

Abstract
Approximately 20% of early-stage breast cancers display amplification or overexpression of the ErbB2/HER2 oncogene, conferring poor prognosis and resistance to endocrine therapy. Targeting HER2+ tumors with trastuzumab or the receptor tyrosine kinase (RTK) inhibitor lapatinib significantly improves survival, yet tumor resistance and progression of metastatic disease still develop over time. While the mechanisms of cytosolic HER2 signaling are well studied, nuclear signaling components and gene regulatory networks that bestow therapeutic resistance and limitless proliferative potential are incompletely understood. Here, we use biochemical and bioinformatic approaches to identify effectors and targets of HER2 transcriptional signaling in human breast cancer. Phosphorylation and activity of the Steroid Receptor Coactivator-3 (SRC-3) is reduced upon HER2 inhibition, and recruitment of SRC-3 to regulatory elements of endogenous genes is impaired. Transcripts regulated by HER2 signaling are highly enriched with E2F1 binding sites and define a gene signature associated with proliferative breast tumor subtypes, cell cycle progression, and DNA replication. We show that HER2 signaling promotes breast cancer cell proliferation through regulation of E2F1-driven DNA metabolism and replication genes together with phosphorylation and activity of the transcriptional coactivator SRC-3. Furthermore, our analyses identified a cyclin dependent kinase (CDK) signaling node that, when targeted using the CDK4/6 inhibitor Palbociclib, defines overlap and divergence of adjuvant pharmacological targeting. Importantly, lapatinib and palbociclib strictly block de novo synthesis of DNA, mostly through disruption of E2F1 and its target genes. These results have implications for rational discovery of pharmacological combinations in pre-clinical models of adjuvant treatment and therapeutic resistance.
Keywords: HER2, SRC-3, E2F, coactivator, phosphorylation, DNA replication
Introduction
The ErbB/HER receptor tyrosine kinase (RTK) family of membrane growth factor receptors activates multiple signaling pathways and is often associated with cancer. Tyrosine kinase activity in this family of receptors is accomplished through homo-or heterodimerization of ErbB family members (EGFR/ERBB1, HER2/ERBB2, ERBB3, and ERBB4) upon ligand binding and/or membrane juxtaposition, and subsequent auto-and cross-phosphorylation of intracellular domains(1). Phosphorylated receptor tyrosine residues serve as docking platforms for various SH2 domain proteins that function, in part, as signal adaptors and amplifiers for downstream kinase activity. Ras/Raf/MAPK and PI3K/AKT pathways are subsequently activated and the cascading substrates of these phospho-signaling events become effectors of cell division, evasion of apoptosis, and general tumorigenicity(2,3).
Steroid Receptor Coactivator-3 (SRC-3/AIB1/NCOA3) is a potent transcriptional coregulator for various nuclear receptors and transcription factors including E2F1, NFκB, and members of the ETS family(4–7). SRC-3 is amplified and/or overexpressed in a wide variety of tumors, including amplification in up to 10% and overexpression in up to 60% of breast tumors(4,8–11). Importantly, SRC-3 is a coactivating partner for estrogen receptor (ERα), the cognate effector for the mitogenic action of estrogenic steroids in the mammary epithelium(12). ERα expression is an important biomarker for prognosis and treatment and is present in ~70% of breast tumors at diagnosis, most of which receive endocrine therapy to block estrogen synthesis or receptor activity(13,14). The function of SRC-3 in breast cancer has been intensively studied since it is a major coactivator for ERα and a prominent, overexpressed oncogene. The coactivation potential of SRC-3 is tightly regulated by post-translational modifications that alter protein activity, stability and sub-cellular localization(15,16). In the context of ERα and NFκB-regulated transcription, SRC-3 is differentially activated via phosphorylation at multiple residues in response to estradiol or TNFα(15,17). Collectively, these studies position SRC-3 as a potential “signal integrator” of various cytokine and hormone elicited cell-signaling events that specify distinct patterns of gene expression(18).
Clinical studies have shown that breast cancer patients with tumors expressing high levels of both HER2 and SRC-3 display resistance to endocrine therapy and have reduced disease-free survival(13,19,20). Furthermore, SRC-3 knockout mice are resistant to HER2/neu-mediated breast tumorigenesis and display significantly reduced phosphorylation of key signaling components, including HER2, AKT, and JNK(21). In MCF-7 breast cancer cells, SRC-3 phosphorylation is influenced by HER2(22). Together, these studies suggest HER2 and SRC-3 are cooperating oncogenes whereby HER2 signaling events influence SRC-3 activity on discrete gene programs. However, the transcriptional targets of HER2 and SRC-3 are largely undescribed and it is unknown whether intrinsic SRC-3 activity and specific phosphorylation sites are affected by HER2. Amplification of HER2 occurs in approximately 20% of all breast tumors and overexpression of HER2 and SRC-3 is associated with endocrine resistance and extremely poor prognosis. Given the link between HER2 signaling and SRC-3 in breast cancer and the importance of SRC-3 phosphorylation for its coactivation of steroid receptors and other transcription factors, the combined effects of HER2 and SRC-3 on gene regulation are understudied in breast cancer. Additionally, nuclear effectors of HER2 signaling that regulate transcription of tumorigenic gene networks are incompletely understood.
Here, we study the effects of HER2 mediated signaling events on SRC-3 activity in breast cancer cells with amplified HER2 and SRC-3 genes. HER2 depletion attenuates site-specific phosphorylation of SRC-3 leading to a reduction in its intrinsic activity. Transcriptomic analysis identifies genes that are regulated by HER2 signaling, with a massive enrichment for transcripts involved in mitotic cell cycle processes and DNA replication. A striking majority of genes downregulated upon HER2 depletion contain canonical and evidence-based binding sites for the E2F1 transcription factor. Moreover, bioinformatics reveals a common HER2/SRC-3/E2F1 target gene (CCND1) that is significantly correlated with sensitivity to RKT inhibitors and identifies a CDK signaling node amendable to pharmacological targeting. Our results suggest that SRC-3 and E2F1 serve a membrane-to-nuclear conduit function for HER2 signaling, resulting in an anabolic DNA gene signature exploitable by combinatorial therapies.
Materials and Methods
Cell culture, growth assays, and reagents
BT-474 and SKBR-3 cells were cultured in DMEM (Gibco/Life Technologies) supplemented with 10% fetal bovine serum (FBS) and 2 mg/L insulin. Cells were obtained from ATCC and cell identity is verified using short tandem repeat (STR) analysis routinely by the Tissue Culture Core at BCM. For growth assays, 5×104 cells were plated per well on 12 or 24-well multiwell plates in complete media and allowed to settle, and the following day were rinsed once with phenol red-free DMEM before treatment in identical conditions supplemented with 5% charcoal-stripped fetal bovine serum. For Figures 1A, 1C, 1E, 6A, and 6B, crystal violet staining with colorimetric absorbance was used to measure cell density. For Figures 6C and 6D, MTS (Promega) assay was used. Detailed descriptions of reagents are listed in Supplementary Table 2.
Figure 1. BT-474 cells are estrogen-responsive and display HER2-dependant SERM resistance.
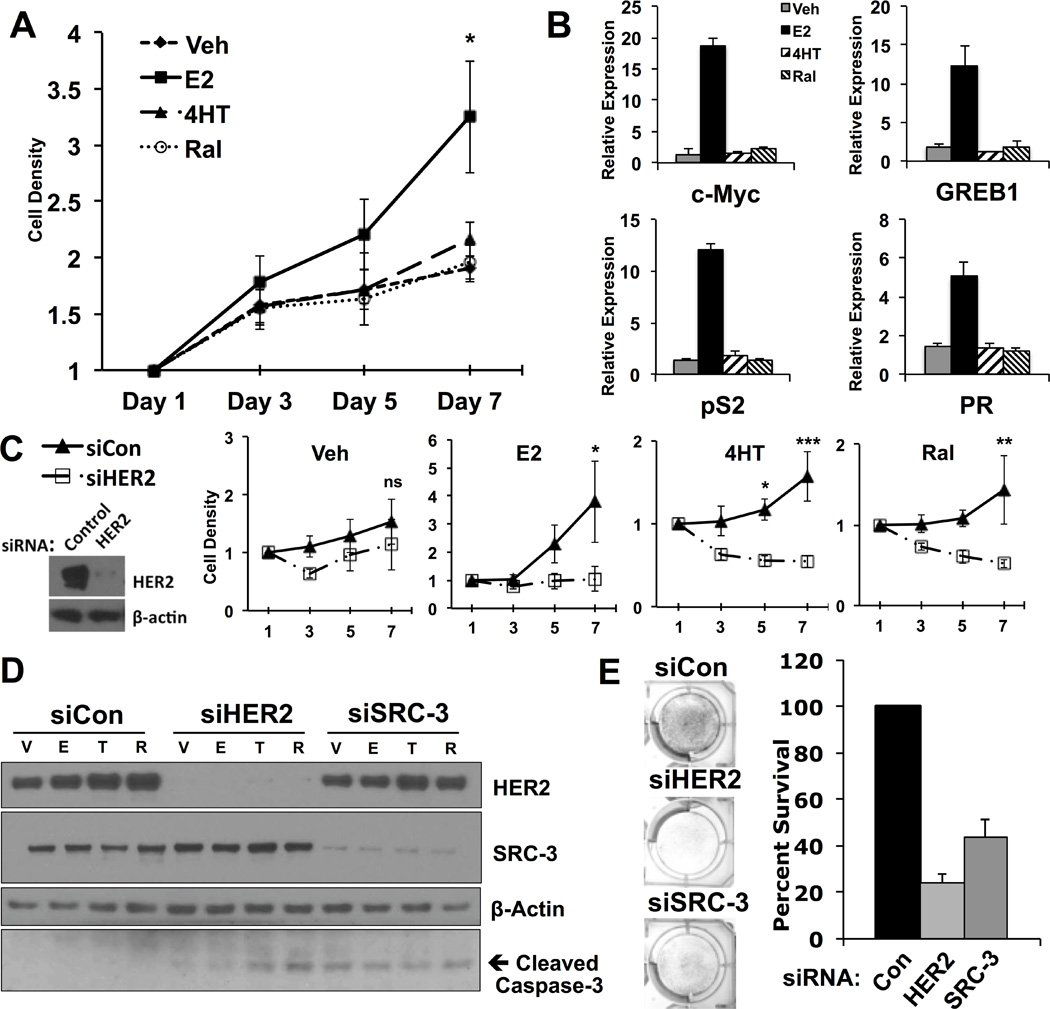
A) BT-474 breast cancer cell growth curve upon treatment with 0.1% ethanol (Veh), 17-β-estradiol (E2), 4-hydroxytamoxifen (4HT) or Raloxifene (Ral).
