 | 10.8 | 8.5 | 9.4 |
| generously allowed | 0.3 | 0.2 | 0.2 | 0.2 |
| disallowed | 0.0 | 0.0 | 0.0 | 0.0 |
| PDB accession code | 5BRD | 5BRE | 5BRF | 5BRH |
Values in parenthesis are for the highest resolution shell.
Rmerge = Σ | I − 〈I〉| / Σ I, where I is the observed intensity, 〈I〉 is the average intensity calculated from replicate data.
R = Σ || Fo | − | Fc || / Σ | Fo | for reflections contained in the working set.
Rfree = Σ || Fo | − | Fc || / Σ | Fo | for 10% of reflections contained in the test set held aside during refinement. | Fo | and | Fc| are the observed and calculated structure factor amplitudes, respectively.
Per asymmetric unit cell.
Calculated using PROCHECK (59).
Calculated using POLYGON (60).
Calculated using MOLEMAN2 (61).
3. Results and discussion
3.1. Structural Comparison between TcGlcK and HsHxKIV
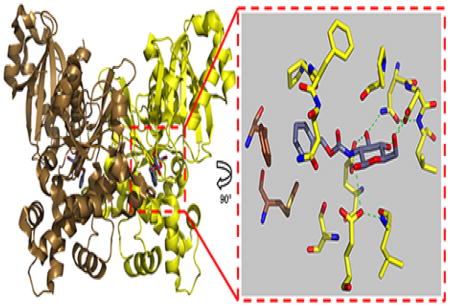
TcGlcK is a dimeric enzyme with two identical subunits. Each monomer has a large α + β domain (residues 145 – 352) and a small α/β domain (residues 1 – 130 and 353 – 367) (Figure 2).
Figure 2.

Dimeric and monomeric representations of the TcGlcK-ADP-D-glucose complex (PDB entry 2Q2R) (31). (a) TcGlcK dimer with N- and C-termini indicated for each subunit and is color-coded as follows: blue for the small domain, green for the large domain, and grey for the linking segments between domains. The same colors as slightly shaded represent the other subunit. (b) TcGlcK monomer revealing α-helices, 310-helices, and β-strands in sequential order. Secondary structure elements were produced by the DSSP program from PDB entry 2Q2R, as exhibited in Figure 1 of reference (31).
TcGlcK selective inhibitor designs were based on structural details of the HsHxKIV and TcGlcK active sites. HsHxKIV has a large loop segment (L165 – G178) and a shorter loop segment (S281 – Q287) found adjacent to the glucose binding site. Various residue side chains stem off of these loops and cluster near each other in addition with some other residues not part of the loops; and they all include residues: K56, P153, N166, T168, and Q286 (Figure 3a). When hydroxyphenyloxopropyl glucosamine (HPOP-GlcN) (Figure 1) is modeled into the glucose binding site (without having any energy minimization performed), the p-hydroxyphenyl moiety is extended by a linker into the clustered side chains region, and the linker is positioned between a narrow channel, which is bordered by P153 and T168 (Figure 3b). From the proximity of all of these residues, we predict a steric clash situation would result. On the other hand, the same region and channel observed in TcGlcK that is next to the glucose binding site (Figure 3c) has some residue side chains, P103 and N105, on the large loop segment (G102 – I110) that are not tightly clustered together; in addition, the side chains of P94 and N105 make up the borders of a much wider channel for the linker to extend through (Figure 3d). Modeling HPOP-GlcN in TcGlcK (by the same method implemented for HsHxKIV) reveals that the linker and p-hydroxyphenyl moiety are not involved in any significant steric clashes, based on our visual assessment.
Figure 3.

Views showing the outer part of the active site for proposed HPOP-GlcN complexes of (a) HsHxKIV and (c) TcGlcK, from PDB entries 4MLE and 2Q2R, respectively. The HPOP-GlcN ligand was modeled into each kinase by superimposing its sugar moiety onto the glucose ligand, followed by removing the original glucose coordinates. Energy minimization was not applied in the modeling of HPOP-GlcN. Non-identical residues are indicated on the outer part of the active sites of both enzymes with residue labels as red for HsHxKIV (a) and black for TcGlcK (c). In these proposed complexes, panel (a) shows that various residue side chains of HsHxKIV (T168, N166, Q286, and K56) have the potential for steric clash with HPOP-GlcN while P153 has the highest likelihood of steric clash for the same ligand because the glucose moiety of HPOP-GlcN is in a fixed position. This situation is predicted to not allow HPOP-GlcN to bind in HsHxKIV. Panel (c) reveals minimum to no steric clashing effects present in the same region for TcGlcK. Panels (b) and (d) show HsHxKIV and TcGlcK on different viewing angles from panels (a) and (c), respectively. Protein residues are shown with very close contact interactions to HPOP-GlcN in HsHxKIV (b) and are essentially distant in TcGlcK (d). Interactions are pending that HPOP-GlcN is able to bind as the two glucose kinase/inhibitor models suggest and are color-coded as follows: orange represents close contacts from “flexible” residue side chains that are expected to move and avoid steric clashing effects; green represents close contacts from a rigid proline (P153) expected to have unavoidable steric clashing effects; purple represents relatively distant contact interactions; and black represents putative hydrogen bonding interactions.
HPOP-GlcN may not favor binding in HsHxKIV, but will most likely favor binding in TcGlcK and thus give rise to selectivity. The clustered side chains in HsHxKIV are mostly flexible, and if HPOP-GlcN truly binds in this active site, as a consequence, we expect the flexible side chains of residues K56, N166, T168, and Q286 to reposition (see orange dashes in Figure 3b). However, an inhibitor with a bulkier tail group such as dioxobenzylthiophenyl glucosamine (DBT-GlcN) (Figure 1) is proposed to have substantial steric effects in that region regardless of side chain repositioning because of little available space. In regard to the channels that the linkers extend through, residue P153 (of HsHxKIV) has a rigid side chain that we predict will not be able to avoid close contact interactions with the linker groups of any of the glucosamine analogues (Figure 1) because there are key glucose-moiety binding interactions that must be maintained at the glucose binding site. P94 (of TcGlcK) is the comparable residue to P153 (Figure 4), but it is positioned differently to avoid steric hindrance of an extending linker (Figure 3d), and thus, gives rise to a wider channel to make easy access for glucosamine analogues in TcGlcK.
Figure 4.

Structure-based sequence alignment of TcGlcK (PDB entry 2Q2R) and HsHxKIV (PDB entry 3IDH) (34), as determined with the web-based protein structure comparison service Fold at European Bioinformatics Institute (PDBeFold) (35) and STRIDE (36, 37). The structural alignment was created with ESPript (version 3.0) (38). The overall sequence identity is 19% based on this alignment. The symbols α, η, and β indicate the secondary structural elements α-helices, 310-helices, and β-strands, respectively. Residues having exact identity are boxed and indicated by white letters on red background; residues with near identity are boxed and indicated by red letters on white background. Active site residues are indicated by brown triangles for TcGlcK and by blue triangles for HsHxKIV. The positions for two important loop segments are indicated by arrows having dashed lines (brown for TcGlcK and blue for HsHxKIV).
The determination of a good competitive inhibitor for hexokinase was previously accomplished by the early work of Anderson et al. (39) and Steitz et al. (40), in which an o-toluoyl glucosamine compound was complexed with yeast hexokinase through X-ray crystallography (PDB entry 2YHX). This finding proceeded to the development of modestly potent benzoyl glucosamine-based inhibitors for TbHxK (22, 41), including the m-bromo-benzoyl glucosamine inhibitor that had a determined Ki of 2.8 μM (22). We also tested one inhibitor that was shorter than the rest, benzoyl glucosamine (BENZ-GlcN), because the chemical structure for this compound was very similar to o-toluoyl glucosamine and this was a class of compound that was never tested on a T. cruzi hexokinase/glucokinase.
