

Abstract
We have previously reported the role of miR-326-HDAC3 loop in anti-cancer drug-resistance. CAGE, a cancer/testis antigen, regulates the response to anti-cancer drug-resistance by forming a negative feedback loop with miR-200b. Studies investigating the relationship between CAGE and HDAC3 revealed that HDAC3 negatively regulated the expression of CAGE. ChIP assays demonstrated the binding of HDAC3 to the promoter sequences of CAGE. However, CAGE did not affect the expression of HDAC3. We also found that EGFR signaling regulated the expressions of HDAC3 and CAGE. Anti-cancer drug-resistant cancer cell lines show an increased expression of pEGFRY845. HDAC3 was found to negatively regulate the expression of pEGFRY845. CAGE showed an interaction and co-localization with EGFR. It was seen that miR-326, a negative regulator of HDAC3, regulated the expression of CAGE, pEGFRY845, and the interaction between CAGE and EGFR. miR-326 inhibitor induced the binding of HDAC3 to the promoter sequences in anti-cancer drug-resistant Malme3MR cells, decreasing the tumorigenic potential of Malme3MR cells in a manner associated with its effect on the expression of HDAC3, CAGE and pEGFRY845. The down-regulation of HDAC3 enhanced the tumorigenic, angiogenic and invasion potential of the anti-cancer drug-sensitive Malme3M cells in CAGE-dependent manner. Studies revealed that PKCδ was responsible for the increased expression of pEGFRY845 and CAGE in Malme3MR cells. CAGE showed an interaction with PKCδ in Malme3MR cells. Our results show that HDAC3-CAGE axis can be employed as a target for overcoming resistance to EGFR inhibitors.
Keywords: anti-cancer drug-resistance, CAGE, HDAC3, EGFR
INTRODUCTION
Multi drug-resistant phenotypes are under epigenetic regulation (Gao et al., 2009; To et al., 2008). It is known that HDAC3 negatively regulates the expression of MDR1 and confers sensitivity to anti-cancer drugs (Kim et al., 2015a; Park et al., 2014b). MicroRNAs (miRNAs) play essential roles in various cellular processes such as cellular proliferation and anti-cancer drug-resistance (Yoon et al., 2014). HDAC3, through a negative feedback loop with miR-326, regulates the response to the anti-cancer drugs (Kim et al., 2014). Through its interaction with tubulin β3, HDAC3 regulates the response to anti-cancer drugs (Kim et al., 2015a). The expression level of HDAC3 has been shown to be down-regulated in various cancer cell lines that are resistant to anti-cancer drugs (Kim et al., 2014). The expression of HDAC3 is controlled via ubiquitination (Kim et al., 2015b). miR-335 increases the expression of HDAC3 by preventing SIAH2 from inducing ubiquitination of HDAC3 (Kim et al., 2015b). The selective inhibition of HDAC3 protects beta cells from cytokine-induced apoptosis (El-Khoury et al., 2007). Previous studies have shown that HDAC3 mediates beta cell apoptosis (Lundh et al., 2012). SAHA, an inhibitor of HDAC(s), induces MDR to confer anti-apoptotic effects (Xu et al., 2012).
The transcriptional repression of HDAC3 enhances tumorigenicity and invasiveness of lung cancer cells (Dhar et al., 2014). HDAC3 acts as a negative regulator of angiogenesis (Park et al., 2014a). The role of HDAC3 in cancer development is known to be tissue-specific. In gastric, prostatic, and colorectal cancer samples, HDAC3 overexpression was significantly associated with poor prognosis (Weichert et al., 2008a; 2008b). In agreement with these reports, it has been shown that HDAC3 appeared to be up-regulated and to repress the tumor suppressor gene p21 in colorectal cancer cells (Wilson et al., 2006). Studies have shown that liver-specific HDAC3 knock-out mice develop hepatoma (Bhaskara et al., 2010). In gastric cancers, HDAC3 inhibits the expression of PUMA (p53-upregulated modulator of apoptosis) and the down-regulation of HDAC3 promotes interaction of p53 with the promoter sequences of PUMA (Feng et al., 2013).
HDAC3 inhibition by vorinostat reduces the EGFR expression level and attenuates cellular proliferation (Gilbert et al., 2011). HDAC inhibition decreases the expression of EGFR and causes dissociation of HDAC3 from the promoter sequences of EGFR in colorectal cancer cells (Chou et al., 2011). TSA, an inhibitor of HDAC activity, inhibits the activation of ERGFR by HIF1β (Robertsson et al., 2012). EGFR signaling down-regulates the expression of Runx2 by up-regulating the expression of HDAC4 and HDAC-6 (Zhu et al., 2011). EGFR signaling regulates the multi drug-resistant phenotypes (Shi et al., 2009). The targeting EGFR in cancers are largely limited due to the status of KRAS mutation (Van Cutsem et al., 2009). The KRAS mutants bypass EGFR to activate the Ras/Raf/MEK/ERK signals, and significantly weaken the therapeutic effect of cetuximab (Walther et al., 2009). Targeting c-Met enhances the therapeutic effect in malignant colon cancer cells harboring KRAS mutation (Li et al, 2014). Thus, the inhibition of both c-Met and EGFR serves as an effective therapy for hepatocellular carcinoma (Steinway et al., 2015). However, the role of HDAC3 in imparting resistance to EGFR inhibitors has so far not been reported.
CAGE, cancer/testis antigen, was isolated by SEREX (serological analysis of recombinant expression library) from the sera of gastric cancer patients (Cho et al., 2002). CAGE, through interaction with HDAC2, decreases the expression of p53, which in turn regulates the response to anti-cancer drugs (Kim et al., 2010). The expression of CAGE is increased in anti-cancer drug-resistant cancer cell lines (Kim et al., 2013). CAGE, through a negative feedback loop with miR-200b, regulates the response to anti-cancer drugs (Kim et al., 2013). The expression of CAGE is under the epigenetic regulation (Cho et al., 2013). CAGE displays oncogenic potential and increases the expression of Cyclin D1 and - E in AP1 and E2F-depenedent manner (Por et al., 2010). CAGE is present in the sera of various cancer patients (Iwata et al., 2005). The expression of CAGE is seen in most of the cancer tissues (Kim et al., 2010). The role of CAGE in conferring resistance to microtubule-targeting anti-cancer drugs, such as taxol and celastrol, has been reported (Kim et al., 2010; 2013). However, the effect of CAGE on the response to EGFR inhibitors has not been reported.
In this study, we show the direct regulation of CAGE expression by HDAC3. We show that HDAC3-CAGE axis regulates the activation of EGFR. HDAC3 targets CAGE to regulate the tumorigenic potential and angiogenic potential of cancer cells and the response to anti-cancer drugs. Our results show that the HDAC3-CAGE axis serves as a strategy for overcoming resistance to EGFR inhibitors.
MATERIALS AND METHODS
Materials
Anti-mouse and anti-rabbit IgG-horse radish peroxidase conjugate antibodies were purchased from Pierce Company. An ECL (enhanced chemiluminiscence) kit was purchased from Amersham. Lipofectamin and PlusTM reagent were purchased from Invitrogen (USA).
Cell lines and cell culture
Cancer cell lines made resistant to microtubule-targeting drugs were established by stepwise addition of the respective drug. Cells surviving drug treatment (attached fraction) were obtained and used throughout this study. SNU387/SNU387R or Malme3M/Malme3MR cells that stably express anti-sense HDAC3 cDNA or HDAC3-Flag were selected by G418 (400 μg/ml).
Western blot analysis
Western blot analysis, immunoprecipitation and cellular fractionation were performed according to the standard procedures (Kim et al., 2013). For analysis of proteins from tumor tissues, frozen samples were grounded to a fine powder using a mortar and pestle over liquid nitrogen. Proteins were solubilized in RIPA buffer containing protease inhibitors, and insoluble material removed by centrifugation.
Cell viability determination
The cells were assayed for their growth activity using the 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT; Sigma). Viable cell number counting was carried out by trypan blue exclusion assays.
