

SUMMARY
The development of pancreatic cancer requires the acquisition of oncogenic Kras mutations and up-regulation of growth factor signaling, but the relationship between these is not well established. We here show that mutant Kras alters mitochondrial metabolism in pancreatic acinar cells, resulting in increased generation of mitochondrial reactive oxygen species (mROS). Mitochondrial ROS then drives the dedifferentiation of acinar cells to a duct-like progenitor phenotype and progression to PanIN. This is mediated via the ROS-receptive kinase Protein Kinase D1 and the transcription factors NF-κB1 and NF-κB2, which up-regulate expression of the epidermal growth factor, its ligands and their sheddase ADAM17. In vivo, intercepting Kras-mediated generation of mROS reduced the formation of pre-neoplastic lesions. Hence, our data provide insight into how oncogenic Kras interacts with growth factor signaling to induce the formation of pancreatic cancer.
Keywords: pancreatic cancer, PanIN, Kras, oxidative stress, mitochondria, growth factor signaling
Graphical Abstract
INTRODUCTION
Due to its late diagnosis and limited treatment options, pancreatic ductal adenocarcinoma (PDA) has one of the poorest prognoses among human neoplasms. A better understanding of key signaling mechanisms driving initiation and progression of this cancer may lead to both early diagnosis and intervention. An initial event in the development of pancreatic preneoplastic lesions is the acquisition of activating Kras mutations (Krasmut) such as KrasG12D or KrasG12V (Hruban et al., 2001; Jaffee et al., 2002). However, the presence of an activating Kras mutation in the pancreas can be occasionally detected in individuals with non-malignant disease and additional events are needed for the progression of Kras-caused lesions to pancreatic cancer. Increasing evidence suggests that Krasmut crosstalks with signaling cascades that activate wildtype Kras (KrasWT); and that the elevation of Kras activity to the pathological levels that are required for development of PDA is mediated by activation of both pathways (Ardito et al., 2012; Bardeesy et al., 2006; Huang et al., 2014; Ji et al., 2009; Sarkisian et al., 2007). Activation of mutant and wildtype Kras synergize and can converge at signaling pathways leading to activation of ERK1/2 or Notch (Ardito et al., 2012; Liou et al., 2015a).
Both inflammation and increased epidermal growth factor (EGFR) signaling have been demonstrated to be essential for KrasG12D-driven development of PDA (Guerra et al., 2007 Navas, 2012 #45; Liou et al., 2015b). While normal pancreatic acinar cells do not express significant levels of EGFR, overexpression of EGFR and its ligands has been shown to occur frequently in the early development process of PDA (Korc, 1998). However, the molecular mechanisms by which Krasmut cooperates with EGFR/KrasWT signaling pathways to initiate pancreatic cancer remain unclear.
Cancer cells have a need for high flux through glycolysis and the pentose phosphate pathway, and many tumors undergo metabolic changes associated with an increase in aerobic glycolysis, known as the Warburg effect (Vander Heiden et al., 2009). Additionally, recent work has established an equally important role for increased mitochondrial metabolism in tumor progression (Ahn and Metallo, 2015). A hallmark of the metabolic functional adaptations common to many cancers, including PDA, is an increase in reactive oxygen species (ROS) (Son et al., 2013). Oxidative stress has been shown to cooperate with mutant Kras to initiate and promote pancreatic carcinogenesis in the murine pancreas (Al Saati et al., 2013). Under pathophysiological conditions mROS can enhance cell proliferation (Shidara et al., 2005; Weinberg et al., 2010), down-regulate tumor suppressors such as p16 and SMAD4 (Mishra et al., 2014), and initiate mitochondrial and nuclear DNA mutations (Ishikawa et al., 2008). A key issue for cancer cells is to keep ROS at levels where they are beneficial for cell proliferation, but below the threshold that leads to induction of senescence or cell death (Liou and Storz, 2010). This is often mediated by an additional increase in antioxidant systems. For example, in Ras-driven neoplasia aberrantly increased ROS levels are accompanied by an Nrf2 (nuclear respiratory factor 2)-dependent antioxidant signature, and both together are required for Ras-induced tumorigenesis (DeNicola et al., 2011; Weinberg et al., 2010). Despite ROS dysregulation being central to cancer proliferation, as exemplified by Ras-mediated tumorigenesis, the mechanisms that drive ROS-dependent neoplasia remain little characterized.
We here demonstrate that oncogenic Kras mutations lead to an increase in mROS to drive the development of pancreatic cancer. This is facilitated by the ROS-activated transcription factors NF-κB1 and NF-κB2 which up-regulate expression of EGFR and its ligands. Thus, our data provide a functional link between oncogenic Kras mutations and induction of EGFR/KrasWT signaling.
RESULTS
Increased levels of oxidative stress in PanIN and regions of ADM are caused by mutant Kras
Approximately 95% of all PDA show acquisition of a Kras mutation (Hruban et al., 2001) and many of these tumors also have increased cellular oxidative stress levels (Kodydkova et al., 2013), as confirmed by staining for protein adducts formed from ROS-generated reactive lipids such as 4-hydroxy-2-nonenal (4HNE) (Fig. 1A). To investigate if activating Kras mutations can increase oxidative stress levels in pancreatic acinar cells, we stained normal pancreas and pancreata of mice expressing oncogenic Kras (KrasG12D mutant) in acinar cells (p48cre;LSL-KrasG12D) for 4HNE adducts (Fig. 1B). Expression of active Kras, as previously described, induced acinar-to-ductal metaplasia (labeled ADM in Figs. 1B2 and 1B5) as well as formation of PanIN1 lesions (indicated by asterisks in Figs. 1B3 and 1B6). We observed strong staining for 4HNE in both of these structures, suggesting increased oxidative stress levels (Fig. 1B and Supplemental Figure S1A). Moreover, IHC staining for 4HNE also was increased in acinar cells of mice expressing active Kras, when compared to acinar cells of control mice (Figs. 1B4 and 1B6, inserts, labeled AC). Taken together, our data indicate that expression of oncogenic Kras induces oxidative stress in acinar cells, which is even more increased when acinar cells undergo ADM or in PanIN. Interestingly, all abnormal structures (including regions of ADM, PanIN1 and PanIN2) in p48cre;LSL-KrasG12D mice were not only positive for 4HNE, but also showed increased expression of Nrf2 (Supplemental Figs. S1B and S1C), as previously described (DeNicola et al., 2011). This indicates that antioxidant systems are also upregulated, most likely to keep ROS at levels where they are pro-proliferative or pro-tumorigenic, but not toxic.
Figure 1. Oxidative stress levels are increased in pancreatic cells expressing oncogenic Kras.

A: Patient samples (adjacent normal and PDA) were analyzed by immunohistochemistry using anti-4HNE (4-Hydroxy-2-nonenal) as a marker for oxidative stress. Shown is a characteristic area of the pancreas. The bar indicates 50 μm. B: Pancreatic sections from control mice (n=5; shown are LSL-KrasG12D) or mice (n=5) expressing the LSL-KrasG12D allele under the p48 promoter (p48cre;LSL-KrasG12D) were analyzed by immunohistochemistry (1–3: H&E staining, 4–6: staining with anti-4HNE). Shown is a characteristic area of the pancreas. AC = acinar cells, ADM = region of acinar-to-ductal metaplasia and PanIN lesions. The asterisk indicates an area with PanIN1 lesions with increased oxidative stress. The insert in 6 demonstrates that acinar cells of p48cre;LSL-KrasG12D mice already have increased levels of intracellular oxidative stress. The bar indicates 50 μm. C: Primary mouse pancreatic acinar cells were isolated either from LSL-KrasG12D mice and adenovirally-infected with adeno-null or adeno-cre. Briefly, 48 hours after infection, the oxygen consumption rate (OCR) over time was measured as described in Materials & Methods. Shown is a representative experiment (out of 3 independent experiments) performed with triplicates for each condition. Oligomycin (O; 2 μM), FCCP (F; 1 μM), and rotenone (R; 0.5 μM) were added at indicated time points. Error bars represent variations between triplicates. * indicates statistical significance (p<0.05) as compared to the control (adeno-null). D, E: Primary mouse pancreatic acinar cells were isolated either from LSL-KrasG12D mice (D), or wildtype mice (E). Cells were then adenovirally-infected with adeno-null or adeno-cre (D), or lentivirally-infected with control virus or virus harboring oncogenic KrasG12V (E). 48 hours after infection, cells were labeled with H2DFFDA and generation of intracellular ROS (fluorescent DCF) was measured over a time period of 500 minutes. * indicates statistical significance (p<0.05) as compared to the control (adeno-null in D; control virus in E).
