

Abstract
The cJun NH2-terminal kinase (JNK) signaling pathway is implicated in metabolic syndrome, including dysregulated blood glucose concentration and insulin resistance. Fibroblast growth factor 21 (FGF21) is a target of the hepatic JNK signaling pathway and may contribute to the regulation of glycemia. To test the role of FGF21, we established mice with selective ablation of the Fgf21 gene in hepatocytes. FGF21-deficiency in the liver caused marked loss of FGF21 protein circulating in the blood. Moreover, the protective effects of hepatic JNK-deficiency to suppress metabolic syndrome in high fat diet-fed mice were not observed in mice with hepatocyte-specific FGF21-deficiency, including reduced blood glucose concentration and reduced intolerance to glucose and insulin. Furthermore, we show that JNK contributes to the regulation of hepatic FGF21 expression during fasting /feeding cycles. These data demonstrate that the hepatokine FGF21 is a key mediator of JNK-regulated metabolic syndrome.
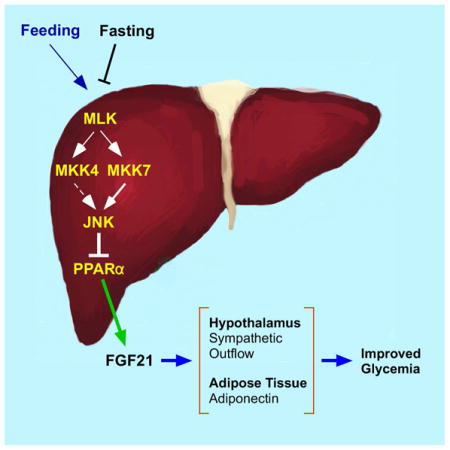
Graphical abstract
INTRODUCTION
The cJun NH2-terminal kinase (JNK) signaling pathway is activated by metabolic stress and contributes to the development of metabolic syndrome in response to the consumption of a high fat diet (HFD), including obesity, hyperglycemia, and insulin resistance (Sabio and Davis, 2010). JNK plays different roles in multiple tissues to cause these phenotypes. Thus, JNK in the hypothalamus and pituitary gland suppress energy expenditure to cause obesity (Belgardt et al., 2010; Sabio et al., 2010a; Vernia et al., 2013) and JNK in macrophages promotes chronic inflammation to cause insulin resistance (Han et al., 2013). JNK in peripheral tissues also contributes to the development of insulin resistance, including adipose tissue, liver, and muscle (Sabio et al., 2008; Sabio and Davis, 2010; Sabio et al., 2010b). Interestingly, hepatic JNK-deficiency causes systemic protection against insulin resistance in HFD-fed mice (Vernia et al., 2014). The mechanism of systemic protection may be caused by JNK-mediated repression of the peroxisome proliferator-activated receptor α (PPARα) - fibroblast growth factor 21 (FGF21) signaling axis (Vernia et al., 2014).
FGF21 circulates in the blood and is a potent regulator of metabolism (Owen et al., 2015). The effects of FGF21 are mediated by binding to FGF receptors (FGFR1c, 2c, or 3c) together with the obligate co-receptor βKlotho (Owen et al., 2015). Clinical trials using FGF21 analogs demonstrate improved dyslipidemia in humans (Gaich et al., 2013; Dong et al., 2015). Murine studies demonstrate that the liver is a target of the metabolic actions of FGF21 by regulating fatty acid oxidation, ketogenesis, gluconeogenesis, and lipogenesis (Potthoff et al., 2012). Indeed, FGF21-deficiency is associated with increased hepatic steatosis, fibrosis, and inflammation (Fisher et al., 2014; Tanaka et al., 2015). Direct effects of FGF21 on the liver may contribute to these hepatic responses, but recent studies indicate that FGF21 primarily mediates these effects by indirect mechanisms that initally target other tissues, including the central nervous system and adipose tissue (Owen et al., 2015). The major hepatic action of FGF21 mediated by adipose tissue is the PPARγ-induced expression of the adipokine adiponectin (Holland et al., 2013; Lin et al., 2013). FGF21 also acts on the hypothalamus-pituitary axis to regulate adrenal glucocorticoid secretion during starvation-induced hepatic gluconeogenesis (Liang et al., 2014). Moreover, FGF21 acts on the hypothalamus within the suprachiasmatic nucleus (Owen et al., 2014) and the paraventricular nucleus (Douris et al., 2015) to increase sympathetic outflow to peripheral tissues, including the liver. This sympathetic activity also targets brown fat and beige/brite cells in subcutaneous white fat depots (Douris et al., 2015) and may contribute to the blood glucose lowering actions of FGF21 by causing increased UCP1-dependent energy expenditure (Kwon et al., 2015).