3.2. X-ray Crystallography of TcGlcK Inhibitor Complexes
The glucosamine analogues all share a common glucose moiety that preserves key enzyme-substrate hydrogen bonding interactions with the monosaccharide hydroxyl groups from C1, C3, C4, and C6. The C2 hydroxyl is replaced by a NH group and also participates in the key hydrogen bonding. However, each compound differs in its linker and tail group designated to interact with the intersubunit region adjacent to the glucose binding site. Crystal growth of TcGlcK in the absence of D-glucose followed by soaks of glucosamine analogues at room temperature allowed inhibitors to bind in the active site as confirmed by X-ray crystallography. All of the TcGlcK inhibitor complexes in this work crystallized as a homodimer (in the asymmetric unit) in the P21 space group and were found in a near fully-closed conformation between the large and small domains. The homodimeric subunit-subunit interface is stabilized by a large contact surface area of 1,912 Å2 (~12% of the total surface area) as determined by the Protein Interfaces, Surfaces, and Assemblies (PISA) server (42). This is in contrast to the loose tetramer that is observed for the TcGlcK-D-Glucose-ADP complex (PDB entry 2Q2R), which crystallizes in a P21212 space group and was found as a fully-closed conformation (31). This tetramer is stabilized by having large contact surface areas as well as by having intermolecular ionic interactions (e.g., salt bridges) and hydrogen bonds. In the P21 space group the intermolecular ionic interactions that normally result from residues I219, D220, and Q223 are essentially absent. A superposition of the TcGlcK P21 crystal form (dimer) onto the P21212 crystal form (tetramer) reveals that the dimers having higher surface area contact are essentially identical (results not shown).
For structural comparisons, all four ligand complexes (including subunits) are well conserved. With the TcGlcK-CBZ-GlcN complex (PDB entry 5BRE) as a reference structure for superposition, the overall fold of the dimer is essentially identical to that of the TcGlcK-BENZ-GlcN complex (PDB entry 5BRD), the TcGlcK-HPOP-GlcN complex (PDB entry 5BRF), and the TcGlcK-DBT-GlcN complex (PDB entry 5BRH). This resulted in a median root-mean-square deviation (r.m.s.d.) of 0.75 Å (range 0.43 – 0.78 Å, N=3) for 726 Cα atoms. The dimer of the TcGlcK-CBZ-GlcN complex superimposes onto the dimer (in the asymmetric unit) of the TcGlcK-Glc-ADP complex (PDB entry 2Q2R) with a r.m.s.d. of 1.02 Å for 720 Cα atoms, indicating that their overall folds are similar. A superposition of the crystal structures reveals conserved features within the active site (Figure 5a). The monomeric structures of each TcGlcK-glucosamine analogue complex are generally similar. The r.m.s.d. values, when monomers of each enzyme are superimposed, are as follows: (1) TcGlcK-BENZ-GlcN vs. TcGlcK-D-glucose-ADP complex (r.m.s.d. of 0.92 Å for 352 Cα atoms); (2) TcGlcK-CBZ-GlcN vs. TcGlcK-BENZ-GlcN complex (r.m.s.d. of 0.29 Å for 367 Cα atoms); (3) TcGlcK-HPOP-GlcN vs. TcGlcK-CBZ-GlcN complex (r.m.s.d. of 0.40 Å for 364 Cα atoms); (4) TcGlcK-DBT-GlcN vs. TcGlcK-D-glucose-ADP complex (r.m.s.d. of 1.00 Å for 362 Cα atoms). R.m.s.d. values were determined through the MacPyMOL software (43).
Figure 5.

Four crystal structures of TcGlcK bound to glucosamine analogue inhibitors in the active site. (a) Superposition of the four structures with BENZ-GlcN (yellow), CBZ-GlcN (red), HPOP-GlcN (blue), and DBT-GlcN (green); hydrogen bond interactions are represented by dashed lines. Simulated annealing omit maps of TcGlcK:inhibitor complexes for (b) BENZ-GlcN (orange map, contoured at 3.1σ), (c) CBZ-GlcN (red map, contoured at 2.6σ), (d) HPOP-GlcN (brown map, contoured at 3.0σ), and (e) DBT-GlcN (blue map, contoured at 3.0σ). Atoms are color-coded in panels b – e as follows: yellow (arbitrary monomeric subunit), brown (opposite monomeric subunit), or grey (inhibitor) for carbon; blue for nitrogen; red for oxygen; and red spheres for solvent. Green dashed lines represent hydrogen bond interactions.
Simulated annealing omit maps are shown for each glucosamine analogue inhibitor in Figure 5 (panels b – e) with an occupancy of 100% for all ligand atoms. BENZ-GlcN, CBZ-GlcN, and DBT-GlcN prefer the β anomer (orientation of the C1 hydroxyl of the glucose moiety) for active site binding in TcGlcK; however, HPOP-GlcN binds as the α anomer (Figure 5a). TcGlcK has a 4-fold higher binding preference for the β anomer of D-glucose over the α anomer based on a KM value comparison (31). In an attempt to refine the β anomer of HPOP-GlcN (as 100% occupancy for all ligand atoms in the A-chain), negative electron density (contoured at −3.6σ) was observed by the C1 hydroxyl O-atom (in the equatorial position) and positive electron density (contoured at +4.5σ) was found below the C1 O-atom (in the axial position) from an almost final |Fo| − |Fc| map (data not shown). By refining the α anomer of HPOP-GlcN (C1 hydroxyl O-atom in the axial position) instead of the β anomer, the negative and positive electron density peaks are completely absent from the final |Fo| − |Fc| map surrounding the same O-atom.
Hydroxyls of the glucose moiety from all four glucosamine analogues interact the same way, in that hydrogen bonds are donated to E236, E207, and D131 (interaction with the C4 and C6 hydroxyls); additionally, the C3 hydroxyl of the glucose moiety accepts a hydrogen bond from the side chain NH2 group (Nδ) of N130 (Figure 5a). From the linker component of each inhibitor that stems from the glucose moiety C2-atom, a hydrogen bond is donated from the NH group (of the peptide bond) to E207. The benzene moiety of BENZ-GlcN makes van der Waals contact with P94, M295, P103, S210, and F337 (residue of the other subunit) (Figure 5b). The phenyl tail group of CBZ-GlcN and the phenol moiety tail group of HPOP-GlcN adopt an orientation for favorable van der Waals interactions with F337 and M334 side chains (residues from the other subunit of the protein dimer (Figure 5, panels c and d)) along with P103 and N105 side chains in the active site cavity. The aromatic groups of these inhibitors fit in the outer part of the active site hydrophobic pocket and engage in a π-stacking interaction with the phenyl group of F337, which may give rise to the enhanced inhibitor binding strength compared to BENZ-GlcN that lacks this particular π-stacking interaction. DBT-GlcN has a larger aromatic tail group than CBZ-GlcN or HPOP-GlcN, but the 1,1-dioxobenzo[b]thiophene moiety also fits in the outer part of the active site hydrophobic pocket (Figure 5e) and there are some notable similarities and differences. The similarities include tail group orientation to maximize van der Waals contact with residues P103 and N105 (in the active site region) and residues F337 and M334 of the other subunit. The phenyl group of F337 and the aromatic 1,1-dioxobenzo[b]thiophene group on DBT-GlcN are similarly positioned in a π-stacking orientation. Finally, the side chains of F337 and M334 are moved away from their regular positions as observed in all of the other complexes in order to help situate the bulky 1,1-dioxobenzo[b]thiophene group. The key difference to the TcGlcK-CBZ-GlcN complex includes the bending region angle. The 1,1-dioxobenzo[b]thiophene group is much more bulky compared to the phenyl group on CBZ-GlcN; and, DBT-GlcN is positioned at the interface of the small and large domains. In order for the N105 and P103 side chains to be in ideal van der Waals contact distance (i.e., 3.4 – 3.6 Å), the small domain positions itself in more of a semi-open conformation by 7.7° (average bending angle from the A- and B- chains) from the fully closed conformation structure (PDB entry 2Q2R) as determined by the program DynDom (44). However, the TcGlcK-CBZ-GlcN complex only has an average bending angle of 4.1° from the fully closed conformation, revealing an even smaller semi-open conformation of the enzyme.