We next tested if expression of KrasG12D in acinar cells can alter the metabolic activity of mitochondria, a major source of intracellular oxidative stress. Therefore, we performed a mitochondrial stress test to determine key parameters (basal respiration, ATP turnover, proton leak and maximal respiration) by sequential use of oligomycin, FCCP and rotenone (Fig. 1C). Acinar cells from LSL-KrasG12D mice were isolated and expression of mutant Kras was initiated by infection with adeno-cre virus. At basal conditions, the oxygen consumption rate (OCR) of acinar cells expressing mutant Kras was identical to control cells. Treatment with oligomycin indicates the extent at which the cells are using mitochondria to generate ATP, and the remaining OCR defines the proton leak across the mitochondrial inner membrane. Acinar cells expressing mutant Kras showed an increase in the proton leak, indicating that mitochondria were less functional. Treatment with the protonophore FCCP showed that KrasG12D-expressing cells had a higher maximal OCR resulting in an approximately 8.5-fold increase in their respiratory reserve, the capacity of cells to generate ATP via oxidative phosphorylation in response for increasing demand for energy. Eventually, treatment with the complex-I inhibitor rotenone caused a similar suppression of OCR in both conditions.
The mitochondrial proton leak can be increased by the damage to inner membrane lipids and proteins caused by oxidative stress. Therefore, we next investigated if oncogenic Kras mutants can induce the generation of oxidative stress in acinar cells. To test this we isolated acinar cells from pancreata of LSL-KrasG12D mice and then treated these cells with either control adenovirus (adeno-null) or adenovirus that transduces cre recombinase (adeno-cre). Induction of KrasG12D expression in acinar cells by adeno-cre led to a significant increase in cellular ROS levels (Fig. 1D; controls in Supplemental Figs. S1D, S1E). We also infected acinar cells isolated from normal mouse pancreas with lentivirus harboring an oncogenic Kras mutant (KrasG12V) and obtained similar increase in ROS production (Fig. 1E; controls in Supplemental Figs. S1D, S1F). The accumulation of intracellular ROS with both experimental systems occurred with similar time kinetics.
Oncogenic Kras induces ADM and formation of duct-like structures through mitochondrial reactive oxygen species
Previously, 3D explant (organoid) culture models were established in which the transdifferentiation of primary pancreatic acinar cells to a duct-like phenotype can be induced in vitro by expression of oncogenic Kras (Liou et al., 2015a; Means et al., 2005). We utilized this model system to investigate if Kras-caused oxidative stress can contribute to ADM. Therefore, we infected mouse primary pancreatic acinar cells with lentivirus carrying oncogenic Kras, before they were seeded in 3D explant culture. To reduce cellular oxidative stress levels, we first used the common antioxidant N-acetylcysteine (NAC) which reduces ROS by increasing glutathione levels. Treatment of acinar cells in 3D culture with NAC reduced KrasG12V-caused oxidative stress (Supplemental Fig. S2A) and efficiently blocked KrasG12V-induced ADM (Fig. 2A).
Figure 2. Oncogenic Kras induces ADM through generation of mitochondrial oxidative stress.

A: Primary mouse pancreatic acinar cells were isolated from wildtype mice, infected with lentivirus harboring control (null) or KrasG12V, and then seeded in 3D collagen explant culture in presence of the ROS scavenger N-Acetyl-Cysteine (NAC, 5 mM). At day 5, bright field pictures were taken (4x magnification, show is a representative picture) and ducts formed (ADM events; number of ducts per field) were counted. The bar indicates 100 μm. B: Primary mouse pancreatic acinar cells were isolated from wildtype mice, double-infected with adeno-null (control, empty virus) or adeno-mCatalase (catalase targeted to the mitochondria), and lentivirus harboring control (null) or KrasG12V. Cells were then seeded in 3D collagen explant culture. At day 5, brightfield pictures were taken (4x magnification, show is a representative picture) and ducts formed (ADM events; number of ducts per field) were counted. The bar indicates 100 μm. C: Primary mouse pancreatic acinar cells were isolated from wildtype mice, infected with lentivirus harboring control (null) or KrasG12V, and then seeded in 3D collagen explant culture in presence of the mitochondria-targeted antioxidant mitoQ (500 nM). At day 5, bright field pictures were taken (4x magnification, show is a representative picture) and ducts formed (ADM events; number of ducts per field) were counted. The bar indicates 100 μm. In A–C, * indicates statistical significance (p<0.05) as compared to control; ** as compared to KrasG12V.
We next determined the source of cellular ROS that serves as effector of oncogenic Kras. Because oncogenic Kras alters the metabolic activity of mitochondria (Fig. 1C), and in lung cancer can regulate mitochondrial ROS production (Weinberg et al., 2010), we first focused on the mitochondria as a potential source. The expression of mitochondria-targeted catalase to reduce mitochondrially-generated hydrogen peroxide effectively blocked ADM (Fig. 2B; controls in Supplemental Figs. S2B, S2C), indicating that mitochondria are indeed the source for KrasG12D-induced ROS. To confirm this, we treated cells with MitoQ, a mitochondria-targeted antioxidant (Smith and Murphy, 2010), which also blocked ADM either when wildtype acinar cells were infected with KrasG12V (Fig. 2C; controls in Supplemental Fig. S2D) or when expression of KrasG12D was induced in acinar cells from LSL-KrasG12D mice (Supplemental Fig. S2E). We also tested if ROS generation by NADPH oxidases (NOX) could contribute to Kras-caused ADM. Therefore, we depleted isolated primary acinar cells of p22phox, a central component of all NADPH oxidase complexes in mice, using lentivirally-delivered specific shRNA (Supplemental Figs. S2F–S2H). A knockdown of p22phox did not affect KrasG12D-induced ADM. Taken together, our data indicate mitochondria as the source for oxidative stress that drives ADM induced by oncogenic Kras. Moreover, the inhibition of ADM by mCatalase suggests hydrogen peroxide as the source of oxidative stress causing ADM. In fact, treatment of freshly-isolated primary acinar cells with hydrogen peroxide, although weakly, and with a time delay (day 9), induced their transdifferentiation (Supplemental Fig. S2I).
Kras-induced ROS can drive ADM through Nuclear Factor-κB
Next, we focused on identifying the mechanisms that mROS use to induce ADM. Since NF-κB, a ROS-sensing transcription factor that also can be activated by oncogenic Kras (Ling et al., 2012), had been shown to be upregulated in pancreatic cancer (Doppler et al., 2013; Pan et al., 2008), we first confirmed its presence in ADM/PanIN lesions that are caused by oncogenic Kras (Supplemental Figures S3A, S3B). We then tested if NF-κB activity can be induced by oncogenic Kras-caused ROS. Increased nuclear NF-κB binding activity was detected in primary acinar cells lentivirally-infected with oncogenic Kras, and this was blocked when cells were treated with MitoQ (Fig. 3A). To test if this correlates with increased NF-κB transcriptional activity, we infected primary acinar cells with adenovirus encoding a NF-κB luciferase gene reporter and lentiviral oncogenic Kras, as indicated, and then treated cells with MitoQ or vehicle (Fig. 3B). Active Kras significantly increased NF-κB activity and this was completely blocked when mitochondrial oxidative stress was decreased using MitoQ.
Figure 3. Oncogenic Kras-induced ROS drive ADM through nuclear factor-κB.