B) Transcript levels of canonical ERα target genes are elevated upon treatment with E2.
C) Depletion of HER2 using siRNA results in loss of estrogen mitogenic activity and SERM resistance.
D) Induction of SERM-induced apoptosis in BT-474 cells upon knockdown of HER2.
E) Depletion of either HER2 or SRC-3 in BT-474 cells grown in complete media decreases cell viability.
Figure 6. Combinatorial effects of lapatinib and palbociclib on cell survival and E2F1-driven DNA synthesis.
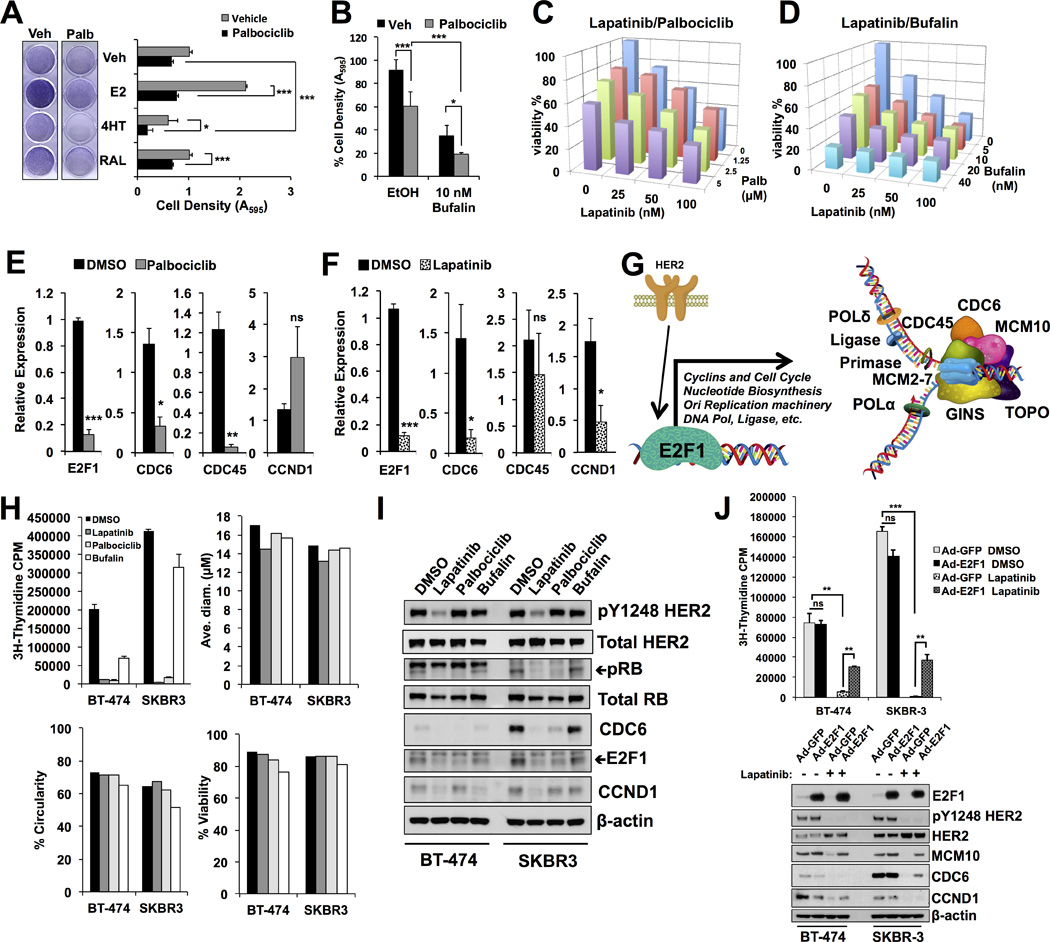
A) BT-474 cells treated with SERMs in the presence of vehicle or palbociclib. Palbociclib reduced the mitogenic effects of estrogen and sensitized the cells to tamoxifen.
B) Cells were treated with palbociclib and the SRC-3 inhibitor bufalin. Palbociclib and bufalin have additive effects. Asterisks denote significance tested using 2-way ANOVA with Bonferroni posttest, *<0.05, **<0.01, ***<0.001.
C) Palbociclib cooperatively sensitizes cells to lapatinib treatment.
D) Bufalin cooperatively sensitizes cells to lapatinib treatment.
E) E2F1 and cell cycle transcripts are reduced upon treatment with palbociclib.
F) Lapatinib reduces E2F1 and cell cycle transcripts analogous to HER2 knockdown.
G) Schematic representation of HER2/E2F1 signaling axis affects on cell cycle, nucleotide metabolism, and DNA replication gene transcription. DNA replication fork genes significantly downregulated upon HER2 knockdown are depicted.
H) Tritiated thymidine (3H) incorporation (DNA replication) in BT-474 and SKBR-3 cells after 24 hour treatment with lapatinib, palbociclib, or bufalin. Note the sharp decrease in DNA synthesis in both cell lines despite any significant effect on cell size, shape, or viability at this time point.
I) Cell cycle proteins E2F1, CCND1, and CDC6 are affected by 24h lapatinib or palbociclib treatment. Phosphorylation of RB and HER2 serve as markers of drug efficacy.
J) Ectopic expression of E2F1 rescues MCM10, CDC6, and CCND1 protein expression and DNA synthesis in the presence of lapatinib in HER2+ breast cancer cells.
Sample preparation, immunoblotting, and RT-PCR
For protein analysis, cells were lysed (20 mM Tris-HCl pH8.0, 150 mM NaCL, 1mM EDTA, and 0.5% NP-40) for one hour at 4 °C, followed by centrifugation at 12,000 × g for ten minutes. The supernatant was removed to a fresh tube and diluted for quantitation using the Bradford method. Samples were separated using NuPAGE 4–12% Bis-Tris gels (Life Technologies) and transferred to nitrocellulose using methanol transfer buffer. Membranes were blocked in 5% fat-free milk reconstituted in PBS plus 0.5% Tween-20 (PBST) for one hour at room temperature. Primary antibodies were diluted in 1% fat-free milk in PBST and blots incubated for several hours at room temperature or overnight at 4°C. Species-specific secondary antibodies conjugated with horseradish peroxidase were used with traditional enhanced chemiluminescense and X-Ray film. Densitometric quantification was performed using ImageJ and numbers in figures represent the area under curve for each band, normalized to actin in the identical lane. TRIzol reagent (low pH phenol, Life Technologies) was used to collect and extract total RNA. RNA concentration was determined using a NanoDrop spectrometer and integrity evaluated using Lab-on-chip (Agilent). Reverse transcription using random hexamer priming (BioRad iScript) was performed before quantitative polymerase chain reaction (qPCR) on ABI OneStep with Roche Universal Library hydrolysis probes. Data were analyzed using the ΔΔCT method (see www.nursa.org/qpcr_tutorials/ for review). Detailed descriptions of primer sequences are listed in Supplementary Table 2.
RNAi, transfections, and luciferase assays
Small-interfering RNA (siRNA) was custom designed and/or ordered as pre-validated duplexes (Life Technologies, see Supplementary Table 2 for details). BT-474 cells were plated on 6-well plates at a density of 4×105 per well one day prior to transfection in reduced serum media (OPTI-MEM, Life Technologies) at a final concentration of 40 nM siRNA. Luciferase reporter and plasmid DNA transfections with siRNA are performed similarly with Lipofectamine 2000 (Life Technologies). Cells were harvested in Tris-EDTA-NaCl (TEN) buffer, pelleted at 3500 × g, and suspended in reporter lysis buffer (Promega, Wisconsin, USA). Reporter activity was measured using a Thermo Luminoskan Ascent. RLU was calculated relative to total protein quantity. Detailed descriptions of reagents and catalog numbers are listed in Supplementary Table 2.