3.3. Inhibition Studies of TcGlcK and HsHxKIV by Glucosamine Analogues
The coupled hexokinase – glucose 6-phosphate dehydrogenase enzymatic colorimetric assay that produces NADPH (45) was utilized to obtain catalytic activity measurements and inhibition constants (Ki) for TcGlcK and HsHxKIV. All kinetic measurements are with respect to substrates D-glucose (used at variable concentrations) and ATP (used at a constant concentration). For catalytic activity at room temperature (22 °C) and pH 7.6, we report for His6-TcGlcK a KM of 0.948 ± 0.295 mM for D-glucose, which is essentially identical to the KM of 1.00 mM observed by Cáceres et al. (28) at room temperature and pH 8.5. However, with respect to His6-HsHxKIV, we obtain a 7-fold higher KM of 41.8 ± 8.7 mM for D-glucose compared to the measurement of 6.0 mM exhibited by the non-His-tagged wt-HsHxKIV (46). Our construct for His6-HsHxKIV is based off of a design used in X-ray crystallography experiments to promote protein crystal growth (34) and results in an enzyme having 11 residues truncated from the N-terminus (with an N-terminal 6x His-tag included) along with 8 residues truncated from the C-terminus. This difference in the KM values for D-glucose is likely resultant from the truncations. Table 2 reveals a comparison of our kinase kinetic parameters (KM, kcat, and kcat/KM) to reported literature values. Inhibition constants for glucosamine analogues are provided in Table 3 for both TcGlcK and HsHxKIV. Representative Lineweaver-Burk plots for all inhibitors tested on both kinases are found in the Supplementary Information section. The glucosamine analogues are revealed as competitive inhibitors through X-ray crystallography because they all bind in the active site; however, the analogues showed mixed-type inhibition from their corresponding Lineweaver-Burk plots. We suspect that the mixed-type inhibition may be a consequence of the glucosamine analogue inhibitor preventing the fully closed conformation of the glucokinase being examined. We were not able to measure the Ki for HsHxKIV and DBT-GlcN because DBT-GlcN at concentrations needed for inhibition caused an interference with the colorimetric assay by absorbing light at 340 nm. The strongest TcGlcK inhibitor known to date is CBZ-GlcN, which has a Ki of 0.71 ± 0.05 μM. HPOP-GlcN was observed as a weaker inhibitor by 1.8-fold to TcGlcK compared to CBZ-GlcN, but that is not surprising since HPOP-GlcN binds as the less-preferred α anomer (31). The trend for inhibitor affinity of TcGlcK is as follows: CBZ-GlcN > HPOP-GlcN > DBT-GlcN > BENZ-GlcN. All of the analogues tested had higher inhibitor affinity toward TcGlcK compared to HsHxKIV. The most selective inhibitors of the series were CBZ-GlcN and HPOP-GlcN with selectivity ratios of 245 and 186, respectively, and the least selective inhibitor was BENZ-GlcN with a selectivity value of 12 (Table 3).
Table 2.
Kinetic Parameters for TcGlcK and HsHxKIV.
| Enzyme | Constructd | KM (mM) | kcat (min−1) | kcat/KM (mM−1 min−1) |
|---|---|---|---|---|
| His6-TcGlcKa | pET-TcGlcK-xtale | 0.948 ± 0.295 | (4.67 ± 2.15) × 102 | (4.93 ± 2.26) × 102 |
| His6-TcGlcKb | NNAf | 1.00 ± 0.06 | 8.95 × 104 | 8.95 × 104 |
| His6-HsHxKIVa | pET-HsHxKIV-xtale | 41.8 ± 8.7 | (1.30 ± 1.73) × 104 | (3.11 ± 4.14) × 102 |
| wt-HsHxKIVc | pEhgk-WT | 6.0 ± 0.4 | 3.98 × 103 | 6.63 × 102 |
From this study; assay was performed at pH 7.6 and 22 °C; measurements performed in triplicate.
From ref (28); assay was performed at pH 8.5 and room temperature.
From ref (46); wild-type human pancreatic β-cell hexokinase IV; assay was performed at pH 7.5 and 30 °C.
Refer to corresponding references for construct details.
Extension “-xtal” signifies that the construct has a custom design to allow the enzyme to be suitable for X-ray crystallography.
Name not available.
Table 3.
| Ki versus substrate (μM) | |||
|---|---|---|---|
| TcGlcK | |||
| Inhibitor | TcGlcK | HsHxKIV | selectivity ratioc |
| BENZ-GlcN | 32 ± 26 | 376 ± 196 | 12 |
| CBZ-GlcN | 0.71 ± 0.05 | 174 ± 50 | 245 |
| HPOP-GlcN | 1.3 ± 0.6 | 242 ± 97 | 186 |
| DBT-GlcN | 4.1 ± 0.3 | ndd | ndd |
All compounds were tested as their racemic mixtures (n ≥ 2).
Colorimetric assay; measurements in triplicate.
Selectivity ratio = Ki (HsHxKIV) / Ki (TcGlcK).
Not determined.
3.4. Analysis of Inhibitor Binding in TcGlcK
The glucosamine analogues are generally in compliance with the Lipinski’s Rule of Five (Ro5) (47, 48), from their physiochemical parameters (Table 4). BENZ-GlcN, CBZ-GlcN, and DBT-GlcN each had a Lipinski score of 4, whereas HPOP-GlcN had a score of 3. The criteria evaluated for the Ro5 included the following: (1) molecular weight (MW) ≤ 500 g/mol, (2) CLogP ≤ 5, (3) number of H-bond donors ≤ 5, and (4) number of H-bond acceptors ≤ 10. In addition, the inhibitors were also in agreement with other key physiochemical parameters that are common amongst many successful drugs and were previously described by Luzina and Popov (49), as follows: (1) polar surface area (PSA) < 140 Å2, (2) molar refractivity (MR) in the range of 40 – 130 cm3/mol, and (3) the number of atoms in the molecule (including hydrogen atoms) in the range of 20 – 70 atoms. All of the inhibitors follow these criteria except for DBT-GlcN, which has a slightly higher PSA of 162.62 Å2.
Table 4.