The purpose of the study reported here was to test the role of FGF21 in the metabolic response to hepatic JNK activation. We show that hepatic JNK-deficiency in HFD-fed mice causes reduced hyperglycemia and improved tolerance to glucose and insulin. These effects of JNK-deficiency were not detected in mice with hepatocyte-specific ablation of the Fgf21 gene. Collectively, these data demonstrate that FGF21 is required for glycemic regulation by hepatic JNK.
RESULTS AND DISCUSSION
JNK signaling suppresses Fgf21 gene expression
Studies of mice with compound ablation of the Mapk8 plus Mapk9 genes (also known as Jnk1 and Jnk2) in the liver demonstrate increased expression of PPARα target genes, including Fgf21 (Vernia et al., 2014). The JNK signaling pathway can therefore repress PPARα signaling. To obtain additional evidence to support this conclusion, we examined the effect of a specific small molecule inhibitor of JNK protein kinase activity (JNK-in-8; (Zhang et al., 2012)) on PPARα-dependent gene expression, including Acox1, Ehhadh, and Fgf21. We found that treatment of wild-type hepatocytes, but not Ppara−/− hepatocytes, with JNK-in-8 caused increased PPARα-dependent gene expression in response to the PPARα agonist WY14043 (Figure 1A). These data support the conclusion that JNK signaling represses PPARα target gene expression, including Fgf21. This PPARα pathway is important for starvation-induced FGF21 expression (Badman et al., 2007; Inagaki et al., 2007), while additional pathways (e.g. ATF4, ChREBP, GCN2, NRF2, and SIRT1) play key roles in FGF21 expression caused by other dietary stresses, including low protein and high carbohydrate diets (Iizuka et al., 2009; Chartoumpekis et al., 2011; De Sousa-Coelho et al., 2012; Li et al., 2014).
Figure 1. Inhibition of the hepatic JNK signaling pathway promotes FGF21 expression.

(A) Primary hepatocytes were treated without or with JNK inhibitor (JNK-in-8) or the PPARα agonist WY14043 (Fibrate). The expression of the PPARα target genes Acox1, Ehhadh, and Fgf21 mRNA was examined by measurement of mRNA using quantitative PCR (mean ± SEM; n=6). PPARα-deficiency suppressed gene expression, including in cells treated with fibrate plus JNK-in-8 (***, p<0.001).
(B,C) Primary hepatocytes prepared from control mice and mice with liver-specific deficiency of MKK4, MKK7, or JNK1/2 were prepared. Genomic DNA was examined by PCR to detect gene ablations (B). Primary hepatocytes treated without or with TNFα were examined by immunoblot analysis (C).
(D) The blood concentration of FGF21 in wild-type and MLK2 plus MLK3-deficient (Map3k10−/ − Map3k11−/ −) mice fed ad-libitum was measured by ELISA (mean ± SEM; n=6; *, p<0.05).
(E) Expression of Acox1, Ehhadh, and Fgf21 mRNA by primary hepatocytes prepared from control mice and mice with liver-specific deficiency of MKK4 or MKK7 was examined by quantitative PCR (mean ± SEM; n=6; *, p<0.05; **, p<0.01; ***, p<0.001).
To obtain independent evidence for repression of Fgf21 expression by the JNK signaling pathway, we examined the effect of defects in the upstream regulatory mechanism that activates JNK. It is established that JNK activation is mediated by the protein kinases MKK4 and MKK7 (Tournier et al., 2001). Disruption of the Map2k4 gene or the Map2k7 gene in hepatocytes partially suppressed JNK signaling, indicated by reduced phosphorylation of JNK and cJun (Figure 1B,C). In contrast, compound ablation of Map2k4 plus Map2k7 (like compound ablation of Mapk8 plus Mapk9) prevented JNK signaling in hepatocytes (Figure 1C). Analysis of Fgf21 mRNA expression demonstrated that both MKK4-deficiency and MKK7-deficiency caused increased Fgf21 expression, but compound MKK4 plus MKK7-deficiency strongly increased Fgf21 expression (Figure 1E). Similarly, compound deficiency of the mixed-lineage protein kinases MLK2 plus MLK3, that function as activators of MKK4 and MKK7 in response to metabolic stress (Kant et al., 2013), causes increased expression of FGF21 circulating in the blood (Figure 1D). Together, these data demonstrate that the JNK signaling pathway acts to suppress FGF21 expression.
