

How motor cortex activity is altered in Parkinson’s disease is poorly understood. Pasquereau et al. report abnormalities in modulation and timing of putative corticospinal neurons in the MPTP model of the disease, implying that dysfunction of such neurons plays a role in the pathophysiology of parkinsonism.
Keywords: MPTP, Parkinson’s disease, kinematics, GLM, loss of specificity, response latency
How motor cortex activity is altered in Parkinson’s disease is poorly understood. Pasquereau et al. report abnormalities in modulation and timing of putative corticospinal neurons in the MPTP model of the disease, implying that dysfunction of such neurons plays a role in the pathophysiology of parkinsonism.
Abstract
Abnormalities in the movement-related activation of the primary motor cortex (M1) are thought to be a major contributor to the motor signs of Parkinson’s disease. The existing evidence, however, variably indicates that M1 is under-activated with movement, overactivated (due to a loss of functional specificity) or activated with abnormal timing. In addition, few models consider the possibility that distinct cortical neuron subtypes may be affected differently. Those gaps in knowledge were addressed by studying the extracellular activity of antidromically-identified lamina 5b pyramidal-tract type neurons (n = 153) and intratelencephalic-type corticostriatal neurons (n = 126) in the M1 of two monkeys as they performed a step-tracking arm movement task. We compared movement-related discharge before and after the induction of parkinsonism by administration of MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) and quantified the spike rate encoding of specific kinematic parameters of movement using a generalized linear model. The fraction of M1 neurons with movement-related activity declined following MPTP but only marginally. The strength of neuronal encoding of parameters of movement was reduced markedly (mean 29% reduction in the coefficients from the generalized linear model). This relative decoupling of M1 activity from kinematics was attributable to reductions in the coefficients that estimated the spike rate encoding of movement direction (−22%), speed (−40%), acceleration (−49%) and hand position (−33%). After controlling for MPTP-induced changes in motor performance, M1 activity related to movement itself was reduced markedly (mean 36% hypoactivation). This reduced activation was strong in pyramidal tract-type neurons (−50%) but essentially absent in corticostriatal neurons. The timing of M1 activation was also abnormal, with earlier onset times, prolonged response durations, and a 43% reduction in the prevalence of movement-related changes beginning in the 150-ms period that immediately preceded movement. Overall, the results are consistent with proposals that under-activation and abnormal timing of movement-related activity in M1 contribute to parkinsonian motor signs but are not consistent with the idea that a loss of functional specificity plays an important role. Given that pyramidal tract-type neurons form the primary efferent pathway that conveys motor commands to the spinal cord, the dysfunction of movement-related activity in pyramidal tract-type neurons is likely to be a central factor in the pathophysiology of parkinsonian motor signs.
Introduction
Dysfunction of the primary motor cortex (M1) has long been thought to play a central role in the impairments of voluntary movement associated with Parkinson’s disease (Berardelli et al., 2001; Wu et al., 2011). As a principal source of corticospinal projections, M1 is essential for the generation of skilled movement (Porter and Lemon, 1993; Dum and Strick, 2002). In addition, M1 is an important site of termination for basal ganglia-thalamocortical projections (Nambu et al., 1988; Hoover and Strick, 1993). Thus, M1 may be a site where the abnormal basal ganglia activity known to be a correlate of parkinsonism (Miller and DeLong, 1987; Filion and Tremblay, 1991; Hutchison et al., 1994; Vila et al., 1997; Wichmann et al., 1999; Raz et al., 2000; Soares et al., 2004) is translated into aberrant motor commands, which result in signs such as impaired movement initiation (akinesia), slowness (bradykinesia), and hypometria. Note that the focus here is on abnormalities in the movement-related activity of M1. Abnormalities in M1 activity in subjects at rest have been addressed in-depth elsewhere (Eidelberg et al., 1994; Steiner and Kitai, 2000; Goldberg et al., 2002; Pasquereau and Turner, 2011; de Hemptinne et al., 2013).
The question of what specific dysfunctions of M1 activity contribute to the impairments of voluntary movement seen in parkinsonism has received considerable attention. Existing evidence, for the most part, is supportive of three possibilities (Fig. 1). First, motor commands are reduced in magnitude compared with those produced by a normal M1. Such a ‘hypoactivation’ of M1 might result from, for example, excessive inhibitory outflow from the parkinsonian basal ganglia (DeLong, 1990; Mink, 1996) or an impairment of the scaling of movement (Berardelli et al., 2001; Desmurget and Turner, 2008) or of the implicit motivation to move (Mazzoni et al., 2007; Baraduc et al., 2013). Second, a loss of functional specificity (‘loss of specificity’) throughout the basal ganglia-thalamo-cortical circuit (Zold et al., 2007; Beck and Hallett, 2011; Bronfeld and Bar-Gad, 2011; Kojovic et al., 2012) may lead to a co-activation in M1 of competing motor programs (Albin et al., 1989; Mink, 1996) and thus motor impairments. Finally, the timing of motor commands in M1 may be altered or dispersed (‘abnormal timing’, Watts and Mandir, 1992; Chen et al., 2001; Hiraoka et al., 2010).
Figure 1.

Hypothetical Parkinson’s-related abnormalities in M1 motor signalling. We tested three general theories concerning the specific dysfunctions in cortical signaling that contribute to abnormal movement in parkinsonism. (i) M1 could be under-activated (i.e. ‘Hypoactivation’), resulting in lower firing rates at rest, a reduced encoding of parameters of movement, smaller-than-normal residual peri-movement activity, and/or an overall decline in the prevalence of movement-related activity. (ii) A loss of functional specificity (‘Loss of Specificity’) could lead to a co-activation in M1 of competing motor programs, inducing increased firing rates at rest, a reduced response selectivity (weaker encoding) and a general increase in the movement-related activation of M1 (i.e. a greater-than-normal residual peri-movement activity and a higher prevalence). (iii) The timing of motor commands in M1 could be dispersed. In that case, response latencies of residual activity relative to movement onset may be altered.
Observations from patients with Parkinson’s disease support each of the three proposed classes of dysfunction. Several imaging studies have reported that the movement-related activation of M1 is abnormally-low in Parkinson’s disease (Rascol et al., 1992; Catalan et al., 1999; Turner et al., 2003; Tessa et al., 2010, 2012). Others, however, reported no difference from normal subjects (Jenkins et al., 1992) or an abnormally-increased activation of M1 (Sabatini et al., 2000; Haslinger et al., 2001; Yu et al., 2007). Studies using transcranial magnetic stimulation have provided support for both a loss of specificity (Shin et al., 2007; Derejko et al., 2013) and increased temporal dispersion of the movement-related activation of M1 (Chen et al., 2001; Hiraoka et al., 2010). What these results imply for the spiking activity of individual cortical neurons remains unclear.
Few studies have investigated movement-related activity in the parkinsonian M1 at the single-unit level (Doudet et al., 1990; Watts and Mandir, 1992; Parr-Brownlie and Hyland, 2005) and those reached different conclusions regarding the primary dysfunction: two studies reported a reduction in the magnitude of activation (i.e. hypoactivation; Watts and Mandir, 1992; Parr-Brownlie and Hyland, 2005), one described a significant loss of specificity in the encoding of a single movement parameter (e.g. of movement direction; Doudet et al., 1990), and one described a marked increase in the temporal dispersion of movement-related activity (Watts and Mandir, 1992). The relatively sparse and inconsistent data on this topic can be traced to at least two complicating factors. First, parkinsonian animals are reluctant to move (Goldberg et al., 2002), particularly under the operant conditions commonly used for single unit recording (Leblois et al., 2006; Emborg, 2007). We addressed that issue by inducing a hemiparkinsonian syndrome in which appetitive behaviours are partially preserved (Bankiewicz et al., 2001). Second, because M1 activity is known to encode the kinematics of active movement (Fromm and Evarts, 1981; Schwartz et al., 1988; Georgopoulos, 1991; Kalaska and Crammond, 1992; Sergio et al., 2005) and proprioceptive feedback (Rosen and Asanuma, 1972; Lemon and Porter, 1976; Fetz et al., 1980), some of the observed abnormalities in M1 activity may be secondary to rather than a cause of bradykinesia.
To address the second issue, we used a generalized linear model (GLM) to quantify two dissociable components of M1 activity, the spike-rate encoding of kinematics and the ‘residual’ activity reflecting a neuron’s sensitivity to movement per se (i.e. independent of variations in performance). This approach allowed us to compare our results against predictions from the hypoactivation, loss of specificity, and abnormal timing hypotheses (Fig. 1). Hypoactivation might manifest as reduced firing rates at rest (i.e. a general under-activation of M1), reduced spike-rate encoding of parameters of movement, smaller-than-normal residual peri-movement modulations, and/or an overall decline in the fraction of neurons showing movement-related activity (i.e. reduced prevalence). A loss of specificity, in contrast, may increase firing rates at rest (Bronfeld and Bar-Gad, 2011), reduce the selectivity of encoding by individual M1 neurons (a greater fraction of neurons respond, e.g. to all directions of movement rather than selectively to one direction), increase the magnitude of M1 responses to movement per se, and increase the fraction of neurons that respond during any one movement. Finally, the abnormal timing hypothesis predicts an increase in the dispersion of response onset times but makes no other predictions.
In addition, there is growing recognition that different neuronal subtypes in M1 are affected differently in the parkinsonian state (Lefaucheur, 2005; Pasquereau and Turner, 2011, 2013; Brazhnik et al., 2012; Shepherd, 2013). We have shown that lamina 5b pyramidal tract-type neurons (PTNs) are affected strongly by the induction of parkinsonism: PTN activity at rest is depressed, more bursty, and more likely to be rhythmic in the beta frequency range (Pasquereau and Turner, 2011), and PTNs respond to proprioceptive stimulation at shorter latencies with reduced directional specificity (Pasquereau and Turner, 2013). Surprisingly, nearby intratelencephalic-projecting corticostriatal neurons (CSNs; a distinct class of cortical pyramidal neuron; Shepherd, 2013) were largely unaffected by the induction of parkinsonism (Pasquereau and Turner, 2011, 2013). We hypothesized that the movement-related activity of PTNs is also affected differentially with the induction of parkinsonism.