A: Primary mouse pancreatic acinar cells were isolated, lentivirally-infected with control or KrasG12V harboring virus and seeded on collagen in presence of MitoQ (500 nM), as indicated. After 48 hours, cells were isolated, nuclear extracts prepared and an EMSA performed to measure NF-κB binding activity. B: Primary mouse pancreatic acinar cells were isolated, co-infected with lentivirus harboring control or KrasG12V virus and adenovirus harboring a NF-κB-luciferase reporter gene. Cells were seeded on collagen in presence of MitoQ (500 nM), as indicated. After 48 hours, cells were isolated and a luciferase assay performed to measure NF-κB activity. C: Primary mouse pancreatic acinar cells were isolated from LSL-KrasG12D mice, infected with adeno-null or adeno-cre, and then seeded in 3D collagen explant culture in presence of BMS345541 (labeled: BMS) at indicated doses. At day 5, ducts formed (ADM events; number of ducts per field) were counted. D: Primary mouse pancreatic acinar cells were isolated from wildtype mice, infected with adenovirus harboring control (null), NF-κB1 or NF-κB2, and then seeded in 3D collagen explant culture. At day 5, brightfield pictures were taken (4x magnification; show is a representative picture; bar = 100 μm) and newly-formed duct-like structures formed were counted. In B–D, * indicates statistical significance (p<0.05) as compared to control; ** as compared to KrasG12V.
We then tested if Kras mediated ADM is blocked when NF-κB is inhibited. First, we infected primary pancreatic acinar cells with adenovirus encoding superdominant IκBα (IκBα.SD) in combination with lentivirus encoding oncogenic Kras, as indicated (Supplemental Fig. S3C). Inhibition of NF-κB with the superdominant repressor significantly decreased Kras-induced ADM events, but did not fully block it. Therefore, we next utilized acinar cells from LSL-KrasG12D mice, infected them with adeno-cre to induce expression of active Kras, and then treated them with BMS345541, a highly selective inhibitor for the catalytic subunits of IKK1 and IKK2, with IC50 of 4 μM and 0.3 μM, respectively. We used this inhibitor at two different concentrations, 1 and 10 μM, at which it was described to inhibit either IKK2 only (1 μM), or both, IKK1 and IKK2 (10 μM). At both concentrations BMS345541 blocked Kras-induced ADM, and at 10 μM it even decreased basal ADM events (Fig. 3C). This indicated that both NF-κB pathways, the canonical and alternative pathway, may be involved in driving Kras-induced ADM. Indeed, when infecting primary acinar cells with either NF-κB1 or NF-κB2, using an adenoviral system, both were able to induce ADM, though NF-κB1 was the more prominent inducer (Fig. 3D; controls in Supplemental Figs. S3D, S3E).
KrasG12D-induced mROS/NF-κB signaling upregulates the expression of EGFR, its ligands and their sheddase ADAM17
Next we investigated how Kras-induced mROS and activation of NF-κB can connect to known key events that trigger ADM and PanIN progression. We focused on EGFR signaling since transgene expression of EGFR or stimulation of cells with its ligand TGFα can initiate the ADM process in vitro and in vivo (Liou et al., 2015a). Analyses of human samples indicated that occurrence of oxidative stress (as measured by 4HNE) correlates with expression of EGFR and its ligand TGFα (Fig. 4A). Similarly, increased expression and activity (measured by probing for Y1068 phosphorylation) of EGFR and TGFα were detected in regions of ADM and PanIN1 from pancreata from p48cre;LSL-KrasG12D mice, but not in normal acinar cells (Figs. 4B, 4C and Supplemental Figs. S4A–S4D). Expression of these molecules correlated with occurrence of oxidative stress as indicated by co-immunofluorescence staining for 4HNE (Figs. 4B, 4C). Detection of CK-19 served as a ductal marker for cells that underwent ADM to a ductal phenotype (Supplemental Figs. S4B, S4C, S4E, S4F). There was no 100 % overlap in CK-19 and EGFR or pY1068-EGFR expressing regions of ADM or PanIN. This is as expected since EGFR-ligand signaling drives ADM and is upregulated in cells that are undergoing ADM, but not necessarily in newly formed ducts.
Figure 4. Oncogenic Kras upregulates expression of EGFR, its ligands and their sheddase ADAM17 through NF-κB.

A: Pancreatic cancer patient samples were analyzed for 4HNE and EGFR (top panel) or 4HNE and TGFα (bottom panel) using immunofluorescent labeling. Nuclei were visualized by DAPI staining. The first set of pictures shows a composite of 4HNE with EGFR or TGFα. The second and third pictures show EGFR, TGFα or 4HNE combined with DAPI. Rectangular windows show zoom in areas. Scale bar is 50 μm. B, C: Pancreatic sections from control mice (shown are p48cre) or mice expressing the LSL-KrasG12D allele under the p48 promoter (p48cre;LSL-KrasG12D) were analyzed by immunofluorescence for expression of EGFR and 4HNE (A) or TGFα and 4HNE (B). Nuclei were stained with DAPI. Shown is a characteristic area of the pancreas. The bar indicates 25 μm. D: Primary mouse pancreatic acinar cells were isolated from LSL-KrasG12D mice, infected with adeno-null (labeled: control) or adeno-cre (labeled: cre), and then seeded in 3D collagen explant culture in presence of MitoQ (500 nM). At day 5, cells were isolated from collagen and expression of murine EGFR, TGFα, EGF and ADAM17 was analyzed using quantitative PCR. * indicates statistical significance (p<0.05) as compared to PBS-treated control; # as compared to PBS-treated control and KrasG12D-expressing cells. E: Primary mouse pancreatic acinar cells were isolated from LSL-KrasG12D mice, infected with adeno-null (labeled: Control) or adeno-cre (labeled: cre), and then seeded in 3D collagen explant culture in presence of BMS345541 (labeled: BMS) at indicated doses. At day 5, cells were isolated from the collagen and expression of murine EGFR, TGFα, EGF and ADAM17 was analyzed using quantitative PCR. * indicates statistical significance (p<0.05) as compared to untreated control; # as compared to untreated KrasG12D-expressing cells; and $ as compared to untreated control and to KrasG12D-expressing cells. F, G: Primary mouse pancreatic acinar cells were isolated from wildtype mice, infected with adenovirus harboring control (null), NF-κB1 or NF-κB2, and then seeded in 3D collagen explant culture. At day 5, cells were isolated from the collagen and expression of murine EGFR (F), or TGFα, EGF and ADAM17 (G) was analyzed using quantitative PCR. In F and G, * indicates statistical significance (p<0.05) as compared to control.
Next we determined if expression of EGFR, its ligands or their sheddase ADAM17 is mediated by Kras through mROS and NF-κB. Therefore, we isolated acinar cells from LSL-KrasG12D mice, infected them with adeno-cre to induce KrasG12D expression, and then treated them with MitoQ or BMS345541 in 3D explant culture. KrasG12D-induced expression of EGFR, TGFα, EGF and ADMA17 was reduced below basal levels when cells were treated with MitoQ (Fig. 4D), or when cells were treated with BMS345541 (Fig. 4E). In addition, primary acinar cells infected with either NF-κB1 or NF-κB2 showed increased expression of EGFR, its ligands and ADAM17 (Figs. 4F, 4G). EGFR mainly was induced by NF-κB1 (approximately 5-fold by NF-κB1 and 2.5-fold by NF-κB2), and TGFα, EGF and ADAM17 were induced mainly by NF-κB2 (approximately 7-fold by NF-κB2 and 2–3-fold by NF-κB1).