To test the role of JNK signaling during physiological regulation of FGF21 expression, we investigated the effect of fasting and re-feeding mice on hepatic FGF21 expression and JNK activity. It is established that FGF21 expression is strongly induced by starvation and that re-feeding rapidly represses FGF21 expression (Potthoff et al., 2012). Previous studies have demonstrated that hepatic JNK is activated by over-nutrition when mice are fed a HFD (Sabio and Davis, 2010). However, it was not known whether hepatic JNK is acutely regulated by fasting and re-feeding. We therefore examined JNK activation in mice fed a chow diet (CD) ad-libitum, mice fed a CD and then starved overnight, and mice that were starved overnight and subsequently re-fed a CD (1 h). This analysis demonstrated the presence of activated hepatic JNK in fed mice, a reduction in JNK activation in starved mice, and rapid re-activation of JNK in re-fed mice (Figure 2A). The hepatic JNK pathway is therefore acutely regulated by fasting/feeding cycles. These changes in JNK pathway activation appear to reflect the activation state of MKK7 rather than MKK4 (Figure 2A). Interestingly, JNK activation and FGF21 expression are anti-correlated; feeding-induced activation of JNK is associated with down-regulation of FGF21 expression (Figure 2B,C). These data indicate that JNK-mediated repression may contribute to the physiological regulation of FGF21 during cycles of fasting and feeding.
Figure 2. Hepatic JNK regulates FGF21 levels in response to fasting.

(A) Mice were fed a chow diet ad-libitum, fasted overnight, or fasted overnight and then re-refed a chow diet (1 h). Liver extracts were prepared and examined by immunoblot analysis (left panel). The amount of phospho-JNK and phospho-cJun (right panels) was quantitated (mean ± SEM; n=3; *, p<0.05; **, p<0.01).
(B,C) The amount of hepatic Fgf21 mRNA (B) was measured by quantitative PCR (mean ± SEM; n=6~10). The blood concentration of FGF21 (C) was measured by ELISA (mean ± SEM; n=10~12). Statistically significant differences between LWT and LΔJ1,J2 mice are indicated (**, p<0.01).
Conditional ablation of the Fgf21 gene in mice
We established Fgf21Loxp/LoxP mice to examine the effect of tissue-specific ablation of the Fgf21 gene (Figure S1A,B). To test the role of hepatic FGF21 expression, we obtained LΔFgf21 mice (Alb- cre+ Fgf21LoxP/LoxP) with Fgf21 gene ablation selectively in hepatocytes. Analysis of LΔFgf21 mice demonstrated no hepatic expression of Fgf21 mRNA or FGF21 protein circulating in blood (Figure S1C–E). In contrast, FΔFgf21 mice (Adipoq-cre+ Fgf21LoxP/LoxP mice) with Fgf21 gene ablation selectively in adipose tissue demonstrated the presence of FGF21 protein in blood despite the complete loss of Fgf21 mRNA expression in adipose tissue (Figure S1C–E). These observations confirm that the liver is the major source of FGF21 circulating in blood (Markan et al., 2014).
Hepatic FGF21-deficiency promotes metabolic syndrome
We examined the effect of feeding a HFD to Control mice and mice with FGF21-deficiency in liver (LΔFgf21) or adipose tissue (FΔFgf21). Measurement of body mass demonstrated increased weight gain by LΔFgf21 mice, but not FΔFgf21 mice, compared with Control mice (Figure 3A & S2A). Analysis of total body composition by 1H-MRS demonstrated that the increased body mass was caused by accumulated fat mass rather than increased lean mass (Figure 3B). Examination of organ mass at necropsy indicated that the majority of the increased weight gain was associated with increased mass of the liver, white adipose tissue, and brown adipose tissue (Figure S2B). Sections prepared from the liver demonstrated that LΔFgf21 mice exhibited increased hepatic steatosis compared with FΔFgf21 mice and Control mice (Figure 3D). Similarly, analysis of sections prepared from brown and white adipose tissues demonstrated increased adipocyte hypertrophy in LΔFgf21 mice compared with FΔFgf21 mice and Control mice (Figure 3E,F). The increased obesity of LΔFgf21 mice was associated with increased HFD-induced hyperinsulinemia and circulating concentrations of the adipokines leptin and resistin (Figure S2C–E). This effect of hepatic FGF21-deficiency to promote obesity is similar to observations reported for whole body FGF21 knockout mice (Badman et al., 2009; Hotta et al., 2009).
Figure 3. FGF21-deficiency in liver increases HFD-induced obesity.

(A) The total body mass gain of CD-fed and HFD-fed mice was examined (mean ± SEM; n = 6~10; *, p<0.05; **, p<0.01). FGF21-deficiency in liver or fat was studied by comparing Control mice (Fgf21LoxP/LoxP) with liver-specific FGF21-deficient mice (LΔFgf21) and adipose tissue-specific FGF21- deficient mice (FΔFgf21).
(B) The fat and lean mass of CD-fed and HFD-fed (12 wks) mice was measured by 1H-MRS analysis (mean ± SEM; n = 6~10; *, p<0.05; **, p<0.01).
(C) The blood glucose concentration in CD-fed and HFD-fed (12 wks) mice starved overnight was examined (mean ± SEM; n = 6~10; *, p<0.05; **, p<0.01).
(D–F) Sections of liver, epididymal white adipose tissue (WAT), and interscapular brown adipose tissue (BAT) from CD-fed and HFD-fed (12 wks) mice were stained with hematoxylin & eosin. Bar, 100 μm.