To test these hypotheses, we studied the movement-related activity of identified PTNs and adjacent CSNs in the arm area of M1 in two rhesus monkeys performing a visuomotor step-tracking task. The prevalence of movement-related activity, neural encoding of motor parameters and of movement per se, and the latencies of movement-related activity were compared between normal and MPTP states.
Materials and methods
Animals, apparatus and tasks
Two female monkeys (Macaca mulatta) were used for these experiments [Monkeys V (4.8 kg) and L (6 kg)]. Procedures were approved by the Institutional Animal Care and Use Committee and complied with the Public Health Service Policy on the humane care and use of laboratory animals (amended 2002). Other data from the same animals were used in recent publications describing post-MPTP changes in resting M1 activity (Pasquereau and Turner, 2011) and in neuronal responses to muscle stretch (Pasquereau and Turner, 2013). Many aspects of the experimental approach are described in detail in those reports.
The animals were trained to perform a visuomotor step-tracking task similar to one used in previous studies of cortical and basal ganglia activity (Alexander, 1987; Alexander and Crutcher, 1990; Turner and DeLong, 2000). The animal sat in a primate chair and faced a computer monitor. The right wrist (Monkey L) or elbow (Monkey V) joint was aligned with the axis of rotation of a one-dimensional torquable manipulandum. Flexion and extension movements rotated the manipulandum in the horizontal plane, thereby controlling the horizontal position of an onscreen cursor (Fig. 2A). A trial began when a centre target appeared on the monitor and the monkey aligned the cursor with the target. After holding the cursor at this position for a start-position hold period (2–5 s, uniform random distribution), the target jumped to the left or right (chosen at random), and the animal moved the cursor to capture the lateral target. After a target hold interval (0.75–1.5 s), the animal received a drop of juice or food followed by an intertrial interval (1.2–1.7 s). After MPTP administration, a computer-controlled torque motor (TQ40W, Aerotech Inc.) assisted the repositioning of the manipulandum to the central start position during intertrial intervals.
Figure 2.

The visuomotor step-tracking task and EMG examples. (A) Flexion and extension movements rotated the manipulandum in the horizontal plane and thereby controlled the horizontal position of an onscreen cursor. On each behavioural trial, after a start-position hold period (random duration, 2–5 s), the animal was required to move the cursor toward one lateral target (left or right direction chosen at random). Examples of angular velocity (B) and muscle activity (C) observed during voluntary movements before (black) and after (red) MPTP administration. Single trial velocity traces are plotted relative to the computed time of movement onset (vertical dashed line). Note the accurate detection of movement onset in both pre- and post-MPTP data. In C, peri-movement means of the digitized EMG signals are shows for two muscles for all valid extension (EXT) and flexion (FLX) movements. Note that the y-scale is inverted for FLX movements in B and C.
Surgery
After training, each monkey was prepared for recording by aseptic surgery under isoflurane inhalation anaesthesia. A cylindrical stainless steel chamber was implanted with stereotaxic guidance over a burr hole allowing access to the arm-related regions of the left M1 and the putamen. The chamber was oriented parallel to the coronal plane at an angle of 35° so that electrode penetrations were orthogonal to the cortical surface. The chamber was fixed to the skull with bone screws and dental acrylic. Bolts were embedded in the acrylic to allow fixation of the head during recording sessions.
For EMG recording, pairs of Teflon-insulated multistranded stainless steel wires were implanted into: flexor carpi ulnaris, flexor carpi radialis, biceps longus, brachioradialis and triceps lateralis in Monkey L; and posterior deltoid, trapezius, triceps longus, triceps lateralis and brachioradialis in Monkey V. The wires were led subcutaneously to a connector fixed to the skull implant.
Placement of electrodes for antidromic identification
The method used to implant chronically-indwelling stimulation electrodes has been described previously (Turner and DeLong, 2000; Pasquereau and Turner, 2011). In brief, PTNs and CSNs were identified by antidromic activation from electrodes implanted in the cerebral peduncle and posteriolateral striatum, respectively. Sites for implantation were identified using standard electrophysiological mapping techniques. Three custom-built PtIr microwire electrodes were implanted in the posterior putamen and one electrode was implanted in the arm-responsive portion of the pre-pontine peduncle (for details, see Turner and DeLong, 2000). Histological reconstruction confirmed that the striatal and peduncle electrodes were at sites known to receive the bulk of M1 CSN and PTN projections, respectively (Brodal, 1978; Flaherty and Graybiel, 1991; Takada et al., 1998).
Data acquisition
Areas of M1 related to the primary joint used in the task were identified using microstimulation (<40 µA, 10 biphasic pulses at 300 Hz) and sensorimotor mapping. We preformed transdural extracellular recording using glass-insulated PtIr microelectrodes mounted in a hydraulic microdrive (MO-95, Narishige Intl.). Microelectrode penetrations were performed throughout the targeted cortical area using sequential stimulation of each putamen and peduncle stimulating site as search stimuli (700 µA, 0.2 ms biphasic pulses, >1.5 s between successive shocks). Neurons were selected for collection if they were activated antidromically or were located nearby (see detailed selection criteria below). Standard tests for antidromic identification were used: constant latency (<0.2 ms jitter), reliable following of high-frequency stimuli (three shocks at 200 Hz), and collision with spontaneously occurring spikes (Fuller and Schlag, 1976). Note that responsiveness of a neuron within the behavioural task was not a criterion for selection.
Neuronal activity was collected while the animal performed the step-tracking task. The microelectrode signal was amplified ×104 and bandpass filtered (0.3–10-KHz, DAM-80, WPI Inc.). Action potentials were discriminated on-line using template-based spike-sorting (MultiSpike Detector, Alpha Omega Engineering, Nazareth, Israel). The timing of sorted spikes and task events was digitized (1-kHz) and stored for offline analysis. EMG signals were amplified differentially (gain =10 000), filtered (0.2–5 kHz), rectified and then low-pass filtered (100 Hz). EMG data were collected during only a subset of data recording sessions (n = 20 pre-MPTP; n = 8 post-MPTP). Analog voltages reflecting joint angle and EMGs were digitized at either 200-Hz (monkey L) or 500-Hz (monkey V).
Administration of MPTP
On completion of sampling from the neurologically-normal animal, hemiparkinsonism was induced by infusion of MPTP into the left internal carotid artery (0.5 mg/kg; Bankiewicz et al., 1986; Wu et al., 2007). This procedure was performed under general anaesthesia (1–3% isoflurane) and antibiotics and analgesics were administered prophylactically. Both animals developed stable parkinsonian signs on the right side of the body. Analyses of the severity of parkinsonism and its stability across time are documented in a previous report (Pasquereau and Turner, 2011). Post-MPTP recording sessions started >30 days after MPTP administration. Dopamine replacement therapy was not administered at any time during the post-MPTP recording period.
Histology
After the last recording session, each monkey was given a lethal dose of sodium pentobarbital and was perfused transcardially with saline followed by 10% formalin in phosphate buffer and then sucrose. The brains were processed histologically to localize microelectrode tracks (cresyl violet) and to document the loss of dopaminergic cells in the substantia nigra pars compacta [tyrosine hydroxylase (TH) immunochemistry]. A stereological comparison of the numbers of TH-positive neurons in the substantia nigra pars compacta of MPTP-treated and untreated hemispheres was performed in tissue from Monkey L [see Pasquereau and Turner (2011) for detailed histological results]. Faulty processing of the TH staining prevented quantification in the tissue from monkey V.
Analysis of behavioural data
Digitized signals reflecting manipulandum position were filtered and differentiated (low-pass 25 Hz; Hamming, 1983). Movement initiation, peak velocity, and movement termination were detected automatically using position, velocity, and duration criteria. These criteria were adjusted to work equally well on pre- and post-MPTP movement data (e.g. Fig. 2B). Reaction time (RT), movement duration (MD), peak velocity (Velmax), movement amplitude (Ampl), and movement acceleration (Acc) were computed. Session-by-session means were entered into two-way ANOVAs to test for effects of MPTP administration and movement direction.
Analysis of neuronal data
Neuronal data were accepted for analysis if they met the following criteria: (i) the recording was obtained from a location within 3-mm of the anterior bank of the central sulcus from which movements of the task-involved part of the arm were evoked by microstimulation (i.e. <40 µA, 10 pulses at 300 Hz); (ii) adequate single unit isolation was maintained throughout the recording; (iii) neurons were either antidromically identified or were encountered along the same track within 0.5 mm of an antidromically activated neuron; and (iv) five or more successful trials for each movement direction were performed while the neuron was being recorded from.
For each trial, a continuous neuronal activation function [spike density function (SDF)] was generated by convolving each discriminated action potential with a Gaussian kernel (20-ms variance). Mean peri-movement SDFs (averaged across trials) were constructed for each task condition (±1000 ms relative to movement onset). A neuron’s baseline firing rate was calculated as the mean of the SDFs across a 500-ms epoch beginning 800 ms before movement onset. Movement-related changes in firing rate were detected by comparing SDF values (millisecond-by-millisecond) during a peri-movement epoch (500-ms centred on movement onset) relative to the cell’s baseline firing rate (P < 0.002, 2-tailed t-test). A neuron was judged to have movement-related activity if a significant increase or decrease in firing rate was found for at least one movement direction. Response onset and offset were defined as the times at which the SDF first and last crossed the P = 0.002 threshold, respectively. The relative time to peak was measured as the time interval between response onset and maximal firing rate deviation. Response magnitude was defined as the absolute difference between maximal deviation and the mean resting firing rate.