Microarray analysis, motif scanning and bioinformatics
RNA was prepared from BT-474 cells as described above and hybridized to Affymetrix GeneChip® Human Genome U133A 2.0 Array chips by the Genomic and RNA Profiling (GARP) core facility at Baylor College of Medicine and analysis was performed using BRB Array Tools (http://linus.nci.nih.gov/BRB-ArrayTools.html), dChip (http://www.hsph.harvard.edu/cli/complab/dchip), and ArrayAnalyzer (http://www.solutionmetrics.com.au/products/s-plus_arrayanalyzer/default.html). Motif scanning was performed using P-scan Ver. 1.2.2 (http://159.149.160.51/pscan/) with −950 to +50 criteria of matched probesets that were at least two-fold downregulated upon HER2 depletion. Bioinformatics were performed using a collection of resources in the Dan L. Duncan Cancer Center and Department of Molecular and Cellular Biology. Briefly, the Galaxy/Cistrome suite, NCBI Gene ID, and existing data deposits were used in combination with SQL-based database organization to queue and display integrated transcriptome and cistrome data based on expression and protein binding, respectively. Gene ontology was displayed using DAVID and pathway mapping by KEGG. Gene signature analysis involving the Kessler et al(23) and TCGA(24) datasets was carried out as previously described(25). Microarray data is deposited in the Gene Expression Omnibus as accession GSE71347.
Chromatin Immunoprecipitation (ChIP)
ChIP was performed as described with minor modifications (York et al 2012(26)). Briefly, cells were fixed in 1% formaldehyde before quenching with 125 mM glycine. Cells were washed twice with ice-cold TEN buffer and scraped into tubes on ice. Cell pellets were either flash frozen and stored at −80°C or resuspended in 500µL of Tris-EDTA (TE)/1% SDS. All steps beyond this point were done on ice or at 4°C. This suspension was sonicated for 8 cycles of 10 pulses at amplitude 90 and duty cycle 3.5 on a Branson Sonifier 250. Chromatin extracts were cleared by centrifugation and diluted 1:10 in a buffer containing 20 mM Tris-HCl (pH8.0), 150 NaCl, 2 mM EDTA, and 1% Triton-X-100 to reduced SDS concentration. Four micrograms of normal rabbit IgG antibody (Santa Cruz) was used with 50 µL Protein A/G Agarose plus salmon sperm DNA (Millipore) to preclear the diluted chromatin for one hour. The cleared supernatant was incubated with 4 µg SRC-3 antibody (Cell Signaling 5E11) or control IgG overnight. Agarose beads were used to precipitate immune complexes for one hour before washing in buffers of increasing stringency as follows: 1X in 20 mM Tris-HCl (pH 8.0), 150 mM NaCl, 2 mM EDTA, and 1% Triton X-100; 1X in 20 mM Tris-HCl (pH 8.0), 500 mM NaCl, 2 mM EDTA, and 1% Triton X-100; 1X in 10 mM Tris-HCl (pH 8.0), 1 mM EDTA, 1% deoxycholate, 1% NP-40, and 0.25 M LiCl; and 2X with TE. Beads were centrifuged at 1000 × g for one minute to pellet. DNA was eluted from beads in 100 mM NaHCO3 and 1% SDS at 65°C overnight. Extraction was performed using PCR product purification columns (Qiagen).
Tritium(3H)-labeled thymidine incorporation
Cells were plated on 6-well plates at a density of 2×105 per well and treated with lapatinib, palbociclib, or bufalin for 24 hours in normal growth media. Four µC of 3H-labeled thymidine (Perkin Elmer) was added to the media and cells were grown for two additional hours. Media was removed and cells were fixed for 5 minutes with 25% acetic acid in methanol, rinsed once with methanol, and protein precipitated using 5% trichloric acid for 5 minutes. Wells were washed an additional 3 times with methanol and allowed to dry. Radioactivity was recovered using 1N NaOH and equilibrated with an equal volume of 1N HCl before addition of biodegradable counting scintillant and liquid scintillation using a Beckman scintillation counter. For E2F1 rescue experiments, cells were plated on 12-well plates at a density of 5×104 per well and infected using 25 virions per cell for 24 hours, then treated with lapatinib for 24 hours followed by 2 µC of 3H–thymidine treatment for two hours and processing as described.
Results
BT-474 cells display HER2-dependent estrogen responsiveness and resistance to anti-estrogens
Resistance to selective estrogen receptor modulators (SERMs) in HER2+ breast cancer is incompletely understood. Specifically, whether HER2 expression is required for sensitivity to estrogen and SERMs in HER2-amplified cells remains unanswered. The BT-474 breast cancer cell line represents the ideal model system to study effects of endogenously amplified HER2 and SRC-3 oncogenes on estrogen signaling since it is also positive for ERα expression (Anzick et al(4) and data not shown). We first tested the mitogenic activity of 17β-estradiol (E2) and effects of the SERMs 4-hydroxytamoxifen (4HT) and raloxifene (Ral) on cell growth and gene expression. Cell density was significantly increased by E2 relative to vehicle treatment, while tamoxifen and raloxifene failed to decrease cell density relative to the vehicle control, indicating resistance (Figure 1A). Likewise, transcript levels of canonical ERα-target genes were significantly induced upon E2 treatment, but not affected by treatment with either SERM (Figure 1B). Upon HER2 knockdown, E2 is no longer mitogenic in BT-474 cells (Figure 1C, E2 panel). Moreover, siRNA against HER2 significantly sensitized cells to treatment with 4HT or Ral (Figure 1C, Figure 4HT and Ral panels, note the decrease in cell density over time in siHER2 transfected cells). These results confirm that BT-474 cell growth requires HER2 expression for estrogen sensitivity and SERM resistance, and indicate that HER2 protects cells from SERM-induced apoptosis. Indeed, when HER2 is depleted using siRNA, caspase-3 is cleaved (activated) upon treatment with 4HT or Ral, but not vehicle or estrogen (Figure 1D, compare T and R to V or E in siHER2 lanes). The action of SERMs is postulated to occur, at least in part, through disruption of the interaction between ERα and its transcriptional coactivators, such as SRC-3(27). Depletion of SRC-3 also induced caspase-3 activation (Figure 1D) and decreased survival of breast cancer cells grown in steroid replete media (Figure 1E), demonstrating a functional dependency on both HER2 and SRC-3 survival pathways in this cell model.
Figure 4. Cooperative regulation of Cyclin D1 by HER2 and SRC-3.
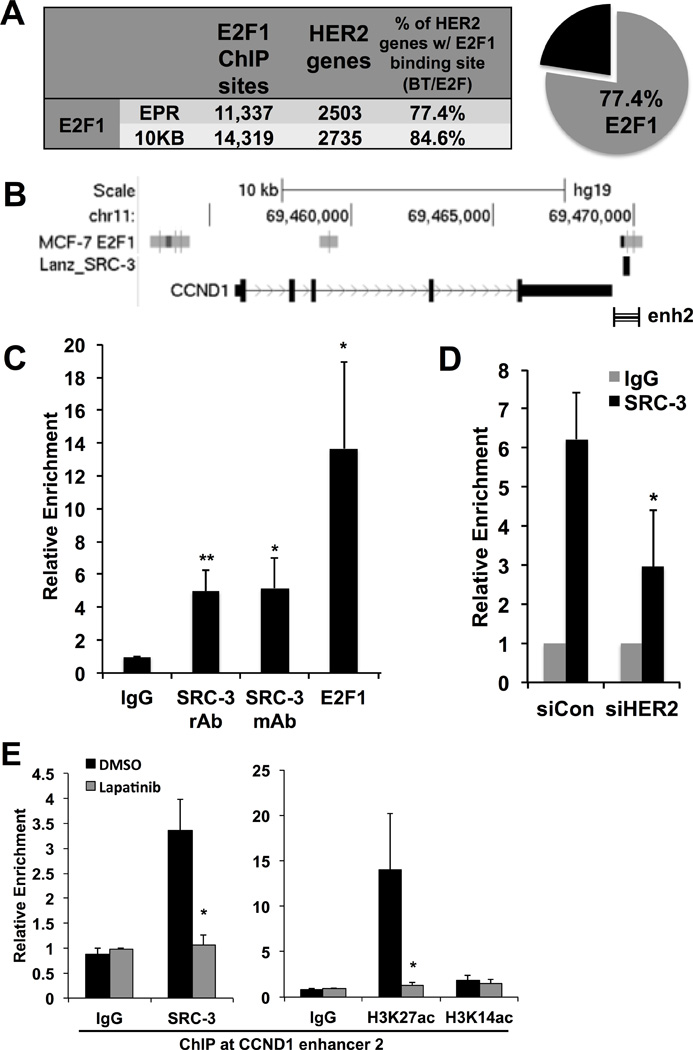
A) E2F1 ChIP-seq peaks were called from published resources and aligned to hg19 for parsing with HER2-regulated genes from the microarray. The pie graph indicates a majority of HER2-regulated genes contained at least one E2F1 binding site in the extended promoter region (EPR).
B) E2F1 and SRC-3 binding from published ChIP-seq at the enhancer 2 (enh2) region of CCND1 (striped bar).
C) E2F1 and SRC-3 binding by ChIP assay to the CCND1 enhancer region.
D) HER2 depletion decreases SRC-3 binding to the CCND1 enhancer.
E) Lapatinib treatment reduces CCND1 transcript levels and dismisses SRC-3 and H3K27ac from the CCND1 enhancer.
HER2 signaling affects SRC-3 phosphorylation and activity
Steroid Receptor Coactivator-3 is a potent coactivator for numerous transcription factors involved in breast cancer and the preference of SRC-3 for, and activity with, different transcription factors is influenced by combinatorial phosphorylation(15). To determine if HER2 signaling impacts SRC-3 phosphorylation and function, siRNA was used to deplete HER2 in BT-474 cells. Knockdown of HER2 results in decreased SRC-3 phosphorylation at T24, S543, S857, and S860 (Figures 2A–C). Importantly, this loss of phosphorylation does not appear to be uniform for SRC-3 protein since S505 phosphorylation remains unchanged upon HER2 depletion (Figure 2A). These data are consistent with a lack of change in total SRC-3 protein levels upon HER2 knockdown, as S505 has been described to function as part of a phospho-degron(28).