Physiochemical parameters of glucokinase inhibitors.a
| Compound | Molecular Formula | MW | CLogPb | H-bond | H-bond | Lipinski | MRb | PSA (Å2)b |
|---|---|---|---|---|---|---|---|---|
| (number of atoms) | (g/mol) | donor | acceptor | Scorec | (cm3/mol) | |||
| BENZ-GlcN | C13H17NO6 (37) | 283.28 | −0.4686 | 5 | 7 | 4 | 68.00 | 119.25 |
| CBZ-GlcN | C14H19NO7 (41) | 313.30 | 0.1253 | 5 | 8 | 4 | 74.33 | 128.48 |
| HPOP-GlcN | C15H21NO7 (44) | 327.33 | −0.3431 | 6 | 8 | 3 | 78.73 | 139.48 |
| DBT-GlcN | C16H19NO9S (46) | 401.39 | −0.4239 | 5 | 10 | 4 | 92.41 | 162.62 |
From an assessment of ligand accessible surface area (ASA) in the active site of TcGlcK and the Ki value trend of the glucosamine analogue inhibitors was that as ligand ASA contact increases from 243 – 365 Å2 (Table 5), CBZ-GlcN had not required the upper limit of the ASA to maintain its relative strong inhibition. This suggests that even though van der Waals contact and glucose-moiety binding interactions served key roles in the inhibitor’s binding strength, the π-stacking interaction of the phenyl tail group with the phenyl group of F337 made another key interaction. This is surprising because DBT-GlcN makes similar interactions as CBZ-GlcN and DBT-GlcN has a much higher ligand ASA contact in the TcGlcK active site (365 Å2). It was expected that DBT-GlcN would have stronger inhibition because of more van der Waals contact in the region (i.e., 365 Å2 vs. 275 Å2, see Table 5). The 6-fold weaker inhibition of DBT-GlcN appears to be explained by the larger bending region angle of 7.7° vs. 4.1° (Table 5). When the two domains are more open from the closed conformation, more solvent access in the active site is evident and will give rise to weaker inhibition. This may occur by the destabilization of the hydrophobic pocket, less optimal hydrogen bonding geometries (especially with glucose-moiety binding residues, N130 and D131), and due to an increased dielectric constant within the active site.
Table 5.
Key parameters for analysis of TcGlcK ligand binding.a
| Ligand | Ligand | Ligand ASA | PDB | Ki (M) | LE | Bending Region |
|---|---|---|---|---|---|---|
| SA (Å2)b | Contact (Å2)c,d | Entry | (of TcGlcK) | (kcal/mol)e | Angle (°)f,g,h | |
| β-D-Glucose | 392 | 169 | 2Q2R | n/a | n/a | 0.0 |
| BENZ-GlcN | 677 | 243 | 5BRD | 3.2 × 10−5 | 0.30 | 4.9 |
| CBZ-GlcN | 742 | 275 | 5BRE | 7.1 × 10−7 | 0.38 | 4.1 |
| HPOP-GlcN | 778 | 315 | 5BRF | 1.3 × 10−6 | 0.35 | 7.6 |
| DBT-GlcN | 913 | 365 | 5BRH | 4.1 × 10−6 | 0.27 | 7.7 |
SA is surface area; ASA is accessible surface area; Ki is inhibition constant; and LE is ligand efficiency.
Calculated using MacPyMOL (43).
Values represent average ligand contact area (interactions from A- and B- subunits of the TcGlcK active site, as closed conformation).
Calculated using the CCP4 program AREAIMOL (62).
LE = −RTlnKi/N, where N is the number of non-hydrogen atoms and R = 0.001987 kcal mol−1 K−1; equation from ref (63).
Average bending region angle between the large and small domains of TcGlcK from the A- and B- subunits.
Reference closed conformation structure is PDB entry 2Q2R.
Calculated using DynDom (44).
CBZ-GlcN has a reasonably low molecular weight of 313 g/mol and is an efficient lead compound with a ligand efficiency (LE) of 0.38 kcal/mol for TcGlcK inhibition. The compound therefore has the possibility of further optimization to be a better inhibitor. Table 5 shows LEs for the entire set of glucosamine analogue compounds tested in this study. HPOP-GlcN had the second highest LE of 0.35 kcal/mol. DBT-GlcN was not as effective, having an LE of 110 cal/mol less than CBZ-GlcN, primarily due to its MW of 401 g/mol and slightly higher Ki value. The LE of BENZ-GlcN was also as low as DBT-GlcN (even though it has a lower MW of 283 g/mol) with a value of 0.30 kcal/mol, but this is explained by the fact that its benzene ring does not engage in the key π-stacking interaction as the other three inhibitors. Although BENZ-GlcN has a lower LE with respect to the other analogues, it has the possibility for optimization because of its lower MW. The benzene ring of BENZ-GlcN simply adds to the non-hydrogen atoms (N) and thereby lowers the efficiency term.
In order to advance glucosamine analogues for the purpose of crafting stronger TcGlcK inhibitors, the following parameters should be maintained in the inhibitor design: (1) the sugar moiety should have the glucose stereochemistry and exhibit the β anomer, (2) the inhibitor should maintain a bending region angle in TcGlcK to be less than 5.0° from the closed conformation (as demonstrated by an X-ray crystal structure of a TcGlcK complex), (3) the ligand ASA in the active site should be ≥ 275 Å2 to help maximize van der Waals interactions, (4) a linker having an aromatic tail group, as in the case of CBZ-GlcN, should be installed, (5) the aromatic tail group should not be too bulky (a phenyl group appears to be a good size), and (6) maintain a Lipinski score of 4 for the Ro5. Development of glucosamine analogues for the purpose of medicinal quality compounds against T. cruzi amastigotes requires a focus on the compound BENZ-GlcN, as follows: (1) maintain the set of guidelines for strong TcGlcK inhibitors (see above) except for guideline number 5 and (2) maintain a short linker length (as it is in the case of BENZ-GlcN). By maintaining these parameters and performing structure-activity relationship (SAR) studies, the best inhibitor optimization possibilities will be anticipated.
3.5. Effects of Glucokinase Inhibitors on In Vitro Amastigote Viability
The in vitro treatment of T. cruzi amastigotes in mammalian NIH-3T3 fibroblasts using glucosamine analogues, such as BENZ-GlcN, CBZ-GlcN, HPOP-GlcN, or DBT-GlcN reveals a trend that does not correlate entirely well with the TcGlcK inhibitor affinity trend. The in vitro IC50 values were measured 4 days after incubation using a glucose-rich DMEM medium. The strongest TcGlcK inhibitor in the study, CBZ-GlcN (Ki = 0.71 μM), was the second most inhibitory for in vitro culture with an IC50 of 48.73 ± 0.69 μM (results not shown). On the other hand, BENZ-GlcN, which was the least potent of the compound series as a TcGlcK inhibitor (Ki = 32 μM) had the highest inhibitory effect against T. cruzi cells with an IC50 of 16.08 ± 0.16 μM (Figure 6a). Benznidazole has a T. cruzi IC50 value of 1.12 ± 0.095 μM (Figure 6b) that compares similarly to the reported values of 1.43 μM (50) and 1.5 μM (51). Amphotericin B has an IC50 value of 0.48 ± 0.0032 μM (results not shown). BENZ-GlcN has a 14-fold weaker inhibition effect and a 34-fold weaker inhibition effect compared to benznidazole and amphotericin B, respectively.
Figure 6.

Dose response of (a) BENZ-GlcN activity and (b) benznidazole activity on T. cruzi (Tulahuen strain) intracellular amastigote growth inhibition in NIH-3T3 fibroblasts, by the method of Andriani et al. (52). The curve and the IC50 were calculated from measurements determined in triplicate and expressed as the % inhibition of parasite growth at each concentration.
The least effective inhibitors against parasites were HPOP-GlcN and DBT-GlcN with IC50 values > 50 μM (results are not shown because the highest concentration of inhibitors tested was 50 μM and changes in the percent activity at 50 μM were not significant). The observations for BENZ-GlcN are suggestive that BENZ-GlcN is either a promiscuous inhibitor that binds to more than one T. cruzi target and leads to a T. cruzi cytotoxic effect or that this inhibitor prevents parasite invasion into mammalian host cells. In either case, this is a surprising result because Willson and colleagues determined that for highly similar analogues of BENZ-GlcN, the compounds (intended to bind to the drug-target TbHxK) were incapable of producing significant biological activity through in vitro cultures of T. brucei using a human physiological concentration of glucose (5 mM) in the growth medium with a 3-day incubation period of parasites (22).