We used a GLM approach to quantify: (i) M1 encoding of multiple parameters of motor performance; and (ii) the ‘residual’ activity after regressing out all activity attributable to motor performance. A sliding window procedure determined the strength and peri-movement timing of neuronal encoding of direction (Dir), position (Pos), speed, acceleration (Acc) and reaction time (RT). For each neuron, we counted the spikes trial-by-trial that fell within a 200-ms test window that was stepped in 25-ms increments from −500 ms to +800 ms relative to movement onset. For each bin, we applied the following model:
| (1) |
where SCi is the number of spikes for the ith time bin, and E(x) is the expected value for x. The exponential function reflects an assumption that spike counts were distributed according to a Poisson distribution (Hatsopoulos et al., 2007). Kinematic data, including position, speed and acceleration, was sampled every 25 ms from −500 ms to +800 ms relative to movement onset. Movement directions were represented by dummy variables (1 and −1). All variables were normalized (Z-scored in standard deviation units) to allow comparison of the strength of encoding between variables. The β coefficients were estimated using the ‘glmfit’ function in Matlab 7 (Mathworks). To test whether individual coefficients were significant, we shuffled spike counts 1000 times across trials and compared actual coefficients to the confidence intervals yielded by shuffling [P = 0.05/(52 independent time bins) to compensate for multiple comparisons].
The residual spike count for the ith time bin (RSCi) was calculated as the difference between the observed and GLM-predicted discharge rates. In other words,
| (2) |
Only neurons showing a significant peri-movement change in residual activity [determined using the same method as used with SDFs (P < 0.002)] were included in the analysis of residual activity.
Results
Behavioural effects of MPTP
Intracarotid administration of MPTP rendered the animals moderately hemiparkinsonian as evidenced by the presence of bradykinesia, rigidity and abnormal posture in the limbs contralateral to the affected hemisphere. Both animals also displayed strong biases to turn to the left, toward the side of the MPTP-lesioned hemisphere (Bankiewicz et al., 2001). These signs were observed during clinical examinations that were performed at regular intervals throughout the post-MPTP data collection period. No sign of tremor was observed at any time post-MPTP in either animal, as would be predicted from previous reports that macaques seldom exhibit parkinsonian tremor following MPTP treatment (Bankiewicz et al., 2001).
The number of TH-positive neurons in the substantia nigra pars compacta was reduced by 67% in the MPTP-treated hemisphere of the animal for which TH staining was available. Performance of the step-tracking task was consistently impaired following MPTP administration (Supplementary Table 1). Reaction times and movement durations were lengthened in both animals (two-way ANOVA; F > 180, P < 0.001). Movement velocities (Fig. 2B) and amplitudes were also altered post-MPTP (F > 15 and P < 0.001). These impairments persisted throughout the post-MPTP recording period, although a subset of measures showed relative recovery over time (for details, see Pasquereau and Turner, 2011).
Recording of EMG from the normal state exhibited the characteristic triphasic pattern of activation (Basmajian and De Luca, 1985) (Fig. 2B). After MPTP-administration, EMG activity showed a slower rate of rise and a later peak in activity during movement. For example, with extension movements (EXT in Fig. 2C top, positive-going traces) activation of the triceps longus, the agonist muscle, began at approximately −100 ms before movement onset in both the neurologically normal and parkinsonian states (black and red traces, respectively). Before MPTP, triceps activity ramped up steeply to peak sharply at +70 ms after movement onset whereas after MPTP triceps activity increased slowly to a lower but long-lasting activation that continued >500-ms after movement onset. A nearly identical pattern was observed in EMG from the brachioradialis when it acted as an agonist during flexion movements (FLX in Fig. 2C bottom, negative-going traces) except that the maximal level of brachioradialis activation was greater following MPTP than before. In addition, the directional pattern of EMG activity was altered, such that peri-movement signals tended to be similar for both movement directions.
Neuronal database
Single unit recordings were obtained from the arm-related areas of the left M1 of two monkeys (Table 1). A total of 325 neurons were studied in the neurologically normal state. Of these, 93 were identified as PTNs (n = 73 and 20 in Monkeys V and L) and 73 were CSNs (n = 50 and 23 in Monkeys V and L). Among the 192 neurons collected during the post-MPTP period, 60 were PTNs (n = 42 and 18 in Monkeys V and L) and 53 were CSNs (n = 44 and 9 in Monkeys V and L). Only three cells were activated antidromically from both the putamen and the peduncle (0.6% of neurons studied). These three were excluded from ‘PTN’ and ‘CSN’ categories but were included in the ‘general M1’ category (i.e. all cells studied including PTNs, CSNs, and ‘non-activated neurons’). Non-activated neurons (n = 159 pre-MPTP and 79 post-MPTP) were not activated antidromically but were recorded at the same time as an antidromically activated neuron or were sampled within 0.5 mm of one along the same microelectrode track. The population of non-activated neurons was likely a heterogeneous mixture of cortical cell types that included interneurons and pyramidal projection neurons that happened to not be activated from any of the implanted stimulating electrodes.
Table 1.
Effects of MPTP on distinct populations of M1 cells
| M1 | PTN | CNS | ||
|---|---|---|---|---|
| Number of cells | Pre-MPTP | 325 | 93 | 73 |
| Post-MPTP | 192 | 60 | 53 | |
| Firing rate at rest (spikes/s) | Pre-MPTP | 11.6 ± 9.7 | 18.2 ± 7.5 | 3.0 ± 3.1 |
| Post-MPTP | 9.7 ± 10.9* | 14.2 ± 10.3*** | 3.9 ± 4.6 | |
| Movement-related cells | Pre-MPTP | 263 (81%) | 87 (94%) | 36 (49%) |
| Post-MPTP | 143 (74%) | 55 (92%) | 25 (47%) | |
| Kinematic-encoding cells | Pre-MPTP | 137 (52%) | 51 (59%) | 8 (22%) |
| Post-MPTP | 61 (43%) | 25 (45%) | 5 (20%) | |
| Movement-related residual | Pre-MPTP | 162 | 69 | 9 |
| Post-MPTP | 106 | 45 | 11 | |
| Residual increases | Pre-MPTP | 122 (75%) | 35 (51%) | 9 (100%) |
| Post-MPTP | 73 (69%) | 21 (47%) | 9 (82%) |
Mean values ± SD before and after MPTP treatment were calculated for all M1 cells (left), for PTNs (middle), and for CSNs (right). Movement-related cells = the number of cells (and percentage of total sample) that showed a significant peri-movement modulation in firing. Kinematic-encoding cells = the number of cells (and percentage of movement-related cells) that showed a significant encoding of one or more measure of motor performance. Movement-related residuals = the numbers of cells with significant increases or decreases in residual peri-movement activity (i.e. after regressing out relations to kinematics). Residual increases = the number (and percentage) of cells in which the movement-related residual was an increase in firing. Statistical comparisons are between pre- and post-MPTP populations: *P < 0.05, ***P < 0.001 (Mann-Whitney U-test).
Movement-related activity
Consistent with previous findings in normal primates (Turner and DeLong, 2000), modulation of activity around the time of movement onset was much more common among antidromically-identified PTNs than CSNs (χ2 = 68.7, P < 0.001). In neurologically-normal animals, peri-movement changes in activity were found in 94% of PTNs and 49% of CSNs (Table 1 and Fig. 3). The activity of both cell types was influenced by parameters of motor performance such as movement direction (Fig. 3A) and speed (Fig. 3B).
Figure 3.

Representative examples of peri-movement activity in PTNs and CSNs. Mean spike density functions (top), raster diagrams (middle) and overlaid traces of single-trial angular velocities (bottom) were constructed around movement onset (dashed line). The motor-related activity of these exemplar neurons was sorted according to (A) movement direction (flexion versus extension) or (B) movement speed (slow versus fast). In the spike density functions, the horizontal lines represent the baseline firing rate ± threshold for response detection (P = 0.002), and the vertical solid line corresponds to the detected response onset. Inset illustrate the antidromic activation and collision tests performed using stimulating electrodes in the peduncle (PTN) and striatum (CSN). Antidromically elicited action potentials (asterisks) occurred at a constant latency after stimulation (inverted triangle). Antidromic spikes collided (downward arrow) with spontaneous spikes when stimulation was delivered after a spontaneous spike at any delay shorter that the cell’s antidromic latency plus the refractory period.
To clarify how M1 neurons encode multiple facets of movement around the time of movement onset, we calculated time-resolved GLMs (Equation 1) for each movement-related cell (n = 263 in the normal state, Fig. 4). Consistent with previous studies of non-identified M1 neurons (Georgopoulos et al., 1982; Georgopoulos, 1991; Kalaska and Crammond, 1992), movement direction was the major parameter encoded, as evidenced by the fraction of neurons encoding (46% of M1 neurons, P < 0.05/52; Fig. 4B) and the standardized regression coefficients (β-coefficients, green trace in Fig. 4C). Other movement variables were encoded by fewer than 12% of M1 neurons. Also consistent with previous reports (Bauswein et al., 1989; Turner and DeLong, 2000), PTNs encoded parameters of performance more frequently than did CSNs (χ2 = 13.52, P < 0.001). Fully 59% of PTNs encoded one or more performance parameter whereas only 22% of CSNs did so (Table 1). The spike count encoding of movement kinematics was also stronger in PTNs than in CNS. Specifically, PTNs had peak β-coefficients that were 51% larger, on average, than those of CSNs (Mann-Whitney U-test, P < 0.05/52). Movement direction dominated the encoding in both neuronal subpopulations (52% of PTNs and 22% of CSNs).