Caspase-3 activity assays
Caspase-3 activity was measured according to the manufacturer’s instructions (BioVision, USA). Cells were lysed in 0.1 M HEPES buffer, pH 7.4, containing 2 mM dithiothreitol, 0.1% CHAPS, and 1% sucrose. Cell lysates were incubated with a colorimetric substrate, 200 μM Ac-DEVD-p-nitroanilide, for 30 min at 30°C. The fluorescence was measured at 405 nm using a microtiter plate reader.
Immunohistochemistry
Paraffin-embedded tissue sections were immunostained using the Vecta stain ABC Elite Kit (Vector Laboratories). Tissue sections were deparaffinized with xylene and washed in ethanol. Endogenous peroxidase activity is blocked with 3% hydrogen peroxide and H2O for 10min. Slides were then blocked with 5% normal goat serum in TBS containing 0.1%Tween-20 (TBS-T) for 1 h. For immunohistochemistry, a primary antibody to HDAC3 (1:100, Santa Cruz), CAGE (1:100, Santa Cruz), EGFR (1:100, Santa Cruz), pEGFRY845 (1:100, Santa Cruz) or IgG (1:100, Santa Cruz) was added and incubation continued at 4°C for 24 h. After washing with TBS-T, slides were treated with biotinylated secondary antibody for 30 min. After washing, slides were incubated in the ABC complex for 30 min, and then stained with diaminobenzidine (DAB, Sigma). Sections were counterstained with hematoxylin and finally mounted using Fixo gum rubber cement (Mercateo, Germany).
Immunofluorescence staining
Cells were seeded onto glass coverslips in 24-well plates and fixed with 4% paraformaldehyde (v/v) for 10 min and then permeabilized with 0.4% Triton X-100 for 10 min. Nonspecific antibody binding sites were blocked by incubation with 1% BSA in TBST for 30 min. Cells were then incubated with primary antibody specific to CAGE (1:200; BD Biosciences) or pEGFRY845 (1:200; Santa Cruz) for 2 h, followed by washing with TBS-T three times. Anti-goat IgG-FITC (for detection of pEGFRY845) or anti-rabbit Alexa Fluor 586 (for detection of CAGE) secondary antibody (Molecular Probes) was added to cells and incubated for 1 h. Cover slips were then washed and mounted by applying Mount solution (Biomeda, USA). Fluorescence images were acquired using a confocal laser scanning microscope and software (Fluoview version 2.0) with X 60 objective (Olympus FV300, Japan).
Chromatin immunoprecipitation (ChIP) Assays
Assays were performed according to manufacturer’s instruction (Upstate). For detection of binding of HDAC3 to CAGE promoter sequences, specific primers of CAGE promoter-1 sequences [5′-CCTGACAAAGTACTGTATTCACTCCA-3′ (sense) and 5′-TGGCTCAGCTTGAGAGCAAC-3′ (antisense)], CAGE promoter-2 sequences [5′-CGCAGAAGTTAAGGAGGCAG-3′ (sense) and 5′-AAGTTGCCCCAGAAACCAGT-3′ (antisense)] and CAGE promoter-3 sequences [5′-ATGTGACTAGCACC CGGAAA-3′ (sense) and 5′-GGGATAGTGGGAGTATCGG-3′ (antisense)] were used.
Transfection
All transfections were performed according to the manufacturer’s instructions. Lipofectamine and Plus reagents (Invitrogen) were used. The construction of siRNA was carried out according to the instruction manual provided by the manufacturer (Ambion, USA). For miR-326 knockdown, cells were transfected with 200 nM of oligonucleotide (inhibitor) with Lipofectamine 2000 (Invitrogen), according to the manufacturer’s protocol. The sequences used were: 5′-CCUCUGGGCCCUUCUCG-3′ (miR-326 inhibitor); and 5′-GCCUCCGGCUUCGCACCUCU-3′ (control inhibitor).
In vivo tumorigenic potential
Athymic nude mice (BALB/c nu/ nu, 5–6-week-old females) were obtained from Orient Bio Inc. (Korea) and were maintained in a laminar air-flow cabinet under aseptic conditions. Each cancer cells (1 × 106) were injected subcutaneously into the dorsal flank area of the mice. Tumor volume was determined by direct measurement with calipers and calculated by the following formula: length × width × height × 0.5. To determine the effect of HDAC3 on in vivo response to microtubule-targeting drugs, each cancer cell line expressing HDAC3-Flag or anti-sense HDAC3 cDNA were injected subcutaneously into the dorsal flank area of the mice. Following the establishment of sizeable tumor, celastrol (1 mg/kg), taxol (1 mg/kg) or vinblastine (0.5 mg/kg) was administered via tail vein. Tumor volume was measured as describe above. To examine the effect of miR-326, control inhibitor (50 μM/kg) or miR-326 inhibitor (50 μM/kg) was administered via tail vein. To determine the effect of HDAC3 and CAGE on the in vivo tumorigenic potential, scrambled siRNA (100 nM), HDAC3 siRNA (100 nM) or CAGE siRNA (100 nM) was injected following the establishment of sizable tumor by Malme3MR cells, via tail vein 5 times in a total of 30 days.
Chemo invasion assays
The invasive potential was determined by using a transwell chamber system with 8-μm pore polycarbonate filter inserts (CoSTAR, USA). The lower and upper sides of the filter were coated with gelatin and Matrigel, respectively. Trypsinized cells (5 × 103) in the serum-free RPMI 1640 medium containing 0.1% bovine serum albumin were added to each upper chamber of the transwell. RPMI 1640 medium supplemented with 10% fetal bovine serum was placed in the lower chamber and cells were incubated at 37°C for 16 h. The cells were fixed with methanol and the invaded cells were stained and counted. Results were analyzed for statistical significance using the Student’s t test. Differences were considered significant when p < 0.05.
Intravital microscopy
Male BALB/c mice (6–8 week old) were obtained from Daehan Biolink (Korea). In vivo angiogenesis was assessed as follows. The mice were anesthetized with 2.5% avertin (v/v) via intraperitoneal injection (Surgivet, USA), and abdominal wall windows were implanted. Next, a titanium circular mount with eight holes on the edge was inserted between the skin and the abdominal wall. Growth factor-reduced matrigel containing the conditioned medium was applied to the space between the windows, and a circular glass cover slip was placed on top and fixed with a snap ring. After four days, the animals were anesthetized and injected intravenously with 50 μl of 25 ng/ml fluorescein isothiocyanate-labeled dextran (molecular weight, Mr ∼2,000,000) via the tail vein. The mice were then placed on a Zeiss Axiovert 200 M microscope. The epi-illumination microscopy setup included a 100-W mercury lamp and filter set for blue light. Fluorescence images were recorded at random locations of each window using an electron-multiplying charge coupled device camera (Photo Max 512, Princeton Instruments, USA) and digitalized for subsequent analysis using the Metamorph program (Universal Imaging, USA). The assay was scored from 0 (negative) to 5 (most positive) in a double-blinded manner.
Statistical analysis
Statistical differences in this were determined by using the Student’s t test.