There are various possibilities for why BENZ-GlcN has a good inhibition effect against T. cruzi parasites in comparison to T. brucei parasites. One possibility is that BENZ-GlcN is very effective at inhibiting T. cruzi parasite invasion into host cells. Another possibility is that BENZ-GlcN can be imported into the glycosomes of T. cruzi amastigotes through a compatible glucose transporter having strong affinity and thereby acting to kill the parasites. This hypothesis is supported by the findings of Tetaud et al. (53), in which they observed for N-acetyl-D-glucosamine (GlcNAc) (an analogue of BENZ-GlcN), the T. cruzi epimastigote form expresses a hexose transporter that is 71 times more sensitive for GlcNAc compared to the hexose transporter that is expressed in T. brucei (bloodstream form). In these trypanosomal parasites, inhibition constants for GlcNAc against radiolabeled D-glucose uptake were found to be 0.156 mM and 11.11 mM, respectively. BENZ-GlcN is also modestly potent as a glucokinase inhibitor and it most likely binds strongly to TcHxK. Since TcHxK is found at higher concentrations in T. cruzi cells with respect to TcGlcK (as μg of enzyme per mg of glycosome) (28), if TcHxK is being inhibited by BENZ-GlcN in addition to TcGlcK, glycolysis would be expected to be severely impacted. Possible causes for the poorer inhibition effects observed by HPOP-GlcN and DBT-GlcN may be related to a lack of inhibition of T. cruzi invasion into mammalian host cells, T. cruzi glucose transporters not having sufficient affinity for these glucosamine analogue inhibitors, and/or these inhibitors may be very selective to only TcGlcK and will not act as inhibitors to TcHxK, if they are indeed imported by a glucose transporter. The data is also suggestive that since the DMEM medium has a very high glucose concentration, the glucose most likely is not a major factor for enzyme competition (with respect to the inhibitors) because we observe modestly potent inhibition to sub-micromolar inhibition affinity with BENZ-GlcN and CBZ-GlcN, respectively.
3.6. Structural Factors for Selectivity of Glucosamine Analogues in TcGlcK and TcHxK
Crystal structures of TcGlcK complexed with glucosamine analogues, such as CBZ-GlcN or DBT-GlcN, reveal that in addition to their glucose moieties binding in the glucose binding site, the aromatic moieties of these compounds bind in a newly identified hydrophobic cavity found at the intersubunit region adjacent to the glucose binding site. The HsHxKIV structure lacks this particular hydrophobic pocket because the enzyme is a monomer. Furthermore, the region is quite distinct between the two enzymes. For example, by superimposing the monomers of TcGlcK (PDB entry 2Q2R) and HsHxKIV (PDB entry 4MLE) (54), a large 14-residue loop (L165 – G178) is found in the small domain of HsHxKIV, which corresponds to a smaller 8-residue loop region (G102 – I110) in TcGlcK (also part of the small domain), see Figure 4. These loops exhibit different conformations based on their flexibility (Figure 7a). In TcGlcK, the G102 – I110 loop includes residues P103 and F104 for which van der Waals contact is made with some of the glucosamine inhibitors (see Figure 5). With this loop being shorter with respect to the L165 – G178 loop of HsHxKIV, steric hindrance is avoided from the aromatic moieties of the glucosamine inhibitors bound in the active site. The L165 – G178 loop of HsHxKIV interacts weakly with a second loop (S281 – Q287) from its large domain by making only van der Waals interactions. However, the corresponding loop segment (K230 – N234) found in the large domain of TcGlcK is highly different in its orientation, by being completely absent from the intersubunit region by the glucose binding site (Figure 7a). Since the S281 – Q287 loop of HsHxKIV is oriented adjacent to the glucose binding site; side chains from residues N283, Q286, and Q287 have the potential to block this region from the tail groups of glucosamine analogues (especially for inhibitors with longer linker groups than the compounds studied herein) unless the small and large domains of the monomer can break into the open conformation (or a semi-open conformation) to allow binding. The side chains from T168 and N166 on HsHxKIV (found on the large L165 – G178 loop) may also prevent glucosamine analogue tail groups from being able to bind in this region. However, this prediction will need to be confirmed by having X-ray crystal structures of glucosamine analogue complexes of HsHxKIV. The reason behind why the TcGlcK K230 – N234 loop is oriented in a completely different manner from the case of HsHxKIV (and from the cases pertaining to the H. sapiens hexokinase isozymes) relates to a β-turn that is present in HsHxKIV (G261 – G264) but absent in TcGlcK (Figure7a). The β-turn (G261 – G264) of HsHxKIV causes the α7 helix to be positioned ideally in order to permit its S281 – Q287 loop to meet at the region adjacent to the glucose binding site. With a missing β-turn in TcGlcK at this segment that is shown by a multiple sequence alignment (Figure 7b) the TcGlcK K230 – N234 loop cannot be positioned at the outer part of the active site (intersubunit region). All of the HsHxK isozymes have this β-turn and have a similar structural comparison at this region (Figure 7c).
Figure 7.

Primary, secondary, and tertiary structure representations of TcGlcK and HsHxKIV at an outer part of the active site of the monomer. (a) Tertiary structure representations for human hexokinase IV (blue) bound to glucose (PDB entry 4MLE) (54) that is superimposed onto the TcGlcK-HPOP-GlcN complex (brown). (b) Multiple sequence alignment of the TcGlcK H201 – S240 segment by means of the Clustal Omega program (55), where TcGlcK (PDB coordinates from the TcGlcK-DBT-GlcN complex) is aligned with human hexokinase I (PDB entry 4F9O), human hexokinase II (PDB entry 2NZT), human hexokinase III (PDB entry 3HM8), human hexokinase IV (PDB entry 3IDH), and T. cruzi hexokinase (GenBank accession number CAD26835.1). Residues of the HsHxKIV S281 – Q287 loop are highlighted in yellow and are between the two blue triangles. The two red stars indicate the location of the two glycines in the human hexokinases and are highlighted yellow that give rise to the β-turn. All 2° structure elements (α-helices, 310-helices, and β-strands) above the sequence alignment were generated from experimentally determined 3° structures (PDB entries) through the program STRIDE (36, 37) and are color-coded as brown for TcGlcK and blue for the human hexokinases. The 2° structure elements below the sequence alignment (α-helices and β-strands) were generated by prediction from a 1° structure (FASTA entry) through the program PSIPRED (version 3.3) (56) and are color-coded as black for TcHxK. The multiple sequence alignment was created with the program ESPript (version 3.0) (38). (c) Superposition of the A-chains of the human hexokinase isozymes in the closed conformation complexed with glucose in the active site, as follows: hexokinase I (PDB entry 4F9O, brown), hexokinase II (PDB entry 2NZT, magenta), hexokinase III (PDB entry 3HM8, light green), and hexokinase IV (PDB entry 4MLE, grey). The superposition reveals the similarity from the two loops [(L165 – G178); (S281 – Q287)] of hexokinase IV as well as the β-turn, when compared to the other 3 human hexokinase isozymes I–III.
Despite the lack of a crystal structure for TcHxK, secondary structure prediction from primary structure was generated for this enzyme (Figure 7b), to reveal the secondary structure elements in this region. The secondary structure and primary structure both reveal clues for the nature of the outer part of the active site in TcHxK. This prediction shows that TcHxK lacks the β-turn, but it has a 10-residue loop for the corresponding 5-residue TcGlcK K230 – N234 loop. This result appears to suggest that the TcHxK 10-residue loop segment will not interfere with the intersubunit region adjacent to the glucose binding site and our glucosamine inhibitors have the potential to bind to this enzyme. Finally, until the TcHxK structure is solved, there will be the unanswered question as to whether this region has involvement in an interface with another subunit, as is the case of dimeric TcGlcK.