(G–I) Glucose tolerance, insulin tolerance, and pyruvate tolerance tests using HFD-fed (12 wks) mice were performed (mean ± SEM; n = 6~10; *, p<0.05; **, p<0.01). See also Figures S1, S2 & S3.
The phenotype of LΔFgf21 mice suggests that hepatic FGF21-deficiency may promote metabolic syndrome. We therefore examined glycemic regulation in LΔFgf21 mice, FΔFgf21 mice, and Control mice. No significant differences in blood glucose concentration were detected in CD-fed mice. Similarly, studies of glucose, insulin, and pyruvate tolerance of CD-fed mice demonstrated no differences between Control mice, LΔFgf21 mice, and FΔFgf21 mice (Figure S3A–C). In contrast, the blood glucose concentration was increased in HFD-fed LΔFgf21 mice compared with HFD-fed FΔFgf21 mice and HFD-fed Control mice (Figure 3C). Furthermore, HFD-fed LΔFgf21 mice were significantly more glucose intolerant (Figure 3G), more insulin resistant (Figure 3H), and more pyruvate intolerant (Figure 3I) than HFD-fed Control mice or FΔFgf21 mice.
Collectively, these data demonstrate that hepatocyte-specific FGF21-deficiency dramatically suppresses the amount of FGF21 that circulates in the blood. Moreover, hepatocyte-specific FGF21-deficiency promotes obesity and hallmarks of metabolic syndrome in HFD-fed mice, including increased hyperglycemia and hyperinsulinemia together with intolerance to glucose, pyruvate, and insulin.
Fgf21 gene ablation suppresses the metabolic effects of hepatic JNK-deficiency
Hepatic JNK-deficiency causes increased expression of PPARα target genes in the liver, including Fgf21 (Vernia et al., 2014). To test the role of FGF21, we examined the effect of compound deficiency of JNK plus FGF21 in hepatocytes by comparing LΔJ1,J2 mice (Alb-cre+ Mapk8LoxP/LoxP Mapk9LoxP/LoxP) with LΔJ1,J2,Fgf21 mice (Alb-cre+ Mapk8LoxP/LoxP Mapk9LoxP/LoxP Fgf21LoxP/LoxP). No significant differences in total body mass, fat mass, or lean mass between LΔJ1,J2 mice, LΔJ1,J2,Fgf21 mice, and Control mice were detected (Figure 4A,B). However, the HFD-induced hyperglycemia in Control mice that was suppressed by JNK-deficiency (compare Control and LΔJ1,J2 mice; Figure 4C) was not detected in FGF21-deficient mice (compare LΔFgf21 mice and LΔJ1,J2,Fgf21 mice; Figure 4C). These data indicate that FGF21 is essential for the effect of hepatic JNK-deficiency to suppress HFD-induced hyperglycemia.
Figure 4. FGF21 is critically required for the metabolic actions of hepatic JNK.

(A) The total body mass gain of CD-fed and HFD-fed mice was examined. The effect of FGF21-deficiency in liver was examined by comparing Control mice (Mapk8LoxP/LoxP Mapk9 LoxP/LoxP) and mice with liver-specific deficiency of JNK (LΔJ1,J2) or JNK plus FGF21 (LΔJ1,J2,Fgf21). No statistically significant differences were detected between groups (mean ± SEM; n = 8~10).
(B) The fat and lean mass of CD-fed and HFD-fed (12 wks) mice was measured by 1H-MRS analysis (mean ± SEM; n = 8~10).
(C) Blood glucose concentration was measured in overnight fasted CD-fed and HFD-fed (12 wks) mice (mean ± SEM; n = 6~10; *, p<0.05; ***, p<0.001).
(D) Glucose tolerance (GTT), insulin tolerance (ITT), and pyruvate tolerance (PTT) tests using HFD-fed (12 wks) mice were performed (mean ± SEM; n = 6~10; *, p<0.05; **, p<0.01; ***, p<0.001).
(E) The blood concentration of insulin, leptin, and resistin in overnight fasted CD-fed and HFD-fed (12 wks) mice was measured (mean ± SEM; n = 6~10; *, p<0.05; **, p<0.01).
(F) The expression of Acox1, Ehhadh, and Fgf21 mRNA in the liver of CD-fed and HFD-fed (12 wks) mice was measured by quantitative PCR (mean ± SEM; n=6; *, p<0.05; **, p<0.01; ***, p<0.001).
(G) Sections of liver from CD-fed and HFD-fed (12 wks) Control, LΔJ1,J2, and LΔJ1,J2,Fgf21 mice were stained with hematoxylin & eosin. Bar, 100 μm. See also Figures S3 and S4.