RESULTS
HDAC3 directly regulates the expression of CAGE
We have previously reported that miR-326-HDAC3 loop regulates the response to anti-cancer drugs. We showed that the expression of HDAC3 is lower in cancer cells resistant to anti-cancer drugs than those that are sensitive to anti-cancer drugs (Kim et al., 2014). By forming a negative feedback loop with miR-200b, CAGE regulates the response to anti-cancer drugs (Kim et al., 2013). The expression of CAGE is higher in resistant cancer cells than those that are sensitive to anti-cancer drugs (Kim et al., 2010). Because HDAC3 and CAGE show an inverse relationship in resistant cancer cells (Kim et al., 2013; 2014), we were interested in examining the relationship between HDAC3 and CAGE. Wild type, but not the catalytically inactive mutant HDAC3 (HDAC3S424A), decreased the expression of CAGE in drug-resistant SNU387R and Malme3MR cells (Fig. 1A), suggesting that HDAC3 activity is necessary for regulating the expression of CAGE by HDAC3. Unlike wild type, the HDAC3S424A mutant does not show nuclear localization (Park et al., 2014b). CAGE promoter sequences contain putative biding sites for DNMT1, GATA-1 and SP1 (Fig. 1B). We know that the decreased the expression of PSalpha gene involves the recruitment of HDAC3 by Sp1 in ER-positive HepG2 cells (Suzuki et al., 2010). This led us to hypothesize that HDAC3 may be exerting a direct regulation on the expression of CAGE. ChIP assays showed binding of HDAC3 to site 1, but not site 2 or site 3, of the CAGE promoter sequences in SNU387 and Malme3M cells (Fig. 1B). Wild type, but not the mutant HDAC3, showed binding to the site 1 of the promoter sequences of CAGE (Fig. 1B), suggesting that HDAC3 activity is necessary for direct regulation of CAGE by HDAC3. The down-regulation of CAGE or overexpression of CAGE did not affect the expression of HDAC3 in Malme3MR or Malme3M cells (Fig. 1C). Taken together, these results suggest the role of HDAC3 in regulating the expression of CAGE.
Fig. 1.

HDAC3 regulates the expression of CAGE. (A) The indicated cancer cells were transiently transfected with wild type HDAC3 or mutant HDAC3 (HDAC3S424A). At 48 h post transfection, cell lysates were subjected to Western blot analysis. Cell lysates from SNU387 or Malme3M cells were also subjected to Western blot analysis. (B) Cell lysates isolated from the indicated cancer cells were subjected to ChIP assays (left panel). The indicated cancer cells were transiently transfected with the indicated siRNA (10 nM) or construct (1 μg). At 48 h post transfection, cell lysates were isolated and subjected to ChIP assays (right panel). (C) The indicated cancer cells were transiently transfected with the indicated siRNA (10 nM) or construct (1 μg). At 48 h post transfection, cell lysates were subjected to Western blot analysis.
HDAC3 interacts with DNMT1 to regulate the expression of CAGE
In our studies, we reported the decreased expression of DNMT1 in SNU387R and Malme3MR cells (Kim et al., 2010). DNMT1 regulates the sensitivity to cisplatin (Xiang et al., 2014). The expression of CAGE is also known to be under epigenetic regulation (Cho et al., 2013). Site 1 of the CAGE promoter contains putative binding site for DNMT1 (Fig. 1B). It is thus reasonable that DNMT1 may regulate the expression of CAGE. We first examined the possibility of an interaction between HDAC3 and DNMT1. HDAC3 interacts with DNMT1 in SNU387 and Malme3M cells (Fig. 2A). Our studies revealed that the down-regulation of HDAC3 decreases the expression of DNMT1 in SNU387 and Malme3M cells (Fig. 2A). This implies that the down-regulation of HDAC3 may regulate the expression of the transcriptional factor that regulates the expression of DNMT1 either directly or indirectly. The inhibition of HDACs reduces the expression of DNMT1 (Brodie et al., 2014). HDAC3 activity is necessary for the interaction with DNMT1 (Fig. 2B). Taken together, these results suggest that HDAC3 regulates the expression of CAGE through interaction with DNMT1.
Fig. 2.
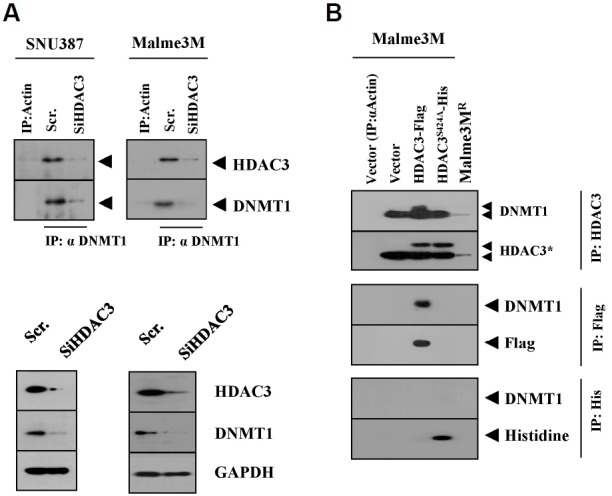
HDAC3 interacts with DNMT1. (A) SNU387 or Malme3M cells were transfected with scrambled siRNA (10 nM) or HDAC3 siRNA (10 nM). At 48 h after transfection, cell lysates were immunoprecipitated with the indicated antibody, followed by Western blot analysis (upper panel). Cell lysates were also subjected to Western blot analysis (lower panel). (B) Malme3M cells were transfected with the indicated construct (1 μg each). At 48 h after transfection, cell lysates were immunoprecipitated with the indicated antibody, followed by Western blot analysis.
EGFR signaling regulates the expression of HDAC3 and CAGE
CAGE increases the expression of Cyclin D1 (Por et al., 2010), and EGFR signaling regulates the expression of Cyclin D1 (Lee et al., 2010). Over-expression of EGFR is correlated with the resistance to taxol (Itamochi et al., 2012). EGFR tyrosine kinase inhibitors overcome this resistance to taxol (Kuang et al., 2010). Over-expression of ATP-binding cassette (ABC) transporters such as ABCB1, ABCC1 and ABCG2 is one of the main causes of multidrug resistance (MDR). AG1478, an inhibitor of EGFR tyrosine kinase, inhibits the function of ABCB1 and ABCG2 (Itamochi et al., 2009). These reports suggest that the MDR phenotype is closely related with EGFR signaling. P53 modulates resistance to EGFR inhibitors (Huang et al., 2011). CAGE, through interaction with HDAC2, regulates he expression of p53 to confer resistance to anti-cancer drugs (Kim et al., 2010). miR-126 regulates EGFR-Akt signaling through miR-126-DNMT1 circuit (Liu et al., 2015). These reports led us to hypothesize that the interaction between the HDAC3-CAGE axis and EGFR signaling regulates the response to anti-cancer drugs. We first examined the relationship between HDAC3-CAGE axis and EGFR signaling. We saw that the EGF treatment increased the expression of CAGE and pEGFRY845 while decreasing the expression of HDAC3 (Fig. 3A). EGFR inhibitors, such as cetuximab (CTX) and gefitinib, prevented EGF from regulating the expression of HDAC3 and CAGE in SNU387 and Malme3M cells (Fig. 3A). These results suggest that EGFR signaling may regulate the expression of HDAC3 and CAGE. ChIP assays also showed that EGF treatment prevented HDAC3 from binding to the promoter sequences of CAGE (Fig. 3B). Furthermore, we found that cetuximab prevented EGF from inhibiting the binding of HDAC3 to the promoter sequences of CAGE (Fig. 3B), and also EGF treatment prevented Snail from binding to the promoter sequences of CAGE (Fig. 3B). Taken together, these results suggest that EGFR signaling, in association with HDAC3-CAGE axis, regulates the response to anti-cancer drugs.
Fig. 3.
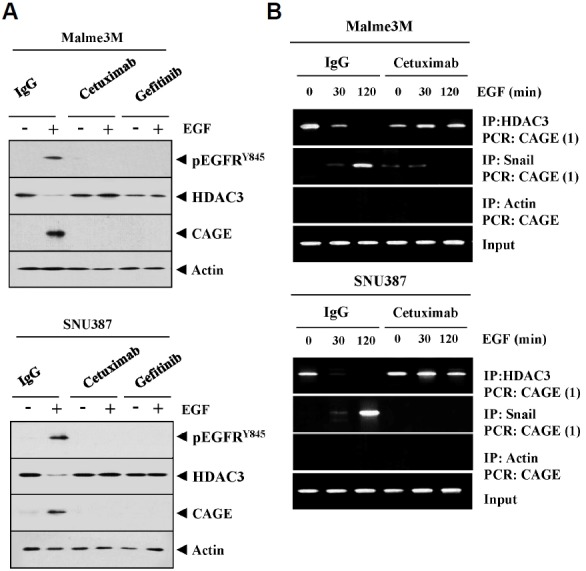
EGFR signaling regulates the expression of HDAC3 and CAGE. (A) Malme3M or SNU387 cells were pretreated with IgG (2 μg/ml), cetuximab (2 μg/ml) or gefitinib (10 μM) for 24 h. The next day, cells were then treated with EGF (50 ng/ml) for 1 h, followed by Western blot analysis. (B) Malme3M or SNU387 cells were pre-treated with IgG (2 μg/ml), cetuximab (2 μg/ml) or gefitinib (10 μM) for 24 h. The next day, cells were then treated with EGF (50 ng/ml) for various time intervals, followed by ChIP assays.