The mitochondria-targeted antioxidant MitoQ decreases Kras-caused formation of pancreatic abnormal structures in vivo
In order to test if the inhibition of mitochondrial oxidative stress has effects on Kras-initiated processes that lead to the development of pancreatic cancer, we treated p48cre;LSL-KrasG12D mice with MitoQ, over 12 weeks (treatment schedule in Supplemental Fig. S5A). Treatment with MitoQ led to a significant reduction (approximately 50%) of KrasG12D-caused abnormal pancreatic structures as shown by H&E, alcian blue and Claudin-18 staining (Figs. 5A, 5B and Supplemental Fig. S5B). A more detailed analysis (Fig. 5B) in which we determined occurrence of ADM, ADM-PanIN (developing PanIN) and PanIN1A/B, indicated that MitoQ decreased the overall numbers of all these structures. However depletion of mROS has a more profound effect on occurrence of ADM and less on PanIN progression as indicated by the relative distribution among ADM, ADM-PanIN and PanIN1A/B. The decrease of mitochondrial oxidative stress in pancreatic tissue after treatment with MitoQ was confirmed by staining for 4HNE (Figs. 5A, 5C). In addition, treatment of mice with MitoQ led to a significant decrease in p65-positive cells (Fig. 5D), decrease in EGFR expression (Fig. 5E), as well as decrease in Nrf2 expression (Supplemental Fig. S5C).
Figure 5. The mitochondrially-targeted antioxidant MitoQ decreases Kras-caused formation of pancreatic abnormal structures in vivo.

A, B: Control mice (LSL-KrasG12D) or p48cre;LSL-KrasG12D mice at an age of 3 weeks were treated with MitoQ or vehicle every other day over a time period of 12 weeks (treatment schedule shown in Supplemental Fig. S5A). At the endpoint, pancreata were analyzed by H&E staining and IHC for 4HNE as a marker for oxidative stress. A representative area of the pancreas tissue under each condition is shown in A, a quantitation of abnormal structures (and subdivision into ADM, ADM-PanIN and PanIN1A/B) per section is shown in B. Scale bar is 50 μm. * indicates statistical significance (p<0.05) as compared to vehicle-treated mice. C, D: The bar graphs show quantifications of 4HNE and p65 positive cells per field (region containing abnormal pancreatic structures). E: Samples were analyzed by immunofluorescence for expression of EGFR. Nuclei were stained with DAPI. Shown is a characteristic area of the pancreas. The bar indicates 50 μm. The bar graph shows a quantification of EGFR positive cells per field (region containing abnormal pancreatic structure).
Kras-mROS signaling toward NF-κB is mediated by Protein Kinase D1
Next we investigated how Kras-caused in mROS could lead to activation of NF-κB. We recently have demonstrated that the serine/threonine kinase protein kinase D1 (PKD1) is involved in Kras-induced PanIN formation by activating Notch1 signaling (Liou et al., 2015a). Interestingly, in cervix carcinoma cell lines PKD1 had been implicated to be a sensor of oxidative stress (Storz and Toker, 2003) that links mROS generation to activation of NF-κB (Storz et al., 2005). Therefore, we investigated the possibility if PKD1 relays KrasG12D-mROS signaling to NF-κB. First, we analyzed p48cre;KrasG12D mice either control treated or treated with MitoQ for expression of PKD1 or phosphorylations that indicate mROS-mediated activation of PKD1 (anti-pY95-PKD antibody; described in (Doppler and Storz, 2007)), or general PKD activity (anti-pS744/748-PKD antibody). Our data shown in Figs. 6A and 6B indicate that active PKD1 indeed is downstream of KrasG12D-induced mROS. To verify our in vivo observations we performed NF-κB reporter gene assays. Therefore, we adenovirally-introduced a NF-κB luciferase reporter into primary acinar cells and additionally infected cells with control or KrasG12V lentivirus as indicated (Supplemental Fig. S6). The introduction of oncogenic Kras in acinar cells induced the NF-κB reporter activity approximately 3.25 fold; and this was blocked with a PKD inhibitor (CRT0066101). To further determine the role of PKD1 in regulating NF-κB and expression EGFR signaling molecules downstream of oncogenic Kras we compared p48cre;KrasG12D mice to p48cre;KrasG12D;PKD−/− mice in which PKD1 was knocked-out in acinar cells (generation and analyses of PKD1fl/fl mice described in (Liou et al., 2015a)). A conditional knockout of PKD1 in acinar cells of p48cre;KrasG12D mice decreased expression of NF-κB (Figs. 6C and 6D) in abnormal pancreatic structures. This correlated with a decrease in expression of EGFR (Fig. 6E) and its ligand TGFα (Fig. 6F), indicating that PKD1 indeed is involved in mediating mROS-NF-κB signaling downstream of oncogenic Kras.
Figure 6. Protein Kinase D1 is downstream of KrasG12D-mediated ROS.

A, B: Control mice (LSL-KrasG12D) or p48cre;LSL-KrasG12D mice treated with MitoQ or vehicle (from experiment shown in Fig. 5A) were analyzed by IHC for expression of PKD1, or PKD phosphorylations indicative for its activation by oxidative stress (pY95-PKD) or general activity (pS744/748-PKD). Scale bar in A is 50 μm. B shows quantifications (% of positive cells per PanIN1). * indicates statistical significance (p<0.05) as compared to vehicle-treated mice. C, E: Pancreas sections from p48cre;KrasG12D;PKD1−/− and control mice at an age of 8 weeks were analyzed by immunofluorescence staining for p65 (C) or EGFR and TGFα (E). Nuclei were visualized by DAPI staining. Scale bar: 50 μm. D, F: Quantifications of cells expressing p65 (D); and EGFR or TGFα (F) in abnormal structures (ADM, PanIN). * indicates statistical significance (p<0.05) as compared to p48cre;KrasG12D (KC).
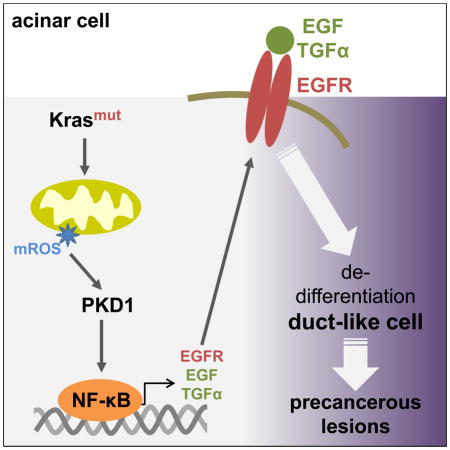
Taken together, our in vitro and in vivo data suggest that acquisition of an activating Kras mutation in pancreatic acinar cells increases mitochondrial oxidative stress levels leading to activation of PKD1 and NF-κB, which then can contribute to ADM and the formation of PanIN by regulating the expression of EGFR and its ligands (Fig. 7). Thus our data provide mechanistic insight of how mutant Kras crosstalks with growth factor signaling to induce the formation of pancreatic cancer.
Figure 7.

Scheme of how oncogenic Kras signals through mROS/PKD1 and NF-κB to induce the expression of EGFR and its ligands in pancreatic acinar cells.
DISCUSSION
Recent evidence suggests that mutant Kras and EGFR signaling cooperate to mediate the development of pancreatic cancer (Ardito et al., 2012; Liou et al., 2015a; Navas et al., 2012). Here we show that mutant Kras can initiate the transcription of the EGFR and its ligands TGFα and EGF by increasing mitochondrial generation of ROS and activation of NF-κB signaling through PKD1. Hence, we provide a direct functional relationship between two major pathways driving the development of PDA, and also provide additional insight how mROS generated by mutant Kras links to NF-κB.
In pancreatic cancer it was shown that catalase is down-regulated, while superoxide dismutase (SOD) is highly expressed. The resulting presence of high levels of hydrogen peroxide correlated with an increase in oxidized low density lipoproteins (LDL) (Kodydkova et al., 2013). Analyses of tissue samples from p48cre;LSL-KrasG12D mice indicate that ROS already are induced in acinar cells and gradually-increased during ADM and PanIN formation and progression (Fig. 1 and Supplemental Fig. S1). Increased ROS can lead to occurrence of aldehydic products of lipid peroxidation, such as 4HNE, which have been implicated in the etiology of pathological changes under oxidative stress. 4HNE can be detected in the cytosol as well as the nucleus. This is because ROS-generated reactive lipids such as 4HNE form adducts with other cellular macromolecules including proteins or nuclear and mitochondrial DNA. For cancer the generation of 4HNE-protein and 4HNE-DNA adducts have been shown to contribute to pathogenesis and progression (Barrera et al., 2015).