To examine the requirement of FGF21 for glycemic regulation, we performed glucose, insulin, and pyruvate tolerance tests. Studies of CD-fed mice demonstrated no significant differences between groups (Figure S3D–F), but JNK-deficiency in HFD-fed mice caused improved performance in each of these tests (Figure 4D). This effect of JNK-deficiency was not detected in FGF21-deficient mice (compare HFD-fed LΔFGF21 mice and HFD-fed LΔJ1,J2,FGF21 mice; Figure 4D). Moreover, hepatic JNK-deficiency did not suppress HFD-induced hyperinsulinemia or circulating concentrations of the adipokines leptin and resistin in liver-specific FGF21-deficient mice (Figure 4E). Together, these data confirm that FGF21 is required for improved glycemic regulation caused by hepatic JNK-deficiency.
Interestingly, the effect of hepatic JNK-deficiency to increase the expression of the PPARα target genes Acox1 and Ehhadh in HFD-fed mice was suppressed in hepatic FGF21-deficient mice (Figure 4F). This observation suggests that FGF21 plays a key role in the promotion of fatty acid oxidation caused by hepatic JNK-deficiency, consistent with the established physiological role of FGF21 (Badman et al., 2007). Indeed, the reduction in HFD-induced hepatic steatosis caused by JNK-deficiency was not detected in the liver of mice with hepatic FGF21-deficiency (Figure 4G). These data highlight the crucial role of FGF21 in hepatic JNK signal transduction.
One pathway that mediates the hepatic actions of FGF21 is represented by the adipokine adiponectin (Holland et al., 2013; Lin et al., 2013). We therefore examined the expression of adiponectin in Control, LΔFgf21, and FΔFgf21 mice; these data demonstrated that hepatic FGF21-deficiency, but not adipocyte FGF21-deficiency, caused reduced expression of Adipoq mRNA in epididymal white adipose tissue (Figure S4A). Moreover, studies of Control, LΔJ1,J2, and LΔJ1,J2,Fgf21 mice indicated that hepatic JNK-deficiency caused increased adiponectin expression that was suppressed in mice with compound hepatic deficiency of JNK plus FGF21 (Figure S4B). Together, these data demonstrate that the increased circulating concentration of FGF21 caused by hepatic JNK-deficiency promotes adipose tissue expression of adiponectin. The FGF21-promoted adiponectin expression, together with FGF21-promoted sympathetic stimulation of brown and beige/brite adipose tissue, may contribute to the improved hepatic function of mice with hepatic JNK-deficiency.
In summary, the requirement of FGF21 for the metabolic actions of hepatic JNK provides a mechanism for metabolic regulation by the JNK signaling pathway in the liver. FGF21 plays a major role in the response to starvation, including increased hepatic gluconeogenesis, ketogenesis, and fatty acid oxidation (Potthoff et al., 2012). Feeding terminates this response by activation of the hepatic JNK signaling pathway and repression of FGF21 expression. JNK activation therefore provides a key regulatory mechanism that contributes to the termination of the starvation response. Conversely, starvation-induced inhibition of the JNK pathway contributes to de-repression of FGF21 expression and promotes adaptive responses to starvation. These data establish a key role played by FGF21 in the metabolic response to hepatic JNK activation caused by cycles of fasting and re-feeding.
The interplay between hepatic JNK and FGF21 contributes to the development of metabolic syndrome in response to diet-induced obesity (Vernia et al., 2014). Indeed, the protective effects of hepatic JNK-deficiency on insulin resistance were not observed in mice with hepatic FGF21-deficiency (Figure 4). This finding is similar to a report of autophagy-deficient mice that exhibit improved metabolic syndrome that was also dependent on increased FGF21 expression (Kim et al., 2013). Similarly, FGF21 improves hyperglycemia in mice with hepatic-deficiency of the insulin receptor (Emanuelli et al., 2014). FGF21 can also provide protection against some forms of drug-induced hepatotoxicity (Ye et al., 2014). Together, these data demonstrate that FGF21 can play strong protective roles in models of metabolic and cellular dysfunction.
EXPERIMENTAL PROCEDURES
Blood analysis
Blood glucose was measured with an Ascensia Breeze 2 glucometer (Bayer). FGF21 was measured using the Rat/Mouse Fibroblast Growth Factor 21 ELISA kit (EZRMFGF21-26K) (Millipore). Adipokines and insulin in plasma were measured by multiplexed ELISA using a Luminex 200 machine (Millipore).
Glucose and insulin tolerance tests
Glucose, insulin and pyruvate tolerance tests were performed by intraperitoneal injection of mice with glucose (1g/kg), insulin (0.5 U/kg) or pyruvate (1g/kg) using methods described previously (Sabio et al., 2008).