HDAC3 targets CAGE to regulate the response to EGFR inhibitors and the invasion potential of cancer cells
The down-regulation of HDAC3 increased the expression of CAGE and MDR1 in CAGE-dependent manner (Fig. 4A). It is apparent that CAGE does not regulate the expression of HDAC3 (Fig. 4A). We studied the relationship between the expression of HDAC3, CAGE and MDR1 in Malme3MR cells. Over-expression of HDAC3 decreased the expression of CAGE and MDR1 in Malme3MR cells (Fig. 4B). HDAC3 decreased the expression of MDR1 while CAGE restored the expression of MDR1 in Malme3MR cells transfected with HDAC3 (Fig. 4B). The down-regulation of HDAC3 prevented cleavage of PARP in response to EGFR inhibitors such as CTX and gefitinib in CAGE-dependent manner (Fig. 4C) and also prevented CTX and gefitinib from increasing caspase-3 activity in CAGE-dependent manner in Malme3M cells (Fig. 4D). We also observed that CAGE prevented HDAC3 from increasing caspase-3 activity in Malme3MR cells (Fig. 4D). The down-regulation of HDAC3 conferred resistance to gefitinib in CAGE-dependent manner in Malme3M cells (Fig. 4E). CAGE prevented HDAC3 from increasing sensitivity to gefitinib in Malme3MR cells (Fig. 4E). The down-regulation of HDAC3 enhanced the invasion potential of Malme3M cells in CAGE-dependent manner (Fig. 4F). The overexpression of CAGE restored the invasion potential of Malme3MR cells transfected with HDAC3 (Fig. 4F). Taken together, these results suggest that HDAC3 targets CAGE to regulate the response to EGFR inhibitor and the invasion potential of cancer cells.
Fig. 4.

HDAC3 targets CAGE to regulate the sensitivity to EGFR inhibitors and invasion potential of cancer cells. (A) Malme3M cells were transiently transfected with the indicated siRNAs (each at 10 nM). At 48 h after transfection, cell lysates were subjected to Western blot analysis. (B) Malme3MR cells were transiently transfected with the indicated constructs (each at 1 μg). At 48 h after transfection, cell lysates were subjected to Western blot analysis. (C) Malme3M cells were transiently transfected with the indicated siRNAs (each at 10 nM). Next day, cells were then treated with cetuximab (2 μg/ml) or gefitinib (10 μM) for 24 h, followed by Western blot analysis. (D) Malme3M cells were transiently transfected with the indicated siRNAs (each at 10 nM). The following day, cells were then treated with cetuximab (2 μg/ml) or gefitinib (10 μM) for 24 h, followed by caspase-3 activity assays (left panel). Malme3MR cells were transiently transfected with the indicated construct (each at 1 μg) and similarly treated (right panel). *p < 0.05; **p < 0.005. (E) Malme3M cells were transiently transfected with the indicated siRNAs (each at 10 nM). The next day, cells were then treated with various concentrations of gefitinib for 24h, followed by MTT assays (left panel). Malme3MR cells were transiently transfected with the indicated construct (each at 1 μg). The next day, cells were then treated with various concentrations of gefitinib for 24 h, followed by MTT assays (right panel). (F) The indicated cancer cells were transfected with the indicated siRNAs (each at 10 nM). At 48 h after transfection, cells were then subjected to chemoinvasion assays. *p < 0.05; ** p < 0.005; ***p < 0.0005.
HDAC3 regulates the expression of pEGFRY845 and an interaction between CAGE and EGFR
Given the fact that p53 negatively regulates the activation of EGFR and CAGE acts as a negative regulator of p53, we hypothesized that the HDAC3-CAGE axis regulates EGFR activation.
Using Malme3M cells as a control, the following observations were made: Malme3MR cells and Malme3MAs-HDAC3 that stably express anti-sense HDAC3 showed higher expression of CAGE and pEGFRY845 (Fig. 5A); Malme3MR-HDAC3-Flag cells that stably express HDAC3 showed a lower expression level of CAGE and pEGFRY845 than Malme3MR cells (Fig. 5A). The down-regulation of HDAC3 increased the expression of pEGFRY845 and induced interaction of EGFR with FAK and Src (Fig. 5B). Some reports show the effect of HDAC inhibition on the expression of EGFR (Chou et al., 2011). However, in our studies, the down-regulation of HDAC3 did not affect the expression of EGFR (Fig. 5B). The down-regulation of EGFR did not affect the expression of HDAC3 (data not shown). Malme3MAs-HDAC3 and Malme3MR cells showed an interaction between CAGE and EGFR (Fig. 5C). Malme3M cells that stably express anti-sense HDAC3 (Malme3MAs-HDAC3) showed resistance to gefitinib (Fig. 5D), whereas the Malme3MR cells that stably express HDAC3 (Malme3MR-HDAC3-Flag) showed sensitivity to gefitinib (Fig. 5D). Malme3MR and Malme3MAs-HDAC3 cells showed higher expression of CAGE and pEGFRY845 and lower expression of HDAC3, as compared to the Malme3M cells (Fig. 5E). However, Malme3MR-HDAC3-Flag cells showed lower expression of CAGE and pEGFRY845 than the Malme3MR and Malme3MAs-HDAC3 cells (Fig. 5E). Taken together, these results suggest that the HDAC3-CAGE axis regulates EGFR signaling in relation to response to anti-cancer drugs.
Fig. 5.

HDAC3 regulates the activation of EGR and induces an interaction between CAGE and EGFR. (A) Cell lysates isolated from the indicated cancer cells were subjected to Western blot analysis. (B) Malme3M cells were transiently transfected with the indicated siRNA (each at 10 nM). At 48 h after transfection, cell lysates were immunoprecipitated with the indicated antibody (2 μg/ml), followed by Western blot analysis. Cell lysates were also subjected to Western blot analysis. (C) Cell lysates isolated from the indicated cancer cells were immunoprecipitated with the indicated antibody (2 μg/ml), followed by Western blot analysis. (D) The indicated cancer cells were treated with various concentrations of gefitinib for 48 h, followed by MTT assays. (E) The indicated cancer cells were treated with gefitinib (1 μM) for 48 h, followed by Western blot analysis (lower panel).
miR-326, a negative regulator of HDAC3, regulates the activation of EGFR and an interaction of EGFR with CAGE and the tumorigenic potential of cancer cells
We reported that miR-326-HDAC3 negative feedback loop regulates the response to anti-cancer drugs (Kim et al., 2014). We therefore examined the effect of miR-326 on the activation of EGFR. We observed that miR-326 specifically decreased the expression of HDAC3 while increasing the expression of CAGE and pEGFRY845 in Malme3M cells (Fig. 6A). The down - regulation of miR-326 by miR-326 inhibitor increased the expression of HDAC3 while decreasing the expression of CAGE and pEGFRY845 in Malme3MR cells (Fig. 6B). miR-326 inhibitor inhibited an interaction between CAGE and EGFR (Fig. 6B), and also decreased the tumorigenic potential of Malme3MR cells (Fig. 6C). Western blot of tumor tissue lysates showed that miR-326 inhibitor increased the expression of HDAC3 while decreasing the expression of CAGE and pEGFRY845 (Fig. 6C). Immunohistochemical staining of tumor tissue showed that miR-326 inhibitor also showed similar results (Fig. 6D). ChIP assay showed that miR-326 inhibitor induced the binding of HDAC3 to the promoter sequences of CAGE (Fig. 6E). Taken together, these results suggest that miR-326 inhibitor decreases the tumorigenic potential of cancer cells by regulating the expression of HDAC3, CAGE and pEGFRY845.