Figure 4.

Time course of kinematic encoding in the neurologically-normal state. (A) Population-averaged spike density functions were aligned on the movement onset (flexion or extension) for all of the movement-related M1 neurons. (B) The fraction of M1 neurons that modulated their activity (permutation test, P < 0.05/54; Equation 1) according to movement direction, hand position, movement speed, acceleration, and reaction time (RT). (C) The population averages of the standardized regression coefficients (β1∼5) at different lags relative to the time of movement onset. The vertical width of each curve represents its standard error of the mean (SEM). (D) The population-averaged residual activity for all movement-related M1 neurons. Residual activity reflects a neuron’s kinematic-independent relationship to movement per se.
After regressing away these performance-related components of peri-movement activity, a substantial residual modulation in activity remained (Fig. 4D). This kinematics-independent modulation in activity was considered to reflect a neuron’s sensitivity to movement per se.
MPTP effects on rest and peri-movement activity
The mean baseline firing rate of general M1 cells was decreased by 16% after MPTP treatment (Mann-Whitney U-test, P < 0.05). MPTP had markedly different effects on the two antidromically-identified populations (Table 1). The resting rate of PTNs was reduced by 22% following MPTP treatment (Mann-Whitney U-test, P < 0.001) whereas that of CSNs remained unchanged (Mann-Whitney U-test, P = 0.5). These results confirm previous comparisons using different analysis epochs from the same general dataset (Pasquereau and Turner, 2011, 2013).
The fraction of M1 neurons with significant movement-related activity declined slightly following MPTP administration (a change of −6.4%; Table 1). The size of this change did not reach statistical significance (χ2 = 4.9, P = 0.08) but also did not permit definitive rejection of the hypothesis that MPTP reduced the prevalence of movement-related activity. There was 95% confidence that the change in prevalence laid between −14.4% and +0.8% (as estimated using the procedure outlined in Newcombe (1998). Thus, any change in the overall prevalence of movement-related activity, if present, was unlikely to be an increase or to be a large decrease (i.e. a >14.4% reduction). For both PTN and CSN populations, the proportions of neurons with peri-movement activity declined only slightly following MPTP (−2% in each population; χ2 < 0.8, P > 0.6).
MPTP effects on encoding of movement kinematics
Next, we tested whether the induction of parkinsonism altered the neuronal encoding of kinematic parameters of movement (Fig. 5). Although the fraction of M1 neurons with kinematics-related activity remained unmodified following MPTP (∼50% of M1 neurons, Table 1), the strength of encoding decreased significantly (mean −29% change in β-coefficients; Mann-Whitney U-test, P < 0.05/52). In neurologically-normal animals, the general M1 population showed a mean β-coefficient of 0.52, whereas that index dropped to 0.31 in the equivalent population post-MPTP. This decoupling of M1 activity and motor parameters was attributable to a weakened encoding of movement direction (−22%), position (−33%), speed (−40%), and acceleration (−49%; Mann-Whitney U-test, P < 0.05/52; Fig. 5 left column). Consistent with previous work (Riehle and Requin, 1989; Merchant et al., 2004), M1 encoding of reaction times was infrequent, appearing in only 7% of all neurons studied and at nearly identical rates in both states (20/263 neurons pre-MPTP and 10/143 neurons post-MPTP; χ2 = 0.39, P = 0.8). Among these few neurons, the β-coefficients were small (Fig. 5, bottom) and did not differ between pre- and post-MPTP conditions (P > 0.05/52).
Figure 5.

MPTP effects on neuronal encoding of task performance. The time-dependent neuronal encoding of motor performance (i.e. direction, position, speed, acceleration and reaction time; reflected by means of the regression coefficients: β1∼5) was compared between neuronal populations sampled before (black) and after (red) MPTP administration. The vertical width of each curve reflects the SEM. This analysis was performed separately for the general population of M1 cells, and then for the PTNs and the CSNs. Grey-shaded boxes indicate time periods in which the coefficients differed significantly between states (Mann-Whitney U-test, P < 0.05/52).
Overall, MPTP affected encoding more strongly in PTNs than in CSNs (Fig. 5). For PTNs, the encoding of performance was weakened markedly (mean −37% change in standardized β’s; 95% confidence: −32% to −42%) and significant reductions were observed for all GLM coefficients except that for reaction time (Mann-Whitney U-test, P < 0.05/52; Fig. 5 centre column). For CSNs, encoding of performance was also attenuated following MPTP but to a lesser degree than in PTNs (mean −18% change; 95% confidence: −12% to −24%) and only significantly for the encoding of movement direction (Fig. 5 right column). Despite the differential attenuation of encoding in PTNs, PTNs continued to encode kinematics more strongly than did CSNs (i.e. following MPTP, mean peak β’s were 30% larger in PTNs than in CSNs; Mann-Whitney U-test, P < 0.05/52).
MPTP effects on kinematics-independent M1 activity
We then examined how parkinsonism altered the neuronal encoding of movement itself, independent of the MPTP-induced changes in kinematics. The components of a neuron’s peri-movement activity attributable to kinematics were regressed out and the residual modulations in firing rate were compared between neurons sampled before and after MPTP administration (Fig. 6). Significant (t-test, P < 0.002) modulations in residual activity were found in 66% of all movement-related M1 neurons (Table 1). Notably, the incidence of increases and decreases in residual activity did not change with the induction of parkinsonism (all χ2 < 0.5, P > 0.05; ‘Residual increases’ in Table 1). Thus, the balance of peri-movement increases versus decreases in M1 activity was not altered with the induction of parkinsonism.
Figure 6.

MPTP effects on residual activity. The mean population-averaged residual activities were aligned on the time of movement onset for neurons sampled before (black) and after (red) MPTP administration. Peri-movement residual responses are shown separately for the general population of M1 cells, then for PTNs and CSNs. Population averages were constructed separately for neurons with residual increases (positive peaks) and decreases (negative peaks) in activity.
Movement-related increases in (residual) activity were smaller in magnitude and altered in timing following the induction of parkinsonism. For the general M1 population, the mean magnitude of peri-movement increases was reduced by 36% following MPTP (−8 spikes/s; Mann-Whitney U-test, P < 0.01; Fig. 7 left column). This effect was accentuated in PTNs, in which increases were reduced by 50% (−12 spikes/s; Mann-Whitney U-test, P < 0.001; Fig. 7 centre column). In contrast, in CSNs the mean magnitude of increases was statistically unchanged (Mann-Whitney U-test, P = 0.38; Fig. 7 right column).
Figure 7.

MPTP effects on kinematic-independent (‘residual’) increases in firing. The distributions of response magnitude (top), onset latency (middle) and relative time to peak (bottom) are plotted for increase-type response residuals sampled before (black) and after (red) MPTP administration. Inset plots show the means (± SEM) and significance of each pre- versus post-MPTP comparison (*P < 0.05, **P < 0.01, ***P < 0.001, Mann-Whitney U-test). Horizontal box plots show the middle two quartiles of the distributions. The central white line within each box corresponds to the median value and horizontal ‘whiskers’ outside each box show the extent of the overall distribution (excluding outliers). n-values indicate the numbers of neurons included in each comparison as determined by presence of a significant increase in peri-movement residual activity (P < 0.002, t-test). Results are shown separately for all M1 neurons, PTNs, and CSNs. Grey shaded boxes in the onset latency plots indicate the 150-ms time window immediately preceding movement onset during which few response onsets were found in the post-MPTP neuronal populations.
Movement-related increases in activity also began earlier relative to movement onset and lasted longer following MPTP. For general M1 cells, increases in firing began earlier (−42 ms change in mean onset latencies; Mann-Whitney U-test, P < 0.01; Fig. 7 left column), increased more gradually (+87 ms change in mean time to peak; Mann-Whitney U-test, P < 0.01; Fig. 7 left column), and lasted longer in the parkinsonian state than the normal condition, resulting in an overall broadening of the population-averaged SDFs (Fig. 6). There was no evidence for an altered latency of M1 response onsets relative to appearance of the movement trigger signal (+16 ms change; Mann-Whitney U-test, P > 0.5). A roughly similar pattern was observed in PTNs, where the mean time to peak was increased by 42% following MPTP (+75 ms change; Mann-Whitney U-test, P < 0.05; Fig. 7). PTN responses also tended to begin earlier relative to movement onset following MPTP administration (–28 ms) although this shift in mean timing was not significant.