Figure 2. HER2 affects phosphorylation and activity of SRC-3.
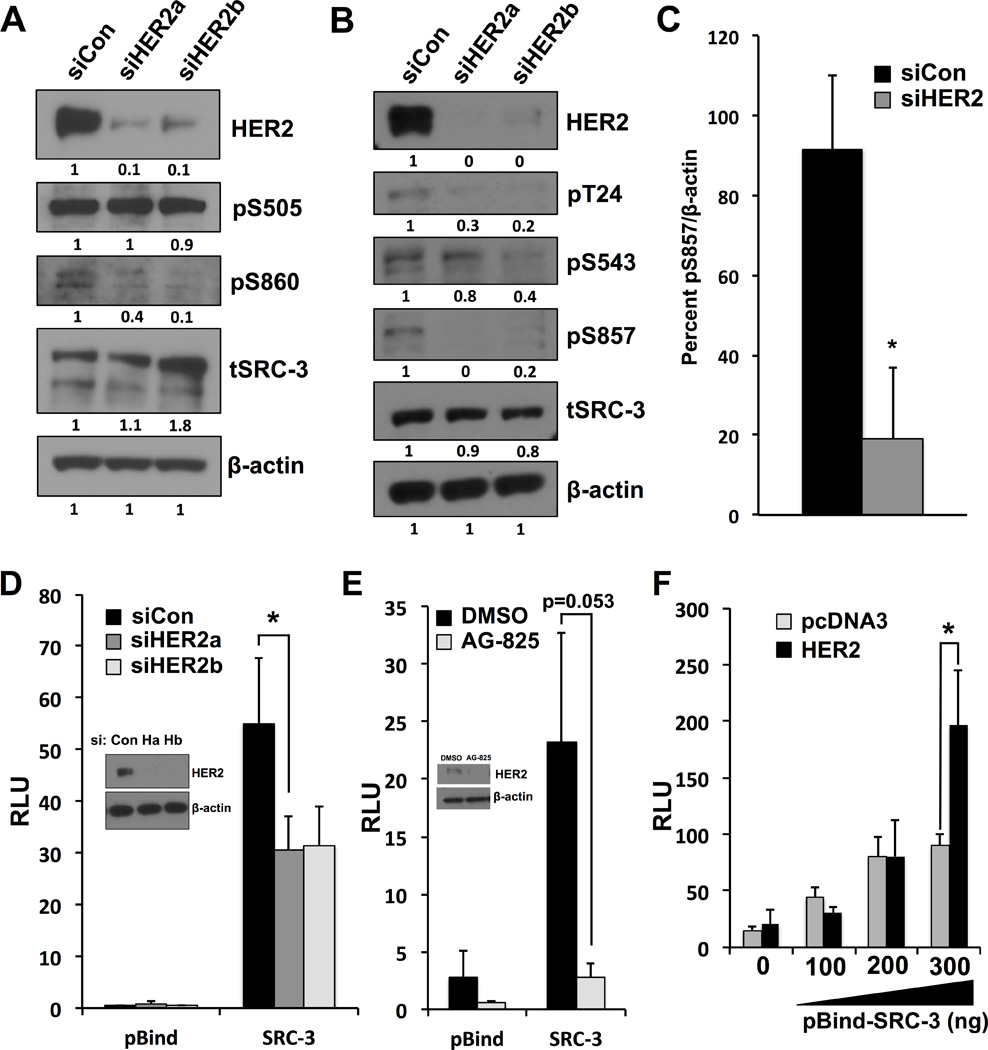
A) and B) BT-474 cells were transfected with siRNA targeting different regions of HER2 transcript and cell lysates were immunoblotted using phospho-specific SRC-3 antibodies. SRC-3 phosphorylation is decreased upon HER2 depletion at T24, S543, S860 and S857, while phosphorylation of S505 is unaltered.
C) Densitometry quantification of S857 phosphorylation.
D) BT-474 cells transfected with pBind-SRC-3/GAL4-Luciferase reporter system and siRNA targeting HER2. Knockdown of HER2 using two separate siRNAs decreases intrinsic SRC-3 activity.
E) AG-825 (Tyrphostin) inhibition of HER2 decreases pBind-SRC-3 activity.
F) Cotransfection of HER2 relieves the activation plateau of pBind-SRC-3 in HeLa cells. Asterisks denote p values <0.05 for all experiments.
Since SRC-3 does not directly bind DNA, a system employing a GAL4-DNA binding domain fused to the N-terminus of SRC-3 (pBIND-SRC-3) and a pGL5-Luciferase reporter was used to interrogate whether HER2 influences intrinsic SRC-3 activity. Upon HER2 knockdown, intrinsic SRC-3 activity is significantly reduced (Figure 2D). These results can be pharmacologically recapitulated using an inhibitor of HER2, AG-825 (tyrphostin) (Figure 2E). Additionally, transient transfection of HER2 is sufficient to enhance SRC-3 transcriptional activity (Figure 2F, compare 200 vs 300 ng of pBind-SRC-3). These data corroborate previous studies suggesting a role for SRC-3 in HER2 tumorigenesis and highlight the ability of this oncogenic signaling pathway to elicit strong effects on transcriptional processes. Taken together, our results show that HER2 signaling has specific effects on SRC-3 phosphorylation and is capable of increasing the intrinsic activity of SRC-3.
HER2 depletion drastically alters the transcriptional landscape
The transcriptional consequences of HER2 action undoubtedly play a significant role in its oncogenic activity, but relatively few studies have comprehensively investigated the transcriptional targets of HER2 signaling. To define the HER2-regulated transcriptome, we performed microarray analysis on cells transfected with control or HER2 siRNA (Figure 3A). HER2 knockdown decreased AKT and Raf phosphorylation, indicating disruption of the downstream HER2 signaling cascade (Figure 3B). Substantial transcriptional changes upon HER2 depletion are evident in the heat map shown in Figure 3C. Gene Ontology analysis of transcripts down-regulated upon HER2 depletion indicated an impressive decrease of transcripts involved in cell cycle and mitotic processes (Figure 3D), and RT-PCR confirmed loss of transcripts involved in S-phase DNA replication, Ras signaling, and transcription factor genes (Supplementary Fig. S1A). Moreover, parsing this HER2-regulated transcriptome with gene signatures associated with Ras/Raf/MAPK or PI3K/AKT signaling revealed significant overlap and divergence between these signaling pathways (Supplementary Fig. S1B–D). Confirming a role for cell cycle disruption when HER2 is depleted, a reduction in G1 to S phase transition is observed using flow cytometry to analyze DNA content (Supplementary Fig. S2).
Figure 3. The HER2-regulated transcriptome reveals loss of genes involved in cell cycle regulation downstream of HER2/SRC-3/E2F signaling axis.
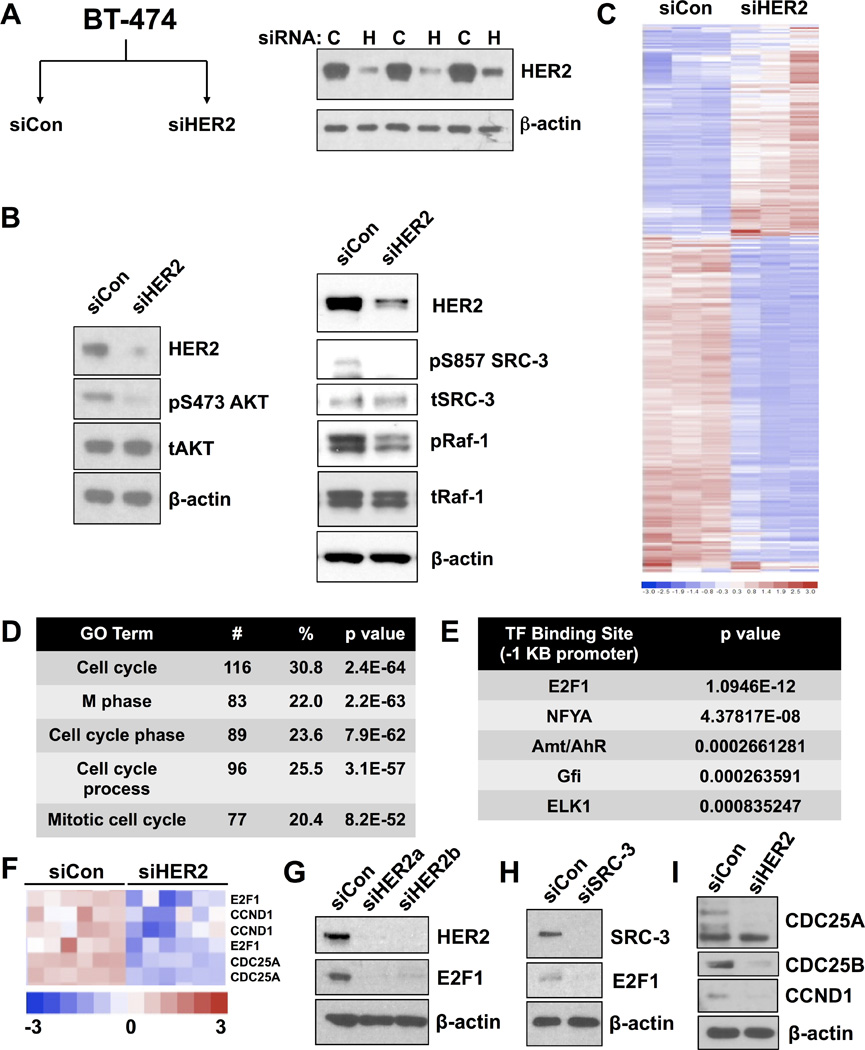
A) Schematic of microarray experimental groups and HER2 knockdown in biological triplicates (inset immunoblot).