4. Conclusions
The work described in this study reveals four glucosamine analogue inhibitor complexes of TcGlcK. X-ray crystallography has revealed an important hydrophobic pocket adjacent to the glucose binding site of TcGlcK (at the interface of the protein dimer), but is absent in HsHxK isoenzymes. This hydrophobic pocket contributes significantly to TcGlcK glucosamine analogue inhibitor affinity and selectivity, particularly when the inhibitor tail groups are aromatic and form a π-stacking interaction with residue F337 (of the opposite subunit of the dimer). We propose that the glucosamine analogues of this study will have difficulty binding in HsHxKIV because of residue P153 along with other residue side chains (T168, N166, Q286, and K56) that contribute to steric clash effects. However, these types of steric clashes were not evident in TcGlcK based on having crystal structures of glucosamine analogue complexes. Enzyme inhibition constants were determined for both TcGlcK and HsHxKIV, and from this glucosamine analogue series, CBZ-GlcN was determined to be the strongest glucokinase inhibitor known to date (Ki = 0.71 μM for TcGlcK). The inhibitors also had good selectivity of inhibition over HsHxKIV. Additionally, inhibitors CBZ-GlcN and BENZ-GlcN showed effective in vitro biological activity against T. cruzi amastigotes co-cultured in mammalian NIH-3T3 fibroblasts.
The trend for TcGlcK inhibitor affinity did not correlate well with the in vitro biological assay growth inhibition results for the small number of compounds tested. This loss in correlation does not permit a firm assignment of TcGlcK as a chemically-validated drug target, one of the objectives we pursued in this study. The biological effectiveness of CBZ-GlcN and BENZ-GlcN may be a consequence of enzyme inhibitor promiscuity; specifically, being able to bind to both TcGlcK and TcHxK that severely diminishes the needed glycolytic and/or PPP metabolic flux. Therefore, we are left with the unsolved challenge of drug-target validation. It is possible, however, to carry out genetic drug-target validation for TcHxK and TcGlcK given the unfortunate case that genetic validation by RNAi in T. cruzi cannot be performed because the organism lacks a functional RNAi pathway (29, 30). Drug-target identification by a method involving in vitro reduced susceptibility analysis by candidate compounds (in this case, the TcGlcK glucosamine analogues) in T. cruzi isolates followed by genome sequencing of the resistant parasites could be implemented (57, 58). However, this method is usually employed in those instances where selective pressure exerted by decades of extensive use of parasite growth inhibitors (on human populations) has resulted in the evolution of resistant strains carrying genetic polymorphisms in the inhibitor target genes (e.g., Plasmodium falciparum has drug resistance in certain strains developed after a ten-year period of usage of chloroquine, quinine, mefloquine, and artesunate (58)). This scenario is less likely to have occurred in the case of TcGlcK glucosamine analogues developed in this study, since natural populations of T. cruzi have never been exposed to these candidate compounds before. The best case for genetic drug-target validation will be until the challenges of RNAi are overcome in T. cruzi.
Supplementary Material
Highlights.
The strategy of structure-based drug design allowed for the determination of selective T. cruzi glucokinase inhibitors.
Crystal structures of T. cruzi glucokinase and its complexes with novel glucosamine analogues were determined.
The inhibitor CBZ-GlcN is the strongest glucokinase inhibitor known to date.
Inhibitors BENZ-GlcN and CBZ-GlcN are highly effective against T. cruzi amastigotes co-cultured in mammalian cells.
Acknowledgments
We thank Dr. Amy E. Sears, Dr. Paul D. Swartz, and Danielle A. Lehman for helpful discussions. The work was supported by the US National Institutes of Health Grant GM103499 (SC INBRE) to E.L.D. and by the University of South Carolina Office of the Vice President for Research (Magellan Program) to M.S.D. and S.P.K. We thank the beamlines of the Northeastern Collaborative Access Team (NE-CAT), which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P41 GM103403). The Pilatus 6M detector on beamline 24-ID-C is funded by a NIH-ORIP HEI grant (S10 RR029205). This research used resources from the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Mass spectra and NMR spectra were obtained at North Carolina State University in the mass spectrometry facility and the nuclear magnetic resonance center, respectively, located in the department of chemistry.
Abbreviations
- ASA
Accessible surface area
- BENZ-GlcN (Benzoyl glucosamine)
2-benzamido-2-deoxy-D-glucopyranose
- CBZ-GlcN (Carboxybenzyl glucosamine)
2-N-carboxybenzyl-2-deoxy-D-glucosamine
- DBT-GlcN (Dioxobenzylthiophenyl glucosamine)
2-[[1,1-dioxobenzo[b]thiophen-2-yl]methyloxocarbonylamino]-2-deoxy-D-glucosamine
- DMEM
Dulbecco’s modified Eagle’s medium
- HPOP-GlcN (Hydroxyphenyloxopropyl glucosamine)
2-[[3-(4-hydroxyphenyl)-1-oxopropyl]amino]-2-deoxy-D-glucosamine
- HsHxKIV
Homo sapiens hexokinase IV
- LE
Ligand efficiency
- PDB
Protein Data Bank
- r.m.s.d
root-mean-square deviation
- RNAi
RNA interference
- TbHxK
Trypanosoma brucei hexokinase
- TcGlcK
Trypanosoma cruzi glucokinase
- TcHxK
Trypanosoma cruzi hexokinase
Footnotes
Accession Codes
The atomic coordinates and structure factors of the TcGlcK-BENZ-GlcN complex, the TcGlcK-CBZ-GlcN complex, the TcGlcK-HPOP-GlcN complex, and the TcGlcK-DBT-GlcN complex have been deposited in the Protein Data Bank (www.rcsb.org) with accession codes 5BRD, 5BRE, 5BRF, and 5BRH, respectively.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.World Health Organization. [accessed Apr, 2015];Media Centre Fact Sheets Homepage. http://www.who.int/mediacentre/factsheets/fs340/en/index.html. Fact Sheet No. 340, Updated Mar, 2015.
- 2.Centers for Disease Control and Prevention. [accessed Apr, 2015];Parasites Homepage. http://www.cdc.gov/parasites/chagas/. Updated Jul, 2013.