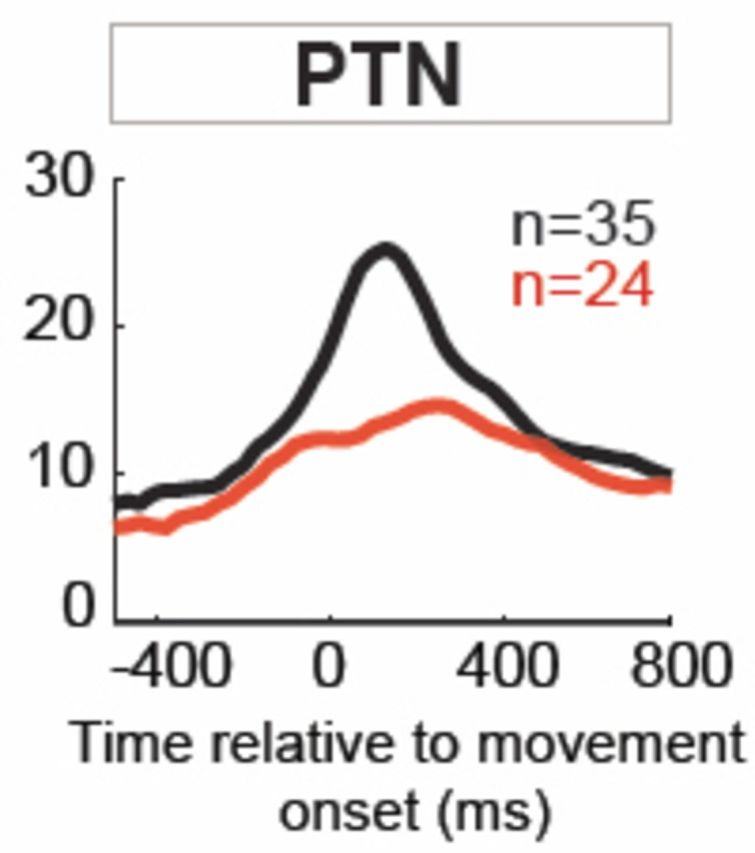
The onset latencies for increases also developed a clear bimodal distribution following MPTP administration. More specifically, the fraction of neurons with response onsets during the 150-ms period immediately preceding movement onset (Fig. 7) was reduced markedly following MPTP administration. This was true both for general M1 neurons (χ2=16.8, P < 0.001; Fig. 7 left) and for PTNs (χ2 = 19.7, P < 0.001; Fig. 7 centre). Fully 51% (18/35) of responses began during this period in the pre-MPTP PTN population, while that fraction fell to 8% (2/24) in the post-MPTP population.
A parallel analysis of peri-movement decreases revealed only a significant shift in onset latencies in general M1 neurons (Supplementary Fig. 1). The magnitudes of decreases did not differ significantly between pre- and post-MPTP conditions for M1 neurons in general or the PTN subpopulation (Mann-Whitney U-test, all P’s > 0.05). Decreases began earlier relative to movement onset for general M1 neurons (−53 ms change in mean onset latencies; Mann-Whitney U-test, P < 0.05; Supplementary Fig. 1). A similar, yet non-significant, shift in onset latencies was observed for the PTN subpopulation (−47 ms; Mann-Whitney U-test, P > 0.05). The relative time to peak did not differ between pre- and post-MPTP conditions for either M1 or PTN populations (Mann-Whitney U-test, P > 0.05).
Recall that few CSNs showed any significant movement-related residual activity and in only two CSNs, both recorded post-MPTP, was that change a decrease in firing (Table 1). Thus, comparisons of the timing of CSN increases in activity (e.g. Figs 6 and 7, right column) were unreliable because of small numbers and a comparison of the timing of CSN decreases was impossible.
Discussion
In these studies we investigated the effects of MPTP-induced parkinsonism on the movement-related activity of neurons in the primate M1 including antidromically identified PTNs and CSNs. We found that parkinsonism was associated with a weakened encoding of movement kinematics, a reduction in neural signalling of movement per se, and a modification of the temporal organization of movement-related activity. These abnormalities were found almost solely in the PTN population and largely spared the CSNs. Given that the primary efferent pathway that conveys motor commands from M1 to segmental motor centres originates in PTNs, the dysfunction of movement-related activity in PTNs may be an important factor in the pathophysiology of motor symptoms in Parkinson’s disease.
Resting and movement-related activity in parkinsonism
We observed a 16% reduction in the resting firing rate of the general population of M1 neurons (Table 1; i.e. during the period of immobility that immediately preceded arm movement). This result agrees with previous analyses of other time epochs from the same dataset (Pasquereau and Turner, 2011, 2013) and is consistent with classical pathophysiological models (Albin et al., 1989; DeLong, 1990; Wichmann and DeLong, 1996). The results were equivocal on whether the prevalence of movement-related activity decreased slightly or did not change. There was no support for the idea that MPTP increased the prevalence of movement-related activity. The potential implications of these findings for theories of M1 dysfunction in Parkinson’s disease are discussed below.
Kinematic encoding in the parkinsonian M1
It is a well-established fact that individual M1 neurons can encode in their firing rates a variety of kinematic parameters including movement direction, position, speed, acceleration, and distance (Georgopoulos et al., 1982, 1984; Fu et al., 1993; Ashe and Georgopoulos, 1994; Moran and Schwartz, 1999). No study to date, however, has investigated whether that form of encoding is disturbed in parkinsonism. A major finding of the current study is that parkinsonism is associated with a widespread weakening of the encoding of kinematics in M1 (Fig. 5). The standardized β-coefficients, which estimate the strength of the relationship between firing rate and kinematic parameters, were reduced by a mean of 29% for the general M1 population. The weakened encoding of movement direction observed here (−22% for β1; Fig. 5) is consistent with Doudet et al. (1990) who reported a marked decrease in the fraction of neurons showing a reciprocal encoding of movement direction. Significant reductions in encoding, however, were also observed for other kinematic measures such as position, speed, and acceleration.
Reductions in kinematics encoding were observed in both PTN and CSN subpopulations, but encoding by PTNs was affected far greater than by CSNs. The mean reduction in β’s was greater for PTNs than for CSNs (−37% versus −18%, respectively), and significant reductions in β’s were found for a greater number of kinematic parameters (four of five parameters for PTNs versus one of five for CSNs). This observation reinforces the idea that PTNs are affected more severely in parkinsonism than are other neuronal subtypes (e.g. CSNs, Pasquereau and Turner, 2011, 2013) and extends that concept to the encoding of movement parameters. Implications of these observations for theories of M1 dysfunction in parkinsonism are discussed below.
Motor commands in the parkinsonian M1
There is little consensus on how movement-related activity in M1 is altered in parkinsonism. Of the three previous single unit recording studies, two demonstrated significantly reduced movement-related activity (Watts and Mandir, 1992; Parr-Brownlie and Hyland, 2005), while another described only a broadening of the motor response duration (Doudet et al., 1990). The lack of consistency might be explained by differences between studies in the severity of motor symptoms induced, in combination with the known sensitivity of M1 neurons to parameters of movement. An MPTP-induced reduction in M1 activity may be either a consequence of motor slowing (via proprioceptive feedback to M1) or a cause of that slowing (via attenuation of efferent motor commands).
The present study controlled for differences in motor performance between normal and parkinsonian states by use of a GLM, thereby allowing us to compare kinematics-independent residual activities between the neurologically normal and parkinsonian states. A second major finding of the current study is that this kinematics-independent activity is reduced significantly in the parkinsonian state (Fig. 6). More specifically, the mean magnitude of movement-related signals was decreased by 36% for the general population of M1 neurons. In addition, kinematics-independent motor commands were attenuated strongly in PTNs (–50%) but were not affected in CSNs. This result adds further support for the functional distinction between PTNs and CSNs in M1 (Pasquereau and Turner, 2011; Shepherd, 2013; Li et al., 2015) and emphasizes the importance of PTN activity in the pathophysiology of Parkinson’s disease.
It is important to acknowledge that our parsing of M1 activity into two relatively simplistic component categories, ‘kinematics-related’ and ‘kinematics-independent’, likely glosses over many of the subtleties of how M1 activity is thought to both recruit and reflect movement (Churchland et al., 2012; Scott, 2012; Sadtler et al., 2014). The specific abnormalities in M1 activity that cause impaired movement are a subject of future work.
Timing of M1 motor commands in parkinsonism
A third major finding of the current study is that the timing of M1 movement-related activity was altered with the induction of parkinsonism (Fig. 7). In the general population of M1 cells, motor commands began earlier, on average, increased more gradually, and lasted longer in parkinsonian state than in normal condition. Similar changes in response timing were observed in the PTN subpopulation, although the shift in latencies did not reach significance. These changes resulted in an increased total duration of movement-related activity in M1 (Fig. 6). Similar abnormalities in the timing of movement-related activity have been described previously for the parkinsonian M1 (Doudet et al., 1990) and globus pallidus interna (Leblois et al., 2006).
Importantly, following MPTP, a reduced fraction of motor commands began during the 150-ms long window that immediately preceded movement onset (Fig. 7). For instance, the fraction of PTN responses that began during this epoch fell from 51% before MPTP to only 8% following MPTP administration. This led to distinctly bimodal distributions for the onset latencies of responses in the general population of M1 neurons and in PTNs (Fig. 7). What mechanism(s) cause such an effect is a matter of conjecture. One possibility is that an exaggerated movement-related activation of basal ganglia output neurons, which has been described for parkinsonian animals (Leblois et al., 2006), causes excessive inhibition of the motor thalamo-cortical circuit during this time window.
Implications for theories of M1 dysfunction in parkinsonism
Our results speak to general theories of M1 dysfunction in parkinsonism (see ‘Introduction’ section and Fig. 1). The hypoactivation hypothesis was upheld in three of four of its predictions: we observed reductions in resting firing rates (Table 1), in kinematic encoding (Fig. 5) and in residual movement-related activity (Figs 6 and 7). Results were equivocal on whether the fraction of cells with movement-related activity was reduced (Table 1). In contrast, the loss of specificity hypothesis was not supported in any of its four predictions; we did not observe increases in resting rates, in residual activity, or in the prevalence of movement-related or residual activity. Finally, our results are consistent with the sole prediction of the abnormal timing hypothesis: the onset times and durations of movement-related responses were indeed altered significantly in the parkinsonian condition.