B) HER2 knockdown results in disruption of AKT and Ras/Raf signaling cascades, as evidenced by decreased AKT and c-Raf phosphorylation. Decrease of SRC-3 phosphorylation was consistently observed upon HER2 knockdown.
C) Heat map of significantly altered probe sets across treatment groups.
D) Top GO terms returned from HER2-downregulated gene list specifies cell cycle and mitotic processes.
E) Predicted E2F1 binding sites are enriched in the promoter region of HER2-downregulated genes.
F) Enlarged heat map showing decreased levels of E2F1CCND1, and CDC25A upon HER2 knockdown.
G) Knockdown of HER2 reduces E2F1 protein expression.
H) Knockdown of SRC-3 reduces E2F1 protein expression.
I) Protein levels of genes from (F) with control and HER2 siRNA transfection in BT-474 cells.
Next, we compared genes that were down-regulated upon HER2 knockdown with publicly available breast cancer gene expression patterns in The Cancer Genome Atlas (TCGA) in relation to HER2 and SRC-3 (NCOA3) expression levels (Supplementary Fig. S3). Strikingly, tumor samples with the highest expression of HER2 (Supplementary Fig. S3, top) or SRC-3 (Supplementary Fig. S3, bottom) also displayed high expression of HER2-sensitive genes identified by microarray upon HER2 depletion. Jointly considered, these data indicate that loss of HER2 in breast cancer cells results in a substantial decrease of transcripts involved in cellular proliferation that relates to expression of HER2 and SRC-3 in a large dataset of breast tumor samples.
Since HER2 is present at extracellular membranes and thus acts as an indirect effector of nuclear transcription, we next investigated transcription factor binding motifs in the proximal promoter regions of down-regulated genes. Interrogation of one kilobase upstream promoter sequences of putative target genes identified an E2F1 consensus sequence as the most significantly enriched transcription factor motif (Figure 3E). Transcript levels for E2F1 and its target genes CCND1 and CDC25A have similar expression patterns in our microarray analysis (Figure 3F) and HER2 knockdown depletes the protein levels of these gene products (Figures 3G–I). Upon HER2 knockdown, E2F1 protein levels were abolished (Figure 3G), which also was observed upon SRC-3 knockdown, indicating that HER2 and SRC-3 signaling likely converge on E2F1 target genes (Figure 3H). Quantitative RT-PCR screening of HER2 target genes revealed that transcripts for additional mitogenic transcription factors FOXM1 and ETV4, as well as the G1 to S check point gene CCND1, are significantly decreased in cells transfected with siRNA against SRC-3 or HER2 (Supplementary Fig. S4C). These results suggest that depletion of HER2 or SRC-3 reduces the expression of multiple transcription factors, including E2F1, which are known to regulate the transcription of genes involved in mitotic progression and proliferation(29–31).
A majority of genes regulated by HER2 contain E2F1 binding sites
All E2F family gene transcripts except E2F4 and E2F6 were decreased upon HER2 knockdown in the microarray, and E2F7 and E2F8 were among the most depleted transcripts in the entire array (Table 1 and Supplementary Fig.S3). Given the large number of cell cycle and mitotic genes downregulated upon HER2 depletion and the enrichment for E2F1 motifs, we investigated E2F1 transcription factor binding to DNA regulatory elements of HER2-regulated genes. Binding sites for E2F1 from published chromatin immunoprecipitation-sequencing (ChIP-seq) in breast cancer cells(32) were compared to transcripts from our BT-474 siHER2 microarray. Briefly, chromosomal coordinates of significantly down-regulated genes were compared to E2F1 binding site intervals within the extended promoter region (EPR, −7.5 to +2.5 Kb) or anywhere within the gene and including 10 Kb gene-flanking regions (10Kb). A striking majority of genes regulated by HER2 (>75%) in our microarray are bound by E2F1 (Figure 4A). Atypical E2F7 and E2F8 ChIP-seq binding in cancer cells also shows significant overlap with the HER2 gene signature, and E2F8 may be a HER2 and SRC-3 target gene (Supplementary Fig. S5A–C). Transcript and protein levels of E2F7 and E2F8 were reduced with siRNA transfection of siHER2 or siSRC-3 (Supplementary Fig. S5D–F). These results are intriguing considering a proposed role of atypical E2Fs in maintenance of genome size and cancer, but E2F7 and E2F8 function was not further explored in this study(33,34). Additionally, HER2-regulated genes with E2F1 promoter binding sites displayed overlap with PI3K/AKT and Ras/Raf/MAPK gene signatures (Supplementary Fig. S6A).
Table 1. HER2 regulates DNA anabolism and replication.
Selection of genes from enriched gene ontology terms involved in nucleotide synthesis, DNA replication fork formation, and replication processivity. Numbers represent log2 fold-change with negative values indicating decrease upon HER2 knockdown and positive numbers indicating increased expression. Boxed gene symbols are validated targets representative of a larger network of transcriptional targets downstream of HER2 signaling.
 |
The CCND1 gene is flanked by E2F1 binding sites including a known SRC-3 binding element downstream, colloquially termed enhancer 2 (enh2)(35) (Figure 4B). This enhancer region is bound by E2F1 and SRC-3 in BT-474 cells (Figure 4C) and upon HER2 knockdown, SRC-3 recruitment to CCND1 enh2 is significantly reduced (Figure 4D). Moreover, treatment with the HER2 inhibitor lapatinib results in decreased SRC-3 recruitment and H3K27 acetylation (but not H3K14ac) is lost from the enhancer region (Figure 4E). Taken together, these data show that HER2-mediated gene regulation involves mechanisms of E2F1 and SRC-3 recruitment to gene regulatory elements involved in mitogenic expression of CCND1.
The HER2 transcriptome signature correlates with E2F1 and proliferative breast cancer subtypes
BT-474 cells have previously been characterized as a Luminal B subtype model for breast cancer owing to HER2 amplification and ERα expression(36), and we observe responsiveness to estrogen but HER2-dependent resistance to anti-estrogens (see Figure 1A). As suggested from cistromic and transcriptomic analyses, comparing our HER2 gene signature expression scores with two large breast tumor datasets reveals a strong correlation with E2F1 expression (Table insets, Figures 5A–B). Additionally, the HER2-regulated gene signature correlates with the most proliferative breast cancer subtypes: basal, HER2+, and luminal B (Figures 5A–B, histograms). Tumors considered to be slow growing (luminal A and normal-like) display a negative correlation with our HER2 gene signature, as do the predictors of luminal A subtype, ERα and PR (Figure 5). Collectively, gene signatures from patient samples are consistent with our in vitro findings and indicate that HER2 signaling mediates a proliferative regulatory gene network in breast cancer.
Figure 5. HER2-regulated transcriptional signature of BT-474 cells strongly correlates with E2F1 and proliferative breast cancer subtypes.
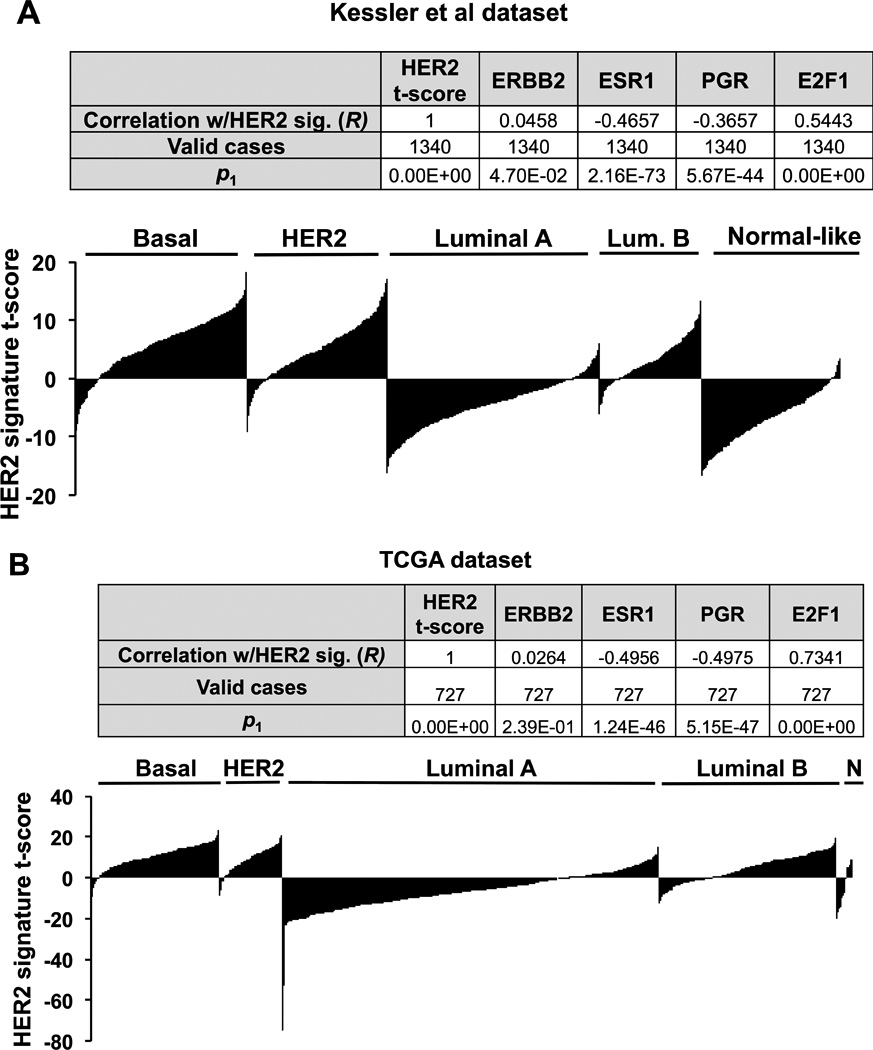
Genes up or down regulated by HER2 depletion were inversely correlated with expression signatures from two large breast cancer datasets from Kessler et al (A) and TCGA (B). ER and PR are negatively correlated with the HER2 signature and E2F1 displays a highly significant positive correlation, as expected from motif analysis in Figure 4. Breast cancer subtypes that correlated with the HER2 gene signature (Basal, HER2+, and Luminal B) are generally considered to have a high proliferative index.