In primary acinar cells oncogenic Kras induces oxidative stress; and inhibition of mitochondrial ROS in organoid explant culture and in mice reduced ADM and PanIN formation (Figs. 2 and 5). On first view, this seems in conflict with published data showing that Nrf2, a key regulator of the inducible antioxidant program, is upregulated in pre-neoplastic lesions (DeNicola et al., 2011). Although, we confirmed that Nrf2 expression is increased in nuclei of acinar cells and PanIN cells caused by oncogenic Kras (Supplemental Figs. S1B, S1C), these abnormal pancreatic structures still show a significant increase in oxidative damage (as indicated by 4HNE staining). Induction of Nrf2 expression seems to be a response to oxidative stress, since it is lost in pancreata of p48cre;LSL-KrasG12D mice that were treated with MitoQ (Supplemental Fig. S5C). Enhanced expression of oncogenic Kras can also increase ROS to levels where they induce senescence (Lee et al., 1999). Therefore, in acinar cells the oncogene-mediated increase in ROS is probably balanced by upregulation of Nrf2 to facilitate levels that promote proliferation and growth of PanIN lesions rather than inducing senescence or DNA damage. Thus, increased Nrf2 levels can go along with increased ROS production. Similar was shown with Pdx1cre;KrasG12D;TP53INP1−/− mice, in which an increase in Rac1-caused oxidative stress at the PanIN1B/PanIN2 stage promotes pancreatic carcinogenesis (Al Saati et al., 2013). A relationship between ROS and Nrf2 signaling has been described for development and progression of other cancers (Perera and Bardeesy, 2011), in which increased expression of antioxidant systems mediates resistance to chemotherapeutical therapies that increase ROS levels (Singh et al., 2010; Zhang et al., 2010). It also should be noted that besides its antioxidant functions Nrf2 targets drug-metabolizing enzymes, efflux pumps, and regulates heat shock proteins and growth factors to mediated pro-tumorigenic signaling (Hayes and McMahon, 2009). Based on this knowledge, one treatment option would be to decrease Nrf2 levels in cells with the net effect of decreasing pro-tumorigenic signaling and elevating ROS to toxic levels.
The contribution of Krasmut targets such as STAT3 to the regulation of metabolic functions has been shown to be involved in oncogenic transformation (Gough et al., 2009). However, it remains to be determined how mutant Kras initiates the generation of mitochondrial ROS. There are reports showing that Kras can induce suppression of respiratory chain complex-I and cause mitochondrial dysfunction and generation of reactive oxygen species (Hu et al., 2012). Our data supports that Kras alters the metabolism of cells in a fashion where mitochondrial efficiency is decreased resulting in an increased production of ROS (Fig. 1C–E). A possible mechanism is the formation of 4HNE adducts with macromolecules leading to inhibition of mitochondrial proteins or damage of mtDNA.
Cellular ROS have been shown to activate both canonical and non-canonical NF-κB pathways (Storz et al., 2005; Storz and Toker, 2003; Zhang et al., 2001). NF-κB levels are increased in PDA cell lines and patient samples, and both NF-κB pathways have been implicated in the development of PDA (Doppler et al., 2013; Pan et al., 2008). One mechanism of action of increased NF-κB activity is that it elevates activation of Kras to pathological levels (Daniluk et al., 2012). We here show that both canonical and alternative pathways, downstream of Kras-induced ROS, upregulate the expression of EGFR, its ligands and their sheddase ADAM17 (Fig. 4 and Supplemental Fig. S4). The importance of this is that activation of EGFR signaling has been identified as essential for Krasmut-driven PDA (Ardito et al., 2012; Navas et al., 2012). By showing that oncogenic Kras can induce expression of EGFR and its ligands via mROS-NF-κB signaling, we provide a direct functional link between two key signaling pathways that drive the development of PDA. Intracellular ROS can activate a multitude of signaling pathways regulating cell survival and proliferation. One of the characteristics of pancreatic acinar cells, when isolated and kept in culture, is that they are senescent and eventually undergo cell death. One function of Krasmut-induced upregulation of EGFR signaling may be to allow pancreatic acinar cells to acquire proliferative potential (Ardito et al., 2012).
Eventually, we have investigated the mechanism by which generation of mROS can relay to activation of NF-κB. The involvement of PKD1 downstream of Kras/mROS to activate NF-κB/EGFR signaling (Fig. 6 and Supplemental Fig. 6) is exciting because PKD1 also can regulate Notch (Liou et al., 2015a); and Notch and NF-κB signaling pathways can co-operate in mediating ADM (Maniati et al., 2011). Thus our data showing that PKD1 acts downstream of KrasG12D-mROS to activate NF-κB, and recently published data demonstrating a KrasG12D-PKD1-Notch signaling axis, bring together two important pathways that drive the formation of precancerous lesions with PKD1 possibly as key mediator downstream of Kras.
In summary, our data highlight a cellular signaling mechanism of how mutant Kras crosstalks with EGFR signaling. We show that such signaling is mediated by Krasmut-induced generation of mitochondrially-generated reactive oxygen species. Our data also suggest that mitochondria-targeted antioxidants such as MitoQ, an orally-active antioxidant that mimics the role of the endogenous mitochondrial antioxidant coenzyme Q10, reduces the development of pancreatic pre-neoplastic lesions in vivo.
EXPERIMENTAL PROCEDURES
Antibodies, viral constructs and reagents
Anti-4-Hydroxy-2-nonenal (4HNE) was from Alpha Diagnostic International (San Antonio, TX), anti-PKD1 was from Acris Antibodies (San Diego, CA), anti-CK-19 and anti-EGFR (for human samples) from Santa Cruz Biotechnology (Dallas, TX), anti-claudin-18 from Invitrogen (Carlsbad, CA), anti-TGFα from RayBiotech (Norcross, GA), anti-p65 from Cell Signaling Technology (Danvers, MA), anti-Nrf2, anti-EGFR (for mouse samples), anti-pY1068-EGFR and anti-pS744/748-PKD antibodies were from Abcam (Cambridge, MA). The anti-pY95-PKD antibody is described in detail in (Doppler and Storz, 2007) and (Zheng et al., 2014). Lentiviral Plasmids to knock down mouse p22phox were purchased from Sigma-Aldrich. pLenti6.3/V5-Flag-TdTomato-KrasG12V has been described before (Liou et al., 2015a). Adenovirus to express IκBα.S32A/S36A (IκBα.SD), NF-κB1/p105, NF-κB2/p100, cre recombinase, the NF-κB-luciferase reporter (Ad-NF-κB-luc), or control (null) was purchased from Vector Biolabs (Philadelphia, PA). Adenovirus to express mitochondrial-localized catalase (mCatalase) was obtained from the Gene Transfer Vector Core of the University of Iowa. N-acetyl-L-cysteine (NAC) was from Sigma-Aldrich (St. Louis, MO). Hoechst 33342 and H2DFFDA were from Invitrogen (Carlsbad, CA). MitoTracker Red and H2O2 were from Fisher Scientific (Hampton, NH). BMS-345541 was from EMD Millipore (Billerica MA). Soybean trypsin inhibitor and collagenase I were from Affymetrix (Santa Clara, CA). Rat tail collagen I was from BD Biosciences (San Jose, CA). CRT0066101 was from TOCRIS (Ellisville, MO). MitoQ was a gift from Robin Smith (Department of Chemistry, University of Otago, Dunedin, New Zealand).