Our findings are largely consistent with the concept of hypoactivation (i.e. that in Parkinson’s disease, M1 receives reduced excitatory drive and thus generates deficient commands for voluntary movements) (Albin et al., 1989; DeLong, 1990; Berardelli et al., 2001; Wichmann and DeLong, 2003). What might cause this reduced excitatory drive? Abnormal output from the parkinsonian basal ganglia is a likely proximate cause (DeLong and Wichmann, 2007; Rubin et al., 2012). The original impetus for the hypoactivation hypothesis came from observations of elevated firing rates in the (inhibitory) output neurons of the parkinsonian basal ganglia (Crossman et al., 1985; Miller and DeLong, 1987; Albin et al., 1989; Filion and Tremblay, 1991). More recent work, however, has emphasized abnormalities in the pattern or rhythm of basal ganglia activity instead (Nini et al., 1995; Raz et al., 2000; Sanders et al., 2013). Regardless of the specific abnormality, the non-linear characteristics of basal ganglia-thalamo-cortical communication (Kojima et al., 1997; Person and Perkel, 2007; Nakamura et al., 2014) make it possible that abnormally-patterned basal ganglia output is translated into reduced excitatory drive at the cortical level. Note that abnormalities in the peri-movement activity of basal ganglia output neurons are a potentially-key contributor to M1 hypoactivation, but these have rarely been investigated [however, see Leblois et al. (2006) and below].
Pathological changes in cortex itself may also contribute to the abnormalities described here (Jan et al., 2003; Moore et al., 2008). Future studies are required to determine the importance of these local changes, which include reduced dopaminergic innervation of M1 (Pifl et al., 1990; Jan et al., 2003; Hosp et al., 2009), reorganization of intrinsic cortical circuits (Ferrer, 2009; Guo et al., 2015) and altered cortical plasticity (Molina-Luna et al., 2009; Suppa et al., 2010; Huang et al., 2011; Kojovic et al., 2012).
Many studies have described a loss of specificity in the responses of cortical and subcortical neurons to proprioceptive stimulation in MPTP-intoxicated monkeys (Filion et al., 1988; Boraud et al., 2000; Goldberg et al., 2002; Pessiglione et al., 2005). Increased fractions of cells respond to experimenter-imposed limb movements and often to movements at multiple joints of more than one body segment. Some investigators have hypothesized that a similar loss of specificity in the motor commands issued from M1 contributes to the genesis of parkinsonian bradykinesia, presumably by way of a co-selection of antagonist or competing commands (Mink, 1996; Bergman et al., 1998; Beck and Hallett, 2011; Bronfeld and Bar-Gad, 2011). Our results, as summarized above, are not consistent with that idea. It appears unlikely that a loss of specificity in the motor commands issued from M1 contributed to the impairments in voluntary movement observed in our MPTP model of parkinsonism.
Our results also support predictions of the abnormal timing hypothesis, but they provide little insight to the potential causal mechanisms. The one known study of movement-related activity in the parkinsonian basal ganglia (Leblois et al., 2006) also described earlier than normal onset latencies and extended movement-related response durations. It remains to be determined whether the deranged timing of motor commands at the cortical level is a product or a cause of the abnormally-timed movement-related signals in the basal ganglia. Our results suggest that parkinsonism is associated with a temporal de-coupling between the occurrence of motor commands in M1 and the onset of movement. Abnormalities in the timing of M1 activity like the ones described here may be central determinants of the impairments in timing of agonist and antagonist EMG activity that have been described for patients with Parkinson’s disease (Hallett and Khoshbin, 1980; Pfann et al., 2001).
Conclusion
In summary, our results are consistent with the idea that the movement deficits of Parkinson’s disease are mediated by a hypoactivation and abnormal timing of motor commands in M1. This conclusion must be accompanied with the significant caveat that MPTP-induced hemi-parkinsonism, however useful (Bankiewicz et al., 2001; Emborg, 2007), is an imperfect model of the slow, progressive, and widespread process of neurodegeneration that constitutes idiopathic Parkinson’s disease (Braak et al., 2003). We found that MPTP-induced hemi-parkinsonism is associated with a weakened encoding of kinematics, reduced encoding of movement per se, and a derangement in the timing of movement-related activity in M1. Loss of specificity appears unlikely to play a major role. As a population, PTNs were affected far more severely than were CSNs. The activity of PTNs is closely tied to and predictive of motor performance, unlike the activity of other cortical neuron subtypes such as CSNs (Turner and DeLong, 2000; Li et al., 2015). Consequently, an under-modulation and temporal dispersion of PTN movement-related activity may be a central factor in the pathophysiology of parkinsonian bradykinesia.
Supplementary Material
Acknowledgements
Andrew Zimnik provided helpful editorial comments in the manuscript.
Glossary
Abbreviations
- CSN
corticostriatal neuron
- GLM
generalized linear model
- M1
primary motor cortex
- MPTP
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- PTN
pyramidal tract-type neuron
- SDF
spike density function
Funding
This work was supported by National Institute of Neurological Disorders and Stroke at the National Institutes of Health (grant numbers R01NS044551 and R01NS070865 to R.S.T.) and the Center for Neuroscience Research in Non-human primates (CNRN, 1P30NS076405-01A1).
Supplementary material
Supplementary material is available at Brain online.
References
- Albin RL, Young AB, Penney JB. The functional anatomy of basal ganglia disorders. Trends Neurosci 1989; 12: 366–75. [DOI] [PubMed] [Google Scholar]
- Alexander GE. Selective neuronal discharge in monkey putamen reflects intended direction of planned limb movements. Exp Brain Res 1987; 67: 623–34. [DOI] [PubMed] [Google Scholar]
- Alexander GE, Crutcher MD. Preparation for movement: neural representations of intended direction in three motor areas of the monkey. J Neurophysiol 1990; 64: 133–50. [DOI] [PubMed] [Google Scholar]
- Ashe J, Georgopoulos AP. Movement parameters and neural activity in motor cortex and area 5. Cereb Cortex 1994; 4: 590–600. [DOI] [PubMed] [Google Scholar]
- Bankiewicz KS, Oldfield EH, Chiueh CC, Doppman JL, Jacobowitz DM, Kopin IJ. Hemiparkinsonism in monkeys after unilateral internal carotid artery infusion of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Life Sci 1986; 39: 7–16. [DOI] [PubMed] [Google Scholar]
- Bankiewicz KS, Sanchez-Pernaute R, Oiwa Y, Kohutnicka M, Cummins A, Eberling J. Preclinical models of Parkinson's disease. Curr Protoc Neurosci 2001; 9.4: 1–32. [DOI] [PubMed] [Google Scholar]
- Baraduc P, Thobois S, Gan J, Broussolle E, Desmurget M. A common optimization principle for motor execution in healthy subjects and parkinsonian patients. J Neurosci 2013; 33: 665–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basmajian JV, De Luca CJ. Muscles alive, their functions revealed by electromyography. 5th edn Baltimore: Williams & Wilkins; 1985. [Google Scholar]
- Bauswein E, Fromm C, Preuss A. Corticostriatal cells in comparison with pyramidal tract neurons: contrasting properties in the behaving monkey. Brain Res 1989; 493: 198–203. [DOI] [PubMed] [Google Scholar]
- Beck S, Hallett M. Surround inhibition in the motor system. Exp Brain Res 2011; 210: 165–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berardelli A, Rothwell JC, Thompson PD, Hallett M. Pathophysiology of bradykinesia in Parkinson's disease. Brain 2001; 124: 2131–46. [DOI] [PubMed] [Google Scholar]
- Bergman H, Feingold A, Nini A, Raz A, Slovin H, Abeles M, et al. Physiological aspects of information processing in the basal ganglia of normal and parkinsonian primates. Trends Neurosci 1998; 21: 32–8. [DOI] [PubMed] [Google Scholar]
- Boraud T, Bezard E, Bioulac B, Gross CE. Ratio of inhibited-to-activated pallidal neurons decreases dramatically during passive limb movement in the MPTP-treated monkey. J Neurophysiol 2000; 83: 1760–3. [DOI] [PubMed] [Google Scholar]
- Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging 2003; 24: 197–211. [DOI] [PubMed] [Google Scholar]
- Brazhnik E, Cruz AV, Avila I, Wahba MI, Novikov N, Ilieva NM, et al. State-dependent spike and local field synchronization between motor cortex and substantia nigra in hemiparkinsonian rats. J Neurosci 2012; 32: 7869–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodal P. The corticopontine projection in the rhesus monkey. Origin and principles of organization. Brain 1978; 101(pt. 2): 251–83. [DOI] [PubMed] [Google Scholar]
- Bronfeld M, Bar-Gad I. Loss of specificity in Basal Ganglia related movement disorders. Front Syst Neurosci 2011; 5: 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catalan MJ, Ishii K, Honda M, Samii A, Hallett M. A PET study of sequential finger movements of varying length in patients with Parkinson's disease. Brain 1999; 122: 483–95. [DOI] [PubMed] [Google Scholar]
- Chen R, Kumar S, Garg RR, Lang AE. Impairment of motor cortex activation and deactivation in Parkinson's disease. Clin Neurophysiol 2001; 112: 600–7. [DOI] [PubMed] [Google Scholar]