Perturbation analysis(37) of HER2 signature genes with E2F1 binding sites using functional annotation with the Genomic Regions Enrichment Annotation Tool (GREAT(37)) reveals a significant enrichment of oncogenic gene networks in diverse cancer types, including resistance to gefitinib (Supplementary Fig. S6B). Importantly, CCND1 is involved in breast cancer sensitivity to the HER/EGFR family inhibitors gefitinib, lapatinib, and afatinib (Genomics of Drug Sensitivity in Cancer (GDSC(38))). Taken in context, these results suggest that HER2/E2F1 signaling regulates Cyclin D1 and other genes that may predict sensitivity to therapy in proliferative breast cancer subtypes.
Cyclin D1/CDK crosstalk with HER2 converges on transcriptional regulation of cell cycle genes
Proliferation is critical in the etiology and associated pathogenicity of cancer. Individual oncogenes and tumor suppressors do not easily define the batteries of genes that regulate proliferation per se, since cell mitosis, DNA replication, and escape from differentiation pathways involve large gene networks working in concert. Importantly, since the tumor has become dependent on cellular events resulting from amplification or mutation of such genes to maintain a proliferative advantage, these genes represent fragility in the network of cancer cell signaling(2,39). Indeed, this has become the basis for directed therapies targeting oncogenic signaling from tyrosine kinases HER2 and EGFR(2).
We used network analysis of CCND1 and another HER2/E2F1 regulated gene from the microarray, CDC6, to identify a fragile component of this signaling network amendable to pharmacological targeting. Network analysis of CCND1 and CDC6 revealed strong overlap with the HER2 transcriptome. Remarkably, 50% of genes in the CCND1-associated network, and 59% of CDC6 network genes are regulated by HER2 in our microarray (Supplementary Table 1). Not surprisingly, members of the Cyclin Dependent Kinase (CDK) family and their regulators are enriched in the CCND1 and CDC6 networks (Supplementary Fig. S7 (40)), which is consistent with KEGG pathway analysis. Since CDK4 and CDK6 function with CCND1 is well described and recent clinical trials have reported optimistic results and recent FDA approval of the CDK4/6 specific inhibitor palbociclib(41) (PD 0332991, Pfizer), we questioned whether this drug could similarly alter cell cycle gene transcription and sensitize breast cancer cells to other pharmacological agents. As shown in Figure 6A, palbociclib prevented the mitogenic effects of estradiol similar to knockdown of HER2 (see Figure 1C). Additionally, combination treatment of palbociclib with 4HT significantly reduced cell survival (Figure 6A).
Integrators of cellular signaling, such as transcriptional coactivators, may represent an additional “weak link” (and therefore a therapeutic target) in cancer gene regulation networks owing to their pleiotropic effects on multiple proliferative pathways. When palbociclib treatment is used in combination with bufalin, a recently described inhibitor of SRC-3(42), we observe a significant combinatorial effect on the decrease in cell survival (Figure 6B). Similarly, cellular sensitivity to lapatinib is augmented by palbociclib or bufalin in a dose-dependent manner (Figures 6C–D). Notably, bufalin is active in the nanomolar range and sensitizes cells to lower concentrations of lapatinib (Figure 6D). Similar results are observed in the HER2+/ERα- breast cancer cell line SKBR-3, suggesting these pathways signal cooperatively in other cell models (Supplementary Fig. S8). Palbociclib treatment reduced transcript levels of cell cycle genes E2F1, CDC6, CDC45, but not CCND1 (Figure 6E). Transcript levels of E2F1, CDC6, CCND1, but not CDC45 are depleted upon lapatinib treatment, suggesting HER2 pathway converges on similar transcriptional targets as CDK signaling (Figure 6F). Additional genes involved in cell cycle regulation were decreased upon palbociclib (i.e. E2F7, E2F8, and MCM10) or lapatinib (i.e. E2F7, E2F8, and ETV4) treatment, indicating that these compounds influence broad cell cycle transcriptional networks (Supplementary Fig. S9A–B). Treatment with lapatinib also decreases protein levels of CCND1, CDC6, CDC45, MCM10, and ribonucleotide reductase (RRM2), a rate-limiting enzyme in DNA nucleoside metabolism (Supplementary Fig. S9C).
Acute lapatinib or palbociclib treatment restricts DNA synthesis in HER2+ breast cancer cells
Given the broad effect of HER2 and CDK signaling pathways on transcriptional regulation of genes involved in cell cycle and DNA replication (Figure 6G), we reasoned that pharmacologically blocking HER2 or CDK should also block replicative synthesis of DNA in HER2+ breast cancer cells. Indeed, lapatinib potently restricts DNA synthesis at low nanomolar concentrations after 24 hours of treatment, before any major changes occur to cellular health (Supplementary Fig. S9D–G) but concomitant with decreased phosphorylation of HER2 and cell cycle protein expression (Supplementary Fig. S9H). Tritiated (3H)-thymidine incorporation to directly measure DNA synthesis revealed that acute (24 hour) treatment with lapatinib or palbociclib blocks de novo DNA replication, without affecting cellular size, shape, or viability (Figure 6H). Bufalin treatment blocks DNA synthesis to a lesser extent (Fig. 6H), suggesting the combined effect of bufalin with lapatinib or palbociclib on cellular viability (see Figs. 6B and 6D) is consistent with its mechanistic role of inducing apoptosis(42). Consistent with the effect of lapatinib and palbociclib on transcription, protein levels of E2F1, CDC6, and CCND1 are reduced in BT-474 and SKBR-3 breast cancer cells after acute treatment (Figure 6I). Moreover, the lapatinib-induced reduction of replication fork proteins and block of DNA replication can be rescued by ectopic expression of E2F1 (Figure 6J). Collectively, these results demonstrate that HER2 and CCND1/CDK signaling pathways regulate DNA replication through E2F1 and its associated transcriptional targets, many of which are directly involved in nucleotide biosynthesis and replicative processivity. Moreover, our findings demonstrate the potential for combined targeting of HER2 and CDK signaling pathways or DNA replication may be a rational model that warrants further investigation in preclinical studies.
Discussion
We describe an E2F1 transcriptional network downstream of HER2 signaling that is influenced by SRC-3 phosphorylation in breast cancer cells. Overexpression of SRC-3 contributes to the etiology and progression of breast cancer and mice lacking SRC-3 are resistant to oncogene and carcinogen-induced breast tumorigenesis(21,43,44). Moreover, SRC-3 is a coactivator for E2F1 transcription resulting in breast cancer cell proliferation and anti-estrogen resistance(6). However, the involvement of E2F1 in HER2-driven transcriptional programs in breast cancer is relatively understudied. Recently, several studies have reported E2F activators (typically considered to be E2F1–3) in the context of HER2/neu, c-Myc, or Ras oncogene-induced breast tumors in mouse models(45,46). Andrechek and Wu et al found that E2f1 and E2f3 (and to a lesser extent E2f2) knockout mice are protected from HER2/neu tumorigenesis and E2F1–3 transcripts are coordinately elevated in human tumors with high HER2 expression(45,46). Moreover, E2F1 is known to be involved in a feed forward auto-regulatory loop with SRC-3(47,48). These reports are in agreement with our findings and highlight the intricacies of a proliferative and anti-apoptotic E2F1 transcriptional network downstream of oncogenic HER2 activity in breast cancer cells that has been underappreciated. More research is needed to understand auto-regulatory mechanisms and crosstalk between all members of the E2F family. Moreover, it will be critical to investigate how oncogenes such as SRC-3 and HER2 hijack E2F-mediated gene regulation to achieve wanton DNA replication and cellular proliferation.
Crosstalk and overlap involving HER2 and estrogen signaling have been extensively studied with implications for resistance to endocrine and adjuvant therapies in breast cancer(49). Mechanisms proposed include phosphorylation-mediated export of ERα from the nuclear compartment, reduction of ERα expression, and high expression of SRC-3(14). Importantly, the genomic loci for HER2 and SRC-3 in the BT-474 cells used in this study are amplified, while maintaining ERα expression and estrogen sensitivity despite SERM resistance (Anzick et al(4) and Figure 1). This resistance is dependent on HER2 expression, and cells undergo caspase-mediated apoptosis upon HER2 depletion and treatment with tamoxifen or raloxifene. Interestingly, BT-474 cells devoid of HER2 fail to respond to the proliferative estrogen signal, indicating substantial coordination between these two pathways in breast cancer cells. Considering that 70% of breast tumors are ER+, future studies should focus on determining the factors regulated by estrogen in the context of HER2 signaling, and what gene programs are responsible for proliferation and cellular immortality afforded by HER2 and ERα expression. Indeed, it is likely that the HER2/E2F1 gene signature described here may relate to estrogen activity(50).