Fig. 6.

miR-326, a negative regulator of HDAC3, regulates the activation of EGFR and an interaction between CAGE and EGFR. (A) Malme3M cells were transiently transfected with control vector or miR-326 construct (each at 1 μg) along with the indicated inhibitor (each at 10 nM). At 48 h after transfection, cell lysates were immunoprecipitated with the indicated antibody (2 μg/ml), followed by Western blot analysis. Cell lysates were also subjected to Western blot analysis. (B) Malme3MR cells were transiently transfected with the indicated inhibitor (each at 10 nM). At 48 h after transfection, cell lysates were subjected to immunoprecipitation and Western blot analysis. (C) Malme3MR cells (1 × 106) were injected into the dorsal flank area of athymic nude mouse. Control inhibitor (40 μg/kg or 50 μM/kg) or miR-326 inhibitor (40 μg/kg or 50 μM/kg) was injected into each nude mouse after the tumor reached a certain size. Tumor volume was measured on the same day as injection of inhibitor. Each value represents an average obtained from five mice of each group. Tumor tissue lysates from each mouse of the experimental group were subjected to Western blot. (D) immunohistochemical staining of tumor tissue derived from Malme3MR cells treated with the indicated inhibitor was performed as described. Immunohistochemistry staining employing secondary antibody alone served as a negative control. Representative images from five animals from each experimental group are shown (magnification, 400X; Olympus). H&E staining was performed to check structural integrity. (E) Malme3MR cells were transiently transfected with the indicated inhibitor (each at 10 nM). At 48 h after transfection, cell lysates were subjected to ChIP assays.
miR-326 inhibitor enhances sensitivity to EGFR inhibitor
miR-326 inhibitor increased the expression of HDAC3 while decreasing the expression of CAGE and MDR1 in Malme3MR cells (Fig. 7A). miR-326 inhibitor decreased the expression of pEGFRY845 (Fig. 7A). miR-326 inhibitor enhanced the sensitivity to gefitinib by targeting HDAC3 in Malme3MR cells (Fig. 7B). miR-326 inhibitor increased caspase-3 activity (Fig. 7C) and PARP cleavage (Fig. 7D) in response to gefitinib in Malme3MR cells. Because miR-326 inhibitor regulated the expression of HDAC3, CAGE and pEGRY845 (Fig. 6B), we examined whether EGFR signaling would affect the expression of miR-326. EGF treatment decreased the expression of HDAC3 while increasing the expression of CAGE and pEGFRY845 in Malme3M cells (Fig. 7E). EGF treatment increased the expression of miR-326 in Malme3M cells (Fig. 7E). Gefitinib prevented EGF from increasing the expression of miR-326 in Malme3M cells (Fig. 7E). This suggests that EGFR signaling regulates the expression of miR-326. Taken together, these results suggest that miR-326 inhibitor enhances sensitivity to EGFR inhibitor by targeting HDAC3.
Fig. 7.

miR-326 inhibitor enhances sensitivity to EGFR inhibitors. (A) Malme3MR cells were transfected with the indicated inhibitor (each at 10 nM) along with the indicated siRNA (each at 10 nM). At 48 h after transfection, cell lysates were subjected to Western blot analysis. (B) Malme3MR cells were transfected with the indicated inhibitor (each at 10 nM) along with the indicated siRNA (each at 10 nM). At 24 h after transfection, cells were treated with various concentrations of gefitinib for 24 h, followed by MTT assays. (C) Malme3MR cells were transfected with the indicated inhibitor (each at 10 nM) along with the indicated siRNA (each at 10 nM). At 24 h after transfection, cells were then treated with gefitinib (10 μM) for 24 h, followed by caspase-3 activity assays. **p < 0.005. (D) Malme3MR cells were transfected with the indicated inhibitor (each at 10 nM) along with the indicated siRNA (each at 10 nM). At 24 h after transfection, cells were then treated with gefitinib (10 μM) for 24 h, followed by Western blot analysis. (E) Malme3M cells were treated with EGF (50 ng/ml) for various time intervals. Cell lysates prepared at each time point were subjected to Western blot analysis and qRT-PCR analysis. Malme3M cells were pretreated with gefitinib (10 μM). The next day, cell were then treated with EGF (50 ng/ml) for 2 h, followed by qRT-PCR analysis (right panel). *p < 0.05; **p < 0.005.
The down-regulation of HDAC3 enhances the tumorigenic potential and angiogenic potential of cancer cells by targeting CAGE
The in vivo down-regulation of HDAC3 enhanced the tumorigenic potential of Malme3M cells in CAGE-dependent manner (Fig. 8A). The down-regulation of HDAC3 enhances the tumorigenic potential of lung cancer cells (Dhar et al., 2014). Western blot of tumor tissue lysates showed that the down-regulation of HDAC3 increased the expression of pEGFRY845 and MDR1 in CAGE-dependent manner (Fig. 8B). The conditioned medium of Malme3M cells transfected with HDAC3 siRNA showed enhanced angiogenic potential based on intravital microscopy (Fig. 8C). Taken together, these results suggest that HDAC3 regulates the tumorigenic and angiogenic potential of cancer cells in a manner associated with its effect on the expression of CAGE and pEGFRY845.
Fig. 8.

The down-regulation of HDAC3 enhances the tumorigenic potential and angiogenic potential of cancer cells by targeting CAGE. (A) Malme3M cells (1 × 106) were injected into the dorsal flank area of athymic nude mouse. The indicated siRNA (each at 10 nM) was injected into each nude mouse after the tumor reached a certain size. Tumor volume was measured on the same day as injection of siRNA. Each value represents an average obtained from five mice of each group. (B) Tumor tissue lysates from each mouse of the experimental group were subjected to Western blot. (C) Malme3M cells were transiently transfected with the indicated siRNAs. Concentrated conditioned medium (10 μl) obtained from the indicated cancer cell line was mixed with 100 μl matrigel and intravital microscopy was performed as described.
PKCδ interacts with CAGE and regulates the expression of CAGE and pEGFRY845
Since the Malme3MR cells showed higher expression of pEGFRY845 than Malme3M cells (Fig. 5A), we investigated the mechanisms associated with the increased expression of pEGFRY845. EGF treatment increased the expression of pPKCδT505 in SNU387 and Malme3M cells (Fig. 9A). The inactivation of PKCδ prevented EGF from regulating the expression of CAGE, HDAC3 and pEGFGRY845 in SUN387 and Malme3M cells (Fig. 9B). The inactivation of PKCδ increased the expression of HDAC3 while decreasing the expression of CAGE and pEGFRY845 (Fig. 9C). CAGE also showed an interaction with PKCδ in Malme3MR cells (Fig. 9D). Taken together, these results suggest that the CAGE-PKCδ-EGFR axis regulates the response to anti-cancer drugs.
Fig. 9.

PKCδ interacts with CAGE and regulates the expression of CAGE and pEGFRY845. (A) The indicated cancer cells were treated with EGF (50 ng/ml) for various time intervals. Cell lysates prepared at each time point were subjected to Western blot analysis. (B) The indicated cancer cells were transfected with the indicated construct (each at 1 μg). At 48 h after transfection, cells were treated with EGF (50 ng/ml) for 1h, followed by Western blot analysis. (C) The indicated cancer cells were transfected with the indicated construct (each at 1μg). At 48 h after transfection, cell lysates were subjected to Western blot analysis. (D) Cell lysates isolated from the indicated cancer cells were subjected to immunoprecipitation, followed by Western blot analysis.
DISCUSSION
Celastrol and taxol increase the expression of pEGFRY845 in SNU387 and Malme3M cells (personal observations). This suggests that resistance to these anti-cancer drugs is closely related to the resistance to EGFR inhibitors. Taxol, in combination with EGFR-targeted nanoparticle, regulates multi drug-resistant phenotype (Milane et al., 2011). Previously, we reported the identification of HDAC3 domain that regulates the expression of MDR1 (Park et al., 2014b). It will be interesting to further examine whether this domain regulates the expression of CAGE and pEGFGRY845.