- 3.Barrett MP, Burchmore RJS, Stich A, Lazzari JO, Frasch AC, Cazzulo JJ, Krishna S. The trypanosomiases. Lancet. 2003;362:1469–1480. doi: 10.1016/S0140-6736(03)14694-6. [DOI] [PubMed] [Google Scholar]
- 4.Tarleton RL, Reithinger R, Urbina JA, Kitron U, Gürtler RE. The challenges of Chagas disease - grim outlook or glimmer of hope? PLoS Med. 2007;4:e332. doi: 10.1371/journal.pmed.0040332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Urbina JA. Specific chemotherapy of Chagas disease: relevance, current limitations and new approaches. Acta Trop. 2010;115:55–68. doi: 10.1016/j.actatropica.2009.10.023. [DOI] [PubMed] [Google Scholar]
- 6.Gascon J, Bern C, Pinazo M-J. Chagas disease in Spain, the United States and other non-endemic countries. Acta Trop. 2010;115:22–27. doi: 10.1016/j.actatropica.2009.07.019. [DOI] [PubMed] [Google Scholar]
- 7.Bern C, Kjos S, Yabsley MJ, Montgomery SP. Trypanosoma cruzi and Chagas’ disease in the United States. Clin Microbiol Rev. 2011;24:655–681. doi: 10.1128/CMR.00005-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Croft SL, Barrett MP, Urbina JA. Chemotherapy of trypanosomiases and leishmaniasis. Trends Parasitol. 2005;21:508–512. doi: 10.1016/j.pt.2005.08.026. [DOI] [PubMed] [Google Scholar]
- 9.Cançado JR. Long term evaluation of etiological treatment of Chagas disease with benznidazole. Rev Inst Med Trop Sao Paulo. 2002;44:29–37. [PubMed] [Google Scholar]
- 10.Rassi AJ, Dias JCP, Marin-Neto JA, Rassi A. Challenges and opportunities for primary, secondary, and tertiary prevention of Chagas’ disease. Heart. 2009;95:524–534. doi: 10.1136/hrt.2008.159624. [DOI] [PubMed] [Google Scholar]
- 11.Urbina JA, Docampo R. Specific chemotherapy of Chagas disease: controversies and advances. Trends Parasitol. 2003;19:495–501. doi: 10.1016/j.pt.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 12.Moraes CB, Giardini MA, Kim H, Franco CH, Araujo-Junior AM, Schenkman S, Chatelain E, Freitas-Junior LH. Nitroheterocyclic compounds are more efficacious than CYP51 inhibitors against Trypanosoma cruzi: implications for Chagas disease drug discovery and development. Sci Rep. 2014;4:1–11. doi: 10.1038/srep04703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jäger T, Koch O, Flohé L, editors. Trypanosomatid diseases: molecular routes to drug discovery. Vol. 4. Wiley-Blackwell; Weinheim (Germany): 2013. [Google Scholar]
- 14.Verlinde CL, Hannaert V, Blonski C, Willson M, Périé JJ, Fothergill-Gilmore LA, Opperdoes FR, Gelb MH, Hol WG, Michels PAM. Glycolysis as a target for the design of new anti-trypanosome drugs. Drug Resist Updates. 2001;4:50–65. doi: 10.1054/drup.2000.0177. [DOI] [PubMed] [Google Scholar]
- 15.Coley AF, Dodson HC, Morris MT, Morris JC. Glycolysis in the African trypanosome: targeting enzymes and their subcellular compartments for therapeutic development. Mol Biol Int. 2011;2011:1–10. doi: 10.4061/2011/123702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cáceres AJ, Portillo R, Acosta H, Rosales D, Quiñones W, Avilán L, Salazar L, Dubourdieu M, Michels PAM, Concepción JL. Molecular and biochemical characterization of hexokinase from Trypanosoma cruzi. Mol Biochem Parasitol. 2003;126:251–262. doi: 10.1016/s0166-6851(02)00294-3. [DOI] [PubMed] [Google Scholar]
- 17.Hudock MP, Sanz-Rodríguez CE, Song Y, Chan JMW, Zhang Y, Odeh S, Kosztowski T, Leon-Rossell A, Concepción JL, Yardley V, Croft SL, Urbina JA, Oldfield E. Inhibition of Trypanosoma cruzi hexokinase by bisphosphonates. J Med Chem. 2006;49:215–223. doi: 10.1021/jm0582625. [DOI] [PubMed] [Google Scholar]
- 18.Sanz-Rodríguez CE, Concepción JL, Pekerar S, Oldfield E, Urbina JA. Bisphosphonates as inhibitors of Trypanosoma cruzi hexokinase: kinetic and metabolic studies. J Biol Chem. 2007;282:12377–12387. doi: 10.1074/jbc.M607286200. [DOI] [PubMed] [Google Scholar]
- 19.Chambers JW, Fowler ML, Morris MT, Morris JC. The anti-trypanosomal agent lonidamine inhibits Trypanosoma brucei hexokinase 1. Mol Biochem Parasitol. 2008;158:202–207. doi: 10.1016/j.molbiopara.2007.12.013. [DOI] [PubMed] [Google Scholar]
- 20.Dodson HC, Lyda TA, Chambers JW, Morris MT, Christensen KA, Morris JC. Quercetin, a fluorescent bioflavanoid, inhibits Trypanosoma brucei hexokinase 1. Exp Parasitol. 2011;127:423–428. doi: 10.1016/j.exppara.2010.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sharlow ER, Lyda TA, Dodson HC, Mustata G, Morris MT, Leimgruber SS, Lee KH, Kashiwada Y, Close D, Lazo JS, Morris JC. A target-based high throughput screen yields Trypanosoma brucei hexokinase small molecule inhibitors with antiparasitic activity. PLoS Neglected Trop Dis. 2010;4:e659. doi: 10.1371/journal.pntd.0000659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Willson M, Sanejouand YH, Perie J, Hannaert V, Opperdoes F. Sequencing, modeling, and selective inhibition of Trypanosoma brucei hexokinase. Chem Biol. 2002;9:839–847. doi: 10.1016/s1074-5521(02)00169-2. [DOI] [PubMed] [Google Scholar]
- 23.Bringaud F, Rivière L, Coustou V. Energy metabolism of trypanosomatids: adaptation to available carbon sources. Mol Biochem Parasitol. 2006;149:1–9. doi: 10.1016/j.molbiopara.2006.03.017. [DOI] [PubMed] [Google Scholar]
- 24.Albert MA, Haanstra JR, Hannaert V, Van Roy J, Opperdoes FR, Bakker BM, Michels PAM. Experimental and in silico analyses of glycolytic flux control in bloodstream form Trypanosoma brucei. J Biol Chem. 2005;280:28306–28315. doi: 10.1074/jbc.M502403200. [DOI] [PubMed] [Google Scholar]
- 25.Cazzulo JJ. Aerobic fermentation of glucose by trypanosomatids. FASEB J. 1992;6:3153–3161. doi: 10.1096/fasebj.6.13.1397837. [DOI] [PubMed] [Google Scholar]
- 26.Engel JC, Franke de Cazzulo BM, Stoppani AOM, Cannata JJB, Cazzulo JJ. Aerobic glucose fermentation by Trypanosoma cruzi axenic culture amastigote-like forms during growth and differentiation to epimastigotes. Mol Biochem Parasitol. 1987;26:1–10. doi: 10.1016/0166-6851(87)90123-x. [DOI] [PubMed] [Google Scholar]
- 27.Tielens AGM, Van Hellemond JJ. Differences in energy metabolism between trypanosomatidae. Parasitol Today. 1998;14:265–271. doi: 10.1016/s0169-4758(98)01263-0. [DOI] [PubMed] [Google Scholar]
- 28.Cáceres AJ, Quiñones W, Gualdrón M, Cordeiro A, Avilán L, Michels PAM, Concepción JL. Molecular and biochemical characterization of novel glucokinases from Trypanosoma cruzi and Leishmania spp. Mol Biochem Parasitol. 2007;156:235–245. doi: 10.1016/j.molbiopara.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 29.Barnes RL, Shi H, Kolev NG, Tschudi C, Ullu E. Comparative genomics reveals two novel RNAi factors in Trypanosoma brucei and provides insight into the core machinery. PLoS Pathog. 2012;8:e1002678. doi: 10.1371/journal.ppat.1002678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.DaRocha WD, Otsu K, Teixeira SMR, Donelson JE. Tests of cytoplasmic RNA interference (RNAi) and construction of a tetracycline-inducible T7 promoter system in Trypanosoma cruzi. Mol Biochem Parasitol. 2004;133:175–186. doi: 10.1016/j.molbiopara.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 31.Cordeiro AT, Cáceres AJ, Vertommen D, Concepción JL, Michels PAM, Versées W. The crystal structure of Trypanosoma cruzi glucokinase reveals features determining oligomerization and anomer specificity of hexose-phosphorylating enzymes. J Mol Biol. 2007;372:1215–1226. doi: 10.1016/j.jmb.2007.07.021. [DOI] [PubMed] [Google Scholar]