The transcriptional consequences of oncogenic signaling pathways have increasingly become a target of study since downstream gene regulation appears to define tumor subtypes and pathogenicity. We present a model whereby HER2 and E2F1 cooperate, in part through SRC-3, to regulate DNA replication and cellular proliferation (Supplementary Fig. S10). HER2 signaling affects SRC-3 phosphorylation and activity, as well as a vast E2F1 transcriptional program that grants promiscuous DNA replication and induces the hallmarks of cellular transformation. An individual oncogene (or drug target) responsible for affecting tumor cell proliferation, evasion of apoptosis, and resistance to anti-estrogens or therapeutic drugs is unlikely. In contrast, coregulators and associated transcription factors serve as nodal integrators of cellular signaling pathways, and drive transcription of gene networks required for oncogenesis and tumorigenicity. It is likely that diverse oncogenic signaling pathways employ discrete coactivators as critical signaling integrators, perhaps in a tissue-specific manner. Importantly, these coactivators may represent a unique class of targets to sensitize tumors to existing therapies.
Therapy options for breast and other cancers are ever evolving and preclinical investigations in tractable models will be informative for hypothesis-driven clinical trials. The data presented in the current study unveils previously unappreciated interplay between known oncogenic and proliferative genes and an anabolic DNA replication pathway employed to drive cell growth. Importantly, we show that combinations of treatments (siRNA, SERMs, small-molecule inhibitors) can increase sensitivity to inhibitors of alternate pathways of gene regulation that normally lead to treatment-resistant growth and cell survival. Moreover, we identify anabolic metabolism of DNA as an important downstream effect of the HER2 oncogene. Future studies should be directed at defining transcriptional and cistromic mechanisms involved in therapy resistance and benefit (or lack thereof) in targeting multiple pathways.
Supplementary Material
Acknowledgments
We thank Jung-Sun Kim, Ed Bingman, Nancy Weigel, Charles Foulds, Ping Yi, Qin Feng, Sophia Tsai, and Ming-Jer Tsai for technical assistance and helpful discussion. Additionally, we are grateful to Cheryl Parker and the Cell Culture Core facility at BCM for technical assistance and Travis Willis of Sivart Graphics (Houston, TX) for graphic design. This project was supported in part by the Genomic and RNA Profiling Core at Baylor College of Medicine with funding from the NIH NCI grant (P30CA125123) and the expert assistance of Dr. Lisa D. White, Ph.D. and the Cytometry and Cell Sorting Core at Baylor College of Medicine with funding from the NIH (P30 AI036211, P30 CA125123, and S10 RR024574) and the expert assistance of Joel M. Sederstrom. BCN was supported by pre-doctoral training grants through the Endocrine Society and Welch Foundation. NIH (HD8818) grant to B.W. O’Malley provided the primary support for this work.
Financial support: This work was supported by NIH grant (HD8818) and Cancer Prevention and Research Institute of Texas grants (RP100348 and RP101251) to B.W. O’Malley.
Footnotes
Conflict of interest disclosure statement: The authors have nothing to declare.
References
- 1.Arteaga CL, Sliwkowski MX, Osborne CK, Perez EA, Puglisi F, Gianni L. Treatment of HER2-positive breast cancer: current status and future perspectives. Nat Rev Clin Oncol. Nature Publishing Group, a division of Macmillan Publishers Limited. All Rights Reserved. 2012;9:16–32. doi: 10.1038/nrclinonc.2011.177. [DOI] [PubMed] [Google Scholar]
- 2.Citri A, Yarden Y. EGF-ERBB signalling: towards the systems level. Nat Rev Mol Cell Biol. 2006;7:505–516. doi: 10.1038/nrm1962. [DOI] [PubMed] [Google Scholar]
- 3.Arteaga CL, Sliwkowski MX, Osborne CK, Perez EA, Puglisi F, Gianni L. Treatment of HER2-positive breast cancer: current status and future perspectives. Nat Rev Clin Oncol. 2012;9:16–32. doi: 10.1038/nrclinonc.2011.177. [DOI] [PubMed] [Google Scholar]
- 4.Anzick SL, Kononen J, Walker RL, Azorsa DO, Tanner MM, Guan X-Y, et al. AIB1, a Steroid Receptor Coactivator Amplified in Breast and Ovarian Cancer. Science. American Association for the Advancement of Science. 1997;277:965–968. doi: 10.1126/science.277.5328.965. [DOI] [PubMed] [Google Scholar]
- 5.Goel A, Janknecht R. Concerted activation of ETS protein ER81 by p160 coactivators, the acetyltransferase p300 and the receptor tyrosine kinase HER2/Neu. J Biol Chem. 2004;279:14909–14916. doi: 10.1074/jbc.M400036200. [DOI] [PubMed] [Google Scholar]
- 6.Louie MC, Zou JX, Rabinovich A, Chen H-W. ACTR/AIB1 functions as an E2F1 coactivator to promote breast cancer cell proliferation and antiestrogen resistance. Mol Cell Biol. 2004;24:5157–5171. doi: 10.1128/MCB.24.12.5157-5171.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu R-C, Qin J, Hashimoto Y, Wong J, Xu J, Tsai SY, et al. Regulation of SRC-3 (pCIP/ACTR/AIB-1/RAC-3/TRAM-1) Coactivator activity by I kappa B kinase. Mol Cell Biol. 2002;22:3549–3561. doi: 10.1128/MCB.22.10.3549-3561.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xie D, Sham JST, Zeng W-F, Lin H-L, Bi J, Che L-H, et al. Correlation of AIB1 overexpression with advanced clinical stage of human colorectal carcinoma. Hum Pathol. 2005;36:777–783. doi: 10.1016/j.humpath.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 9.He L-R, Zhao H-Y, Li B-K, Zhang L-J, Liu M-Z, Kung H-F, et al. Overexpression of AIB1 negatively affects survival of surgically resected non-small-cell lung cancer patients. Ann Oncol. 2010;21:1675–1681. doi: 10.1093/annonc/mdp592. [DOI] [PubMed] [Google Scholar]
- 10.Gnanapragasam VJ, Leung HY, Pulimood AS, Neal DE, Robson CN. Expression of RAC 3, a steroid hormone receptor co-activator in prostate cancer. Br J Cancer. Cancer Research Campaign. 2001;85:1928–1936. doi: 10.1054/bjoc.2001.2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Henke RT, Haddad BR, Kim SE, Rone JD, Mani A, Jessup JM, et al. Overexpression of the Nuclear Receptor Coactivator AIB1 (SRC-3) during Progression of Pancreatic Adenocarcinoma. Clin Cancer Res. American Association for Cancer Research. 2004;10:6134–6142. doi: 10.1158/1078-0432.CCR-04-0561. [DOI] [PubMed] [Google Scholar]
- 12.Xu J, Liao L, Ning G, Yoshida-Komiya H, Deng C, O’Malley BW. The steroid receptor coactivator SRC-3 (p/CIP/RAC3/AIB1/ACTR/TRAM-1) is required for normal growth, puberty, female reproductive function, and mammary gland development. Proc Natl Acad Sci U S A. National Acad Sciences. 2000;97:6379–6384. doi: 10.1073/pnas.120166297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kirkegaard T, McGlynn LM, Campbell FM, Muller S, Tovey SM, Dunne B, et al. Amplified in Breast Cancer 1 in Human Epidermal Growth Factor Receptor–Positive Tumors of Tamoxifen-Treated Breast Cancer Patients. Clin Cancer Res. American Association for Cancer Research. 2007;13:1405–1411. doi: 10.1158/1078-0432.CCR-06-1933. [DOI] [PubMed] [Google Scholar]
- 14.Osborne CK, Schiff R. Mechanisms of Endocrine Resistance in Breast Cancer. Annu Rev Med. 2011;62:233–247. doi: 10.1146/annurev-med-070909-182917. Annual Reviews. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu RC, Qin J, Yi P, Wong J, Tsai SY, Tsai MJ, et al. Selective phosphorylations of the SRC-3/AIB1 coactivator integrate genomic reponses to multiple cellular signaling pathways. Mol Cell. (2004) 2004;15:937–949. doi: 10.1016/j.molcel.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 16.Amazit L, Pasini L, Szafran AT, Berno V, Wu R-C, Mielke M, et al. Regulation of SRC-3 intercompartmental dynamics by estrogen receptor and phosphorylation. Mol Cell Biol. 2007;27:6913–6932. doi: 10.1128/MCB.01695-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zheng FF, Wu R-C, Smith CL, O’Malley BW. Rapid estrogen-induced phosphorylation of the SRC-3 coactivator occurs in an extranuclear complex containing estrogen receptor. Mol Cell Biol. 2005;25:8273–8284. doi: 10.1128/MCB.25.18.8273-8284.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu R-C, Smith CL, O’Malley BW. Transcriptional Regulation by Steroid Receptor Coactivator Phosphorylation. Endocr Rev. The Endocrine Society. 2005;26:393–399. doi: 10.1210/er.2004-0018. [DOI] [PubMed] [Google Scholar]
- 19.Su Q, Hu S, Gao H, Ma R, Yang Q, Pan Z, et al. Role of AIB1 for tamoxifen resistance in estrogen receptor-positive breast cancer cells. Oncology. 2008;75:159–168. doi: 10.1159/000159267. [DOI] [PubMed] [Google Scholar]