It is established that p53 regulates the activation of EGFR (Dong et al., 2009). In this study, we found that the down-regulation of p53 increases the expression of pEGFRY845 in Malme3M cells (personal observations). It would be interesting to examine whether HDAC3, through interaction with DNMT1 and p53, binds to the promoter sequences of CAGE.
Inhibition of class I HDACs suppresses EGFR phosphorylation as well as reduces its expression (Tang et al., 2014). In this study, we showed that the down-regulation of HDAC3 increases the expression of pEGFRY845 (Fig. 5A). Thus, further studies should be done to investigate whether HDAC2, increased in Malme3MR cells (Kim et al., 2010), regulates the expression of pEGFRY845.
In this study, we found that CAGE confers resistance to trastuzumab, an inhibitor of HER2 (personal observations). It would be interesting to examine the effect of CAGE on the expression of HER2 and on the sensitivity to HER2 inhibitors. HDAC3 acts as a substrate of Src (Longworth and Laimins, 2006). C-Src-HDAC3 interaction decreases the expression of CXCR4 in highly invasive breast tumor cells (Matteucchi et al., 2007). Src kinase pathway is involved in trastuzumab-resistance in HER2-amplified breast cancers (Boyer et al., 2013). EGFR and HER2 are transactivated by c-Src and MMPs (Garcia-Recio et al., 2015). Thus, studying the role of Src in the increased phosphorylation of EGFR by HDAC3-CAGE axis, and the possible interaction between CAGE and Src, would reveal relevant information.
The down-regulation of HDAC3 enhances the angiogenic potential of Malme3M cells (Fig. 8C). We previously showed that recombinant CAGE protein enhances the angiogenic potential of cancer cells (Kim et al., 2013). HDAC3 negatively regulates the expression of PAI-1, a downstream target of TGFβ (Park et al., 2014a). We found that CAGE increases the expression of PAI-1 (personal observations). It would be interesting to examine whether PAI-1 affects the activation of EGFR. It is probable that PAI-1 confers resistance to EGFR inhibitors by activating EGFR.
TGF-β1/SMAD3 pathway activation in renal fibrosis (induced by ureteral ligation) is correlated with epidermal growth factor receptor (Y845) (EGFR (Y845)) and p53 (Ser15) phosphorylation and is responsible for the induction of PAI-1(Samarakoon et al., 2013). HDACs, and in particular HDAC3, are required for activation of the ERK and PI3K signaling pathways by TGF-β and for the subsequent gene induction dependent on these signaling pathways (Barter et al., 2010). It is probable that the resistance to EGFR inhibitors involves a cross-talk between EGFR and TGFβR. In this study, we found an activation of TGFβR in Malme3MR cells (personal observations). It would be necessary to examine the effect of HDAC3 on the activation of TGFβR.
Given the fact that CAGE interacts with EGFR (Fig. 6A), it would be necessary to examine whether CAGE displays kinase activity toward EGFR. PKCα mediates resistance to EGFR inhibitor (erlotinib) and enhances EMT in lung cancer cells (Abera and Kazanietz, 2015). Atypical protein kinase C (aPKC) confers resistance to EGFR inhibitors in gliomas (Kusne et al., 2014). Our studies revealed that Malme3MR cells show the activation of PKCδ (Fig. 9C), but not PKCα, and also an interaction between CAGE and PKCδ (Fig. 9D). It is probable that CAGE, through interaction with PKCδ, regulates the expression of EGFR.
In summary, the decreased expression of HDAC3 lead to the increased expression of miR-326 in Malme3MR cells (Fig. 10). HDAC3-miR326 negative feedback loop regulates the expression of CAGE and pEGFRY845 to regulate the response to anti-cancer drugs (Fig. 10). Therefore, we can conclude that the HDAC3-CAGE-EGFR axis serves as a target for overcoming resistance to EGFR inhibitors.
Fig. 10.
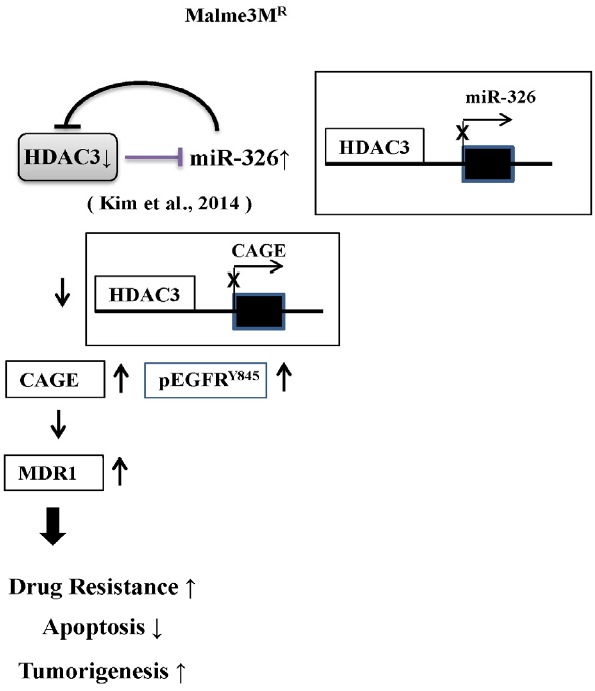
The proposed mechanism of anti-cancer drug-resistance regulated by HDAC-EGFR-CAGE axis.
Acknowledgments
This work was supported by National Research Foundation Grants (2014R1A2A2A01002448, 2015R1A1A3A04001339, and 2015R1A2A1A15051678), a grant from the BK21 plus Program, and by National R&D Program for Cancer Control, Ministry for Health and Welfare, Republic of Korea Grant 1320160. This work was also supported by a grant from Kangwon National University.