Animals and treatments
FVB mice for isolation of primary pancreatic acinar cells were purchased from Harlan Laboratories (Tampa, FL). Ptf1a/p48Cre/+, LSL-KrasG12D/+ and PKD1fl/fl mouse strains and genotyping of mice have been described previously (Liou et al., 2015a; Liou et al., 2015b). For treatment with MitoQ, 3 week old animals were intraperitoneally (IP) injected with MS010 (MitoQ adsorbed to cyclodextrin) at a dose of 20 mg/kg or phosphate-buffered saline solution as control vehicle every 2nd day for 12 weeks. At week 15 mice were sacrificed and tissues harvested (see time line Supplemental Figure S5A). All animal experiments were approved by the Mayo Clinic IACUC committee and were performed in accordance with relevant institutional and national guidelines and regulations.
Human pancreatic tissue samples
Patient tissues were obtained from archival materials in accordance with institutional guidelines and prior institutional review board (IRB) approval.
Isolation of primary pancreatic acinar cells
The protocol for isolation of primary pancreatic acinar cells has been described in detail previously (Liou et al., 2015b; Means et al., 2005). In brief, the pancreas was removed, washed twice with ice-cold HBSS media, incised into 1–5 mm pieces and digested with collagenase I (37 °C, shaker). The collagen digestion was terminated by addition of an equal volume of ice-cold HBSS media containing 5% FBS. The digested pancreatic pieces were washed twice with HBSS media containing 5% FBS, pipetted through 500 μm, and then 105 μm meshes. The supernatant of the cell suspension containing acinar cells was dropwise added to 20 ml HBSS media supplemented with 30% FBS. Acinar cells were pelleted (1000 rpm, 2 minutes, at 4 °C) and re-suspended in 10 ml Waymouth complete media (1% FBS, 0.1 mg/ml trypsin inhibitor, 1 μg/ml dexamethasone).
3D explant culture of primary pancreatic acinar cells and quantification
This method was described in detail before (Liou et al., 2015b; Means et al., 2005). In short, cell culture plates were coated with collagen I in Waymouth media without supplements. Freshly isolated primary pancreatic acinar cells were added on top of this mixture and overlaid with Waymouth complete media (replaced every other day). Inhibitors or compounds were added to both, the cell/gel mixture and the media on top. Acinar cells were infected with virus of interest for 3–5 hours before embedding in the collagen/media mixture. After 48 hours, an aliquot of cells for each condition was analyzed by qPCR to control expression or knockdown. Viability of cells after viral infection was confirmed using Hoechst 33342. All samples/experimental conditions were performed in triplicates and after indicated time (usually day 5), numbers of ducts per field (whole well) for each condition were determined and photos were taken to document cellular structures. Each experiment was performed in three to five replicates using pancreata of different individual mice.
DAB immunohistochemistry of tissues
Samples were deparaffinized for 1 hour at 60 °C, de -waxed in xylene (five times for 4 minutes) and gradually re-hydrated with enthanol (100%, 95%, 75%, twice with each concentration for 3 minutes). The rehydrated samples were rinsed in water and subjected to antigen retrieval in citrate buffer (pH 6.0). Samples were treated with 3% hydrogen peroxide for 5 minutes, washed with PBS containing 0.5% Tween 20, and blocked with protein block serum-free solution (DAKO, Carpinteria, CA) for five minutes at room temperature. Anti-4HNE antibody (1:500), anti-Nrf2 (1:500), anti-claudin-18 (1:500), anti-PKD1 (1:50), anti-pS744/748-PKD (1:50), or anti-pY95-PKD (1:50) were diluted in Antibody Diluent Background Reducing Solution (DAKO) and visualized using the EnVision Plus Anti-Rabbit Labelled Polymer Kit (DAKO). H&E staining was performed as previously described (Liou et al., 2015a). Images were captured using ScanScope XT scanner and ImageScope software (Aperio, Vista, CA).
Immunofluorescence analyses of tissues
For immunofluorescence analysis, pancreas tissue was prepared by perfusion with 10 ml 0.1 M PBS, followed by 40 ml 4 % paraformaldehyde (PFA) in 0.1 M PBS. Tissues then were fixed for 3 hours in 4 % PFA, followed by three washes with 0.1 M PBS and an overnight float in 30 % sucrose. Samples were incubated in a 1:1 mixture of 30 % sucrose and OCT for 30 minutes, embedded in OCT embedding media (Sakura Tissue-Tek, Torrance, CA) and frozen at −80 °C. 7 μm tissue sections were permeabilized with 0.1% Triton X-100 in 10 mM PBS and blocked in 10 mM PBS containing 5% donkey serum/1% BSA for 1 hour at RT. Tissue samples were then incubated with primary antibodies (4HNE 1:1000, TGFα 1:1500, 1:2500, EGFR (human) 1:250, EGFR (mouse) 1:100, pY1068-EGFR 1:100, NF-κB p65 1:100, CK-19 1:100, Nrf2 1:500) diluted in 10 mM PBS containing 1% BSA, 0.1% Triton X-100 overnight at RT. Slides were washed three times with 0.1% Triton X-100/PBS, followed by an incubation of Alexa Fluor 488- or 594-conjugated secondary antibody (Invitrogen). Stained slides were washed three times as previously described and blocked again in 10 mM PBS containing 5% donkey serum, 1% BSA for 1 hour at RT. For the tissue samples that were co-stained with 4HNE, 4HNE antiserum was incubated with Alexa Fluor 488 through overnight rotating at 4 °C for pre-conjugation and applied to sample slides that were already labeled with TGFα or EGFR overnight. Slides were then washed three times with 0.1% Triton X-100/PBS, rinsed with deionized water and mounted in Vectashield containing DAPI (Vector Laboratories, Ontario, Canada). The fluorescent images were collected with consistent exposure time among samples using ScanScope FL and ImageScope software (Aperio).
RNA Isolation and quantitative PCR
Cells were harvested from 3D collagen culture by digestion in a 1 mg/ml collagenase solution at 37 °C for 30 minutes on a shaker. Cells were washed once with HBSS and twice with PBS and total RNA isolation was performed using the miRCURY™ RNA isolation kit (Exiqon, Woburn, MA) and the TURBO DNA-free kit (Ambion, Austin, TX). Quantitative reverse transcriptase real-time PCR (qPCR) was performed using the high capacity cDNA reverse transcriptase kit (Applied Biosystems, Bedford, MA), the TaqMan Universal PCR master mix (Applied Biosystems) and below primer sets in a 7900HT Fast real-time thermocycler (Applied Biosystems). Thermocycler program: 95 °C for 20 seconds; 40 cycles of 95 °C for one second and 60 °C for 20 seconds. Probe/primer sets were from Applied Biosystems (Mm00438696_m1 for EGF, Mm00446232_m1 for TGFα, Mm00433023_m1 for EGFR, Mm00456428_m1 for ADAM17, Mm00514478_m1 for p22phox, Hs00765730_m1 for NF-κB1, Hs01028901_g1 for NF-κB2, Hs00270666_g1 for human Kras; Hs00156308_m1 for Catalase). Amplification data were collected by a Prism 7900 sequence detector and analyzed with Sequence Detection System software (Applied Biosystems). Data were normalized to mouse 18S rRNA or GAPDH, and mRNA abundance was calculated using the ΔΔCT method.
Measurement of oxygen consumption
The oxygen consumption rate (OCR) of primary pancreatic acinar cells was measured using a XF96 Extracellular Flux Analyzer and the XF Cell Mito Stress Test Kit (Seahorse Bioscience, North Billerica, MA) according to the manufacturer’s protocol. Briefly, 48 hours after infection, primary acinar cells from LSL-KrasG12D mice were seeded in XF96 tissue culture plates. One hour prior to the mito stress test assays cells were washed and transferred to XF96 tissue culture plates. The stress test was performed as suggested in the manufacturer’s protocol. Oligomycin (O; 2 μM), FCCP (F; 1 μM), and rotenone (R; 0.5 μM) were added at indicated time points.
Measurement of ROS Generation
48 hours post infection, primary pancreatic acinar cells were labeled with H2DFFDA (20 μM) in phenol red-free Waymouth complete media at 37 °C for 20 minutes. The cells were washed with HBSS and transferred to phenol red-free Waymouth media. ROS generation was determined using a SpectraMax M5 fluorescent microplate reader (Molecular Devices, Sunnyvale, CA) at 495/529 (Ex/Em) nm.