RNA analysis
Tissue isolated from mice starved overnight was used to isolate total RNA with the RNAeasy mini kit (Qiagen). Total RNA (500ng) was converted into cDNA using the high capacity cDNA reverse transcription kit (Life Technologies). The diluted cDNA was used for real-time quantitative PCR analysis using a Quantstudio PCR machine (Life Technologies). TaqMan® assays (Life Technologies) were used to quantify Acox1 (Mm01246831_m1), Adipoq (Mm00456425-m1), and Ehhadh (Mm00619685_m1). Fgf21 expression was quantified using the primers FGF21F67 (5’-AGATGGAGCTCTCTATGGATCG-3’) and FGF21R67 (5’-GGGCTTCAGACTGGTACACAT-3’) with universal probe #67 (Universal Probe Library, Roche). The relative mRNA expression was normalized by measurement of the amount of 18S RNA in each sample using Taqman© assays (catalog number 4308329; Life Technologies).
Immunoblot analysis
Liver extracts were prepared using Triton lysis buffer (20 mM Tris-pH 7.4, 1% Triton-X100, 10% glycerol, 137 mM NaCl, 2 mM EDTA, 25 mM β-glycerophosphate, 1 μM sodium orthovanadate, 1 μM PMSF and 10 μg/mL leupeptin plus aprotinin). Extracts (30–50 μg of protein) were examined by immunoblot analysis by probing with antibodies to pSer63-cJUN, pJNK, pMKK4, MKK4, pMKK7, and MKK7 (Cell Signaling Technologies), JNK1/2 (BD Pharmingen), cJUN and GAPDH (Santa Cruz) and β-Tubulin (Covance). Immunocomplexes were detected by fluorescence using anti-mouse and anti-rabbit secondary IRDye antibodies (Li-Cor) and quantitated using the Li-Cor Imaging system.
Analysis of tissue sections
Histology was performed using tissue fixed in 10% formalin (24 h), dehydrated, and embedded in paraffin. Sections (7 μm) were cut and stained using hematoxylin & eosin (American Master Tech Scientific). Sections isolated from 5 mice per group were examined by a board-certified pathologist who was blinded with respect to the identity of each group of mice.
Statistical analysis
Differences between groups were examined for statistical significance using the Student’s test or analysis of variance (ANOVA) with the Fisher’s test.
Supplementary Material
Highlights.
Hepatic JNK activity regulates FGF21 expression during fasting /feeding cycles
Hepatic JNK suppresses circulating FGF21 and promotes metabolic syndrome
Hepatocyte FGF21 is required for the effects of hepatic JNK on metabolic syndrome
Acknowledgments
We thank Dr. David Garlick for the analysis of tissue sections, Kathy Gemme for administrative assistance. These studies were supported by a grant R01 DK107220 from the National Institutes of Health. RJD is an investigator of the Howard Hughes Medical Institute.
Footnotes
AUTHOR CONTRIBUTIONS
SV and RJD designed the study; JC-K and TB performed experiments; and SV, CT, and RJD analyzed data and wrote the paper.
Supplemental information includes Supplemental Experimental Procedures, Supplemental References, and 4 Supplemental Figures and can be found with this article online at http:
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Badman MK, Koester A, Flier JS, Kharitonenkov A, Maratos-Flier E. Fibroblast growth factor 21-deficient mice demonstrate impaired adaptation to ketosis. Endocrinology. 2009;150:4931–4940. doi: 10.1210/en.2009-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badman MK, Pissios P, Kennedy AR, Koukos G, Flier JS, Maratos-Flier E. Hepatic fibroblast growth factor 21 is regulated by PPARalpha and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab. 2007;5:426–437. doi: 10.1016/j.cmet.2007.05.002. [DOI] [PubMed] [Google Scholar]
- Belgardt BF, Mauer J, Wunderlich FT, Ernst MB, Pal M, Spohn G, Bronneke HS, Brodesser S, Hampel B, Schauss AC, et al. Hypothalamic and pituitary c-Jun N-terminal kinase 1 signaling coordinately regulates glucose metabolism. Proc Natl Acad Sci U S A. 2010;107:6028–6033. doi: 10.1073/pnas.1001796107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chartoumpekis DV, Ziros PG, Psyrogiannis AI, Papavassiliou AG, Kyriazopoulou VE, Sykiotis GP, Habeos IG. Nrf2 represses FGF21 during long-term high-fat diet-induced obesity in mice. Diabetes. 2011;60:2465–2473. doi: 10.2337/db11-0112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Sousa-Coelho AL, Marrero PF, Haro D. Activating transcription factor 4-dependent induction of FGF21 during amino acid deprivation. Biochem J. 2012;443:165–171. doi: 10.1042/BJ20111748. [DOI] [PubMed] [Google Scholar]