- Churchland MM, Cunningham JP, Kaufman MT, Foster JD, Nuyujukian P, Ryu SI, et al. Neural population dynamics during reaching. Nature 2012; 487: 51–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossman AR, Mitchell IJ, Sambrook MA. Regional brain uptake of 2-deoxyglucose in N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced parkinsonism in the macaque monkey. Neuropharmacology 1985; 24: 587–91. [DOI] [PubMed] [Google Scholar]
- de Hemptinne C, Ryapolova-Webb ES, Air EL, Garcia PA, Miller KJ, Ojemann JG, et al. Exaggerated phase-amplitude coupling in the primary motor cortex in Parkinson disease. Proc Natl Acad Sci USA 2013; 110: 4780–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLong MR. Primate models of movement disorders of basal ganglia origin. Trends Neurosci 1990; 13: 281–5. [DOI] [PubMed] [Google Scholar]
- DeLong MR, Wichmann T. Circuits and circuit disorders of the basal ganglia. Arch Neurol 2007; 64: 20–4. [DOI] [PubMed] [Google Scholar]
- Derejko M, Rakowicz M, Antczak J, Inglot E, Niewiadomska M. Corticomotor excitability in drug-naive patients with Parkinson disease. Neurol Neurochir Pol 2013; 47: 109–15. [DOI] [PubMed] [Google Scholar]
- Desmurget M, Turner RS. Testing Basal Ganglia motor functions through reversible inactivations in the posterior internal globus pallidus. J Neurophysiol 2008; 99: 1057–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doudet DJ, Gross C, Arluison M, Bioulac B. Modifications of precentral cortex discharge and EMG activity in monkeys with MPTP-induced lesions of DA nigral neurons. Exp Brain Res 1990; 80: 177–88. [DOI] [PubMed] [Google Scholar]
- Dum RP, Strick PL. Motor areas in the frontal lobe of the primate. Physiol Behav 2002; 77: 677–82. [DOI] [PubMed] [Google Scholar]
- Eidelberg D, Moeller JR, Dhawan V, Spetsieris P, Takikawa S, Ishikawa T, et al. The metabolic topography of parkinsonism. J Cereb Blood Flow Metab 1994; 14: 783–801. [DOI] [PubMed] [Google Scholar]
- Emborg ME. Nonhuman primate models of Parkinson's disease. ILAR J 2007; 48: 339–55. [DOI] [PubMed] [Google Scholar]
- Ferrer I. Early involvement of the cerebral cortex in Parkinson's disease: convergence of multiple metabolic defects. Prog Neurobiol 2009; 88: 89–103. [DOI] [PubMed] [Google Scholar]
- Fetz EE, Finocchio DV, Baker MA, Soso MJ. Sensory and motor responses of precentral cortex cells during comparable passive and active joint movements. J Neurophysiol 1980; 43: 1070–89. [DOI] [PubMed] [Google Scholar]
- Filion M, Tremblay L, Bedard PJ. Abnormal influences of passive limb movement on the activity of globus pallidus neurons in parkinsonian monkeys. Brain Res 1988; 444: 165–76. [DOI] [PubMed] [Google Scholar]
- Filion M, Tremblay L. Abnormal spontaneous activity of globus pallidus neurons in monkeys with MPTP-induced parkinsonism. Brain Res 1991; 547: 142–51. [PubMed] [Google Scholar]
- Flaherty AW, Graybiel AM. Corticostriatal transformations in the primate somatosensory sstem. Projections from physiologically mapped body-part representations. J Neurophysiol 1991; 66: 1249–63. [DOI] [PubMed] [Google Scholar]
- Fromm C, Evarts EV. Relation of size and activity of motor cortex pyramidal tract neurons during skilled movements in the monkey. J Neurosci 1981; 1: 453–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu QG, Suarez JI, Ebner TJ. Neuronal specification of direction and distance during reaching movements in the superior precentral premotor area and primary motor cortex of monkeys. J Neurophysiol 1993; 70: 2097–116. [DOI] [PubMed] [Google Scholar]
- Fuller JH, Schlag JD. Determination of antidromic excitation by the collision test: problems of interpretation. Brain Res 1976; 112: 283–98. [DOI] [PubMed] [Google Scholar]
- Georgopoulos AP, Kalaska JF, Caminiti R, Massey JT. On the relations between the direction of two-dimensional arm movements and cell discharge in primate motor cortex. J Neurosci 1982; 2: 1527–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgopoulos AP, Caminiti R, Kalaska JF. Static spatial effects in motor cortex and area 5: quantitative relations in a two-dimensional space. Exp Brain Res 1984; 54: 446–54. [DOI] [PubMed] [Google Scholar]
- Georgopoulos AP. Higher order motor control. Annu Rev Neurosci 1991; 14: 361–77. [DOI] [PubMed] [Google Scholar]
- Goldberg JA, Boraud T, Maraton S, Haber SN, Vaadia E, Bergman H. Enhanced synchrony among primary motor cortex neurons in the 1-methyl-4- phenyl-1,2,3,6-tetrahydropyridine primate model of Parkinson's disease. J Neurosci 2002; 22: 4639–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo L, Xiong H, Kim JI, Wu YW, Lalchandani RR, Cui Y, et al. Dynamic rewiring of neural circuits in the motor cortex in mouse models of Parkinson's disease. Nat Neurosci 2015; 18: 1299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallett M, Khoshbin S. A physiological mechanism of bradykinesia. Brain 1980; 103: 301–14. [DOI] [PubMed] [Google Scholar]
- Hamming RW. Digital filters. 0 ed. Englewood Cliffs, NJ: Prentice Hall; 1983. [Google Scholar]
- Haslinger B, Erhard P, Kampfe N, Boecker H, Rummeny E, Schwaiger M, et al. Event-related functional magnetic resonance imaging in Parkinson's disease before and after levodopa. Brain 2001; 124: 558–70. [DOI] [PubMed] [Google Scholar]
- Hatsopoulos NG, Xu Q, Amit Y. Encoding of movement fragments in the motor cortex. J Neurosci 2007; 27: 5105–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiraoka K, Notani M, Iwata A, Minamida F, Abe K. Premovement facilitation of corticospinal excitability in patients with Parkinson's disease. Int J Neurosci 2010; 120: 104–9. [DOI] [PubMed] [Google Scholar]
- Hoover JE, Strick PL. Multiple output channels in the basal ganglia. Science 1993; 259: 819–21. [DOI] [PubMed] [Google Scholar]
- Hosp JA, Molina-Luna K, Hertler B, Atiemo CO, Luft AR. Dopaminergic modulation of motor maps in rat motor cortex: an in vivo study. Neuroscience 2009; 159: 692–700. [DOI] [PubMed] [Google Scholar]
- Huang YZ, Rothwell JC, Lu CS, Chuang WL, Chen RS. Abnormal bidirectional plasticity-like effects in Parkinson's disease. Brain 2011; 134: 2312–20. [DOI] [PubMed] [Google Scholar]
- Hutchison WD, Lozano AM, Davis KD, Saint-Cyr JA, Lang AE, Dostrovsky JO. Differential neuronal activity in segments of globus pallidus in Parkinson's disease patients. Neuroreport 1994; 5: 1533–7. [DOI] [PubMed] [Google Scholar]
- Jan C, Pessiglione M, Tremblay L, Tande D, Hirsch EC, Francois C. Quantitative analysis of dopaminergic loss in relation to functional territories in MPTP-treated monkeys. Eur J Neurosci 2003; 18: 2082–6. [DOI] [PubMed] [Google Scholar]
- Jenkins IH, Fernandez W, Playford ED, Lees AJ, Frackowiak RSJ, Passingham RE, et al. Impaired activation of the supplementary motor area in Parkinson's disease is reversed when akinesia is treated with apomorphine. Ann Neurol 1992; 32: 749–57. [DOI] [PubMed] [Google Scholar]
- Kalaska JF, Crammond DJ. Cerebral cortical mechanisms of reaching movements. Science 1992; 255: 1517–23. [DOI] [PubMed] [Google Scholar]
- Kojima J, Yamaji Y, Matsumura M, Nambu A, Inase M, Tokuno H, et al. Excitotoxic lesions of the pedunculopontine tegmental nucleus produce contralateral hemiparkinsonism in the monkey. Neurosci Lett 1997; 226: 111–4. [DOI] [PubMed] [Google Scholar]
- Kojovic M, Bologna M, Kassavetis P, Murase N, Palomar FJ, Berardelli A, et al. Functional reorganization of sensorimotor cortex in early Parkinson disease. Neurology 2012; 78: 1441–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leblois A, Meissner W, Bezard E, Bioulac B, Gross CE, Boraud T. Temporal and spatial alterations in GPi neuronal encoding might contribute to slow down movement in Parkinsonian monkeys. Eur J Neurosci 2006; 24: 1201–8. [DOI] [PubMed] [Google Scholar]
- Lefaucheur JP. Motor cortex dysfunction revealed by cortical excitability studies in Parkinson's disease: influence of antiparkinsonian treatment and cortical stimulation. Clin Neurophysiol 2005; 116: 244–53. [DOI] [PubMed] [Google Scholar]
- Lemon RN, Porter R. Afferent input to movement-related precentral neurones in conscious monkeys. Proc R Soc Lond B Biol Sci 1976; 194: 313–39. [DOI] [PubMed] [Google Scholar]
- Li N, Chen TW, Guo ZV, Gerfen CR, Svoboda K. A motor cortex circuit for motor planning and movement. Nature 2015; 519: 51–6. [DOI] [PubMed] [Google Scholar]
- Mazzoni P, Hristova A, Krakauer JW. Why don't we move faster? Parkinson's disease, movement vigor, and implicit motivation. J Neurosci 2007; 27: 7105–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merchant H, Battaglia-Mayer A, Georgopoulos AP. Neural responses during interception of real and apparent circularly moving stimuli in motor cortex and area 7a. Cereb Cortex 2004; 14: 314–31. [DOI] [PubMed] [Google Scholar]
- Miller WC, DeLong MR. Altered tonic activity of neurons in the globus pallidus and subthalamic nucleus in the primate MPTP model of parkinsonism. In: Carpenter MB, Jayaraman A, editors. The Basal Ganglia II. 0 ed. New York: Plenum Press; 1987. p. 415–27. [Google Scholar]
- Mink JW. The basal ganglia: focused selection and inhibition of competing motor programs. Prog Neurobiol 1996; 50: 381–425. [DOI] [PubMed] [Google Scholar]
- Molina-Luna K, Pekanovic A, Rohrich S, Hertler B, Schubring-Giese M, Rioult-Pedotti MS, et al. Dopamine in motor cortex is necessary for skill learning and synaptic plasticity. PLoS One 2009; 4: e7082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore RY, Whone AL, Brooks DJ. Extrastriatal monoamine neuron function in Parkinson's disease: an 18F-dopa PET study. Neurobiol Dis 2008; 29: 381–90. [DOI] [PubMed] [Google Scholar]