NF-κB reporter gene assays
Primary pancreatic acinar cells were infected with adenovirus Ad-NF-κB-luc and lentivirus of pLenti6.3/V5-Flag-TdTomato-KrasG12V or control. 24 hours after infection, media were replaced with fresh Waymouth complete media. MitoQ (500 nM) was added to the media for another 24 hours. The cells were lysed in passive lysis buffer (Promega, Madison, WI). Reporter gene activity was measured using the Dual-Luciferase reporter assay kit (Promega) and a Veritas microplate luminometer (Turner Biosystems, Sunnyvale, CA).
Nuclear extracts
Primary pancreatic acinar cells were rinsed twice with ice cold PBS, resuspended in 0.8 ml lysis buffer (10 mM HEPES pH 7.9, 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM DTT, 1 mM PMSF), and lysates were incubated for 15 minutes on ice. 50 μl 10 % NP-40 was added and samples were placed on a shaker (2 minutes at 4 ºC). Samples were spun down for 1 minute (room temperature; 13,000 rpm), the pellet (nuclei) was re-suspended in 50 μl high salt buffer (20 mM HEPES pH 7.9, 4 M NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, 1mM PMSF) followed by rough shaking for 1 hour at 4 ºC and centrifugation (15,000 rpm) for 5 minutes at 4 ºC. Supernatants were transferred to a new tube and protein concentration was determined.
Electrophoretic mobility shift assay (EMSA)
IRDye 700 NF-κB Consensus Oligonucleotide and Odyssey Infrared EMSA Kit were purchased from LI-COR Biosciences (Lincoln, NE). For EMSA assays, 8 μg of nuclear extract was incubated in 20 μl of a buffer containing 10 mM HEPES (pH 7.5), 50 mM KCl, 0.1 mM EDTA, 1 mM dithiotreitol, 0.1 % NP-40 and 0.05 mg/ml poly(dI-dC) on ice. Samples were resolved on a non-denaturing 5 % polyacrylamide gel in 0.5x TBE. Imaging was performed with an Odyssey (LI-COR Biosciences) using the 700 nm channel.
Statistical Analysis
Data are presented as mean ± SD. P values were acquired with the student’s t-test using Graph Pad software, and p < 0.05 was considered statistically significant.
Supplementary Material
Highlights.
Kras-induced mROS initiate the dedifferentiation of pancreatic acinar cells
Kras/mROS signaling upregulates EGFR and its ligands via PKD1/NF-kB
The antioxidant mitoQ blocks the formation of pancreatic precancerous lesions
Acknowledgments
This work was supported by the NIH grants CA200572, CA140182 (to PS), CA159222 (to HCC), and by grant 197261 from the Norwegian Research Council (to ML). MPM holds patents in the area of mitochondria-targeted antioxidants and is a consultant for Antipodean Pharmaceuticals Inc., which is developing MitoQ as a potential pharmaceutical. We thank the Pancreatic Cancer Action Network (PanCAN) for their support.
Footnotes
AUTHOR CONTRIBUTIONS
Conceived and designed the experiments: PS, GYL, HD, MPM. Performed the experiments: GYL, KED, HD. Analyzed the data: GYL, HD, KED, LZ, HCC, PS. Contributed reagents/materials/analysis tools: GYL, HCC, HD, MPM, ML. Wrote the paper: PS, MPM, GYL, HD.
All other authors have no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahn CS, Metallo CM. Mitochondria as biosynthetic factories for cancer proliferation. Cancer Metab. 2015;3:1. doi: 10.1186/s40170-015-0128-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al Saati T, Clerc P, Hanoun N, Peuget S, Lulka H, Gigoux V, Capilla F, Beluchon B, Couvelard A, Selves J, et al. Oxidative stress induced by inactivation of TP53INP1 cooperates with KrasG12D to initiate and promote pancreatic carcinogenesis in the murine pancreas. Am J Pathol. 2013;182:1996–2004. doi: 10.1016/j.ajpath.2013.02.034. [DOI] [PubMed] [Google Scholar]
- Ardito CM, Gruner BM, Takeuchi KK, Lubeseder-Martellato C, Teichmann N, Mazur PK, Delgiorno KE, Carpenter ES, Halbrook CJ, Hall JC, et al. EGF receptor is required for KRAS-induced pancreatic tumorigenesis. Cancer Cell. 2012;22:304–317. doi: 10.1016/j.ccr.2012.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardeesy N, Aguirre AJ, Chu GC, Cheng KH, Lopez LV, Hezel AF, Feng B, Brennan C, Weissleder R, Mahmood U, et al. Both p16(Ink4a) and the p19(Arf)-p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proc Natl Acad Sci U S A. 2006;103:5947–5952. doi: 10.1073/pnas.0601273103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrera G, Pizzimenti S, Ciamporcero ES, Daga M, Ullio C, Arcaro A, Cetrangolo GP, Ferretti C, Dianzani C, Lepore A, Gentile F. Role of 4-hydroxynonenal-protein adducts in human diseases. Antioxid Redox Signal. 2015;22:1681–1702. doi: 10.1089/ars.2014.6166. [DOI] [PubMed] [Google Scholar]
- Daniluk J, Liu Y, Deng D, Chu J, Huang H, Gaiser S, Cruz-Monserrate Z, Wang H, Ji B, Logsdon CD. An NF-kappaB pathway-mediated positive feedback loop amplifies Ras activity to pathological levels in mice. J Clin Invest. 2012;122:1519–1528. doi: 10.1172/JCI59743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, Mangal D, Yu KH, Yeo CJ, Calhoun ES, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011;475:106–109. doi: 10.1038/nature10189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doppler H, Liou GY, Storz P. Downregulation of TRAF2 mediates NIK-induced pancreatic cancer cell proliferation and tumorigenicity. PLoS One. 2013;8:e53676. doi: 10.1371/journal.pone.0053676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doppler H, Storz P. A novel tyrosine phosphorylation site in protein kinase D contributes to oxidative stress-mediated activation. J Biol Chem. 2007;282:31873–31881. doi: 10.1074/jbc.M703584200. [DOI] [PubMed] [Google Scholar]
- Gough DJ, Corlett A, Schlessinger K, Wegrzyn J, Larner AC, Levy DE. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science. 2009;324:1713–1716. doi: 10.1126/science.1171721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerra C, Schuhmacher AJ, Canamero M, Grippo PJ, Verdaguer L, Perez-Gallego L, Dubus P, Sandgren EP, Barbacid M. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell. 2007;11:291–302. doi: 10.1016/j.ccr.2007.01.012. [DOI] [PubMed] [Google Scholar]
- Hayes JD, McMahon M. NRF2 and KEAP1 mutations: permanent activation of an adaptive response in cancer. Trends Biochem Sci. 2009;34:176–188. doi: 10.1016/j.tibs.2008.12.008. [DOI] [PubMed] [Google Scholar]
- Hruban RH, Iacobuzio-Donahue C, Wilentz RE, Goggins M, Kern SE. Molecular pathology of pancreatic cancer. Cancer J. 2001;7:251–258. [PubMed] [Google Scholar]
- Hu Y, Lu W, Chen G, Wang P, Chen Z, Zhou Y, Ogasawara M, Trachootham D, Feng L, Pelicano H, et al. K-ras(G12V) transformation leads to mitochondrial dysfunction and a metabolic switch from oxidative phosphorylation to glycolysis. Cell Res. 2012;22:399–412. doi: 10.1038/cr.2011.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Daniluk J, Liu Y, Chu J, Li Z, Ji B, Logsdon CD. Oncogenic K-Ras requires activation for enhanced activity. Oncogene. 2014;33:532–535. doi: 10.1038/onc.2012.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa K, Takenaga K, Akimoto M, Koshikawa N, Yamaguchi A, Imanishi H, Nakada K, Honma Y, Hayashi J. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science. 2008;320:661–664. doi: 10.1126/science.1156906. [DOI] [PubMed] [Google Scholar]