- Dong JQ, Rossulek M, Somayaji VR, Baltrukonis D, Liang Y, Hudson K, Hernandez-Illas M, Calle RA. Pharmacokinetics and pharmacodynamics of PF-05231023, a novel long-acting FGF21 mimetic, in a first-in-human study. Br J Clin Pharmacol. 2015 doi: 10.1111/bcp.12676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douris N, Stevanovic DM, Fisher FM, Cisu TI, Chee MJ, Nguyen NL, Zarebidaki E, Adams AC, Kharitonenkov A, Flier JS, et al. Central Fibroblast Growth Factor 21 Browns White Fat via Sympathetic Action in Male Mice. Endocrinology. 2015;156:2470–2481. doi: 10.1210/en.2014-2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emanuelli B, Vienberg SG, Smyth G, Cheng C, Stanford KI, Arumugam M, Michael MD, Adams AC, Kharitonenkov A, Kahn CR. Interplay between FGF21 and insulin action in the liver regulates metabolism. J Clin Invest. 2014;124:515–527. doi: 10.1172/JCI67353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher FM, Chui PC, Nasser IA, Popov Y, Cunniff JC, Lundasen T, Kharitonenkov A, Schuppan D, Flier JS, Maratos-Flier E. Fibroblast growth factor 21 limits lipotoxicity by promoting hepatic fatty acid activation in mice on methionine and choline-deficient diets. Gastroenterology. 2014;147:1073–1083. e1076. doi: 10.1053/j.gastro.2014.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaich G, Chien JY, Fu H, Glass LC, Deeg MA, Holland WL, Kharitonenkov A, Bumol T, Schilske HK, Moller DE. The effects of LY2405319, an FGF21 analog, in obese human subjects with type 2 diabetes. Cell Metab. 2013;18:333–340. doi: 10.1016/j.cmet.2013.08.005. [DOI] [PubMed] [Google Scholar]
- Han MS, Jung DY, Morel C, Lakhani SA, Kim JK, Flavell RA, Davis RJ. JNK expression by macrophages promotes obesity-induced insulin resistance and inflammation. Science. 2013;339:218–222. doi: 10.1126/science.1227568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland WL, Adams AC, Brozinick JT, Bui HH, Miyauchi Y, Kusminski CM, Bauer SM, Wade M, Singhal E, Cheng CC, et al. An FGF21-Adiponectin-Ceramide Axis Controls Energy Expenditure and Insulin Action in Mice. Cell Metab. 2013;17:790–797. doi: 10.1016/j.cmet.2013.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotta Y, Nakamura H, Konishi M, Murata Y, Takagi H, Matsumura S, Inoue K, Fushiki T, Itoh N. Fibroblast growth factor 21 regulates lipolysis in white adipose tissue but is not required for ketogenesis and triglyceride clearance in liver. Endocrinology. 2009;150:4625–4633. doi: 10.1210/en.2009-0119. [DOI] [PubMed] [Google Scholar]
- Iizuka K, Takeda J, Horikawa Y. Glucose induces FGF21 mRNA expression through ChREBP activation in rat hepatocytes. FEBS Lett. 2009;583:2882–2886. doi: 10.1016/j.febslet.2009.07.053. [DOI] [PubMed] [Google Scholar]
- Inagaki T, Dutchak P, Zhao G, Ding X, Gautron L, Parameswara V, Li Y, Goetz R, Mohammadi M, Esser V, et al. Endocrine regulation of the fasting response by PPARalpha-mediated induction of fibroblast growth factor 21. Cell Metab. 2007;5:415–425. doi: 10.1016/j.cmet.2007.05.003. [DOI] [PubMed] [Google Scholar]
- Kant S, Barrett T, Vertii A, Noh YH, Jung DY, Kim JK, Davis RJ. Role of the mixed-lineage protein kinase pathway in the metabolic stress response to obesity. Cell Rep. 2013;4:681–688. doi: 10.1016/j.celrep.2013.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KH, Jeong YT, Oh H, Kim SH, Cho JM, Kim YN, Kim SS, Kim do H, Hur KY, Kim HK, et al. Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat Med. 2013;19:83–92. doi: 10.1038/nm.3014. [DOI] [PubMed] [Google Scholar]
- Kwon MM, O'Dwyer SM, Baker RK, Covey SD, Kieffer TJ. FGF21-Mediated Improvements in Glucose Clearance Require Uncoupling Protein 1. Cell Rep. 2015;13:1521–1527. doi: 10.1016/j.celrep.2015.10.021. [DOI] [PubMed] [Google Scholar]
- Li Y, Wong K, Giles A, Jiang J, Lee JW, Adams AC, Kharitonenkov A, Yang Q, Gao B, Guarente L, et al. Hepatic SIRT1 attenuates hepatic steatosis and controls energy balance in mice by inducing fibroblast growth factor 21. Gastroenterology. 2014;146:539–549. e537. doi: 10.1053/j.gastro.2013.10.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Q, Zhong L, Zhang J, Wang Y, Bornstein SR, Triggle CR, Ding H, Lam KS, Xu A. FGF21 maintains glucose homeostasis by mediating the cross talk between liver and brain during prolonged fasting. Diabetes. 2014;63:4064–4075. doi: 10.2337/db14-0541. [DOI] [PubMed] [Google Scholar]