- 32.Kawai S, Mukai T, Mori S, Mikami B, Murata K. Hypothesis: structures, evolution, and ancestor of glucose kinases in the hexokinase family. J Biosci Bioeng. 2005;99:320–330. doi: 10.1263/jbb.99.320. [DOI] [PubMed] [Google Scholar]
- 33.Lunin VV, Li Y, Schrag JD, Iannuzzi P, Cygler M, Matte A. Crystal structures of Escherichia coli ATP-dependent glucokinase and its complex with glucose. J Bacteriol. 2004;186:6915–6927. doi: 10.1128/JB.186.20.6915-6927.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Petit P, Antoine M, Ferry G, Boutin JA, Lagarde A, Gluais L, Vincentelli R, Vuillard L. The active conformation of human gluokinase is not altered by allosteric activators. Acta Crystallogr. 2011;D67:929–935. doi: 10.1107/S0907444911036729. [DOI] [PubMed] [Google Scholar]
- 35.Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. 2004;D60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 36.Frishman D, Argos P. Knowledge-based protein secondary structure assignment. Proteins: Struct Funct Genet. 1995;23:566–579. doi: 10.1002/prot.340230412. [DOI] [PubMed] [Google Scholar]
- 37.Heinig M, Frishman D. STRIDE: a web server for secondary structure assignment from known atomic coordinates of proteins. Nucleic Acids Res. 2004;32:W500–W502. doi: 10.1093/nar/gkh429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Robert X, Gouet P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014;42:W320–W324. doi: 10.1093/nar/gku316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Anderson CM, Stenkamp RE, Steitz TA. Sequencing a protein by X-ray crystallography: II. Refinement of yeast hexokinase B co-ordinates and sequence at 2.1 Å resolution. J Mol Biol. 1978;123:15–33. doi: 10.1016/0022-2836(78)90374-1. [DOI] [PubMed] [Google Scholar]
- 40.Steitz TA, Anderson WF, Fletterick RJ, Anderson CM. High resolution crystal structures of yeast hexokinase complexes with substrates, activators, and inhibitors. J Biol Chem. 1977;252:4494–4500. [PubMed] [Google Scholar]
- 41.Willson M, Alric I, Perie J, Sanejouand Y-H. Yeast hexokinase inhibitors designed from the 3-D enzyme structure rebuilding. J Enzyme Inhib. 1997;12:101–121. doi: 10.3109/14756369709035812. [DOI] [PubMed] [Google Scholar]
- 42.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 43.DeLano WL. MacPyMOL: A PyMOL-based molecular graphics application for MacOS X. Delano Scientific LLC; Palo Alto, California: 2007. [Google Scholar]
- 44.Hayward S, Berendsen HJC. Systematic analysis of domain motions in proteins from conformational change: new results on citrate synthase and T4 lysozyme. Proteins: Struct Funct, Genet. 1998;30:144–154. [PubMed] [Google Scholar]
- 45.Romero CS, Olmo R, Teijón C, Blanco MD, Teijón JM, Romero A. Structural and functional implications of the hexokinase-nickel interaction. J Inorg Biochem. 2005;99:2395–2402. doi: 10.1016/j.jinorgbio.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 46.Xu LZ, Zhang W, Weber IT, Harrison RW, Pilkis SJ. Site-directed mutagenesis studies on the determinants of sugar specificity and cooperative behavior of human β-cell glucokinase. J Biol Chem. 1994;269:27458–27465. [PubMed] [Google Scholar]
- 47.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Delivery Rev. 1997;23:3–25. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 48.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Delivery Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 49.Luzina EL, Popov AV. Synthesis, evaluation of anticancer activity and COMPARE analysis of N-bis(trifluoromethyl)alkyl-N′-substituted ureas with pharmacophoric moieties. Eur J Med Chem. 2012;53:364–373. doi: 10.1016/j.ejmech.2012.03.026. [DOI] [PubMed] [Google Scholar]
- 50.Alonso-Padilla J, Cotillo I, Presa JL, Cantizani J, Peña I, Bardera AI, Martín JJ, Rodriguez A. Automated high-content assay for compounds selectively toxic to Trypanosoma cruzi in a myoblastic cell line. PLoS Neglected Trop Dis. 2015:1–17. doi: 10.1371/journal.pntd.0003493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Buckner FS, Verlinde CL, La Flamme AC, Van Voorhis WC. Efficient technique for screening drugs for activity against Trypanosoma cruzi using parasites expressing β-galactosidase. Antimicrob Agents Chemother. 1996;40:2592–2597. doi: 10.1128/aac.40.11.2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Andriani G, Chessler ADC, Courtemanche G, Burleigh BA, Rodriguez A. Activity in vivo of anti-Trypanosoma cruzi compounds selected from a high throughput screening. PLoS Neglected Trop Dis. 2011;5:e1298. doi: 10.1371/journal.pntd.0001298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tetaud E, Chabas S, Giroud C, Barrett MP, Baltz T. Hexose uptake in Trypanosoma cruzi: structure-activity relationship between substrate and transporter. Biochem J. 1996;317:353–359. doi: 10.1042/bj3170353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hinklin RJ, Boyd SA, Chicarelli MJ, Condroski KR, DeWolf WEJ, Lee PA, Lee W, Singh A, Thomas L, Voegtli WC, Williams L, Aicher TD. Identification of a new class of glucokinase activators through structure-based design. J Med Chem. 2013;56:7669–7678. doi: 10.1021/jm401116k. [DOI] [PubMed] [Google Scholar]
- 55.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD, Higgins DG. Fast, scalable generation of high-quality protein multiple sequence alignments using clustal omega. Mol Syst Biol. 2011;7:1–6. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jones DT. Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol. 1999;292:195–202. doi: 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
- 57.Bustamante C, Folarin OA, Gbotosho GO, Batista CN, Mesquita EA, Brindeiro RM, Tanuri A, Struchiner CJ, Sowunmi A, Oduola A, Wirth DF, Zalis MG, Happi CT. In vitro–reduced susceptibility to artemether in P. falciparum and its association with polymorphisms on transporter genes. J Infect Dis. 2012;206:324–332. doi: 10.1093/infdis/jis359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chaijaroenkul W, Wisedpanichkij R, Na-Bangchang K. Monitoring of in vitro susceptibilities and molecular markers of resistance of Plasmodium falciparum isolates from Thai-Myanmar border to chloroquine, quinine, mefloquine and artesunate. Acta Trop. 2010;113:190–194. doi: 10.1016/j.actatropica.2009.10.016. [DOI] [PubMed] [Google Scholar]
- 59.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 60.Urzhumtseva L, Afonine PV, Adams PD, Urzhumtsev A. Crystallographic model quality at a glance. Acta Crystallogr. 2009;D65:297–300. doi: 10.1107/S0907444908044296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kleywegt GJ, Zou JY, Kjeldgaard M, Jones TA, Around O. In: International Tables of Crystallography. Rossman MG, Arnold E, editors. Kluwer Academic Publishers; Dordrecht, The Netherlands: 2001. pp. 353–356.pp. 366–367. [Google Scholar]
- 62.Collaborative Computational Project n. The CCP4 suite: programs for protein crystallography. Acta Crystallogr Sect D: Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 63.Abad-Zapatero C. Ligand efficiency indices for effective drug discovery. Expert Opin Drug Discov. 2007;2:469–488. doi: 10.1517/17460441.2.4.469. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.