- Moran DW, Schwartz AB. Motor cortical representation of speed and direction during reaching. J Neurophysiol 1999; 82: 2676–92. [DOI] [PubMed] [Google Scholar]
- Nakamura KC, Sharott A, Magill PJ. Temporal coupling with cortex distinguishes spontaneous neuronal activities in identified Basal Ganglia-recipient and cerebellar-recipient zones of the motor thalamus. Cereb Cortex 2014; 24: 81–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nambu A, Yoshida S, Jinnai K. Projection on the motor cortex of thalamic neurons with pallidal input in the monkey. Exp Brain Res 1988; 71: 658–62. [DOI] [PubMed] [Google Scholar]
- Newcombe RG. Improved confidence intervals for the difference between binomial proportions based on paired data. Stat Med 1998; 17: 2635–50. [PubMed] [Google Scholar]
- Nini A, Feingold A, Slovin H, Bergman H. Neurons in the globus pallidus do not show correlated activity in the normal monkey, but phase-locked oscillations appear in the MPTP model of Parkinsonism. J Neurophysiol 1995; 74: 1800–5. [DOI] [PubMed] [Google Scholar]
- Parr-Brownlie LC, Hyland BI. Bradykinesia induced by dopamine D2 receptor blockade is associated with reduced motor cortex activity in the rat. J Neurosci 2005; 25: 5700–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquereau B, Turner RS. Primary motor cortex of the parkinsonian monkey: differential effects on the spontaneous activity of pyramidal tract-type neurons. Cereb Cortex 2011; 21: 1362–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquereau B, Turner RS. Primary motor cortex of the parkinsonian monkey: altered neuronal responses to muscle stretch. Front Syst Neurosci 2013; 7: 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Person AL, Perkel DJ. Pallidal neuron activity increases during sensory relay through thalamus in a songbird circuit essential for learning. J Neurosci 2007; 27: 8687–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pessiglione M, Guehl D, Rolland AS, Francois C, Hirsch EC, Feger J, et al. Thalamic neuronal activity in dopamine-depleted primates: evidence for a loss of functional segregation within basal ganglia circuits. J Neurosci 2005; 25: 1523–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfann KD, Buchman AS, Comella CL, Corcos DM. Control of movement distance in Parkinson's disease. Mov Disord 2001; 16: 1048–65. [DOI] [PubMed] [Google Scholar]
- Pifl C, Bertel O, Schingnitz G, Hornykiewicz O. Extrastriatal dopamine in symptomatic and asymptomatic rhesus monkeys treated with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Neurochem Int 1990; 17: 263–70. [DOI] [PubMed] [Google Scholar]
- Porter R, Lemon R. Corticospinal function and voluntary movement. Oxford; New York: Clarendon Press; Oxford University Press; 1993. [Google Scholar]
- Rascol O, Sabatini U, Chollet F, Celsis P, Montastruc J-L, Marc-Vergnes J-P, et al. Supplementary and primary sensory motor area activity in Parkinon's disease. Regional cerebral blood flow changes during finger movements and effects of apomorphine. Arch Neurol 1992; 49: 144–8. [DOI] [PubMed] [Google Scholar]
- Raz A, Vaadia E, Bergman H. Firing patterns and correlations of spontaneous discharge of pallidal neurons in the normal and the tremulous 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine vervet model of parkinsonism. J Neurosci 2000; 20: 8559–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riehle A, Requin J. Monkey primary motor and premotor cortex: single-cell activity related to prior information about direction and extent of an intended movement. J Neurophysiol 1989; 61: 534–49. [DOI] [PubMed] [Google Scholar]
- Rosen I, Asanuma H. Peripheral afferent inputs to the forelimb area of the monkey motor cortex: input-output relations. Exp Brain Res 1972; 14: 257–73. [DOI] [PubMed] [Google Scholar]
- Rubin JE, McIntyre CC, Turner RS, Wichmann T. Basal ganglia activity patterns in parkinsonism and computational modeling of their downstream effects. Eur J Neurosci 2012; 36: 2213–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini U, Boulanouar K, Fabre N, Martin F, Carel C, Colonnese C, et al. Cortical motor reorganization in akinetic patients with Parkinson's disease: a functional MRI study. Brain 2000; 123: 394–403. [DOI] [PubMed] [Google Scholar]
- Sadtler PT, Quick KM, Golub MD, Chase SM, Ryu SI, Tyler-Kabara EC, et al. Neural constraints on learning. Nature 2014; 512: 423–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders TH, Clements MA, Wichmann T. Parkinsonism-related features of neuronal discharge in primates. J Neurophysiol 2013; 110: 720–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz AB, Kettner RE, Georgopoulos AP. Primate motor cortex and free arm movements to visual targets in three-dimensional space. I. Relations between single cell discharge and direction of movement. J Neurosci 1988; 8: 2913–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott SH. The computational and neural basis of voluntary motor control and planning. Trends Cogn Sci 2012; 16: 541–9. [DOI] [PubMed] [Google Scholar]
- Sergio LE, Hamel-Paquet C, Kalaska JF. Motor cortex neural correlates of output kinematics and kinetics during isometric-force and arm-reaching tasks. J Neurophysiol 2005; 94: 2353–78. [DOI] [PubMed] [Google Scholar]
- Shepherd GM. Corticostriatal connectivity and its role in disease. Nat Rev Neurosci 2013; 14: 278–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin HW, Kang SY, Sohn YH. Disturbed surround inhibition in preclinical parkinsonism. Clin Neurophysiol 2007; 118: 2176–9. [DOI] [PubMed] [Google Scholar]
- Soares J, Kliem MA, Betarbet R, Greenamyre JT, Yamamoto B, Wichmann T. Role of external pallidal segment in primate parkinsonism: comparison of the effects of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced parkinsonism and lesions of the external pallidal segment. J Neurosci 2004; 24: 6417–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner H, Kitai ST. Regulation of rat cortex function by D1 dopamine receptors in the striatum. J Neurosci 2000; 20: 5449–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suppa A, Iezzi E, Conte A, Belvisi D, Marsili L, Modugno N, et al. Dopamine influences primary motor cortex plasticity and dorsal premotor-to-motor connectivity in Parkinson's disease. Cereb Cortex 2010; 20: 2224–33. [DOI] [PubMed] [Google Scholar]
- Takada M, Tokuno H, Nambu A, Inase M. Corticostriatal projections from the somatic motor areas of the frontal cortex in the macaque monkey: segregation versus overlap of input zones from the primary motor cortex, the supplementary motor area, and the premotor cortex. Exp Brain Res 1998; 120: 114–28. [DOI] [PubMed] [Google Scholar]
- Tessa C, Lucetti C, Diciotti S, Baldacci F, Paoli L, Cecchi P, et al. Decreased and increased cortical activation coexist in de novo Parkinson's disease. Exp Neurol 2010; 224: 299–306. [DOI] [PubMed] [Google Scholar]
- Tessa C, Lucetti C, Diciotti S, Paoli L, Cecchi P, Giannelli M, et al. Hypoactivation of the primary sensorimotor cortex in de novo Parkinson's disease : a motor fMRI study under controlled conditions. Neuroradiology 2012; 54: 261–8. [DOI] [PubMed] [Google Scholar]
- Turner R, DeLong M. Corticostriatal activity in primary motor cortex of the macaque. J Neurosci 2000; 20: 7096–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner R, Grafton S, McIntosh A, DeLong M, Hoffman J. The functional anatomy of parkinsonian bradykinesia. Neuroimage 2003; 19: 163–79. [DOI] [PubMed] [Google Scholar]
- Vila M, Levy R, Herrero MT, Ruberg M, Faucheux B, Obeso JA, et al. Consequences of nigrostriatal denervation on the functioning of the basal ganglia in human and nonhuman primates: an in situ hybridization study of cytochrome oxidase subunit I mRNA. J Neurosci 1997; 17: 765–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts RL, Mandir AS. The role of motor cortex in the pathophysiology of voluntary movement deficits associated with parkinsonism. Neurol Clin North Am 1992; 10: 451–69. [PubMed] [Google Scholar]
- Wichmann T, DeLong MR. Functional and pathophysiological models of the basal ganglia. Curr Opin Neurobiol 1996; 6: 751–8. [DOI] [PubMed] [Google Scholar]
- Wichmann T, Bergman H, Starr PA, Subramanian T, Watts RL, DeLong MR. Comparison of MPTP-induced changes in spontaneous neuronal discharge in the internal pallidal segment and in the substantia nigra pars reticulata in primates [In Process Citation]. Exp Brain Res 1999; 125: 397–409. [DOI] [PubMed] [Google Scholar]
- Wichmann T, DeLong MR. Pathophysiology of Parkinson's disease: the MPTP primate model of the human disorder. Ann N Y Acad Sci 2003; 991: 199–213. [DOI] [PubMed] [Google Scholar]
- Wu AK, McCairn KW, Zada G, Wu T, Turner RS. Motor cortex stimulation: mild transient benefit in a primate model of Parkinson disease. J Neurosurg 2007; 106: 695–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T, Wang L, Hallett M, Chen Y, Li K, Chan P. Effective connectivity of brain networks during self-initiated movement in Parkinson's disease. Neuroimage 2011; 55: 204–15. [DOI] [PubMed] [Google Scholar]
- Yu H, Sternad D, Corcos DM, Vaillancourt DE. Role of hyperactive cerebellum and motor cortex in Parkinson's disease. Neuroimage 2007; 35: 222–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zold CL, Larramendy C, Riquelme LA, Murer MG. Distinct changes in evoked and resting globus pallidus activity in early and late Parkinson's disease experimental models. Eur J Neurosci 2007; 26: 1267–79. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.