- Lin Z, Tian H, Lam KS, Lin S, Hoo RC, Konishi M, Itoh N, Wang Y, Bornstein SR, Xu A, et al. Adiponectin Mediates the Metabolic Effects of FGF21 on Glucose Homeostasis and Insulin Sensitivity in Mice. Cell Metab. 2013;17:779–789. doi: 10.1016/j.cmet.2013.04.005. [DOI] [PubMed] [Google Scholar]
- Markan KR, Naber MC, Ameka MK, Anderegg MD, Mangelsdorf DJ, Kliewer SA, Mohammadi M, Potthoff MJ. Circulating FGF21 is liver derived and enhances glucose uptake during refeeding and overfeeding. Diabetes. 2014;63:4057–4063. doi: 10.2337/db14-0595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen BM, Ding X, Morgan DA, Coate KC, Bookout AL, Rahmouni K, Kliewer SA, Mangelsdorf DJ. FGF21 acts centrally to induce sympathetic nerve activity, energy expenditure, and weight loss. Cell Metab. 2014;20:670–677. doi: 10.1016/j.cmet.2014.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen BM, Mangelsdorf DJ, Kliewer SA. Tissue-specific actions of the metabolic hormones FGF15/19 and FGF21. Trends Endocrinol Metab. 2015;26:22–29. doi: 10.1016/j.tem.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potthoff MJ, Kliewer SA, Mangelsdorf DJ. Endocrine fibroblast growth factors 15/19 and 21: from feast to famine. Genes Dev. 2012;26:312–324. doi: 10.1101/gad.184788.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabio G, Cavanagh-Kyros J, Barrett T, Jung DY, Ko HJ, Ong H, Morel C, Mora A, Reilly J, Kim JK, et al. Role of the hypothalamic-pituitary-thyroid axis in metabolic regulation by JNK1. Genes Dev. 2010a;24:256–264. doi: 10.1101/gad.1878510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabio G, Das M, Mora A, Zhang Z, Jun JY, Ko HJ, Barrett T, Kim JK, Davis RJ. A stress signaling pathway in adipose tissue regulates hepatic insulin resistance. Science. 2008;322:1539–1543. doi: 10.1126/science.1160794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabio G, Davis RJ. cJun NH2-terminal kinase 1 (JNK1): roles in metabolic regulation of insulin resistance. Trends Biochem Sci. 2010;35:490–496. doi: 10.1016/j.tibs.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabio G, Kennedy NJ, Cavanagh-Kyros J, Jung DY, Ko HJ, Ong H, Barrett T, Kim JK, Davis RJ. Role of muscle c-Jun NH2-terminal kinase 1 in obesity-induced insulin resistance. Mol Cell Biol. 2010b;30:106–115. doi: 10.1128/MCB.01162-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka N, Takahashi S, Zhang Y, Krausz KW, Smith PB, Patterson AD, Gonzalez FJ. Role of fibroblast growth factor 21 in the early stage of NASH induced by methionine- and choline-deficient diet. Biochim Biophys Acta. 2015;1852:1242–1252. doi: 10.1016/j.bbadis.2015.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tournier C, Dong C, Turner TK, Jones SN, Flavell RA, Davis RJ. MKK7 is an essential component of the JNK signal transduction pathway activated by proinflammatory cytokines. Genes Dev. 2001;15:1419–1426. doi: 10.1101/gad.888501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernia S, Cavanagh-Kyros J, Barrett T, Jung DY, Kim JK, Davis RJ. Diet-induced obesity mediated by the JNK/DIO2 signal transduction pathway. Genes Dev. 2013;27:2345–2355. doi: 10.1101/gad.223800.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernia S, Cavanagh-Kyros J, Garcia-Haro L, Sabio G, Barrett T, Jung DY, Kim JK, Xu J, Shulha HP, Garber M, et al. The PPARalpha-FGF21 hormone axis contributes to metabolic regulation by the hepatic JNK signaling pathway. Cell Metab. 2014;20:512–525. doi: 10.1016/j.cmet.2014.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye D, Wang Y, Li H, Jia W, Man K, Lo CM, Wang Y, Lam KS, Xu A. Fibroblast growth factor 21 protects against acetaminophen-induced hepatotoxicity by potentiating peroxisome proliferator-activated receptor coactivator protein-1alpha-mediated antioxidant capacity in mice. Hepatology. 2014;60:977–989. doi: 10.1002/hep.27060. [DOI] [PubMed] [Google Scholar]
- Zhang T, Inesta-Vaquera F, Niepel M, Zhang J, Ficarro SB, Machleidt T, Xie T, Marto JA, Kim N, Sim T, et al. Discovery of potent and selective covalent inhibitors of JNK. Chem Biol. 2012;19:140–154. doi: 10.1016/j.chembiol.2011.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.