- Jaffee EM, Hruban RH, Canto M, Kern SE. Focus on pancreas cancer. Cancer Cell. 2002;2:25–28. doi: 10.1016/s1535-6108(02)00093-4. [DOI] [PubMed] [Google Scholar]
- Ji B, Tsou L, Wang H, Gaiser S, Chang DZ, Daniluk J, Bi Y, Grote T, Longnecker DS, Logsdon CD. Ras activity levels control the development of pancreatic diseases. Gastroenterology. 2009;137:1072–1082. 1082 e1071–1076. doi: 10.1053/j.gastro.2009.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodydkova J, Vavrova L, Stankova B, Macasek J, Krechler T, Zak A. Antioxidant status and oxidative stress markers in pancreatic cancer and chronic pancreatitis. Pancreas. 2013;42:614–621. doi: 10.1097/MPA.0b013e318288360a. [DOI] [PubMed] [Google Scholar]
- Korc M. Role of growth factors in pancreatic cancer. Surg Oncol Clin N Am. 1998;7:25–41. [PubMed] [Google Scholar]
- Lee AC, Fenster BE, Ito H, Takeda K, Bae NS, Hirai T, Yu ZX, Ferrans VJ, Howard BH, Finkel T. Ras proteins induce senescence by altering the intracellular levels of reactive oxygen species. J Biol Chem. 1999;274:7936–7940. doi: 10.1074/jbc.274.12.7936. [DOI] [PubMed] [Google Scholar]
- Ling J, Kang Y, Zhao R, Xia Q, Lee DF, Chang Z, Li J, Peng B, Fleming JB, Wang H, et al. KrasG12D-induced IKK2/beta/NF-kappaB activation by IL-1alpha and p62 feedforward loops is required for development of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21:105–120. doi: 10.1016/j.ccr.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou GY, Doppler H, Braun UB, Panayiotou R, Scotti Buzhardt M, Radisky DC, Crawford HC, Fields AP, Murray NR, Wang QJ, et al. Protein kinase D1 drives pancreatic acinar cell reprogramming and progression to intraepithelial neoplasia. Nat Commun. 2015a;6:6200. doi: 10.1038/ncomms7200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou GY, Doppler H, Necela B, Edenfield B, Zhang L, Dawson DW, Storz P. Mutant KRAS-induced expression of ICAM-1 in pancreatic acinar cells causes attraction of macrophages to expedite the formation of precancerous lesions. Cancer Discov. 2015b;5:52–63. doi: 10.1158/2159-8290.CD-14-0474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou GY, Storz P. Reactive oxygen species in cancer. Free Radic Res. 2010;44:479–496. doi: 10.3109/10715761003667554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniati E, Bossard M, Cook N, Candido JB, Emami-Shahri N, Nedospasov SA, Balkwill FR, Tuveson DA, Hagemann T. Crosstalk between the canonical NF-kappaB and Notch signaling pathways inhibits Ppargamma expression and promotes pancreatic cancer progression in mice. J Clin Invest. 2011;121:4685–4699. doi: 10.1172/JCI45797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Means AL, Meszoely IM, Suzuki K, Miyamoto Y, Rustgi AK, Coffey RJ, Jr, Wright CV, Stoffers DA, Leach SD. Pancreatic epithelial plasticity mediated by acinar cell transdifferentiation and generation of nestin-positive intermediates. Development. 2005;132:3767–3776. doi: 10.1242/dev.01925. [DOI] [PubMed] [Google Scholar]
- Mishra PK, Raghuram GV, Jain D, Jain SK, Khare NK, Pathak N. Mitochondrial oxidative stress-induced epigenetic modifications in pancreatic epithelial cells. Int J Toxicol. 2014;33:116–129. doi: 10.1177/1091581814524064. [DOI] [PubMed] [Google Scholar]
- Navas C, Hernandez-Porras I, Schuhmacher AJ, Sibilia M, Guerra C, Barbacid M. EGF receptor signaling is essential for k-ras oncogene-driven pancreatic ductal adenocarcinoma. Cancer Cell. 2012;22:318–330. doi: 10.1016/j.ccr.2012.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X, Arumugam T, Yamamoto T, Levin PA, Ramachandran V, Ji B, Lopez-Berestein G, Vivas-Mejia PE, Sood AK, McConkey DJ, Logsdon CD. Nuclear factor-kappaB p65/relA silencing induces apoptosis and increases gemcitabine effectiveness in a subset of pancreatic cancer cells. Clin Cancer Res. 2008;14:8143–8151. doi: 10.1158/1078-0432.CCR-08-1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera RM, Bardeesy N. Cancer: when antioxidants are bad. Nature. 2011;475:43–44. doi: 10.1038/475043a. [DOI] [PubMed] [Google Scholar]
- Sarkisian CJ, Keister BA, Stairs DB, Boxer RB, Moody SE, Chodosh LA. Dose-dependent oncogene-induced senescence in vivo and its evasion during mammary tumorigenesis. Nat Cell Biol. 2007;9:493–505. doi: 10.1038/ncb1567. [DOI] [PubMed] [Google Scholar]
- Shidara Y, Yamagata K, Kanamori T, Nakano K, Kwong JQ, Manfredi G, Oda H, Ohta S. Positive contribution of pathogenic mutations in the mitochondrial genome to the promotion of cancer by prevention from apoptosis. Cancer Res. 2005;65:1655–1663. doi: 10.1158/0008-5472.CAN-04-2012. [DOI] [PubMed] [Google Scholar]
- Singh A, Bodas M, Wakabayashi N, Bunz F, Biswal S. Gain of Nrf2 function in non-small-cell lung cancer cells confers radioresistance. Antioxid Redox Signal. 2010;13:1627–1637. doi: 10.1089/ars.2010.3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RA, Murphy MP. Animal and human studies with the mitochondria-targeted antioxidant MitoQ. Ann N Y Acad Sci. 2010;1201:96–103. doi: 10.1111/j.1749-6632.2010.05627.x. [DOI] [PubMed] [Google Scholar]
- Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, Perera RM, Ferrone CR, Mullarky E, Shyh-Chang N, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. 2013;496:101–105. doi: 10.1038/nature12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storz P, Doppler H, Toker A. Protein kinase D mediates mitochondrion-to-nucleus signaling and detoxification from mitochondrial reactive oxygen species. Mol Cell Biol. 2005;25:8520–8530. doi: 10.1128/MCB.25.19.8520-8530.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storz P, Toker A. Protein kinase D mediates a stress-induced NF-kappaB activation and survival pathway. EMBO J. 2003;22:109–120. doi: 10.1093/emboj/cdg009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, Kalyanaraman B, Mutlu GM, Budinger GR, Chandel NS. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci U S A. 2010;107:8788–8793. doi: 10.1073/pnas.1003428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Johnston G, Stebler B, Keller ET. Hydrogen peroxide activates NFkappaB and the interleukin-6 promoter through NFkappaB-inducing kinase. Antioxid Redox Signal. 2001;3:493–504. doi: 10.1089/15230860152409121. [DOI] [PubMed] [Google Scholar]
- Zhang P, Singh A, Yegnasubramanian S, Esopi D, Kombairaju P, Bodas M, Wu H, Bova SG, Biswal S. Loss of Kelch-like ECH-associated protein 1 function in prostate cancer cells causes chemoresistance and radioresistance and promotes tumor growth. Mol Cancer Ther. 2010;9:336–346. doi: 10.1158/1535-7163.MCT-09-0589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Shen M, Zha YL, Li W, Wei Y, Blanco MA, Ren G, Zhou T, Storz P, Wang HY, Kang Y. PKD1 phosphorylation-dependent degradation of SNAIL by SCF-FBXO11 regulates epithelial-mesenchymal transition and metastasis. Cancer Cell. 2014;26:358–373. doi: 10.1016/j.ccr.2014.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.