REFERENCES
- Abera M.B., Kazanietz M.G. Protein kinase Cα mediates erlotinib resistance in lung cancer cells. Mol. Pharmacol. 2015;87:832–841. doi: 10.1124/mol.115.097725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barter M.J., Pybus L., Litherland G.J., Rowan A.D., Clark I.M., Edwards D.R., Cawston T.E., Young D.A. HDAC-mediated control of ERK- and PI3K-dependent TGF-β-induced extracellular matrix-regulating genes. Matrix Biol. 2010;29:602–612. doi: 10.1016/j.matbio.2010.05.002. [DOI] [PubMed] [Google Scholar]
- Bhaskara S., Knutson S.K., Jiang G., Chandrasekharan M.B., Wilson A.J., Zheng S., Yenamandra A., Locke K., Yuan J.L., Bonine-Summers A.R., et al. Hdac3 is essential for the maintenance of chromatin structure and genome stability. Cancer Cell. 2010;18:436–447. doi: 10.1016/j.ccr.2010.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer A.P., Collier T.S., Vidavsky I., Bose R. Quantitative proteomics with siRNA screening identifies novel mechanisms of trastuzumab resistance in HER2 amplified breast cancers. Mol. Cell. Proteomics. 2013;12:180–193. doi: 10.1074/mcp.M112.020115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodie S.A., Li G., El-Kommos A., Kang H., Ramalingam S.S., Behera M., Gandhi K., Kowalski J., Sica G.L., Khuri F.R., Vertino P.M., Brandes J.C. Class I HDACs are mediators of smoke carcinogen-induced stabilization of DNMT1 and serve as promising targets for chemoprevention of lung cancer. Cancer Prev. Res. (Phila). 2014;7:351–361. doi: 10.1158/1940-6207.CAPR-13-0254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho B., Lim Y., Lee D.Y., Park S.Y., Lee H., Kim W.H., Yang H., Bang Y.J., Jeoung D.I. Identification and characterization of a novel cancer/testis antigen gene CAGE. Biochem. Biophys. Res. Commun. 2002;292:715–726. doi: 10.1006/bbrc.2002.6701. [DOI] [PubMed] [Google Scholar]
- Cho B., Lee H., Jeong S., Bang Y.J., Lee H.J., Hwang K.S., Kim H.Y., Lee Y.S., Kang G.H., Jeoung D.I. Promoter hypomethylation of a novel cancer/testis antigen gene CAGE is correlated with its aberrant expression and is seen in premalignant stage of gastric carcinoma. Biochem. Biophys. Res. Commun. 2003;307:52–63. doi: 10.1016/s0006-291x(03)01121-5. [DOI] [PubMed] [Google Scholar]
- Chou C.W., Wu M.S., Huang W.C., Chen C.C. HDAC inhibition decreases the expression of EGFR in colorectal cancer cells. PLoS One. 2011;6:e18087. doi: 10.1371/journal.pone.0018087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhar S.S., Alam H., Li N., Wagner K.W., Chung J., Ahn Y.W., Lee MG. Transcriptional repression of histone deacetylase 3 by the histone demethylase KDM2A is coupled to tumorigenicity of lung cancer cells. J. Biol. Chem. 2014;289:7483–7496. doi: 10.1074/jbc.M113.521625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong P., Xu Z., Jia N., Li D., Feng Y. Elevated expression of p53 gain-of-function mutation R175H in endometrial cancer cells can increase the invasive phenotypes by activation of the EGFR/PI3K/AKT pathway. Mol. Cancer. 2009;8:103. doi: 10.1186/1476-4598-8-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Khoury V., Breuzard G., Fourré N., Dufer J. The histone deacetylase inhibitor trichostatin A downregulates human MDR1 (ABCB1). gene expression by a transcription-dependent mechanism in a drug-resistant small cell lung carcinoma cell line model. Br. J. Cancer. 2007;97:562–573. doi: 10.1038/sj.bjc.6603914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L., Pan M., Sun J., Lu H., Shen Q., Zhang S., Jiang T., Liu L., Jin W., Chen Y., et al. Histone deacetylase 3 inhibits expression of PUMA in gastric cancer cells. J. Mol. Med (Berl). 2013;91:49–58. doi: 10.1007/s00109-012-0932-x. [DOI] [PubMed] [Google Scholar]
- Gao P., Yang X., Xue Y.W., Zhang X.F., Wang Y., Liu W.J., Wu X.J. Promoter methylation of glutathione S-transferase pi1 and multidrug resistance gene 1 in bronchioloalveolar carcinoma and its correlation with DNA methyltransferase 1 expression. Cancer. 2009;115:3222–3232. doi: 10.1002/cncr.24369. [DOI] [PubMed] [Google Scholar]
- Garcia-Recio S., Pastor-Arroyo E.M., Marín-Aguilera M., Almendro V., Gascón P. The Transmodulation of HER2 and EGFR by Substance P in Breast Cancer Cells Requires c-Src and Metalloproteinase Activation. PLoS One. 2015;10:e0129661. doi: 10.1371/journal.pone.0129661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert R.E., Huang Q., Thai K., Advani S.L., Lee K., Yuen D.A., Connelly K.A., Advani A. Histone deacetylase inhibition attenuates diabetes-associated kidney growth: potential role for epigenetic modification of the epidermal growth factor receptor. Kidney Int. 2011;79:1312–1321. doi: 10.1038/ki.2011.39. [DOI] [PubMed] [Google Scholar]
- Huang S., Benavente S., Armstrong E.A., Li C., Wheeler D.L., Harari P.M. p53 modulates acquired resistance to EGFR inhibitors and radiation. Cancer Res. 2011;71:7071–70709. doi: 10.1158/0008-5472.CAN-11-0128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itamochi H., Kato M., Nishimura M., Oishi T., Shimada M., Sato S., Naniwa J., Sato S., Nonaka M., Kudoh A., et al. Establishment and characterization of a novel ovarian serous adenocarcinoma cell line, TU-OS-4, that overexpresses EGFR and HER2. Hum. Cell. 2012;25:111–115. doi: 10.1007/s13577-012-0048-1. [DOI] [PubMed] [Google Scholar]
- Iwata T., Fujita T., Hirao N., Matsuzaki Y., Okada T., Mochimaru H., Susumu N., Matsumoto E., Sugano K., Yamashita N., et al. Frequent immune responses to a cancer/testis antigen, CAGE, in patients with microsatellite instability-positive endometrial cancer. Clin. Cancer Res. 2005;11:3949–3957. doi: 10.1158/1078-0432.CCR-04-1702. [DOI] [PubMed] [Google Scholar]
- Kim Y., Park H., Park D., Lee Y.S., Choe J., Hahn J.H., Lee H., Kim Y.M., Jeoung D. Cancer/testis antigen CAGE exerts negative regulation on p53 expression through HDAC2 and confers resistance to anti-cancer drugs. J. Biol. Chem. 2010;285:25957–25968. doi: 10.1074/jbc.M109.095950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y., Park D., Kim H., Choi M., Lee H., Lee Y.S., Choe J., Kim Y.M., Jeoung D. miR-200b and cancer/testis antigen CAGE form a feedback loop to regulate the invasion and tumorigenic and angiogenic responses of a cancer cell line to microtubule-targeting drugs. J. Biol. Chem. 2013;288:36502–36518. doi: 10.1074/jbc.M113.502047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y., Kim H., Park H., Park D., Lee H., Lee Y.S., Choe J., Kim Y.M., Jeoung D. miR-326-histone deacetylase-3 feedback loop regulates the invasion and tumorigenic and angiogenic response to anti-cancer drugs. J. Biol. Chem. 2014;289:28019–28039. doi: 10.1074/jbc.M114.578229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y., Kim H., Jeoung D. Tubulin beta3 serves as a target of HDAC3 and mediates resistance to microtubule-targeting drugs. Mol. Cells. 2015a;38:705–714. doi: 10.14348/molcells.2015.0086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y., Kim H., Park D., Jeoung D. miR-335 targets SIAH2 and confers sensitivity to anti-cancer drugs by increasing the expression of HDAC3. Mol. Cells. 2015b;38:562–572. doi: 10.14348/molcells.2015.0051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuang Y.H., Shen T., Chen X., Sodani K., Hopper-Borge E., Tiwari A.K., Lee J.W., Fu L.W., Chen Z.S. Lapatinib and erlotinib are potent reversal agents for MRP7 (ABCC10).-mediated multidrug resistance. Biochem. Pharmacol. 2010;79:154–161. doi: 10.1016/j.bcp.2009.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusne Y., Carrera-Silva E.A., Perry A.S., Rushing E.J., Mandell E.K., Dietrich J.D., Errasti A.E., Gibbs D., Berens M.E., Loftus J.C., et al. Targeting aPKC disables oncogenic signaling by both the EGFR and the proinflammatory cytokine TNFα in glioblastoma. Sci. Signal. 2014;7:ra75. doi: 10.1126/scisignal.2005196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C.H., Hung H.W., Hung P.H., Shieh Y.S. Epidermal growth factor receptor regulates beta-catenin location, stability, and transcriptional activity in oral cancer. Mol. Cancer. 2010;9:64. doi: 10.1186/1476-4598-9-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Wang J., Gao X., Han W., Zheng Y., Xu H., Zhang C., He Q., Zhang L., Li Z., Zhou D. c-Met targeting enhances the effect of irradiation and chemical agents against malignant colon cells harboring a KRAS mutation. PLoS One. 2014;9:e113186. doi: 10.1371/journal.pone.0113186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R., Gu J., Jiang P., Zheng Y., Liu X., Jiang X., Huang E., Xiong S., Xu F., Liu G, et al. DNMT1-MicroRNA126 Epigenetic Circuit Contributes to Esophageal Squamous Cell Carcinoma Growth via ADAM9-EGFR-AKT Signaling. Clin. Cancer Res. 2015;21:854–863. doi: 10.1158/1078-0432.CCR-14-1740. [DOI] [PubMed] [Google Scholar]