- 20.Osborne CK, Bardou V, Hopp TA, Chamness GC, Hilsenbeck SG, Fuqua SAW, et al. Role of the Estrogen Receptor Coactivator AIB1 (SRC-3) and HER-2/neu in Tamoxifen Resistance in Breast Cancer. J Natl Cancer Inst. Oxford University Press. 2003;95:353–361. doi: 10.1093/jnci/95.5.353. [DOI] [PubMed] [Google Scholar]
- 21.Fereshteh MP, Tilli MT, Kim SE, Xu J, O’Malley BW, Wellstein A, et al. The Nuclear Receptor Coactivator Amplified in Breast Cancer-1 Is Required for Neu (ErbB2/HER2) Activation, Signaling, and Mammary Tumorigenesis in Mice. Cancer Res. American Association for Cancer Research. 2008;68:3697–3706. doi: 10.1158/0008-5472.CAN-07-6702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shou J, Massarweh S, Osborne CK, Wakeling AE, Ali S, Weiss H, et al. Mechanisms of Tamoxifen Resistance: Increased Estrogen Receptor-HER2/neu Cross-Talk in ER/HER2-Positive Breast Cancer. J Natl Cancer Inst. 2004;96:926–935. doi: 10.1093/jnci/djh166. [DOI] [PubMed] [Google Scholar]
- 23.Kessler JD, Kahle KT, Sun T, Meerbrey KL, Schlabach MR, Schmitt EM, et al. A SUMOylation-Dependent Transcriptional Subprogram Is Required for Myc-Driven Tumorigenesis. Science. American Association for the Advancement of Science. 2012;335:348–353. doi: 10.1126/science.1212728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koboldt DC, Fulton RS, McLellan MD, Schmidt H, Kalicki-Veizer J, McMichael JF, et al. Comprehensive molecular portraits of human breast tumours. Nature. Nature Publishing Group, a division of Macmillan Publishers Limited. All Rights Reserved. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feng Q, Zhang Z, Shea MJ, Creighton CJ, Coarfa C, Hilsenbeck SG, et al. An epigenomic approach to therapy for tamoxifen-resistant breast cancer. Cell Res. Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences. 2014;24:809–819. doi: 10.1038/cr.2014.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.York B, Yu C, Sagen JV, Liu Z, Nikolai BC, Wu RC, et al. Reprogramming the posttranslational code of SRC-3 confers a switch in mammalian systems biology. Proc Natl Acad Sci U S A. (2010) 107:11122–11127. doi: 10.1073/pnas.1005262107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shiau AK, Barstad D, Loria PM, Cheng L, Kushner PJ, Agard DA, et al. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell. 1998;95:927–937. doi: 10.1016/s0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- 28.Wu R-C, Feng Q, Lonard DM, O’Malley BW. SRC-3 coactivator functional lifetime is regulated by a phospho-dependent ubiquitin time clock. Cell. 2007;129:1125–1140. doi: 10.1016/j.cell.2007.04.039. [DOI] [PubMed] [Google Scholar]
- 29.Wang X, Kiyokawa H, Dennewitz MB, Costa RH. The Forkhead Box m1b transcription factor is essential for hepatocyte DNA replication and mitosis during mouse liver regeneration. Proc Natl Acad Sci U S A. National Acad Sciences. 2002;99:16881–16886. doi: 10.1073/pnas.252570299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hollenhorst PC, Paul L, Ferris MW, Graves BJ. The ETS gene ETV4 is required for anchorage-independent growth and a cell proliferation gene expression program in PC3 prostate cells. Genes Cancer. 2011;1:1044–1052. doi: 10.1177/1947601910395578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mudryj M, Hiebert SW, Nevins JR. A role for the adenovirus inducible E2F transcription factor in a proliferation dependent signal transduction pathway. EMBO J. 1990;9:2179–2184. doi: 10.1002/j.1460-2075.1990.tb07387.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Khatun J, Epstein CB, Shoresh N, Ernst J, Kheradpour P, Ward LD, et al. An integrated encyclopedia of DNA elements in the human genome. Nature. Nature Publishing Group, a division of Macmillan Publishers Limited. All Rights Reserved. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen H-Z, Ouseph MM, Li J, Pecot T, Chokshi V, Kent L, et al. Canonical and atypical E2Fs regulate the mammalian endocycle. Nat Cell Biol. Nature Publishing Group, a division of Macmillan Publishers Limited. All Rights Reserved. 2012;14:1192–1202. doi: 10.1038/ncb2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pandit SK, Westendorp B, Nantasanti S, van Liere E, Tooten PCJ, Cornelissen PWA, et al. E2F8 is essential for polyploidization in mammalian cells. Nat Cell Biol. 2012;14:1181–1191. doi: 10.1038/ncb2585. [DOI] [PubMed] [Google Scholar]
- 35.Eeckhoute J, Carroll JS, Geistlinger TR, Torres-Arzayus MI, Brown M. A cell-type-specific transcriptional network required for estrogen regulation of cyclin D1 and cell cycle progression in breast cancer. Genes Dev. Cold Spring Harbor Lab. 2006;20:2513–2526. doi: 10.1101/gad.1446006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006;10:515–527. doi: 10.1016/j.ccr.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, et al. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol. Nature Publishing Group. 2010;28:495–501. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang W, Soares J, Greninger P, Edelman EJ, Lightfoot H, Forbes S, et al. Genomics of Drug Sensitivity in Cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. Oxford University Press. 2013;41:D955–D961. doi: 10.1093/nar/gks1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rambaldi D, Giorgi FM, Capuani F, Ciliberto A, Ciccarelli FD. Low duplicability and network fragility of cancer genes. Trends Genet. 2008;24:427–430. doi: 10.1016/j.tig.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 40.An O, Pendino V, D’Antonio M, Ratti E, Gentilini M, Ciccarelli FD. NCG 4.0: the network of cancer genes in the era of massive mutational screenings of cancer genomes. Database. Oxford University Press. 2014;2014 doi: 10.1093/database/bau015. bau015–bau015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Turner NC, Ro J, Andre F, Loi S, Verma S, Iwata H, et al. Palbociclib in Hormone-Receptor-Positive Advanced Breast Cancer. N Engl J Med. 2015;373:209–219. doi: 10.1056/NEJMoa1505270. [DOI] [PubMed] [Google Scholar]
- 42.Wang Y, Lonard DM, Yu Y, Chow DC, Palzkill TG, O’Malley BW. Small molecule inhibition of the steroid receptor coactivators, SRC-3 and SRC-1. Mol Endocrinol. 25:2041–2053. doi: 10.1210/me.2011-1222. 2011 ed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kuang S-Q, Liao L, Zhang H, Lee AV, O’Malley BW, Xu J. AIB1/SRC-3 Deficiency Affects Insulin-Like Growth Factor I Signaling Pathway and Suppresses v-Ha-ras-induced Breast Cancer Initiation and Progression in Mice. Cancer Res. American Association for Cancer Research. 2004;64:1875–1885. doi: 10.1158/0008-5472.can-03-3745. [DOI] [PubMed] [Google Scholar]
- 44.Kuang S-Q, Liao L, Wang S, Medina D, O’Malley BW, Xu J. Mice Lacking the Amplified in Breast Cancer 1/Steroid Receptor Coactivator-3 Are Resistant to Chemical Carcinogen-Induced Mammary Tumorigenesis. Cancer Res. 2005;65:7993–8002. doi: 10.1158/0008-5472.CAN-05-1179. [DOI] [PubMed] [Google Scholar]
- 45.Andrechek ER. HER2/Neu tumorigenesis and metastasis is regulated by E2F activator transcription factors. Oncogene. Macmillan Publishers Limited. 2013 doi: 10.1038/onc.2013.540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu L, de Bruin A, Wang H, Simmons T, Cleghorn W, Goldenberg LE, et al. Selective roles of E2Fs for ErbB2- and Myc-mediated mammary tumorigenesis. Oncogene. Macmillan Publishers Limited. 2013 doi: 10.1038/onc.2013.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mussi P, Yu C, O’Malley BW, Xu J. Stimulation of steroid receptor coactivator-3 (SRC-3) gene overexpression by a positive regulatory loop of E2F1 and SRC-3. Mol Endocrinol. 2006;20:3105–3119. doi: 10.1210/me.2005-0522. [DOI] [PubMed] [Google Scholar]
- 48.Louie MC, Revenko AS, Zou JX, Yao J, Chen H-W. Direct control of cell cycle gene expression by proto-oncogene product ACTR, and its autoregulation underlies its transforming activity. Mol Cell Biol. 2006;26:3810–3823. doi: 10.1128/MCB.26.10.3810-3823.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schiff R, Massarweh SA, Shou J, Bharwani L, Arpino G, Rimawi M, et al. Advanced concepts in estrogen receptor biology and breast cancer endocrine resistance: implicated role of growth factor signaling and estrogen receptor coregulators. Cancer Chemother Pharmacol. Springer-Verlag. 2005;56:10–20. doi: 10.1007/s00280-005-0108-2. [DOI] [PubMed] [Google Scholar]
- 50.Miller TW, Balko JM, Fox EM, Ghazoui Z, Dunbier A, Anderson H, et al. ERalpha-dependent E2F transcription can mediate resistance to estrogen deprivation in human breast cancer. Cancer Discovery. 2011;1:338–351. doi: 10.1158/2159-8290.CD-11-0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.