- Longworth M.S., Laimins L.A. Histone deacetylase 3 localizes to the plasma membrane and is a substrate of Src. Oncogene. 2006;25:4495–4500. doi: 10.1038/sj.onc.1209473. [DOI] [PubMed] [Google Scholar]
- Lundh M., Christensen D.P., Damgaard Nielsen M., Richardson S.J., Dahllöf M.S., Skovgaard T., Berthelsen J., Dinarello C.A., Stevenazzi A., Mascagni P., et al. Histone deacetylases 1 and 3 but not 2 mediate cytokine-induced beta cell apoptosis in INS-1 cells and dispersed primary islets from rats and are differentially regulated in the islets of type 1 diabetic children. Diabetologia. 2012;55:2421–2431. doi: 10.1007/s00125-012-2615-0. [DOI] [PubMed] [Google Scholar]
- Matteucci E., Ridolfi E., Maroni P., Bendinelli P., Desiderio M.A. c-Src/histone deacetylase 3 interaction is crucial for hepatocyte growth factor dependent decrease of CXCR4 expression in highly invasive breast tumor cells. Mol. Cancer Res. 2007;5:833–845. doi: 10.1158/1541-7786.MCR-07-0054. [DOI] [PubMed] [Google Scholar]
- Milane L., Duan Z., Amiji M. Therapeutic efficacy and safety of paclitaxel/lonidamine loaded EGFR-targeted nanoparticles for the treatment of multi-drug resistant cancer. PLoS One. 2011;6:e24075. doi: 10.1371/journal.pone.0024075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park D., Park H., Kim Y., Kim H., Jeoung D. HDAC3 acts as a negative regulator of angiogenesis. BMB Rep. 2014a;47:227–232. doi: 10.5483/BMBRep.2014.47.4.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H., Kim Y., Park D., Jeoung D. Nuclear localization signal domain of HDAC3 is necessary and sufficient for the expression regulation of MDR1. BMB Rep. 2014b;47:342–347. doi: 10.5483/BMBRep.2014.47.6.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Por E., Byun H.J., Lee E.J., Lim J.H., Jung S.Y., Park I., Kim Y.M., Jeoung D.I, Lee H. The cancer/testis antigen CAGE with oncogenic potential stimulates cell proliferation by up-regulating cyclins D1 and E in an AP-1- and E2F-dependent manner. J. Biol. Chem. 2010;285:14475–14485. doi: 10.1074/jbc.M109.084400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson E.D., Weir L., Romanowska M., Leigh I.M., Panteleyev A.A. ARNT controls the expression of epidermal differentiation genes through HDAC- and EGFR-dependent pathways. J. Cell Sci. 2012;125:3320–3332. doi: 10.1242/jcs.095125. [DOI] [PubMed] [Google Scholar]
- Samarakoon R., Dobberfuhl A.D., Cooley C., Overstreet J.M., Patel S., Goldschmeding R., Meldrum K.K., Higgins P.J. Induction of renal fibrotic genes by TGF-β1 requires EGFR activation, p53 and reactive oxygen species. Cell Signal. 2013;25:2198–2209. doi: 10.1016/j.cellsig.2013.07.007. [DOI] [PubMed] [Google Scholar]
- Shi Z., Tiwari A.K., Shukla S., Robey R.W., Kim I.W., Parmar S., Bates S.E., Si Q.S., Goldblatt C.S., Abraham I., et al. Inhibiting the function of ABCB1 and ABCG2 by the EGFR tyrosine kinase inhibitor AG1478. Biochem. Pharmacol. 2009;77:781–793. doi: 10.1016/j.bcp.2008.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinway S.N., Dang H., You H., Rountree C.B., Ding W. The EGFR/ErbB3 Pathway Acts as a Compensatory Survival Mechanism upon c-Met Inhibition in Human c-Met+ Hepatocellular Carcinoma. PLoS One. 2015;10:e0128159. doi: 10.1371/journal.pone.0128159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A., Sanda N., Miyawaki Y., Fujimori Y., Yamada T., Takagi A., Murate T., Saito H., Kojima T. Down-regulation of PROS1 gene expression by 17beta-estradiol via estrogen receptor alpha (ERalpha).-Sp1 interaction recruiting receptor-interacting protein 140 and the corepressor-HDAC3 complex. J. Biol. Chem. 2010;285:13444–13453. doi: 10.1074/jbc.M109.062430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J., Yan Y., Zhao T.C., Gong R., Bayliss G., Yan H., Zhuang S. Class I HDAC activity is required for renal protection and regeneration after acute kidney injury. Am. J. Physiol. Renal Physiol. 2014;307:303–316. doi: 10.1152/ajprenal.00102.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- To K.K., Polgar O., Huff L.M., Morisaki K., Bates S.E. Histone modifications at the ABCG2 promoter following treatment with histone deacetylase inhibitor mirror those in multidrug-resistant cells. Mol. Cancer Res. 2008;6:151–164. doi: 10.1158/1541-7786.MCR-07-0175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Cutsem E., Köhne C.H., Hitre E., Zaluski J., Chang Chien C.R., Makhson A., D'Haens G., Pintér T., Lim R., Bodoky G., et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N. Engl. J. Med. 2009;360:1408–1417. doi: 10.1056/NEJMoa0805019. [DOI] [PubMed] [Google Scholar]
- Walther A., Johnstone E., Swanton C., Midgley R., Tomlinson I., Kerr D. Genetic prognostic and predictive markers in colorectal cancer. Nature reviews Cancer. 2009;9:489–499. doi: 10.1038/nrc2645. [DOI] [PubMed] [Google Scholar]
- Weichert W., Röske A., Gekeler V., Beckers T., Ebert M.P., Pross M., Dietel M., Denkert C., Röcken C. Association of patterns of class I histone deacetylase expression with patient prognosis in gastric cancer: a retrospective analysis. Lancet Oncol. 2008a;9:139–148. doi: 10.1016/S1470-2045(08)70004-4. [DOI] [PubMed] [Google Scholar]
- Weichert W., Röske A., Gekeler V, Beckers T, Stephan C, Jung K, Fritzsche FR, Niesporek S, Denkert C, Dietel M, et al. Histone deacetylases 1, 2 and 3 are highly expressed in prostate cancer and HDAC2 expression is associated with shorter PSA relapse time after radical prostatectomy. Br. J. Cancer. 2008b;98:604–610. doi: 10.1038/sj.bjc.6604199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson A. J., Byun D. S., Popova N., Murray L.B., L'Italien K., Sowa Y., Arango D., Velcich A., Augenlicht L.H., Mariadason J. M. Histone deacetylase 3 (HDAC3). and other class I HDACs regulate colon cell maturation and p21 expression and are deregulated in human colon cancer. J. Biol. Chem. 2006;281:13548–13558. doi: 10.1074/jbc.M510023200. [DOI] [PubMed] [Google Scholar]
- Xiang Y., Ma N., Wang D., Zhang Y., Zhou J., Wu G., Zhao R., Huang H., Wang X., Qiao Y., et al. MiR-152 and miR-185 co-contribute to ovarian cancer cells cisplatin sensitivity by targeting DNMT1 directly: a novel epigenetic therapy independent of decitabine. Oncogene. 2014;33:378–386. doi: 10.1038/onc.2012.575. [DOI] [PubMed] [Google Scholar]
- Xu Y., Jiang Z., Yin P., Li Q., Liu J. Role for Class I histone deacetylases in multidrug resistance. Exp. Cell Res. 2012;318:177–186. doi: 10.1016/j.yexcr.2011.11.010. [DOI] [PubMed] [Google Scholar]
- Yoon S., Han E., Choi Y.C., Kee H., Jeong Y., Yoon J., Baek K. Inhibition of cell proliferation and migration by miR-509-3p that targets CDK2, Rac1, and PIK3C2A. Mol. Cells. 2014;37:314–321. doi: 10.14348/molcells.2014.2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J., Shimizu E., Zhang X., Partridge N.C., Qin L. EGFR signaling suppresses osteoblast differentiation and inhibits expression of master osteoblastic transcription factors Runx2 and Osterix. J. Cell. Biochem. 2011;112:1749–1760. doi: 10.1002/jcb.23094. [DOI] [PMC free article] [PubMed] [Google Scholar]